

Abstract
Spinal glutamate transporters (GT) have been implicated in the mechanisms of neuropathic pain; however, how spinal GT uptake activity is regulated remains unclear. Here we show that alteration of spinal arachidonic acid (AA) turnover after peripheral nerve injury regulated regional GT uptake activity and glutamate homeostasis. Chronic constriction nerve injury (CCI) in rats significantly reduced spinal GT uptake activity (3H-glutamate uptake) with an associated increase in extracellular AA and glutamate concentration from spinal microdialysates on postoperative day 8. AACOCF3 (a cytosolic phospholipase A2 inhibitor, 30 μg) given intrathecally twice a day for postoperative day 1 – 7 reversed this CCI-induced spinal AA production, prevented the reduced spinal GT uptake activity and increased extracellular glutamate concentration. Conversely, alteration of spinal AA metabolism by diclofenac (a cyclooxygenase 1/2 inhibitor, 200 μg) further reduced spinal GT uptake activity and increased extracellular glutamate concentration in CCI rats. GT uptake activity was also attenuated when AA (10 or 100 nM) was directly added into spinal samples of naïve rats in an in vitro 3H-glutamate uptake assay, indicating a direct inhibitory effect of AA on GT uptake activity. Consistent with these findings, AACOCF3 reduced the development of both thermal hyperalgesia and mechanical allodynia, whereas diclofenac exacerbated thermal hyperalgesia, in CCI rats. Thus, spinal AA turnover may serve as a regulator in CCI-induced changes in regional GT uptake activity, glutamate homeostasis, and neuropathic pain behaviors. These data suggest that regulating spinal AA turnover may be a useful approach to improving the clinical management of neuropathic pain.
Keywords: Glutamate transporter, neuropathic pain, nerve injury, phospholipase A2, Arachidonic acid, glutamate uptake, microdialysis, High pressure liquid chromatography, Gas chromatography
Introduction
Peripheral nerve injury can result in signs and symptoms of neuropathic pain such as allodynia, hyperalgesia, and spontaneous pain (Wall et al. 1974;Devor 1983;Woolf and Mannion 1999;Zimmermann 2001). Primary sensory afferents release glutamate into synaptic clefts within the spinal cord dorsal horn when stimulated by physical and/or chemical sources (al Ghoul et al. 1993). While glutamate plays a key role in a number of physiological functions (Maragakis and Rothstein 2004), excessive and sustained excitation of glutamate receptors has been implicated in the mechanisms of neurotoxicity and neuropathic pain (Yamamoto and Yaksh 1992;Mao et al. 1992a;Mao et al. 1992b;Mao et al. 1995). Regional glutamate homeostasis is maintained mainly through a high capacity glutamate transporter (GT) system that exists perisynaptically with neuronal and glial cells (Robinson and Dowd 1997;Semba and Wakuta 1998;Danbolt 2001;Kanai and Hediger 2003). The activity of GT directly influences synaptic glutamate turnover through removing glutamate from synaptic cleft (Maragakis and Rothstein 2004), and may play a pivotal role in a variety of neurodegenerative diseases (Lievens et al. 2000;Trotti et al. 2001;Urban et al. 2003).
In a previous study (Sung et al. 2003), we have shown that peripheral nerve injury resulted in an initial increase in spinal GT expression followed by a progressive downregulation of spinal GT expression mediated through a mitogen-activated protein kinase pathway, which contributed to the development and maintenance of neuropathic pain behaviors in rats. However, it remains unclear as to how GT activity is regulated by endogenous regulators in relation to the pathogenesis of neuropathic pain.
Arachidonic acid (AA), produced from membranes of both neuronal and glial cells in response to pathological insults (Danbolt 2001), is a cell membrane lipid product and a potent inhibitor of GT activity in in vitro studies (Volterra et al. 1994;Trotti et al. 1995;Manzoni and Mennini 1997;Dorandeu et al. 1998). The inhibitory effect of AA on GT activity is more potent in the cerebellum and spinal cord than in the hippocampus, which is likely due to its direct inhibitory action on GT (Dorandeu et al. 1998). Furthermore, the AA metabolic pathway has been well delineated, which begins with activation of phospholipase A2 (PLA2) resulting in the enzymatic hydrolysis of AA from lipid membranes and is followed by the downstream AA metabolism via several enzymatic pathways including cyclooxygenase 1 and 2 (COX-1/2) (Raja et al. 1988;Sawin et al. 1997;Kirchheiner et al. 2003;Farooqui and Horrocks 2004). If AA were an active endogenous regulator of GT activity, it would be expected that regulation of regional AA turnover through inhibition of either PLA2 or COX-1/2 should alter GT uptake activity and regional glutamate concentration with a concurrent change in neuropathic pain behaviors following peripheral nerve injury. This hypothesis was examined in the present study by utilizing a rat model of chronic constriction nerve injury (CCI) and a combination of spinal 3H-glutamate uptake assay, intrathecal microdialysis, regional glutamate and AA assay by high performance liquid chromatography (HPLC) and gas chromatography (GC), and behavioral testing.
Materials and Methods
Animal surgery and drugs
Adult male Sprague Dawley rats weighing 275–325 gm (Charles River Laboratories, Wilmington, MA) were used. The animal room was artificially lighted from 7:00 A.M. to 7:00 P.M. The surgical procedure was performed aseptically under pentobarbital anesthesia (50 mg/kg, intraperitoneally). CCI rats were produced by loosely ligating one common sciatic nerve according to the method of Bennett and Xie (Bennett and Xie 1988). Briefly, on one side the rat’s sciatic nerve was exposed in the mid-thigh, and four loose ligatures using 4.0 chromic gut sutures were made around the dissected sciatic nerve with a 1.0 –1.5 mm interval between each ligature. Skin wound was closed with wound clips. Sham rats were made following the same surgical procedure except for nerve ligation. The general experimental protocol was approved through our Institutional Animal Care and Use Committee.
A cytosolic PLA2 inhibitor, AACOCF3 (Lucas et al. 2005), and a COX-1/2 inhibitor, diclofenac (Kirchheiner et al. 2003), were used to regulate the AA metabolism. The final dose for either AACOCF3 (30 μg) or diclofenac (200 μg, (Pinardi et al. 2003)) used in this study was determined after a pilot dose-response experiment for each agent. We first examined the effect of each agent (30, 60 μg for AACOCF3 and 200, 400 μg for diclofenac) using a small number (3) of rats. We did not find further increase in the effects of each agent with a higher dose. To avoid possible neurological toxicity such as gait and sensory function, we chose to use AACOCF3 (30 μg) and diclofenac (200 μg) in the experiments. Both agents were purchased from Sigma (St. Louis, MO). AACOCF3 was dissolved in 100% DMSO and diclofenac in 5% DMSO and 95% artificial CSF. The 100 % and 5% DMSO solutions were used as vehicles for the AACOCF3 and the diclofenac, respectively. Vehicles contained the same solution as that for AACOCF3 or diclofenac. Each agent (10 μl volume) was injected intrathecally (i.t.) using a microsyringe injector followed by a saline flush (10 μl). The injections were given twice a day at 9:00 AM and 5:00 PM beginning within 1 min after the operation. Doses were divided into two injections each day for the drugs to be distributed more constantly.
