

Abstract
A combination of in vitro embryonic stem (ES) cell differentiation and targeted gene disruption has defined complex regulatory events underlying oxidative stress-induced cardiac apoptosis, a model of postischemic reperfusion injury of myocardium. ES cell-derived cardiac myocytes (ESCM) having targeted disruption of the MEKK1 gene were extremely sensitive, relative to wild-type ESCM, to hydrogen peroxide-induced apoptosis. In response to oxidative stress, MEKK1−/− ESCM failed to activate c-Jun kinase (JNK) but did activate p38 kinase similar to that observed in wild-type ESCM. The increased apoptosis was mediated through enhanced tumor necrosis factor α production, a response that was positively and negatively regulated by p38 and the MEKK1-JNK pathway, respectively. Thus, MEKK1 functions in the survival of cardiac myocytes by inhibiting the production of a proapoptotic cytokine. MEKK1 regulation of the JNK pathway is a critical response for the protection against oxidative stress-induced apoptosis in cardiac myocytes.
Reperfusion of ischemic myocardium results in cellular injury, which involves apoptosis and necrosis (1, 2). Myocardial reperfusion injury is mediated, in part, by reactive oxygen species (ROS) that are generated during reoxygenation of ischemic tissue (3, 4). ROS damage the myocardium directly by oxidizing cell proteins and indirectly by stimulating the production of inflammatory cytokines such as tumor necrosis factor α (TNFα) in cardiac myocytes (5). TNFα is capable of inducing cardiac apoptosis (6). In separated rat neonatal cardiac myocytes and the perfused rat heart, exposure to oxidative stress such as hydrogen peroxide (H2O2) or hypoxia/reoxygenation activates mitogen-activated protein kinases (MAPKs), including the extracellular signal-response kinase (ERK), c-Jun kinase (JNK), and p38 (7, 8). Using a specific inhibitor of p38 and a dominant-negative mutant of MKK6, a MAPK kinase that activates p38, it was shown that p38 played a proapoptotic role in oxidative cardiac injury by stimulating increased expression of TNFα (5, 9). The roles of other MAPKs in myocardial injury by oxidative stress are still unclear (5, 7, 10). MEKK1 is a MAPK kinase kinase activated in response to specific extracellular stimuli. MEKK1 activates JNK in response to stresses that alter cell shape and microtubule cytoskeleton (11, 12). Activation of MEKK1 and the JNK pathway in response to shape change protects cells from apoptosis (13). ROS can induce changes in cell adherence and shape (14). Furthermore, reperfusion of ischemic cardiac tissue has been shown to induce changes in the cardiomyocyte cytoskeleton (15). The recent targeted disruption of the MEKK1 gene in embryonic stem (ES) cells provided a direct test of the role of MEKK1 signaling in cellular function (12). Using ES cell-derived cardiac myocytes (ESCM), here we demonstrate a protective role of MEKK1 in cardiomyocyte responsiveness to ROS.
Materials and Methods
Cardiac Myocyte Selection Vector.
The pBM20 vector (Boehringer Mannheim), carrying both an α-cardiac myosin heavy chain–aminoglycoside phosphotransferase (MHC-neor) and a phosphoglycerate kinase hygromycin-resistance transgene (pGK-hygror), was provided by Loren J. Field (Indiana University, Indianapolis) (16). The MEKK1 gene-targeted ES cells are resistant to G418 (12); therefore, neor was replaced in the vector with a gene encoding Sh ble (Zeor) to confer resistance to Zeocin. The ES cells were maintained as we described (12). The linearized MHC-Zeor/pGK hygror plasmid was transfected into wild-type and MEKK1−/− ES cells by electroporation (12). Transfected clones were selected by growth in the presence of hygromycin (200 μg/ml). PCR analyses revealed that both the MHC-GFP-Zeor and pGK-hygror sequences were present in the selected cell lines.
In Vitro Differentiation of ESCM.
