

Abstract
Cancer cell genomes contain alterations beyond known etiologic events, but their total number has been unknown at even the order of magnitude level. By sampling colorectal premalignant polyp and carcinoma cell genomes through use of the technique inter-(simple sequence repeat) PCR, we have found genomic alterations to be considerably more abundant than expected, with the mean number of genomic events per carcinoma cell totaling approximately 11,000. Colonic polyps early in the tumor progression pathway showed similar numbers of events. These results indicate that, as with certain hereditary cancer syndromes, genomic destabilization is an early step in sporadic tumor development. Together these results support the model of genomic instability being a cause rather than an effect of malignancy, facilitating vastly accelerated somatic cell evolution, with the observed orderly steps of the colon cancer progression pathway reflecting the consequences of natural selection.
Tumor progression represents a form of somatic evolution, at the ultimate expense of the host organism. But how broad is the pathway of this evolution? Is it a narrow, focused pathway with relatively few extraneous genomic alterations, or is it diffuse and extensive? Five to 10 genetic alterations appear necessary for the generation of the malignant phenotype, with the classical example of a multistep progression pathway being Vogelstein et al.'s model for sporadic colorectal carcinoma (1, 2). Loeb (3) first recognized the inadequacy of normal mutation rates to permit accumulation of the necessary number of contributing mutations and proposed the need for a mutator phenotype to destabilize the genome and enable progression to reach completion. A mutator phenotype is further attractive in that it can generate additional nonpathway mutations, generating the subpopulations of tumor cells characteristic of tumor heterogeneity (4). In hereditary colorectal cancers arising from the mismatch repair defect mutator phenotype, Perucho (5) estimated approximately 100,000 mutations occur, although this is viewed as a special case. The questions we address here are how many genomic events occur in sporadic cancers, when do these occur, and what fraction of these are relevant to tumorigenesis? In other words, how fluid is the genome during tumor progression, and to what degree is evolution accelerated?
Genomic instability in cancer appears in three major forms: (i) aneuploidy, in which entire chromosomes are gained or lost, (ii) intrachromosomal instability, characterized by insertions, deletions, translocations, amplifications, and other forms all sharing the feature of utilizing DNA breakage as an early step, and (iii) point or oligobase mutations, which are rare except for DNA replication error inherited syndromes (hereditary nonpolyposis colorectal cancer) and a small fraction of sporadic cancers (6, 7). Quantitation of genomic instability in tumor biopsy specimens has been severely hampered by limitations of assay methodologies. The N-phosphono-l-aspartate (PALA) selection colony formation assay measuring carbamyl-P-synthetase/aspartate transcarbamylase/dihydroorotase gene amplification rates requires prior establishment of clonogenic tumor cell lines, where such establishment itself may select altered degrees of genomic instability (8). PALA itself enhances DNA breakage, contributing to genomic instability in cell cycle checkpoint-deficient cells (9). Comparative genomic hybridization provides a genome-wide scanning approach revealing amplifications and deletions, but is unable to resolve deletions of less than 10 Mb and amplifications of less than 2 Mb (10). Although the resolution of this technique improves with conversion to chip-based approaches where large arrays of smaller fragments are examined, it remains unable to readily detect insertions, translocations, de novo-synthesized sequences, or small events (11). Restriction landmark genome scanning could be adapted to assess genomic instability, but the tissue requirements, expense, and cumbersome nature of this two-dimensional gel technique prohibit routine use (12, 13).
We have used inter-(simple sequence repeat) PCR (inter-SSR PCR) to simultaneously sample numerous diverse regions of the genome, specifically those regions of the genome present between inverted abundant repetitive elements (14). This process allows us to readily measure the occurrence of genomic events in a tumor, by comparing tumor DNA to normal tissue DNA from the same patient. As we have described, an estimate of the relative degree of genomic instability is readily obtained for each tumor, in turn enabling examination of molecular and clinical correlates of genomic instability, including finding a lack of association with p53 mutation (15). In this report we estimate the total number of genomic events that occur in premalignant and malignant colorectal tumors and find the number for each to be surprisingly large. We present the rigorous sequencing of an altered segment revealed by inter-SSR PCR, which manifests genomic damage. Our results indicate that in sporadic colorectal cancer numerous genomic damage events are likely to fall below the range of detection of many other genome anatomy methodologies.