Behavioral tests
Animals were habituated to the test environment for 60 min per day for two consecutive days before baseline testing. All animals were tested for thermal hyperalgesia and mechanical allodynia. For testing thermal hyperalgesia, the plantar surface of a rat’s hindpaw was exposed to a beam of radiant heat through a transparent perplex glass surface (Hargreaves et al. 1988). The withdrawal latency was averaged from at least two trials separated by a 2-min interval, and the cutoff was set at 25 sec to avoid tissue damage. For examining mechanical allodynia, each rat was placed on a metal mesh floor and covered with a plastic box (15×15×18 cm). Each test began after a 30 min period of habituation. The mechanical stimulation resulting from the bending force of a von Frey filament was applied to the plantar surface of each hindpaw. Each trial consisted of five applications of a von Frey filament given every 4 sec, and the cutoff force was 20 gm. Brisk foot withdrawals (at least three times of five applications) in response to von Frey filament stimulation were considered positive. Depending on the initial response, subsequent filaments were applied in the order of either descending or ascending force to determine the threshold force (Tal and Bennett 1994;Mao et al. 1997).
Construction and placement of a microdialysis catheter
Intrathecal loop dialysis catheters were constructed from hollow fibers (200 μm ID, 216 μm OD, 18,000 M.W. cut-off; Spectra/Por, Part No:132295, www.spectrapor.com) according to the modified method described by Hua et al. (1999). A Nichrome wire (50 μm thick, Part No: 7620, A-M systems, www.a-msystems.com) was then passed through the fiber. The fiber was then bent in the middle to form a “U”-shaped loop. Both ends of this fiber were attached to the two side holes of the triple lumen tubing of 8 cm long (Part No; E100-0353, Spectranetics, 1-800-231-0978, www.spectranetics.com) by a 1 cm-long polycarbonate tubing connector (Part No; 2000021; Polymicro Tech., Inc. 1-602-375-4100, www.polymicro.com). The three holes at the other end of the triple lumen tubing were connected to PE-10 tubing (5 cm in length) by the above-mentioned polycarbonate tubing connector with cyanoacrylate.
To place a microdialysis catheter of which each triple lumen connected to a PE-10 tubing (Yaksh and Rudy 1976), rats were anesthetized with pentobarbital as described above. The animals were then placed in a stereotaxic head holder with the head slightly flexed forward. After the cisternal membrane was exposed and cut open, the end of the U-shaped loop of the catheter was inserted and passed caudally to the lumbar intrathecal space (about 9 cm from the incision site for the size of rats used in the study). The other end of the dialysis catheter was then externalized and secured on the neck. Of the three external tubing holes, the median hole (a PE 10 catheter) was used as a drug injection while the other holes at both sides were reserved for microdialysis study – one end for infusing the artificial cerebrospinal solution and the other end for collecting the infused solution.
All animals were allowed to recover for three days prior to the experiments. Those rats exhibiting postoperative neurological deficits or poor grooming were excluded from the experiments. We only observed fewer than 10 % animals with neurological changes after the catheter insertion with the size of rats described above. We did not experience blockage of the intrathecal catheter for microperfusion and drug injection when the experiments were conducted under 8 days after the implantation.
Intrathecal microdialysis
To initiate dialysis, one of the externalized PE-10 connections was attached to a piece of PE-10 tubing (inflow, 30 cm in length) and the other arm to another PE-10 tubing (outflow, 25 cm in length) in an awake animal gently placed in a container. A syringe pump (Harvard Compact Infusion Pump, Model 975 with a 5 ml plastic syringe) was connected and the dialysis catheter was perfused with artificial cerebrospinal fluid (ASCF) at a rate of 10 μl/min. ACSF contained (mM) Na+ 151.1, K+ 2.6, Mg2+ 0.9, Ca2+ 1.3, C1− 122.7, HCO3− 21.0, HPO4− 2.5 and dextrose 3.5. ACSF was bubbled with 95% O2/5% CO2 before each experiment to adjust the final pH to 7.2. The dialysis procedure typically began with a 30-min washout period followed by two collection periods of 10 min each. The single median PE-10 catheter was used only for intrathecal drug injection in order to avoid potential interference with the dialysis.
Spinal glutamate concentration assay (HPLC)
Glutamate concentration from the intrathecal microdialysates was analyzed using HPLC according to a modified method (Hua et al. 1999). The analysis was accomplished by using the phenylisothiocyanate (PITC) derivatization procedure (Shimadzu HPLC, SCL 10A-VP) with Waters AccQ•Taq amino acid analysis column (3.9×150 mm, 4 μm particle size, silica base bonded with C18) and a UV detector (254 nm, Shimadzu, SPD-10A VP). A volume of 40 μl microdialysates was vacuum-dried after being mixed with 4 μl methionine sulfone (0.2 mM in 0.1 M HCl) as an internal standard. The mixtures were dried again after adding a coupling buffer [20 μl, methanol:water:triethylamine (TEA), 2:2:1 in volume ratio]. The coupled amino acids were derivatized with PITC (Molnar-Perl I 1994). The derivatization reagent (20 μl, methanol:water:TEA:PITC, 7:1:1:1 in volume ratio, freshly made on each day of assay) was reacted with the coupled amino acids for 20 minutes at room temperature. Excessive PITC was removed through the vacuum-dry process. Derivatized amino acid samples were then reconstituted with 70 μl of 0.05 M sodium acetate buffer before being injected (25 μl of reconstituents) by an autosampler (Shimadzu, SIL-10AF).
The solvent system consisted of two eluents: A) an aqueous buffer and B) 60% acetonitrile in water (Bidlingmeyer et al. 1984). A typical aqueous buffer was 0.14 M sodium acetate containing 0.5 ml/L TEA and titrated to pH 6.35 with glacial acetic acid. A gradient, which was run for the separation, consisted of 10% B eluent traversing to 51% B eluent in 10 min. After this step, a washing step was programmed to 100% B eluent so that any residual sample components were cleaned from the column. The accuracy of the measurement was expressed as the ratio of glutamate concentration determined by standard curves to the known concentration of glutamate together with methionine sulfone added as an internal standard. Standard curves were made daily by injections of 1 to 10 μM of L-glutamic acid (M.W. 147.1) from Sigma. These curves, with Correlation Coefficients of 0.990 or greater, were linear within the range between 0.1 and 10 μM of glutamate concentrations. For a given of experiment, the measurement quality control was made using five replicates of quality control samples in which glutamate concentrations ranged from 0.1 to 10 μM. Between days, the measurement quality control was made once a week by using freshly prepared control samples of same concentrations as above. Glutamate Ratio over the methionine sulfone as an internal standard was measured and expressed in the Y-axis in figures.