To induce differentiation, wild-type and MEKK1−/− ES cells were suspended in differentiation medium (IMDM containing 15% FCS, 0.5 mM monothioglycerol, and 50 units/ml penicillin/50 μg/ml streptomycin, lacking supplemental leukemia inhibitory factor). Cells were cultured for 2 days by the hanging-drop method (3 × 102 ES cells per 30 μl in each drop) (17). Embryoid bodies (EB) in hanging drops were transferred to suspension culture in 100-mm dishes and cultured for an additional 3 days. The resulting EB were plated onto plastic 100-mm gelatin-coated dishes and cultured for 1 week. Subsequently, EB were treated with Zeocin (200 μg/ml) for 8 days to select cardiac myocytes. After the selection with Zeocin, cultures of ESCM were treated with 2 mg/ml collagenase B for 30 min at 37°C and gently shaken for 1 h at room temperature, and the suspension of ESCM was plated onto gelatin-coated six-well dishes or cover glasses and cultured in differentiation medium. This medium was changed 24 h after plating to a defined serum-free differentiation medium. All experiments were performed 24 h after transfer of cells to serum-free medium.
Immunofluorescence Analyses of ESCM.
Cultures of ESCM plated on gelatin-coated cover glasses were fixed in 4% paraformaldehyde and treated with 0.2% Triton X-100 (Sigma). MF 20 (anti-sarcomeric myosin; Developmental Studies Hybridoma Bank, Iowa City) was used as the primary antibody. The secondary antibody was Cy3-conjugated purified donkey anti-mouse IgG (Jackson ImmunoResearch). All samples were counterstained with Hoechst 33258 (Molecular Probes) and were visualized by fluorescence microscopy.
Ultrastructural Analysis of Selected ESCM.
Selected ESCM were fixed in 1.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.3) containing 0.1 M sucrose and 0.05% CaCl2, followed by postfixation in 1.0% osmium tetroxide in the same buffer. Samples were stained with 3.0% aqueous uranyl acetate, dehydrated, and embedded in Eponate 12-Araldite 502 resin (Pelco, Redding, CA). After trimming, the block was thin-sectioned and stained with uranyl acetate and Reynold's lead stain. Specimens were viewed on a Phillips 400 T transmission electron microscope.
Measurement of Spontaneous Contracting Rate.
The average contracting rate of cultured ESCM was determined in several randomly selected, spontaneously contracting clusters of wild-type and MEKK1−/− ESCM (n = 10, each) before and 1 h after treatment with 0.1 or 1.0 μM of isoproterenol.
Hypoxia and Reoxygenation.
Hypoxic condition (95% N2 and 5% CO2) was achieved in an anaerobic box. By placing flasks, which contain serum-free differential medium, in an anaerobic box overnight, the medium was balanced with the hypoxic atmosphere. ESCM were incubated in the serum-free differentiation medium for 24 h and subjected to a hypoxic atmosphere by immediately replacing the medium with the hypoxic medium. After incubating in a hypoxic condition for the duration indicated, wild-type and MEKK1−/− ESCM were reoxygenated by immediately replacing the hypoxic medium with a normoxic serum-free differentiation medium.
Agarose Gel Electrophoresis for DNA Fragmentation (DNA Ladder).
Cells (5 × 105) were lysed in 0.2 ml of a lysis buffer (10 mM Tris⋅HCl, pH 7.4/10 mM EDTA/0.5% Triton X-100) and treated with 40 μg of RNase (Boehringer Mannheim) for 1 h at 37°C and with 40 μg of proteinase K (Boehringer Mannheim) for 1 h at 37°C. The extracted DNA was fractionated on 1.5% agarose gel and stained with ethidium bromide.
Terminal Deoxynucleotidyltransferase-Mediated dUTP Nick End Labeling (TUNEL).
Cells cultured on a cover glass were fixed with 4% paraformaldehyde and incubated in a TUNEL reaction mixture containing both terminal deoxynucleotidyltransferase and fluorescein-dUTP for 1 h at 37°C (Boehringer Mannheim). These samples were counterstained with Hoechst 33258 and analyzed by fluorescence microscopy.
Measurement of MAPK Activities.
Activity of JNK, ERK2, and p38 was measured as we described previously (12, 13).
Immunoblot Analysis.
Protein extracts were subjected to immunoblot analysis by using phospho-specific ERK or p38 antibodies as described previously (8).
TNFα Concentration.
TNFα concentrations in both the medium and protein extracts of ESCM were measured by using an ELISA kit for mouse TNFα (Genzyme). The total protein amount of each plate was measured by the Lowry method.
TNFα Promoter Reporter Assay.