Materials and Methods
Tumors and DNA Extraction.
Tissue samples were from 58 consecutive sporadic colorectal cancer and 12 polyp patients who underwent surgery at the Roswell Park Cancer Institute and University of Montreal Hospital Center between 1991 and 1998. All patients gave written consent to obtain tissue specimens, as approved by the Roswell Park Institute Review Board. DNA was extracted from liquid nitrogen-frozen tumor or polyp samples and from adjacent normal mucosa as described (14). Tissue wedges with a volume of approximately 8 mm3 were digested for 3 hr at 65°C with 1 μg/ml proteinase K, followed by a l-hr RNase A treatment at 0.5 μg/ml. The DNA was further purified with two phenol/chloroform/isoamyl alcohol extractions, a chloroform/isoamyl alcohol extraction, and ethanol precipitation.
Inter-SSR PCR.
The elements of inter-SSR PCR methodology and the reproducibility of the technique have been detailed (14, 15). 32P-end labeled primers homologous to dinucleotide repeats and anchored at the 3′ end by two nonrepeat nucleotides were used in a PCR to amplify sequences between the repeat elements. PCR was carried out in a volume of 20 μl containing 1 μM primer (1:5 labeled/unlabeled oligonucleotide), 50 ng genomic DNA, and 0.3 unit of Taq polymerase (Life Technologies, Bethesda, MD) in 1× PCR buffer (10 mM Tris⋅HCI, pH 9.0/2% formamide/50 mM KCI/0.2 mM deoxynucleotide triphosphates/1.5 mM MgCI2/0.01% gelatin/0.01% Triton X-100). The following conditions were used for amplification: 3-min initial denaturation at 94°C, 30 cycles of 30 sec at 94°C/45 sec at 52°C/2 min at 72°C, 7 min at 72°C. The labeled PCR products were resolved by nondenaturing PAGE at 80 W for 22 min, followed by 50 W for 3,400 V-hr for CA-based PCR products and 2,800 V-hr for CG-based PCR products. The gels were dried and autoradiographed. PCR primers were from the Roswell Park Cancer Institute Biopolymer Facility. A genomic instability index was computed by dividing the number of altered bands seen in the PCR products amplified from the tumor DNA by the total number of products generated from the normal tissue DNA.
Results
Inter-SSR PCR uses a single primer based on repetitive DNA sequences, anchored at the 3′ end with unique sequences to prevent slippage. This technique produces amplification of those DNA sequences, typically less than 2 kb in size, which are present between relatively close, inverted primer-binding repeat sequences (16, 17). Exploiting abundant repetitive sequences such as CA repeats generates approximately 40 PCR products when the primer (CA)8RY is used. A comparison of products amplified from tumor DNA and normal tissue DNA from the same patient reveals a reproducible pattern of bands in which most products are identical, although for cases where tumor-specific rearrangements have occurred, a small number of new products also are seen, and a few products disappear. Examples of colorectal tumors whose genomes are relatively stable and unstable are shown in Fig. 1.
Figure 1.

Inter-SSR PCR analyses of colorectal cancers (14). (CA)8 was anchored with either RG or RY, where R is an equal mixture of the purines adenosine and guanine, and Y is an equal mixture of the pyrimidines cytosine and thymidine. (CG)4 was anchored with RY. Patients 3131 and 3213 each were examined with the (CA)- and (CG)-based primers, and products amplified from tumor DNA were compared with those from normal colonic tissue DNA from the same patient. Dots placed alongside lanes indicate bands found at significantly greater (>2 fold) intensity in the tumor or normal tissue DNA. For patient 3213 one new band from the tumor migrated close to the position of a band that disappeared from the normal tissue, and thus may represent a small deletion, indicated by >.
By calculating how much of the entire genome is being sampled in each assay, and comparing this to how many genomic events have occurred within this sample, we are able to estimate the total number of genomic events that have occurred in tumor cells. The inter-SSR PCR products generated by our reaction conditions fell in a broad size range, with the majority being less than 900 bp (Fig. 2). The mean total size of the products generated was 52.3 kbp for the (CA)8RY primer and 31.9 kbp for the (CA)8RG primer, for a combined total of 84.2 kbp being sampled, although small variations occur for each patient. Patterns were highly similar but not uniform for all patients' normal DNA control PCR products, reflecting polymorphisms in the population (14), although multiple disperse normal colonic biopsies from individual patients revealed identical patterns for each patient (14).