Spinal glutamate uptake assay
A spinal synaptosome (GT) preparation was used to assess glutamate uptake activity according to a previously published method (Mitrovic et al. 1999;Azbill et al. 2000;Mu et al. 2000). In brief, animals were sacrificed under pentobarbital after microdialysis. Fresh tissue samples from L4 and L5 spinal dorsal horns were removed as quickly as possible by laminectomy. Dorsal horns ipsilateral and contralateral to CCI were homogenized separately in an ice-cold buffer solution (pH 7.2) containing 0.32 M sucrose plus (in g/ml) 4 pepstatin, 5 aprotinin, 20 trypsin inhibitor (# T1426, Sigma, St. Louis, MO), 4 leupeptin, and (in mM) 0.2 PMSF (phenylmethanesulfonyl fluoride), 2 EDTA, 2 EGTA, and 20 HEPES. The homogenates were centrifuged at 1,500 rpm for 10 min at 4°C and the supernatant was collected. The remaining pellets were resuspended by using the same buffer solution and recentrifuged as above. Both supernatants were combined and again centrifuged at 13,000 rpm for 10 min at 4°C. The so-obtained synaptosomal pellets, which contained both neuronal and glial GT (Mitrovic et al. 1999;Azbill et al. 2000;Mu et al. 2000), were suspended in 2 ml of Locke’s buffer containing (in mM) 154 NaHCO3, 5.6 glucose, 5 HEPES, pH 7.2, and saturated with 95% O2/5% CO2. The protein concentration of final synaptosome pellets was measured by the Bradford method and was adjusted to 200 μg/ml in Locke’s buffer.
Glutamate uptake activity was determined by incubating the synaptosome preparation (in 100 μg protein content) with 0.4 μl [3H]L-glutamate (Perkin-Elmer, Boston, MA) at 37°C for 5 min. The reaction was terminated by filtering synaptosomes through a Whatman (Maidstone, UK) GF/C 2.4 cm filter that was presoaked in the same buffer solution. The filter was then washed three times with ice-cold Locke’s buffer (2 ml) and transferred to a vial containing scintillation mixture (10 ml; Fisher Scientific, Houston, TX). Radioactivity in the final samples was measured by a liquid scintillation counter (Bio-Rad). The basal uptake activity in counts per minute (cpm) was measured in the absence of any sample.
The unit of cpm measured by the instrument was converted to the mole concentration of radioactive glutamate or 3H-glutamate uptaken to GT by using the following conversion units:
Note: the efficiency of the liquid scintillation counter = 0.612 dpm/cpm; 1 Ci = 2.22×1012 disintegrations per minute (dpm); stock concentration of the 3H-glutamate by the manufacturer = 52.0 Ci/mmol; the synaptosome included in each reaction = 10−4g.
Spinal AA concentration assay (GC)
Arachidonic acid in spinal microdialysates was analyzed according to a previously described method (Pound et al. 2001). A volume of 40 μl microdialysate with 2 μg tricosanoic acid as an internal standard was mixed with 1.5 ml BF3/Methanol (Product No: 49370, Pierce Co. Rockford, IL) and 2 ml 100% hexane (Sigma, St. Louis, MO). The mixture in the screw capped-glass tube filled with Nitrogen gas was heated to 100°C for 60 minutes in a heat-block. The tube was cooled to room temperature, 1.0 ml H2O was added and the contents mixed with a vortex for 1 minute. Two phases were separated; the top hexane phase was placed in a new tube for an extraction to be repeated once again. The hexane phase was dried under nitrogen gas in a fume hood and redissolved with 40 μl of hexane. The redissolved sample was transferred to a GC tube and sealed with crimper. Arachidonic acid methyl esters were quantified with GC/MS by using an HP-5890 Series II gas chromatograph equipped with a Supelcowax SP-10 capillary column (Supelco, Bellefonte, PA, USA). The temperatures of the injector and detector were 260°C and 280°C, respectively. The oven program was maintained initially at 150°C for 2 min, ramped to 200°C at 10°C/min and held for 4 min, ramped again at 5°C/min to 240°C, held for 3 min, and finally ramped to 270°C at 10°C/min and maintained for 5 min. The carrier gas-flow rate was held constantly at 0.8 mL/min throughout the process. The total ion was monitored and encompassing mass ranged from 50 to 550 atomic mass units. The fatty acid (AA) mass was determined by comparing various areas of analyzed fatty acids to that of a fixed concentration of internal standard tricosanoic acid. Trichosanoic acid was an internal standard and the ratio of AA over trichosanoic acid was expressed in the Y-axis.
For the AA analysis by GC, the internal standard, tricosanoic acid, and AA were quantified by the peak area analysis. The detector response was linear, with correlation coefficients of 0.990 or greater within the sample concentration ranges for all standards. The accuracy of the method was expressed as the ratio of AA measured by the GC to the known concentration of the AA standard from Sigma. The assay precision was assessed by calculating the standard deviation of repeated measurements as a percentage of the mean value. Within-day and between-day quality controls were made using five replicates of quality control samples and freshly prepared control samples once a week, respectively, as described for HPLC.
Statistical data analysis
Data from both thermal hyperalgesia and mechanical allodynia tests were analyzed by using nonparametric Friedman Repeated Measures ANOVA on Ranks across various time points to detect overall differences in each groups followed by the Dunnett’s method. For comparisons between each group, Mann-Whitney Rank Sum Test was used. For glutamate uptake and dialysis data analysis, either Kruskal-Wallis one-way ANOVA on Ranks followed by the Dunn’s method or Student’s t-test was used according to the normality test. For all data analysis, differences were set as statistically significant at the level of α=0.05.
Results
Decreased spinal GT uptake following CCI: effect of AACOCF3 and diclofenac
Spinal GT uptake activity was significantly reduced within the ipsilateral spinal cord dorsal horn of CCI rats (n=5) as compared to the sham control (n=5), when examined on postoperative day 8 (Fig. 1, P <0.01). In addition, our pilot time course experiments showed a statistically significant change of GT uptake activity on postoperative day 8, which was consistent with the behavioral change on that day after CCI (see below). In our previous study (Sung et al. 2003), we reported that the GT expression showed a biphasic pattern in CCI rats with an initial GT upregulation on postoperative days 1 and 4 and followed by GT downregulation on postoperative days 7 and 14. In the present study, we selected the samples on postoperative day 8 for assays because the GT downregulation was stable at postoperative day 8, which was similar to that on postoperative day 14. There were also no differences in pain behaviors between postoperative days 8 and 14. Thus, the data analysis was focused on the ipsilateral spinal cord dorsal horn on postoperative day 8 following CCI.