The promoter for the mouse TNFα gene (18) was provided by Bruce Beutler (University of Texas Southwestern) and inserted upstream of the luciferase gene in pGL3-luciferase reporter basic vector (Promega). Cells were transfected by using Lipofectamine and, 24 h later, were incubated in the presence or absence of H2O2 (0.1 mM) for 6 h and washed once in PBS before lysis in Passive Lysis Buffer (Promega). Luciferase activities were determined by using a luminometer according to the manufacturer's instructions (Analytical Luminescence Laboratory, San Diego). TNFα promoter activity was normalized for transfection efficiency based on the cotransfected Renilla luciferase reporter construct.
Statistical Analysis.
Changes in contracting rates and kinase activities of wild-type or MEKK1−/− ESCM were analyzed by one factorial-repeated measures of ANOVA followed by Bonfferoni's test. Data are expressed as mean ± SEM.
Results
In Vitro Differentiation of Gene-Targeted ES Cells into Cardiac Myocytes.
It is well established that ESCM have physical and molecular features observed in cardiac myocytes isolated from hearts (16, 17, 19–22). We expressed a transgene having a cardiac-specific promoter controlling the expression of Zeocin drug resistance and successfully isolated highly purified cardiac myocytes derived from wild-type and MEKK1−/− ES cells. Characteristically, the time needed to observe contractions in wild-type and MEKK1−/− ESCM was identical after the initiation of ES cell differentiation. The proportion and morphologic appearance of the contracting cardiac myocytes did not differ between wild-type and MEKK1−/− ESCM. Fig. 1a illustrates staining of DNA and sarcomeric myosin in a cluster of MEKK1−/− ESCM. Greater than 95% Zeocin-resistant cells positively stained with the anti-sarcomere antibody in both wild-type and MEKK1−/− ESCM cultures. In addition, ultrastructural analysis revealed a normal sarcomeric structure and intercalated discs in the selected MEKK1−/− ESCM (Fig. 1b). Fig. 1c shows the change in isoproterenol-induced contraction rate of wild-type and MEKK1−/− ESCM. Isoproterenol increased contraction rate in a dose-dependent manner in both wild-type and MEKK1−/− ESCM. Additionally, angiotensin II induced mRNA expression of atrial natriuretic factor (ANF) in both wild-type and MEKK1−/− ESCM (data not shown).
Figure 1.
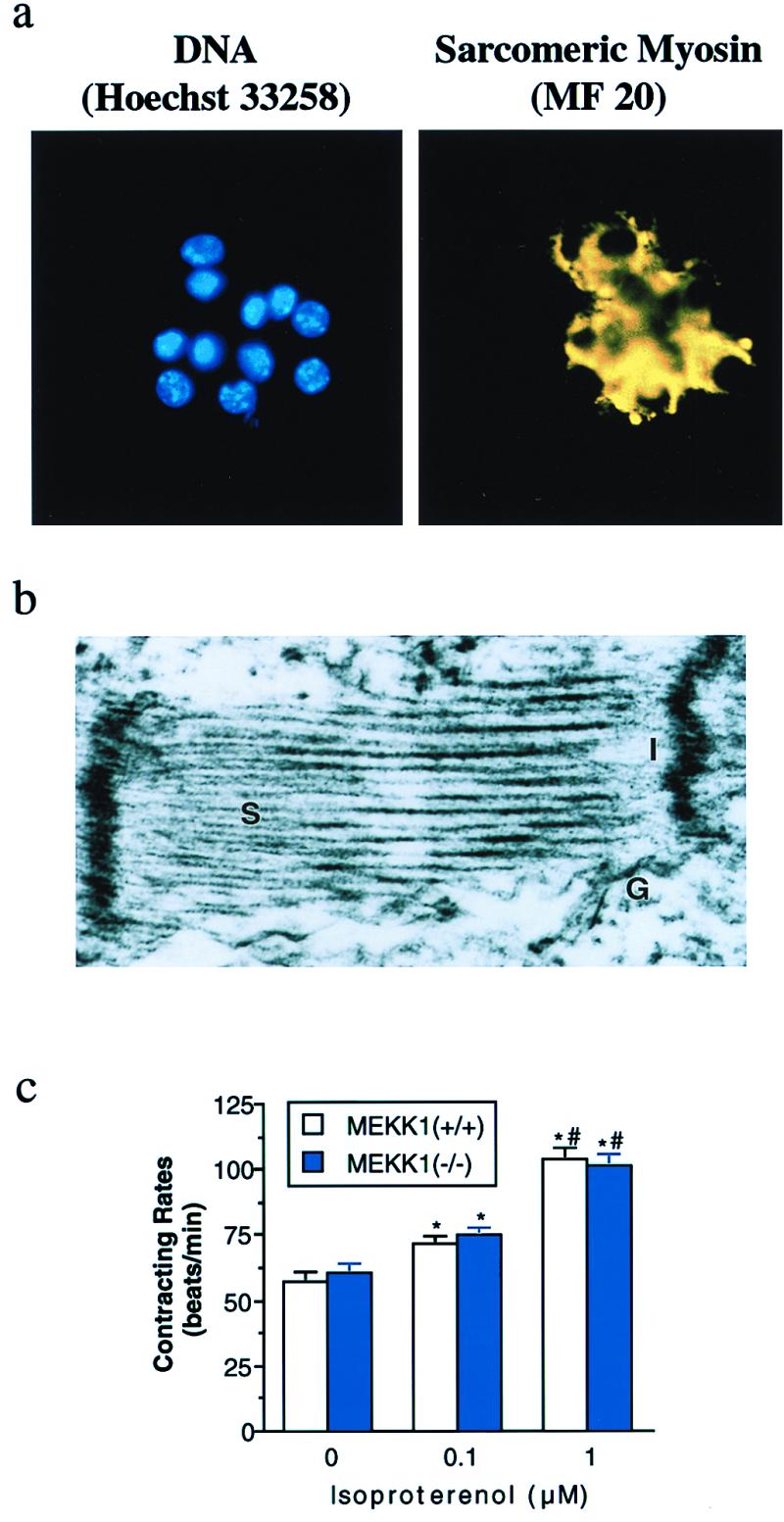