Figure 2.

The size distribution of altered bands is similar to the size distribution of total inter-SSR PCR products. (a) Size distribution of inter-SSR PCR products for 58 human colorectal cancers. The number of inter-SSR PCR products for (CA)8RY-primed (black bars) and (CA)8(RG)-primed (gray bars) reactions was determined for each 100-bp size interval. (b) The number of new or lost inter-SSR PCR bands was determined for each indicated 100-bp size interval. New bands or bands with increased intensity in the tumor are shown by bars above the x axis; bars below the axis indicate lost bands or bands with decreased intensity in the tumor.
Inter-SSR PCR band alterations need not be limited to insertions or deletions between the primer binding sites, nor to the deletion of a single primer binding site itself. Such processes indeed alter or eliminate bands, but larger-scale processes such as deletions of entire chromosomes or large fragments thereof could in one step eliminate more than one band. In contrast, bands newly appearing in tumors must reflect individual events, in turn enabling us to estimate the minimum number of total events. Although amplifications will produce an increase in signal intensity approximating the degree of amplification, the normal counterpart product should in this case still be visible. We therefore determined the number and size of bands with 2-fold or more reduced or increased intensity in our set of 58 colorectal tumors and the number and size of tumor-specific bands with no evident normal tissue counterpart at all. As illustrated in Fig. 2, nearly equivalent numbers of bands are increased and decreased in tumors, and there appear to be no size class preferences of altered bands relative to the total profile. Approximately one-tenth (18 of 174) of the altered PCR products of the 58 sporadic colorectal carcinomas represented new tumor specific bands that had no visible normal tissue counterpart, indicating that in these cases either the genomic event itself had occurred between the two primers or else included the site of one primer.
Altered bands are particularly valuable in that they can reveal where and how genomic instability events have occurred. Cloning of nine altered products revealed events that were widely dispersed in the genome (Table 1). What sorts of events produce altered inter-SSR PCR products? And might altered bands represent artifactual PCR products? We examined these issues by cloning PCR products from multiple independent PCRs, followed by sequencing of the products, preparing new nested primers designed to test for the alteration, and then using these nested primers to specifically amplify the altered sequence from tumor DNA. A thorough analysis of clone RG9 is shown in Fig. 3, where a four-base insertion was detected in tumor 3153. The initial alteration was seen with (CA)8RG as the primer, as well as with (CA)8RG with the R1 linker sequence GGAATTC attached at its 5′ position (Fig. 3A). The difference in electrophoretic mobility remained visible in PCR products cloned from reactions using tumor or normal DNA (Fig. 3B). Sequencing data revealed a four-base insertion had occurred in the tumor (Fig. 3C), even though the (CA)8RG-primed product migrated slightly faster for the tumor. Nested primers then were used to amplify the correspondingly different products from tumor and normal DNA, as illustrated in Fig. 3D. In this case, the tumor-specific nested product ran slightly slower. Sequencing verified the four-base difference in the nested product. The four-base insertion seen in tumor 3153 is not the result of classical microsatellite instability, because only two of 111 microsatellite markers we examined for this tumor showed instability, a value far below the 20% and 40% standards set by Boland et al. (18).
Table 1.
Inter-SSR PCR bands altered in tumors
| Clone | Size | Homology to* | Homology, % | Chromosome |
|---|---|---|---|---|
| RY1 | 556 bp | Twist gene | 99 | 7p21 |
| RY2 | 559 bp | — | — | — |
| RY3 | 336 bp | cDNA/D16S494 | 93 | 16q21 |
| RY4 | 472 bp | — | — | — |
| RY5 | 216 bp | cDNA/STS4–310 | 96 | 4 |
| RY6 | 196 bp | cDNA/STS4–310 | 94 | 4 |
| RY7 | 536 bp | 257 bp w/ D20S227,† | 92 | 20 |
| remainder no homology | ||||
| RY8.1 | 269 bp | Creatine transporter | 87 | X |
| RG9 | 923 bp | — | — | — |
Homology is defined as more than 75% sequence similarity to sequences in GenBank, as determined by fastA search.
†Clone RY7 contained two separate PCR products as manifested by linker and primer sequences.
Figure 3.