Fig. 1. Changes of spinal GT uptake activity following CCI.

The GT uptake activity from the spinal dorsal horn homogenates ipsilateral to CCI was reduced when examined on postoperative day 8. AACOCF3 (a cytosolic PLA2 inhibitor, 30 μg) and diclofenac (a COX-1/2 inhibitor, 200 μg), given intrathecally twice a day for postoperative day 1 – 7, enhanced and further reduced spinal GT uptake activity, expressed as mean and S.E. (standard error) respectively. GT uptake was analyzed by converting the cpm unit into the mole concentration of radioactive 3H-glutamate per 100 μg of proteins. * P< 0.05, as compared to the sham group; + P< 0.05, as compared to both sham and CCI+vehicle groups. cpm: counts per minute;
The GT uptake activity within the ipsilateral spinal cord dorsal horn of CCI rats (n=7) treated with the PLA2 inhibitor AACOCF3 (30 μg, i.t., twice daily for postoperative day 1–7) was nearly seven folds as high as in those CCI rats (n=5) treated with a vehicle, when examined on postoperative day 8 (Fig. 1, P< 0.05). The GT uptake activity also was significantly higher in those CCI rats treated with AACOCF3 than in sham-operated rats (n=5, P< 0.05), while AACOCF3 alone did not significantly change GT uptake activity in sham-operated rats (n=8). In contrast, the GT uptake activity within the ipsilateral spinal cord dorsal horn was further reduced in CCI rats (n=6) treated with diclofenac (a COX-1/2 inhibitor, 200 μg, i.t., twice daily for postoperative day 1–7) as compared to both sham-operated rats (n=5) and CCI rats (n=5) treated with a vehicle (Fig. 1, P< 0.05). These results indicate that AACOCF3 effectively prevented, while diclofenac further exacerbated, the reduced spinal GT uptake activity following CCI.
Increased spinal glutamate concentration following CCI: effect of AACOCF3 and diclofenac
Spinal microdialysates were assayed using HPLC to measure regional glutamate concentration. On postoperative day 8, there was a significant increase (102%) in regional glutamate concentration in CCI rats (n=6) over that of sham rats (n=3) in the absence of overt peripheral stimulation (Fig. 2a,b, P< 0.05). Consistent with the effect of AACOCF3 on spinal GT uptake activity, the same AACOCF3 treatment (30 μg, i.t., twice daily for postoperative day 1–7) in these CCI rats (n=4) resulted in a reduction of regional glutamate concentration in microdialysates by 35% as compared to CCI rats (n=3) treated with a vehicle, when also examined on postoperative day 8 (Fig. 2a, P< 0.05). Furthermore, regional glutamate concentration in the spinal microdialysates was no longer statistically different between CCI rats (n=4) treated with AACOCF3 and sham-operated rats (n=6) (Fig. 2a, P> 0.05).
Fig. 2. Changes of spinal glutamate concentration following CCI.

The regional glutamate concentration from spinal microdialysates was increased when examined on postoperative day 8 in CCI rats. A: AACOCF3 (30 μg) and diclofenac (200 μg), given intrathecally twice a day for postoperative day 1 – 7, reduced and further increased regional glutamate concentration, expressed as mean and S.E., respectively. * P< 0.05, as compared to the sham group; + P< 0.05, as compared to both sham and CCI+vehicle groups. Methionine sulfone is an internal standard and the ratio of glutamate over methionine sulfone was expressed in the Y-axis. B: Representative peaks of glutamate concentration over an internal standard (methionine sulfone) as assayed by HPLC. The glutamate peaks from several samples groups, as labeled, were superimposed to provide a direct comparison.
Similar to the effect of diclofenac on spinal GT uptake activity in CCI rats, CCI rats (n=4) treated with diclofenac (200 μg, i.t., twice daily for postoperative day 1–7) showed an over 200% and 400% increase in regional glutamate concentration as compared to the vehicle-treated CCI rats (n=6) and sham rats (n=3), respectively, when examined on postoperative day 8 (Fig. 2a,b, P< 0.05). Diclofenac alone did not alter the basal spinal glutamate concentration in sham-operated rats. Overall, these results indicate that inhibition of PLA2 with AACOCF3 significantly reduced, whereas inhibition of COX-1/2 with diclofenac dramatically increased, spinal regional glutamate concentration after CCI.
Increased spinal AA concentration following CCI: effect of AACOCF3 and diclofenac
Spinal microdialysates also were assayed using the GC method to measure spinal regional AA concentration. When examined on postoperative day 8, there was a significant increase (more than 4-folds) in spinal regional AA concentration in CCI rats (n=4) over that of sham rats (n=3) (Fig. 3, P< 0.05). Consistent with the role of AACOCF3 as a cytosolic PLA2 inhibitor, the treatment with AACOCF3 (30 μg, i.t., twice daily for postoperative day 1–7) in CCI rats (n=3) resulted in a substantial reduction of regional AA concentration by nearly 80% as compared to CCI rats treated with a vehicle (n=3), when both examined on postoperative day 8 (Fig. 3, P< 0.05). When compared with both sham and vehicle-treated groups, the AACOCF3 treatment effectively prevented the increase in regional AA production following CCI (Fig. 3).
Fig. 3. Changes of spinal AA concentration following CCI.