Differentiation of MEKK1−/− ES cells into cardiac myocytes. (a) Hoechst 33258 DNA staining of the cluster of MEKK1−/− ESCM after Zeocin selection (Left). Anti-sarcomeric myosin immunofluorescence with MF 20 of the same field (Right). (b) Ultrastructural analysis of selected MEKK1−/− ESCM. S, sarcomere; I, intercalated disc; G, gap junction. (c) Changes in contracting rate of ESCM in response to isoproterenol. Open and solid bars indicate wild-type and MEKK1−/− ESCM (n = 10, each), respectively. * and # indicate significant differences (P < 0.05) from the values at corresponding control and at treatment with 0.1 μM isoproterenol, respectively.
Increased Sensitivity of MEKK1−/− ESCM to H2O2-Induced Apoptosis.
We examined the apoptotic response induced by exposure of ESCM to H2O2. When ESCM were exposed to 0.1 mM H2O2 for 6 h, the genomic DNA from wild-type ESCM showed a slight increase in DNA laddering that was characteristic of apoptosis. The same H2O2 treatment of MEKK1−/− ESCM induced markedly higher amounts of fragmented DNA (Fig. 2a). When nuclei of ESCM were stained by using the TUNEL method, few cells were positive in control cultures of wild-type and MEKK1−/− ESCM (Fig. 2b). Treatment with 0.01 or 0.1 mM H2O2 for 6 h increased TUNEL-positive cells significantly in MEKK1−/− but not in wild-type ESCM. These results demonstrate that ESCM having the targeted disruption of MEKK1 expression have an increased sensitivity to ROS-induced apoptosis. It should be noted that there was no significant difference in DNA fragmentation or the number of TUNEL-positive cells between wild-type and MEKK1−/− ESCM when cells were treated with a higher dose (1 mM) of H2O2 for 6 h.
Figure 2.

Apoptosis induced by H2O2. (a) H2O2-induced DNA fragmentation in ESCM. Wild-type and MEKK1−/− ESCM were treated with either 0.01 or 0.1 mM H2O2 for 6 h in the presence or absence of either the vehicle (DMSO 0.1%), PD98059 (50 μM), SB 203580 (10 μM), or anti-TNFα antibody (5 μg/ml, purified goat anti-mTNFα IgG; Research and Diagnostic Antibodies, Berkeley, CA). (b) H2O2-induced apoptotic cell death in ESCM evaluated by TUNEL staining. Wild-type and MEKK1−/− ESCM were treated with either 0.01 or 0.1 mM H2O2 for 6 h. Five hundred nuclei of cardiac myocytes costained with Hoechst 33258 were counted and the number of TUNEL positive cells was presented as a percentage from three independent experiments (mean ± SEM). *, P < 0.05 vs. each corresponding control.