Characterization and verification of an altered inter-SSR PCR product amplified from tumor DNA. (A) Tumor (T) and normal (N) DNA from patient 3153 was used as a template in independent PCRs primed with (CA)8RG alone or with the EcoRI linker-tagged primer GGAATTC (CA)8RG. (B) Clones were prepared in vector pCR 2.1 (Invitrogen) by using their TA cloning kit. The insert size of the clones was the same as that of the original inter-SSR PCR product. (C) Clones were sequenced in both directions by using the T7 primer (forward) and the M13 primer (reverse). Sequence data are shown for a 78/74-bp region that contained the tumor-specific difference in the 923/919-bp sequences of the cloned inter-SSR PCR products. (D) Nested tumor specific primers G19–1P1 and G19–1P2 were used to prime three independent reactions from the original tumor and normal DNA, confirming the presence of a four-base insertion in the tumor DNA. Control PCRs used the clone 9T- and clone 23N-containing plasmids as templates.
Use of 3′ end-anchored (CA)8RG or (CA)8RY primers might be yielding results unrepresentative of the entire genome, particularly in view of the relatively lengthy 16-base repeat sequence in the primer. To evaluate this possibility, we examined the (CG)n-based 3′ anchored primer (CG)4RY where the repeat length was reduced to eight bases, but where its high GC content still allowed it to prime effectively at a PCR-range annealing temperature of 52o. Using this new primer, we examined by inter-SSR PCR 20 sets of tumor and corresponding normal mucosal DNA for which we already had data, using the (CA)8RG and (CA)8RY primers. As was the case for the (CA)n-based primers, the (CG)4RY primer also generated approximately 40 bands in each reaction (Fig. 1). Although the number of bands was similar to that seen with the (CA)n-based primers, the amplified products were slightly smaller (≤1.0 kbp). And the number of tumor-specific altered bands was likewise similar to that seen with the (CA)n-based primers, ranging in number from zero to five of the bands, which corresponded to genomic instability index values of 0–12%. If genomic instability in sporadic colorectal cancer is a random process, then there should be a concordance of instability indices regardless of the specific primer used. Correlation of CG-based instability and CA-based instability was examined by the Pearson method (19) and was found significant with a P value of P = .05.
How many genomic events have occurred in sporadic colorectal cancer cells? The linearly ordered pathway of colorectal tumor progression described by Fearon and Vogelstein (1) has generated the widespread impression that cancer is the end result of fewer than 10 relatively precisely staged genomic events, although comparative genomic hybridization has suggested a somewhat more chaotic scenario (11). We have analyzed our inter-SSR PCR data to estimate the total number of genomic alterations and events that have occurred in the typical sporadic colorectal cancer cell and limited ourselves to events that produced altered size products reflecting insertions, deletions and the like: (i) with the mean number of tumor-specific size-altered bands per inter-SSR PCR equaling three, (ii) with the total size of the PCR fragments sampling the genome equaling 84.2 kb, and (iii) with 18/174 altered bands representing events which must be unique, then
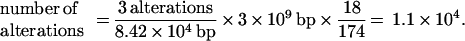 |
Thus we estimate at least 11,000 individual genomic events have occurred in each tumor cell. The actual number may range as high as 1.1 × 105, if every inter-SSR-altered band should represent an independent event. It is important to note this method scores only those events present in most tumor cells in the assay sample; late-stage events producing tumor heterogeneity may enlarge the final number (20). Interestingly, our numbers are consistent with what we calculate from restriction landmark genome scanning data in other studies, where about 20 events are seen in a total of about 1 Mb of the genome being randomly scanned (12, 13).
When does genomic instability first appear during tumor progression? If genomic instability is a significant effector of tumor progression, it must initiate before completion of progression and would be most effective facilitating this process if initiated early. In contrast, if genomic instability is a consequence of malignancy itself, then it should appear only in malignancies. Hereditary nonpolyposis colorectal cancer has revealed that genomic instability in the form of defective DNA mismatch repair is able to facilitate tumor progression, but is sporadic colorectal cancer analogous (21, 22)?