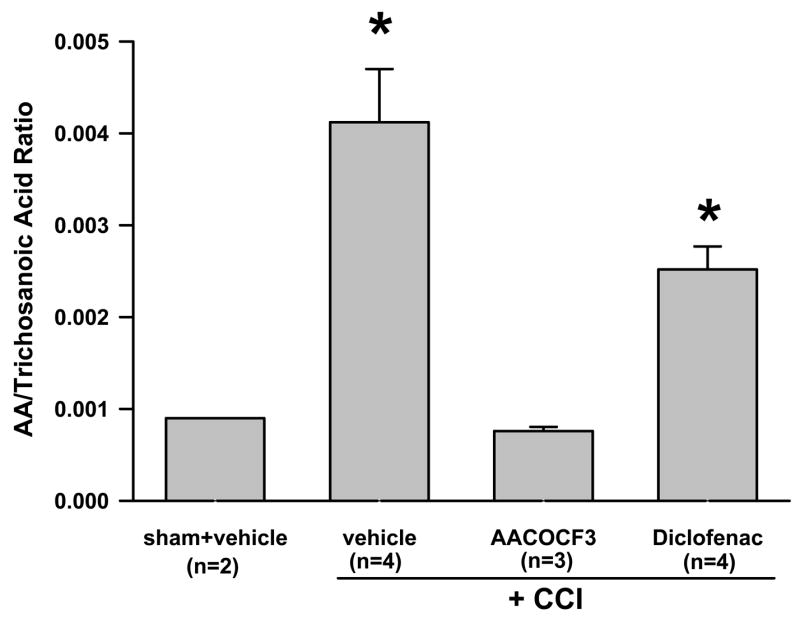
The regional AA concentration from spinal microdialysates was increased when examined on postoperative day 8 in CCI rats. AACOCF3 (30 μg) given intrathecally twice a day for postoperative day 1 – 7 reduced the regional AA concentration. The spinal AA concentration, expressed as mean and S.E., remained elevated in CCI rats treated with diclofenac (200 μg) for postoperative day 1–7. * P< 0.05, as compared to the sham group. Trichosanoic acid is an internal standard and the ratio of AA over trichosanoic acid was expressed in the Y-axis.
While there was a mild decrease in the spinal regional AA concentration in diclofenac-treated CCI rats, the AA concentration was not statistically different between CCI rats (each n=4) treated with either a vehicle or diclofenac (Fig. 3, P> 0.05). Thus, regional AA concentration in the spinal microdialysates from CCI rats treated with diclofenac (200 μg, i.t., twice daily for postoperative day 1–7) remained elevated as compared to both sham rats and AACOCF3-treated CCI rats (Fig. 3, P< 0.05). However, the diclofenac treatment did not further increase regional AA concentration in CCI rats as compared to CCI rats without receiving the diclofenac treatment (Fig. 3, P> 0.05). Thus, inhibition of PLA2 with AACOCF3 effectively prevented the increased AA production after CCI, although regional AA concentration remained unchanged in CCI rats after the diclofenac treatment.
Taken together with the data from measuring spinal GT uptake activity and regional glutamate concentration, these results indicated an overall correlation between the increased regional AA concentration, the decreased GT uptake, and the increased regional glutamate concentration after CCI.
Inhibition of in vitro GT uptake by exogenous AA
In order to confirm the effect of AA on spinal GT uptake activity, lumbar spinal samples of naïve rats were taken and processed to perform the 3H-glutamte uptake assay in the presence and absence of exogenous AA. Compared with the vehicle group (n=3), exogenous AA at the concentration of either 10 or 100 nM (each n=6) significantly reduced GT uptake activity (Fig. 4, each P< 0.01), supporting an inhibitory effect of AA on spinal GT uptake activity. The data also indicated that the effect of AA on spinal GT uptake was saturated at the concentration of 10 nM AA, since there were no differences in the reduction of GT uptake activity between two doses of exogenous AA.
Fig. 4. Inhibition of GT uptake activity by exogenous AA.

Addition of exogenous AA (10 or 100 nM) into the synaptosome preparation obtained from the lumbar spinal segments of naïve rats attenuated GT uptake activity, expressed as mean and S.E. See Figure 1 legend for the description of the GT uptake unit. *P <0.05, as compared to the vehicle control.
Effect of AACOCF3 and diclofenac on thermal hyperalgesia
Thermal hyperalgesia developed over time on the hindpaw ipsilateral to CCI as compared to the sham control. While sham operation did not change the baseline thermal nociceptive response over time (Fig. 5a, ANOVA, P> 0.05, n=6), the ipsilateral hindpaw withdrawal latency to radiant heat was progressively reduced in CCI rats (n=8) and was reliably different from that of sham rats on postoperative day 8 (Fig. 5a, Mann-Whitney, P< 0.05). The same AACOCF3 treatment regimen mentioned above (30 μg, i.t.) attenuated the development of thermal hyperalgesia over the experimental period in CCI rats (Fig. 5a, ANOVA, P< 0.05, n=8), whereas the same diclofenac treatment (200 μg, i.t.) exacerbated the development of thermal hyperalgesia during the same experimental period after CCI (Fig. 5a, ANOVA, P< 0.05, n=8).
Fig. 5. Effect of AACOCF3 and diclofenac on the development of neuropathic pain behaviors in CCI rats.

Thermal hyperalgesia (A) and mechanical allodynia (B) developed over time on the hindpaw ipsilateral to CCI as compared to the sham group. AACOCF3 (30 μg) and diclofenac (200 μg), given intrathecally twice a day for postoperative day 1 – 7, prevented and exacerbated the development of thermal hyperalgesia (A), respectively, over the experimental period in CCI rats. AACOCF3 also prevented the development of
Effect of AACOCF3 and diclofenac on mechanical allodynia
Mechanical allodynia also developed over time on the hindpaw ipsilateral to CCI as compared to the sham control. While sham operation did not change the baseline mechanical nociceptive responses over time against the preoperative (PRE) day (Fig. 5b, ANOVA, Postoperatve day (N) 1: P=0.905; N4: P=0.730; N8: P=0.571, n=6), the ipsilateral hindpaw withdrawal threshold to von Frey filament stimulation was progressively reduced in CCI rats (n=8) beginning on postoperative day 1 and was reliably different from that of sham rats on postoperative day 8 (Fig. 5b, Mann-Whitney, P< 0.05). The AACOCF3 treatment regimen mentioned above (30 μg, i.t.) effectively attenuated the development of mechanical allodynia over the experimental period in CCI rats (Fig. 5b, ANOVA, P< 0.05, n=8), whereas the diclofenac treatment (200 μg, i.t.) had a minimal effect on mechanical allodynia after CCI (Fig. 5b, n=8).
Discussion
The present data demonstrate that 1) CCI significantly reduced spinal GT uptake activity with an associated increase in spinal AA and glutamate concentration when examined on postoperative day 8; 2) reduction of spinal AA production by AACOCF3 (a cytosolic PLA2 inhibitor) prevented the reduced spinal GT uptake activity and increased extracellular glutamate concentration in CCI rats; 3) alteration of spinal AA metabolism by diclofenac (a COX-1/2 inhibitor) further reduced spinal GT uptake activity and increased extracellular glutamate concentration in CCI rats; 4) exogenous AA directly inhibited in vitro GT uptake activity in naïve rats; and 5) consistent with these findings, AACOCF3 attenuated the development of both thermal hyperalgesia and mechanical allodynia, whereas diclofenac exacerbated thermal hyperalgesia, in CCI rats. Thus, spinal AA turnover may serve as a regulator in CCI-induced changes in regional GT uptake activity, glutamate homeostasis, and neuropathic pain behaviors.