Loss of JNK Activation in MEKK1−/− ESCM in Response to Oxidative Stress.
The change in sensitivity to ROS-induced apoptosis in ESCM lacking MEKK1 prompted us to measure the effects of oxidative stress on the activity of MAPK pathways in wild-type and MEKK1−/− ESCM. As shown in Fig. 3, H2O2 stimulated the activation of JNK, p38, and ERK in wild-type ESCM. JNK activity increased within 5 min after addition of 1 mM H2O2 to the culture medium, peaked at 15 min, and decreased thereafter (Fig. 3a). Activation of JNK also was dependent on the concentration of H2O2. Both phosphorylation and activation of p38 (Fig. 3b) and ERK (Fig. 3c) increased within 5 min and peaked at 10 min after exposure of wild-type ESCM to H2O2. Significantly, in contrast to wild-type ESCM, JNK activation was reduced markedly in MEKK1−/− ESCM (Fig. 3a). Phosphorylation and activation of both p38 and ERK in response to H2O2 were similar for wild-type and MEKK1−/− ESCM (Fig. 3 b and c). Thus, loss of MEKK1 expression caused a selective loss of JNK signaling in response to ROS.
Figure 3.

MAP kinase activation by oxidative stress. (a) JNK activation by H2O2. (Top) Time course of JNK activation by H2O2 in ESCM. Wild-type ESCM were treated with 1 mM H2O2 for 0–60 min. (Middle and Bottom) Dose-response of JNK activation by H2O2 in wild-type and MEKK1−/− ESCM (15-min treatment). The data represent the average fold of the controls from three independent experiments (mean ± SEM). * and #, P < 0.05 vs. each corresponding control. (b) p38 activation by H2O2. (Top) Phosphorylation of p38 in wild-type ESCM incubated with 1 mM H2O2 for various periods of time. (Middle) Dose response of p38 phosphorylation by H2O2 in wild-type and MEKK1−/− ESCM. (Bottom) Activation of p38 in wild-type and MEKK1−/−ESCM treated with 1 mM H2O2 for 10 min. (c) ERK activation by H2O2. (Top) Phosphorylation of ERK in wild-type ESCM incubated with 1 mM H2O2 for various periods of time. (Middle) Dose response of ERK phosphorylation by H2O2 in wild-type and MEKK1−/− ESCM. (Bottom) Activation of ERK2 in wild-type and MEKK1−/− ESCM treated with 1 mM H2O2 for 10 min. (d) JNK activation by hypoxia/reoxygenation. Wild-type and MEKK1−/− ESCM were subjected to a hypoxic atmosphere by immediately replacing the medium with the hypoxic medium. Cells were incubated in a hypoxic condition for 10 min (Hypoxia-1) or 60 min (Hypoxia-2). After 60-min treatment of cells in hypoxia, cells were reoxygenated by immediately replacing the hypoxic medium with a normoxic medium and incubated in a normoxic condition for an additional 15 min. The data in the graph represent the average percentage of the controls from three independent experiments (mean ± SEM). * and #, P < 0.05 vs. each corresponding control.
We further examined JNK activation by a second oxidative stress, hypoxia/reoxygenation. In wild-type and MEKK1−/− ESCM, JNK activity increased within 10 min after the initiation of hypoxic stimuli and returned to the basal level within 60 min after sustained hypoxic stimulation (Fig. 3d). However, the magnitude of JNK activation in wild-type ESCM was much greater than that in MEKK1−/− ESCM. Moreover, reoxygenation activated JNK only in wild-type but not in MEKK1−/− ESCM. These findings indicate that MEKK1 contributes to normal JNK activation in response to hypoxia and is required for JNK activation during reoxygenation.
Enhanced TNFα Production in MEKK1−/− ESCM.