We examined the degree of genomic instability in early-stage colorectal tumor progression, with inter-SSR PCR analyses performed on 11 adenomatous polyps, two hyperplastic polyps, and one mixed. Eight were from sporadic colorectal patients, and six were from patients with the cancer predisposing syndrome familial adenomatous polyposis, where the APC gene is mutated. Using (CA)8RY and (CA)8RG individually as primers, we observed genomic instability in all 14 polyps, with a range in genomic instability index of 1.3 to 19.4 (Table 2). In most cases the degree of instability observed in polyps was only slightly less than that in corresponding carcinoma from the same patient, indicating most of the events we see occur early in tumor progression. In one case (patient 3177–8) with tissues both from an adenomatous polyp and a synchronous adenocarcinoma, the polyp showed three altered bands and the carcinoma six, including three migrating at the same positions as those altered in the polyp. In another case (193–4), all three bands altered in the polyp also were altered in the tumor, along with seven new bands. In a third case (199–201), three bands altered in the polyp were included among five in the tumor, and in a fourth case (patient 3021–3), of the four bands altered in the tumor, two were found altered in the polyp, along with one polyp-specific band that was either deleted during tumor progression or evolved separately from the true tumor progenitor cell (Fig. 4). Together, these results indicate that the onset of genomic instability is an early event in colorectal tumor progression, evidently being a facilitator and not a consequence of malignancy.
Table 2.
Genomic instability of colonic polyps
| Patient | Tissue | Type | Sporadic/FAP | Genomic instability index, percent |
|---|---|---|---|---|
| 3021–3 | Polyp | Tubulovillous adenoma | Sporadic | 5.2 |
| Carcinoma | Sporadic | 7.8 | ||
| 6394–5 | Polyp | Hyperplastic | Sporadic | 6.5 |
| Carcinoma | Sporadic | 6.5 | ||
| 3177–8 | Polyp | Tubulovillous adenoma | Sporadic | 3.9 |
| Carcinoma | Sporadic | 7.8 | ||
| 190–1 | Polyp | Tubulovillous adenoma | Sporadic | 1.3 |
| 193–4 | Polyp | Tubulovillous adenoma | Sporadic | 5.0 |
| Carcinoma | 14.1 | |||
| 197–8 | Polyp | Mixed hyperplastic tubulovillous adenoma | Sporadic | 7.5 |
| 199–201 | Polyp | Hyperplastic | Sporadic | 3.7 |
| Carcinoma | 6.1 | |||
| 202–4 | Polyp | Villous adenoma | Sporadic | 1.3 |
| Carcinoma | 2.2 | |||
| 2140–3 | Polyp | Rectal adenoma* | FAP | 1.3 |
| Polyp | Duodenal adenoma | FAP | 5.2 | |
| Polyp | Duodenal adenoma | FAP | 5.2 | |
| Carcinoma | FAP | 2.6 | ||
| 2829 | Polyp | Tubular adenoma | FAP | 1.3 |
| 2831 | Polyp | Tubular adenoma† | FAP | 3.9 |
| 2832 | Polyp | Tubular adenoma | FAP | 19.4 |
FAP, familial adenomatous polyposis. Genomic instability was determined by inter-SSR PCR (14).
Pathology reports showed tubular and tubulovillous adenoma and hyperperplastic polyps at this site, although typing is unknown for the individual polyp.
† Several polyps were examined histologically and were predominantly tubular adenomas, but typing is unknown for this particular polyp.
Figure 4.
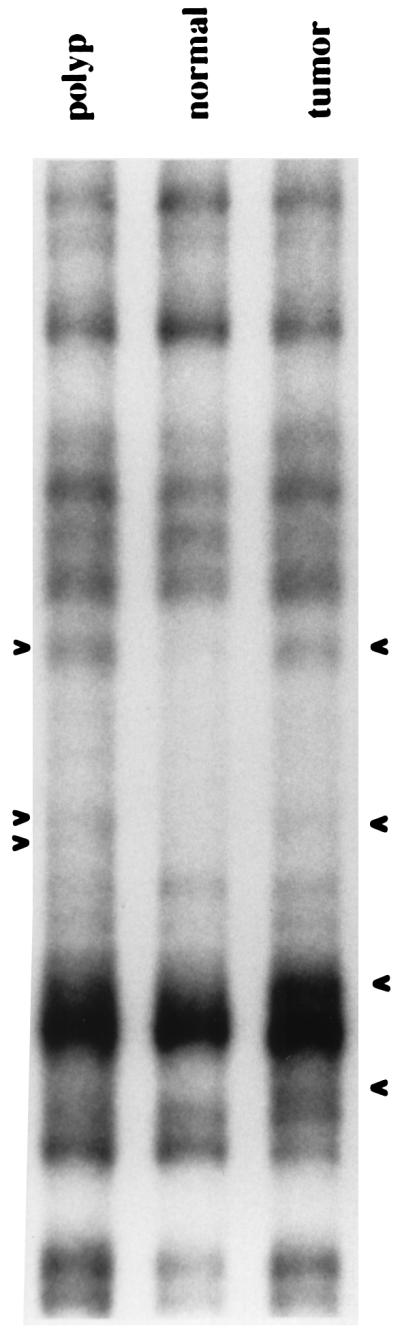
Inter-SSR PCR analyses of a colorectal carcinoma and synchronous adenomatous polyp from patient 3022. Three band alterations are present in the polyp and four in the carcinoma, with two bands common to the benign and malignant tumors.