Several methodological issues should be considered with regard to the data interpretation. In the present study, we selected the samples on postoperative day 8 for assays because the GT downregulation was stable at postoperative day 8, which was similar to that on postoperative day 14 (Sung et al. 2003). There were also no differences in pain behaviors between postoperative days 8 and 14. Thus, the data analysis was focused on the ipsilateral spinal cord dorsal horn on postoperative day 8 following CCI.
In this study, two critical sites of the AA metabolic pathway (PLA2 and COX-1/2) were regulated by AACOCF3 and diclofenac, respectively. However, changes of spinal glutamate homeostasis through regulation of AA metabolism in an in vivo system could be influenced by a number of factors other than AA itself. For instance, changes in spinal glutamate concentration in microdialysates would be a net result of glutamate release and reuptake and the former can be mediated at least in part through spinal prostaglandins that are clearly regulated by both PLA2 and COX-1/2 activity. We consider that the present findings primarily reflect the effect of AA on spinal GT uptake activity for several reasons: 1) there was an overall correlation between changes in AA production, spinal GT uptake activity, and regional glutamate concentration in CCI rats treated with either a vehicle, AACOCF3, or diclofenac; 2) AACOCF3 resulted in the decreased regional glutamate concentration associated with an increased spinal GT uptake activity; 3) diclofenac, which decreases the production of prostaglandins, dramatically increased regional glutamate concentration in association with a worsening reduction of spinal GT uptake activity in CCI rats; and 4) exogenous AA directly inhibited spinal GT uptake activity in an in vitro 3H-glutamate uptake assay, supporting a specific inhibitory effect of AA on spinal GT uptake activity. The AA concentrations used in the in vitro experiment were chosen based on the AA concentration used for the intracerebrovascular or intrathecal injections (Okubo et al. 1984; Okuyama et al. 1985; Lucas et al. 2005). Besides using exogenous AA, the effect of endogenous AA on the GT uptake activity from spinal cord samples was indirectly assessed in our experiments using agents that modulate the production of endogenous AA.
Cytosolic PLA2 (cPLA2) is expressed within the spinal cord dorsal horn (Mabuchi et al. 2004; Lucas et al. 2005) and the main active enzyme in the production of AA, contributing to the development of inflammatory pain (Bazan 1998; Gil et al. 2002; Pompeia et al. 2003). Kuwata et al. (1998) observed that cytosolic PLA2 is required for cytokine-induced expression of type IIa secretory PLA2 (sPLA2). While sPLA2 also enhanced excitatory neurotransmission in rat’s Substantia Gelatinosa (Yue et al. 2005) and inhibition of sPLA2 reduced the response to von Frey hair stimulation after the carrageenan injection (Yeo et al. 2004), it has been suggested that certain metabolites produced by the cPLA2-dependent pathway are essential for the subsequent induction of type II sPLA inhibitor (Morioka et al. 2002) that there is a functional crosstalk between cPLA2 and sPLA2 enzymes. In this study, AACOCF3 was used as a PLA2 inhibitor because it selectively blocks the activity of cPLA2 (Lucas et al. 2005).
AA has been shown to contribute to neuronal toxicity through the production of free radicals as well as to inflammatory pain via its metabolic products including prostaglandins at both peripheral and central sites (Malmberg and Yaksh 1992;Katsuki and Okuda 1995;Samad et al. 2001;Farooqui et al. 2004). In addition, previous studies have shown that AA inhibits both in vitro and in vivo GT uptake thereby enhancing glutamate availability in the synaptic space (Volterra et al. 1992;Trotti et al. 1995;Zerangue et al. 1995;Manzoni and Mennini 1997;Breukel et al. 1997;Kawahara et al. 2001;Binns et al. 2005), which is consistent with our result on the in vitro GT uptake activity using exogenous AA. Our findings indicate that spinal AA played an important role in regulating spinal GT uptake activity, regional glutamate homeostasis and neuropathic pain behaviors after peripheral nerve injury. This is supported by an increased spinal AA production following CCI through the PLA2 activity because AACOCF3 substantially reduced spinal AA production and increased GT uptake activity in CCI rats. This is consistent with a previous report (Lucas et al. 2005) that the cPLA2 inhibitor AACOCF3 administered intrathecally prevented thermal hyperalgesia induced by peripheral inflammation.
It should also be noted that glutamate itself may stimulate AA production through glutamate receptor-mediated changes in cytosolic Ca++ concentration (Oomagari et al. 1991;Zerangue et al. 1995;White et al. 2000). However, since AACOCF3 inhibited spinal AA production with a resultant increase in GT uptake activity and a decrease in regional glutamate concentration in CCI rats, this glutamate-regulated feedback mechanism of AA metabolism, if present in an in vivo setting, is unlikely to have made a substantial contribution to the outcome in our experiment.
Prostaglandins administered intrathecally have shown to result in mechanical allodynia mediated through glutamate receptors (Minami et al. 1994;Minami et al. 2001). Prostaglandins also contribute to the development and maintenance of neuropathic pain behaviors, which is mediated at least in part through prostaglandin-regulated glutamate release from primary afferents (Syriatowicz et al. 1999;Ma et al. 2002; De et al. 2003;Hefferan et al. 2003;Zhu and Eisenach 2003;Broom et al. 2004;Chiechio et al. 2004;Hefferan and Loomis 2004;Muja and DeVries 2004;Schafers et al. 2004). The present data indicate that inhibition of COX-1/2 with diclofenac, a potent water-soluble agent and clinically available non-steroid anti-inflammatory drug (Scholer et al. 1986;Kirchheiner et al. 2003), played a significant role in the mechanisms of neuropathic pain. Diclofenac is likely to modulate the AA metabolism, because diclofenac did increase regional glutamate concentration by several folds with an associated decrease in spinal GT uptake activity in CCI rats. Should the observed change in regional glutamate concentration be due to an altered glutamate release through the effect of prostaglandins, there would have been a decrease in regional glutamate concentration in the spinal microdialysates from CCI rats treated with diclofenac. Thus, the data from diclofenac-treated CCI rats is in agreement with the previous in vitro data indicating that it is the AA but not prostaglandins that regulates GT uptake activity (Danbolt 2001).
Of interest to note is that spinal AA concentration from CCI rats treated with diclofenac was not statistically different from CCI rats receiving a vehicle treatment. That is, diclofenac did not further increase regional AA concentration although diclofenac substantially increased regional glutamate concentration and decreased spinal GT uptake activity. This observation is likely due to the fact that AA is a substrate of multiple metabolic pathways including those mediated by lipooxygenase. In addition, our in vitro assay indicated that the inhibitory effect of AA on GT uptake activity was saturated at a dose range close to the physiological AA concentration (Rychkov et al. 2005). Thus, it is possible that alteration of the AA metabolism through inhibition of its major metabolic pathway (COX-1/2) by diclofenac enhanced the inhibitory effect of AA on spinal GT uptake activity, leading to an overall increase in glutamate concentration in spinal microdialysates from CCI rats.