Recent studies demonstrate that cardiac myocytes themselves can produce cytokines including TNFα in response to ischemia/reperfusion and oxidative stress (5, 23–26). Moreover, TNFα production in hearts was implied to play a role in cardiac injury induced by oxidative stress (5). We therefore examined TNFα production in ESCM. Surprisingly, treatment of ESCM with H2O2 (0.1 mM) for 6 h increased levels of TNFα in the supernatant (Fig. 4a) and ESCM (Fig. 4b) in MEKK1−/− but not in wild-type ESCM. When MEKK1−/− ESCM were pretreated with a TNFα-blocking antibody for 1 h before exposure to H2O2, apoptosis as measured by DNA fragmentation (Fig. 2a) and TUNEL assay (data not shown) was prevented (Fig. 2a), demonstrating that TNFα is required for H2O2-induced apoptosis of ESCM. Pretreatment of MEKK1−/− ESCM with the compound SB203580, an inhibitor of p38, inhibited the H2O2-induced increase in TNFα production (Fig. 4a) and DNA fragmentation (Fig. 2a). In contrast, incubation of MEKK1−/− ESCM with PD98059, an inhibitor of the ERK pathway, did not affect TNFα production. In these studies, incubation of wild-type ESCM with SB203580 before treatment with H2O2 inhibited phosphorylation and activation of p38 by 90% and had little or no effect on JNK or ERK activation. Incubation of ESCM with PD98059 inhibited ERK activation by nearly 95% with little or no effect on JNK or p38 activity, demonstrating the specificity of these inhibitors (not shown).
Figure 4.

TNFα production by H2O2 in ES-derived cardiac myocytes. (a) ELISA assay. Wild-type and MEKK1−/− ESCM were treated with 0.1 mM H2O2 for 6 h in the presence and absence of drugs. TNFα concentration in the supernatant was measured as described in Materials and Methods. (b) Immunoblot. MEKK1−/− ESCM were treated with 0.1 mM H2O2 for 6 h in the presence and absence of drugs. Total cellular proteins were extracted and expression of TNFα in cardiac myocytes was detected by immunoblot analysis by using a specific antibody against TNFα. (c) TNFα promoter reporter assay. pGL3TNF (500 ng) and pRL-TK (50 ng) were cotransfected into wild-type ESCM with 3 μg of either pMEKK1-K1253M, pJNK1-APF, or an empty pCEP4 vector by using a liposome method. Cells were incubated for 24 h and treated with hydrogen peroxide (0.1 mM) for an additional 6 h. Luciferase activities were determined by using a luminometer. TNFα promoter activity was normalized for transfection efficiency based on the cotransfected Renilla luciferase reporter construct.
To substantiate the role of MEKK1-regulated JNK in oxidative stress-induced TNFα production in ESCM, we examined the effects of inhibitory mutants of MEKK1 and JNK on the promoter activity for the TNFα gene in H2O2-treated ESCM. A TNFα promoter–luciferase reporter (pGL3TNF) was transfected into wild-type ESCM in the presence of an expression vector for a kinase-inactive mutant of MEKK1 (MEKK1-K1253M) or an equivalent amount of empty vector (Control). The kinase-inactive MEKK1 mutant has been shown to function as a dominant negative MEKK1 and to suppress endogenous MEKK1 activity (27). As shown in Fig. 4c, expression of the dominant negative MEKK1 increased basal activity of the TNFα promoter. Additionally, H2O2 (0.1 mM) further increased TNFα promoter activity in the cells cotransfected with the dominant negative MEKK1 mutant, whereas it failed to do so in the control ESCM. These data corroborate our other findings that MEKK1 plays an inhibitory role in TNFα transcription in ESCM. The role of JNK in TNFα transcription in ESCM was also determined in these experiments. Kinase-inactive JNK1 (JNK1-APF) is a competitive inhibitory mutant that is capable of partially inhibiting endogenous JNK activation in response to different stimuli (28). Expression of JNK1-APF enhanced the activity of the TNFα promoter induced by addition of H2O2. These data suggest that the inhibitory role of MEKK1 in TNFα transcription may be mediated in part via JNK activity in ESCM.