Discussion
Our results indicate that genomic instability in cancer produces genomic damage at a level of around 10,000 events occurring per cell. At this point, it is difficult to make a more precise estimate with our methodology, which relies on product size differences. The sampling technique of inter-SSR PCR detects only amplifications or deletions present in a large fraction of the tumor cells, although the rarer event of newly arising bands will be visible if present in a smaller fraction. Our procedure is also vulnerable to the possibility that sequences near simple sequence repeats present within a few kilobases of another, or sequences near inverted repeats, may be preferentially susceptible to rearrangement (23). However, by comparing our results to those of Arribas et al. (24), where a mean of 1.1% of the bands amplified by two-primer arbitrarily primed PCR were altered, this level of preference is unlikely to exceed a factor of three (14, 24). In our computation of the total number of events we limited ourselves to newly arising bands and excluded amplifications and deletions that had the potential of affecting multiple bands with a single genomic event. Clustered point mutations from a localized problem during DNA replication might be occurring, and yet not be detected because only size-altered products have been screened. And lastly, we are detecting genomic events that have occurred during development of the tumor; they may not reflect a constant rate of instability throughout tumor progression. Despite all of these caveats, the level of genomic rearrangements in cancer is clearly remarkably high.
The efficacy of the inter-SSR PCR technique in studying the evolution of species first was established by Zietkiewicz et al. (17). Our studies show the utility of this technique for studying the analogous but much more rapid evolution that occurs during tumor progression. Perhaps our most surprising finding is the overall similarity between the extent of tumor cell evolution, as manifested by the degree of tumor genomic rearrangements, and the degree of genomic rearrangements seen during Darwinian evolution of closely related species (25). Zietkiewicz et al. found (CA)8RG primed inter-SSR PCR comparing humans and chimpanzees produces approximately half of all bands exhibiting differences (17); we found up to 19% of bands showing differences between tumor and normal DNA. For sporadic colorectal cancer at least, malignancy thus appears to not simply represent the end result of a few discrete mutations, but instead is the consequence of cells with highly unstable genomes having several years to evolve within the host.
Why do we see the ordered events of the Fearon and Vogelstein pathway (1)? Just as natural selection creates ordered evolution in Darwinian terms, natural selection also must be the central force in cellular evolution during tumor progression. In this latter case, coordinating and regulating genes acquired during hundreds of millions of years of Darwinian evolution are lost as the tumor evolves toward its own ends. The 5,000 genes in yeast may be taken as an estimate of the number of genes required for eukaryotic cell viability; with about 100,000 genes in humans, only a small fraction of genomic events are likely to impact cell viability itself. From genomic chaos a naturally selected order of tumor progression arises, with the tumor itself representing a genomically heterogeneous evolved population. Once an appropriate degree of genomic instability arises in a proliferating cell, cancer becomes the almost inevitable outcome. But how is the genomic instability of sporadic cancer itself arising? Although evidence exists for several mechanisms, including cell cycle checkpoint loss, chromosome bridge-breakage-fusion, and excessive breakage because of nuclease activation, it is not clear that a single mechanism is involved (26–32). Nor is it certain that cancer genomic instability only reflects rearrangements of existing sequences and does not include de novo syntheses.