Our previous study has shown that the expression of spinal glial and neuronal GT after peripheral nerve injury was regulated through the cellular mechanisms involving mitogen-activated protein kinases (Sung et al. 2003). The present findings indicate that modulation of GT uptake activity itself through an AA-mediated endogenous regulatory mechanism may also play a crucial role in the mechanisms of neuropathic pain behaviors in rats. Consistently, AACOCF3 and diclofenac prevented and exacerbated the development of thermal hyperalgesia, respectively. AACOCF3 also prevented the development of mechanical allodynia in CCI rats, whereas diclofenac at the present dose moderately reduced mechanical allodynia. The differential effects of diclofenac on the mechanical allodynia and the thermal hyperalgesia may be explained by separate pathways to develop the allodynia and the hyperalgesia (Ossipov et al. 2000). Nonetheless, the present findings suggest that regulating spinal AA turnover may be a useful approach to improving the medical management of neuropathic pain.
Acknowledgments
This work was supported by US PHS RO1 grants NS42661 and NS45681. We wish to thank Drs. Tony Yaksh and Lee Koetzner from the University of California at San Diego for their technical assistance in microdialysis and Dr. Ashok Khatri from the Massachusetts General Hospital for his generous help in HPLC assay.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- al Ghoul WM, Volsi GL, Weinberg RJ, Rustioni A. Glutamate immunocytochemistry in the dorsal horn after injury or stimulation of the sciatic nerve of rats. Brain Res Bull. 1993;30:453–459. doi: 10.1016/0361-9230(93)90278-j. [DOI] [PubMed] [Google Scholar]
- Azbill RD, Mu X, Springer JE. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res. 2000;871:175–180. doi: 10.1016/s0006-8993(00)02430-6. [DOI] [PubMed] [Google Scholar]
- Bazan NG. Eicosanoids, Platelet-Activating Factor and Inflammation. In: Siegel George J MD, Fisher Stephen K PhD, Albers R Wayne PhD, Agranoff Bernard W MD, Uhler Michael D PhD, editors. Basic Neurochemistry. Lippincott Williams & Wilkins; 1998. [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Bidlingmeyer BA, Cohen SA, Tarvin TL. Rapid analysis of amino acids using pre-column derivatization. J Chromatogr. 1984;336:93–104. doi: 10.1016/s0378-4347(00)85133-6. [DOI] [PubMed] [Google Scholar]
- Binns BC, Huang Y, Goettl VM, Hackshaw KV, Stephens RL., Jr Glutamate uptake is attenuated in spinal deep dorsal and ventral horn in the rat spinal nerve ligation model. Brain Res. 2005;1041:38–47. doi: 10.1016/j.brainres.2005.01.088. [DOI] [PubMed] [Google Scholar]
- Breukel AI, Besselsen E, Lopes da Silva FH, Ghijsen WE. Arachidonic acid inhibits uptake of amino acids and potentiates PKC effects on glutamate, but not GABA, exocytosis in isolated hippocampal nerve terminals. Brain Res. 1997;773:90–97. doi: 10.1016/s0006-8993(97)00918-9. [DOI] [PubMed] [Google Scholar]
- Broom DC, Samad TA, Kohno T, Tegeder I, Geisslinger G, Woolf CJ. Cyclooxygenase 2 expression in the spared nerve injury model of neuropathic pain. Neuroscience. 2004;124:891–900. doi: 10.1016/j.neuroscience.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Chiechio S, Copani A, Melchiorri D, Canudas AM, Storto M, Calvani M, Nicolai R, Nicoletti F. Metabotropic receptors as targets for drugs of potential use in the treatment of neuropathic pain. J Endocrinol Invest. 2004;27:171–176. [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- De S, Trigueros MA, Kalyvas A, David S. Phospholipase A2 plays an important role in myelin breakdown and phagocytosis during Wallerian degeneration. Mol Cell Neurosci. 2003;24:753–765. doi: 10.1016/s1044-7431(03)00241-0. [DOI] [PubMed] [Google Scholar]
- Devor M. Nerve pathophysiology and mechanisms of pain in causalgia. J Auton Nerv Syst. 1983;7:371–384. doi: 10.1016/0165-1838(83)90090-5. [DOI] [PubMed] [Google Scholar]
- Dorandeu F, Antier D, Pernot-Marino I, Lapeyre P, Lallement G. Venom phospholipase A2-induced impairment of glutamate uptake: an indirect and nonselective effect related to phospholipid hydrolysis. J Neurosci Res. 1998;51:349–359. doi: 10.1002/(SICI)1097-4547(19980201)51:3<349::AID-JNR8>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Horrocks LA. Brain phospholipases A2: a perspective on the history. Prostaglandins Leukot Essent Fatty Acids. 2004;71:161–169. doi: 10.1016/j.plefa.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Ong WY, Horrocks LA. Biochemical aspects of neurodegeneration in human brain: involvement of neural membrane phospholipids and phospholipases A2. Neurochem Res. 2004;29:1961–1977. doi: 10.1007/s11064-004-6871-3. [DOI] [PubMed] [Google Scholar]
- Gil A. Polyunsaturated fatty acids and inflammatory diseases. Biomed Pharmacother. 2002;56:388–396. doi: 10.1016/s0753-3322(02)00256-1. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hefferan MP, Carter P, Haley M, Loomis CW. Spinal nerve injury activates prostaglandin synthesis in the spinal cord that contributes to early maintenance of tactile allodynia. Pain. 2003;101:139–147. doi: 10.1016/s0304-3959(02)00322-6. [DOI] [PubMed] [Google Scholar]
- Hefferan MP, Loomis CW. Interaction of spinal nitric oxide and prostaglandins after L5-L6 spinal nerve ligation in the rat: an isobolographic analysis. Anesthesiology. 2004;100:1611–1614. doi: 10.1097/00000542-200406000-00040. [DOI] [PubMed] [Google Scholar]
- Hua XY, Chen P, Marsala M, Yaksh TL. Intrathecal substance P-induced thermal hyperalgesia and spinal release of prostaglandin E2 and amino acids. Neuroscience. 1999;89:525–534. doi: 10.1016/s0306-4522(98)00488-6. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Hediger MA. The glutamate and neutral amino acid transporter family: physiological and pharmacological implications. Eur J Pharmacol. 2003;479:237–247. doi: 10.1016/j.ejphar.2003.08.073. [DOI] [PubMed] [Google Scholar]