Discussion
Our studies demonstrate the powerful combination of in vitro ES cell differentiation and targeted gene disruption to define complex regulatory events. Cardiac myocytes derived from ES cells have been shown to be an excellent model system for physiological and molecular studies (16, 21, 22). Although the analyses of selected ESCM here were of a very limited scope, the results were consistent with the previous reports demonstrating that ESCM have electrophysiological, contractile, and morphological features and cardiac-specific gene expression resembling primary cardiac myocytes isolated from newborn animals (16, 17, 19–22). The isolation of MEKK1−/− ESCM here allowed us to define the role of MEKK1 in cardiac myocyte regulation of MAPK signaling and cell survival. MEKK1 is required for ROS and reoxygenation stimulation of the JNK pathway. Functionally, MEKK1 signaling enhances survival of ESCM during exposure to these stresses. The role of MEKK1 activation of the JNK pathway was to negatively regulate TNFα production via repressing the activity of the TNFα promoter. Our findings demonstrate an opposing coregulation by JNK and p38 in the production of a proapoptotic cytokine, TNFα (Fig. 5). The opposing effect of JNK and p38 on regulating TNFα production allows different stimuli to alter the balance of activation of the two pathways for control of TNFα production. Selective p38 activation or temporally prolonged p38 activation relative to JNK activation could lead to TNFα production in ESCM. It is likely that similar coincident regulation of other genes will be found for MAPK-signaling pathways. In this regard, it is interesting that JNK and p38 frequently are coordinately activated in response to a large number of cytokines and stress stimuli. The disruption or activation of one arm of the two pathways, either JNK or p38, has been correlated with both pro- and antiapoptotic responses in different cell types. Our findings provide a mechanism for this type of observation in ESCM. Studies using the TNFα promoter–reporter gene construct suggest that JNK has a repressor function for controlling TNFα gene expression. The factors in cardiac myocytes controlled by JNK are now being examined.
Figure 5.

A proposed model for the role of MEKK1 in cardiac myocytes under oxidative stress.
It should be stressed that the results in ESCM may not apply directly to primary cardiac myocytes; however, the system developed here enables us to evaluate rapidly the function of targeted molecules, which can be tested further by using primary cardiac myocytes. Finally, the use of in vitro ES cell differentiation is proving to be an invaluable tool in the genetic analysis of signaling in mouse cells. It is possible to differentiate ES cells to functional macrophages, mast cells, erythroid cells, neurons, cardiac myocytes, and fibroblasts (29). In the future it will be possible to extend the number of cell types that can be differentiated from ES cells. Particularly when a gene disruption is embryonic lethal, the ability to differentiate ES cells to specific cell types becomes extremely useful for the identification of differentiated functions regulated by protein for the targeted gene.
Acknowledgments
We are indebted to L. Field (Indianapolis) and B. Beutler (Dallas) for providing the plasmid constructs and G. Keller, E. Gelfand, D. Riches, and J. Routes (Denver) for helpful suggestions and discussions. This work was supported by grants from the National Institutes of Health (CA64685 to N.T., HL03625 to E.D.C., DK37381 and GM30324 to G.L.J.) and from the American Heart Association (9951086Z to N.T.). T.M. is a recipient of the Banyu Fellowship Award in Lipid Metabolism and Atherosclerosis.
Abbreviations
- ES cell
embryonic stem cell
- ESCM
ES cell-derived cardiac myocytes
- TNFα
tumor necrosis factor α
- JNK
c-Jun kinase
- ROS
reactive oxygen species
- MAPK
mitogen-activated protein kinase
- ERK
extracellular-signal response kinase
- TUNEL
terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling
References
- 1.Lucchesi B R. Annu Rev Physiol. 1990;52:561–576. doi: 10.1146/annurev.ph.52.030190.003021. [DOI] [PubMed] [Google Scholar]