Our findings have clinical relevance. Quantitation of the degree of genomic instability in early-stage tumors should produce insights as to their prognosis, which would appear particularly applicable to breast and prostate cancers where early-stage disease is widely detected, but only a fraction progresses. As demonstrated by Arribas et al. (24) with another form of arbitrarily primed PCR, the degree of genomic damage in colon cancer inversely correlates with survival. An inter-SSR fingerprint should be able to readily distinguish independently occurring primary tumors from tumor recurrences, and sequencing of altered products should permit synthesis of specific PCR primers to monitor tumor recurrence with exquisite sensitivity. At the molecular level, definition of the newly gained or lost sequences in inter-SSR PCR products and characterization of junction points can be expected to provide important clues to the mechanistic bases underlying genomic instability in cancer. At the therapeutic level, the multitude of genomic events suggests highly effective means of evading the immune response become obligatory early in tumor progression; immunological curative therapeutic approaches to cancer such as targeting specific tumor antigens may be expected to be fraught with difficulties (33, 34). And as has been widely observed with solid tumor cytotoxic chemotherapy, drug-resistant cell subpopulations and/or evolving populations within the tumor generally lead to an unfavorable outcome. In contrast, more attractive therapeutic approaches in view of our findings would appear to include prevention in the form of stabilizing the genome before progression can be completed (35), and for existing tumors, targeting the genomically stable nontumor cells essential for the tumor's survival, as occurs with the disruption of angiogenesis (36, 37).
Acknowledgments
We acknowledge Cynthia Bates for expert manuscript preparation, Debbie Driscoll for statistical help, and Arnold Mittleman, Bonnie King, Bill Burhans, Lee Hartwell, Joe Gray, and Manuel Perucho for valuable insights. This work was supported by the Charlotte Geyer Foundation, National Institutes of Health Grants ROl CA 74127 and P30-CA16056, the Samuel McLaughlin Foundation, and the Roswell Alliance Foundation.
Abbreviation
- inter-SSR PCR
inter-(simple sequence repeat) PCR
Footnotes
References
- 1.Fearon E R, Vogelstein B. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 2.Kinzler K W, Vogelstein B. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 3.Loeb L A. In: Cancer Surveys, Genetic Instability in Cancer. Lindahl T, editor. Plainview, NY: Cold Spring Harbor Lab. Press; 1996. pp. 329–342. [Google Scholar]
- 4.Alberts D S. Cancer J Sci Am. 1998;4:25–26. [PubMed] [Google Scholar]
- 5.Perucho M. Biol Chem. 1996;377:675–684. [PubMed] [Google Scholar]
- 6.Barrett J C, Tsutsui T, Tlsty T, Oshimura M. In: Genetic Mechanisms in Carcinogenesis and Tumor Progression. Harris C C, Liotta L A, editors. New York: Wiley; 1990. pp. 97–114. [Google Scholar]
- 7.Windle B, Draper B W, Yin Y, O'Gorman S, Wahl G M. Genes Dev. 1991;5:160–174. doi: 10.1101/gad.5.2.160. [DOI] [PubMed] [Google Scholar]
- 8.Livingstone L R, White A, Sprouse J, Livanos E, Jacks T, Tlsty T D. Cell. 1992;70:923–935. doi: 10.1016/0092-8674(92)90243-6. [DOI] [PubMed] [Google Scholar]
- 9.Paulson T G, Almasan A, Brudy L L, Wahl G M. Mol Cell Biol. 1998;18:3089–3100. doi: 10.1128/mcb.18.5.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bentz M, Plesch A, Stilgenbauer S, Dohner H, Lichter P. Genes Chromosomes Cancer. 1998;21:172–175. [PubMed] [Google Scholar]