- 2.Gottlieb R A, Burleson K O, Kloner R A, Babior B M, Engler R L. J Clin Invest. 1994;94:1621–1628. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Werns S W, Lucchesi B R. Trends Pharmacol Sci. 1990;11:161–166. doi: 10.1016/0165-6147(90)90068-J. [DOI] [PubMed] [Google Scholar]
- 4.McCord J M. Fed Proc. 1987;46:2402–2406. [PubMed] [Google Scholar]
- 5.Meldrum D R, Dinarello C A, Cleveland J C, Jr, Cain B S, Shames B D, Meng X, Harken A H. Surgery. 1998;124:291–296. doi: 10.1067/msy.1998.90570. [DOI] [PubMed] [Google Scholar]
- 6.Krown K A, Page M T, Nguyen C, Zechner D, Gutierrez V, Comstock K L, Glembotski C C, Quintana P J, Sabbadini R A. J Clin Invest. 1996;98:2854–2865. doi: 10.1172/JCI119114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Tanaka M, Shiojima I, Hiroi Y, Yazaki Y. J Clin Invest. 1997;100:1813–1821. doi: 10.1172/JCI119709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clerk A, Fuller S J, Michael A, Sugden P H. J Biol Chem. 1998;273:7228–7234. doi: 10.1074/jbc.273.13.7228. [DOI] [PubMed] [Google Scholar]
- 9.Mackay K, Mochly-Rosen D. J Biol Chem. 1999;274:6272–6279. doi: 10.1074/jbc.274.10.6272. [DOI] [PubMed] [Google Scholar]
- 10.Sugden P H, Clerk A. Circ Res. 1998;83:345–352. doi: 10.1161/01.res.83.4.345. [DOI] [PubMed] [Google Scholar]
- 11.Lange-Carter C A, Pleiman C M, Gardner A M, Blumer K J, Johnson G L. Science. 1993;260:315–319. doi: 10.1126/science.8385802. [DOI] [PubMed] [Google Scholar]
- 12.Yujiri T, Sather S, Fanger G R, Johnson G L. Science. 1998;282:1911–1914. doi: 10.1126/science.282.5395.1911. [DOI] [PubMed] [Google Scholar]
- 13.Yujiri T, Fanger G R, Garrington T P, Schlesinger T K, Gibson S, Johnson G L. J Biol Chem. 1999;274:12605–12610. doi: 10.1074/jbc.274.18.12605. [DOI] [PubMed] [Google Scholar]
- 14.Burton K P, McCord J M, Ghai G. Am J Physiol. 1984;246:H776–H783. doi: 10.1152/ajpheart.1984.246.6.H776. [DOI] [PubMed] [Google Scholar]
- 15.Sato H, Hori M, Kitakaze M, Iwai K, Takashima S, Kurihara H, Inoue M, Kamada T. Circ Res. 1993;72:361–375. doi: 10.1161/01.res.72.2.361. [DOI] [PubMed] [Google Scholar]
- 16.Klug M G, Soonpaa M H, Koh G Y, Field L J. J Clin Invest. 1996;98:216–224. doi: 10.1172/JCI118769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metzger J M, Lin W I, Samuelson L C. J Cell Biol. 1994;126:701–711. doi: 10.1083/jcb.126.3.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beutler B, Brown T. J Clin Invest. 1991;87:1336–1344. doi: 10.1172/JCI115137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maltsev V A, Rohwedel J, Hescheler J, Wobus A M. Mech Dev. 1993;44:41–50. doi: 10.1016/0925-4773(93)90015-p. [DOI] [PubMed] [Google Scholar]
- 20.Narita N, Bielinska M, Wilson D B. Development. 1997;124:3755–3764. doi: 10.1242/dev.124.19.3755. [DOI] [PubMed] [Google Scholar]
- 21.Sowell M O, Ye C, Ricupero D A, Hansen S, Quinn S J, Vassilev P M, Mortensen R M. Proc Natl Acad Sci USA. 1997;94:7921–7926. doi: 10.1073/pnas.94.15.7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolossov E, Fleischmann B K, Liu Q, Bloch W, Viatchenko-Karpinski S, Manzke O, Ji G J, Bohlen H, Addicks K, Hescheler J. J Cell Biol. 1998;143:2045–2056. doi: 10.1083/jcb.143.7.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapadia S, Lee J, Torre-Amione G, Birdsall H H, Ma T S, Mann D L. J Clin Invest. 1995;96:1042–1052. doi: 10.1172/JCI118090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gwechenberger M, Mendoza L H, Youker K A, Frangogiannis N G, Smith C W, Michael L H, Entman M L. Circulation. 1999;99:546–551. doi: 10.1161/01.cir.99.4.546. [DOI] [PubMed] [Google Scholar]
- 25.Meldrum D R, Dinarello C A, Shames B D, Cleveland J C, Jr, Cain B S, Banerjee A, Meng X, Harken A H. Circulation. 1998;98, Suppl. II:214–218. [PubMed] [Google Scholar]
- 26.Wang C Y, Naka Y, Liao H, Oz M C, Springer T A, Gutierrez-Ramos J C, Pinsky D J. Circ Res. 1998;82:762–772. doi: 10.1161/01.res.82.7.762. [DOI] [PubMed] [Google Scholar]
- 27.Fanger G R, Johnson N L, Johnson G L. EMBO J. 1997;16:4961–4972. doi: 10.1093/emboj/16.16.4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butterfield L, Storey B, Maas L, Heasley L E. J Biol Chem. 1997;272:10110–10116. doi: 10.1074/jbc.272.15.10110. [DOI] [PubMed] [Google Scholar]
- 29.Keller G, Snodgrass H R. Nat Med. 1999;5:151–152. doi: 10.1038/5512. [DOI] [PubMed] [Google Scholar]