- 11.Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo W-L, Chen C, Zhai Y, et al. Nat Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 12.Nagai H, Hirotsune S, Komatsubara H, Hatada I, Mukai T, Hayashizaki Y, Matsubara K. Cancer Detect Prev. 1993;17:399–404. [PubMed] [Google Scholar]
- 13.Konishi N, Hiasa Y, Nakamura M, Kitahori Y, Matsubara K, Nagai H. Am J Pathol. 1997;150:305–314. [PMC free article] [PubMed] [Google Scholar]
- 14.Basik M, Stoler D L, Kontzoglou K C, Rodriguez-Bigas M A, Petrelli N J, Anderson G R. Genes Chromosomes Cancer. 1997;18:19–29. [PubMed] [Google Scholar]
- 15.Kahlenberg M S, Stoler D L, Basik M, Petrelli N J, Rodriguez-Bigas M, Anderson G R. J Natl Cancer Inst. 1996;88:1665–1670. doi: 10.1093/jnci/88.22.1665. [DOI] [PubMed] [Google Scholar]
- 16.Godwin I D, Aitken E A, Smith L W. Electrophoresis. 1997;18:1524–1528. doi: 10.1002/elps.1150180906. [DOI] [PubMed] [Google Scholar]
- 17.Zietkiewicz E, Rafalski A, Labuda D. Genomics. 1994;20:176–183. doi: 10.1006/geno.1994.1151. [DOI] [PubMed] [Google Scholar]
- 18.Boland C R, Thibodeau S N, Hamilton S R, Sidransky D, Eshleman J R, Burt R W, Meltzer S J, Rodriguez-Bigas M A, Fodde R, Ranzani G N, Srivastava S. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 19.Kendall M G. Rank Correlation Methods. 4th ed. London: Griffin; 1970. [Google Scholar]
- 20.Macintosh C A, Stower M, Reid N, Maitland N J. Cancer Res. 1998;58:23–28. [PubMed] [Google Scholar]
- 21.Fishel R, Lescoe M K, Rao M R S, Copeland N G, Jenkins N A, Garber J, Kane M, Kolodner R. Cell. 1993;75:1027–1103. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 22.Ionov Y, Peinado M A, Malkhosyan S, Shibata D, Perucho M. Nature (London) 1993;363:558–561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 23.Chen C, Umezu K, Kolodner R D. Mol Cell. 1998;2:9–22. doi: 10.1016/s1097-2765(00)80109-4. [DOI] [PubMed] [Google Scholar]
- 24.Arribas R, Capella G, Tortola S, Masramon L, Grizzle W E, Perucho M, Peinado M A. J Clin Oncol. 1997;15:3230–3240. doi: 10.1200/JCO.1997.15.10.3230. [DOI] [PubMed] [Google Scholar]
- 25.Darwin C. The Origin of Species. London: Oxford Univ. Press; 1859. [Google Scholar]
- 26.Donehower L A. Cancer Surv. 1997;29:329–352. [PubMed] [Google Scholar]
- 27.Wahl G M, Linke S P, Paulson T G, Huang L C. Pezcoller Foundation Symp. 1997;8:33–52. [Google Scholar]
- 28.Smith K A, Stark M B, Gorman P A, Stark G R. Proc Natl Acad Sci USA. 1992;89:5427–5431. doi: 10.1073/pnas.89.12.5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Russo C A, Weber T K, Volpe C M, Stoler D L, Petrelli N J, Rodriguez-Bigas M, Burhans W C, Anderson G R. Cancer Res. 1995;55:1122–1128. [PubMed] [Google Scholar]
- 30.Chen R Z, Petterson U, Beard C, Jackson-Grusby L, Jaenisch R. Nature (London) 1998;395:89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- 31.Loquelle A, Toledo F, Stern S, Bieth A, Debatisse M. Cell. 1998;2:259–265. doi: 10.1016/s1097-2765(00)80137-9. [DOI] [PubMed] [Google Scholar]
- 32.Lengauer C, Kinzler K W, Vogelstein B. Nature (London) 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 33.Rabinowich H, Suminami Y, Reichert T E, Crowley-Nowick P, Bell M, Edwards R, Whiteside T L. Int J Cancer. 1996;68:276–284. doi: 10.1002/(SICI)1097-0215(19961104)68:3<276::AID-IJC2>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 34.Becker J C, Brocker E B. Recent Results Cancer Res. 1995;139:205–214. doi: 10.1007/978-3-642-78771-3_15. [DOI] [PubMed] [Google Scholar]
- 35.Kahlenberg M S, Volpe C, Stoler D, Petrelli N J, Anderson G R. Surgical Forum. 1997;48:868–870. [Google Scholar]
- 36.Boehm T, Folkman J, Browder T, O'Reilly M S. Nature (London) 1997;390:404–407. doi: 10.1038/37126. [DOI] [PubMed] [Google Scholar]
- 37.O'Reilly M S, Boehm T, Shing Y, Fukai N, Vasios G, Lane W S, Flynn E, Birkhead J R, Olsen B R, Folkman J. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]