- Katsuki H, Okuda S. Arachidonic acid as a neurotoxic and neurotrophic substance. Prog Neurobiol. 1995;46:607–636. doi: 10.1016/0301-0082(95)00016-o. [DOI] [PubMed] [Google Scholar]
- Kawahara H, Sakamoto A, Takeda S, Onodera H, Imaki J, Ogawa R. A prostaglandin E2 receptor subtype EP1 receptor antagonist (ONO-8711) reduces hyperalgesia, allodynia, and c-fos gene expression in rats with chronic nerve constriction. Anesth Analg. 2001;93:1012–1017. doi: 10.1097/00000539-200110000-00043. [DOI] [PubMed] [Google Scholar]
- Kirchheiner J, Meineke I, Steinbach N, Meisel C, Roots I, Brockmoller J. Pharmacokinetics of diclofenac and inhibition of cyclooxygenases 1 and 2: no relationship to the CYP2C9 genetic polymorphism in humans. Br J Clin Pharmacol. 2003;55:51–61. doi: 10.1046/j.1365-2125.2003.01712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwata H, Nakatani Y, Murakami M, Kudo I. Cytosolic phospholipase A2 is required for cytokine-induced expression of type IIA secretory phospholipase A2 that mediates optimal cyclooxygenase-2-dependent delayed prostaglandin E2 generation in rat 3Y1 fibroblasts. J Biol Chem. 1998;273:1733–1740. doi: 10.1074/jbc.273.3.1733. [DOI] [PubMed] [Google Scholar]
- Lievens JC, Bernal F, Forni C, Mahy N, Kerkerian-Le Goff L. Characterization of striatal lesions produced by glutamate uptake alteration: cell death, reactive gliosis, and changes in GLT1 and GADD45 mRNA expression. Glia. 2000;29:222–232. doi: 10.1002/(sici)1098-1136(20000201)29:3<222::aid-glia4>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Lucas KK, Svensson CI, Hua XY, Yaksh TL, Dennis EA. Spinal phospholipase A(2) in inflammatory hyperalgesia: role of Group IVA cPLA(2) Br J Pharmacol. 2005;144:940–952. doi: 10.1038/sj.bjp.0706116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Du W, Eisenach JC. Role for both spinal cord COX-1 and COX-2 in maintenance of mechanical hypersensitivity following peripheral nerve injury. Brain Res. 2002;937:94–99. doi: 10.1016/s0006-8993(02)02593-3. [DOI] [PubMed] [Google Scholar]
- Mabuchi T, Kojima H, Abe T, Takagi K, Sakurai M, Ohmiya Y, Uematsu S, Akira S, Watanabe K, Ito S. Membrane-associated prostaglandin E synthase-1 is required for neuropathic pain. Neuroreport. 2004;15:1395–1398. doi: 10.1097/01.wnr.0000129372.89000.31. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Yaksh TL. Hyperalgesia mediated by spinal glutamate or substance P receptor blocked by spinal cyclooxygenase inhibition. Science. 1992;257:1276–1279. doi: 10.1126/science.1381521. [DOI] [PubMed] [Google Scholar]
- Manzoni C, Mennini T. Arachidonic acid inhibits 3H-glutamate uptake with different potencies in rodent central nervous system regions expressing different transporter subtypes. Pharmacol Res. 1997;35:149–151. doi: 10.1006/phrs.1997.0129. [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Hayes RL, Lu J, Mayer DJ. Differential roles of NMDA and non-NMDA receptor activation in induction and maintenance of thermal hyperalgesia in rats with painful peripheral mononeuropathy. Brain Res. 1992a;598:271–278. doi: 10.1016/0006-8993(92)90193-d. [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Hayes RL, Lu J, Mayer DJ. Intrathecal GM1 ganglioside and local nerve anesthesia reduce nociceptive behaviors in rats with experimental peripheral mononeuropathy. Brain Res. 1992b;584:28–53. doi: 10.1016/0006-8993(92)90874-9. [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ. Mechanisms of hyperalgesia and morphine tolerance: a current view of their possible interactions. Pain. 1995;62:259–274. doi: 10.1016/0304-3959(95)00073-2. [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Zhu J, Lu J, Mayer DJ. The inhibition of nitric oxide-activated poly(ADP-ribose) synthetase attenuates transsynaptic alteration of spinal cord dorsal horn neurons and neuropathic pain in the rat. Pain. 1997;72:355–366. doi: 10.1016/s0304-3959(97)00063-8. [DOI] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD. Glutamate transporters: animal models to neurologic disease. Neurobiol Dis. 2004;15:461–473. doi: 10.1016/j.nbd.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Minami T, Matsumura S, Okuda-Ashitaka E, Shimamoto K, Sakimura K, Mishina M, Mori H, Ito S. Characterization of the glutamatergic system for induction and maintenance of allodynia. Brain Res. 2001;895:178–185. doi: 10.1016/s0006-8993(01)02069-8. [DOI] [PubMed] [Google Scholar]
- Minami T, Nishihara I, Uda R, Ito S, Hyodo M, Hayaishi O. Involvement of glutamate receptors in allodynia induced by prostaglandins E2 and F2 alpha injected into conscious mice. Pain. 1994;57:225–231. doi: 10.1016/0304-3959(94)90227-5. [DOI] [PubMed] [Google Scholar]
- Mitrovic AD, Maddison JE, Johnston GA. Influence of the oestrous cycle on L-glutamate and L-aspartate transport in rat brain synaptosomes. Neurochem Int. 1999;34:101–108. doi: 10.1016/s0197-0186(98)00066-7. [DOI] [PubMed] [Google Scholar]
- Molnar-Perl I. Advances in the high-performance liquid chromatographic determination of phenylthiocarbamyl amino acids. Journal of Chromatography A. 1994;661:43–50. [Google Scholar]
- Morioka N, Takeda K, Kumagai K, Hanada T, Ikoma K, Hide I, Inoue A, Nakata Y. Interleukin-1beta-induced substance P release from rat cultured primary afferent neurons driven by two phospholipase A2 enzymes: secretory type IIA and cytosolic type IV. J Neurochem. 2002;80:989–997. doi: 10.1046/j.0022-3042.2002.00722.x. [DOI] [PubMed] [Google Scholar]
- Mu X, Azbill RD, Springer JE. Riluzole improves measures of oxidative stress following traumatic spinal cord injury. Brain Res. 2000;870:66–72. doi: 10.1016/s0006-8993(00)02402-1. [DOI] [PubMed] [Google Scholar]
- Muja N, DeVries GH. Prostaglandin E(2) and 6-keto-prostaglandin F(1alpha) production is elevated following traumatic injury to sciatic nerve. Glia. 2004;46:116–129. doi: 10.1002/glia.10349. [DOI] [PubMed] [Google Scholar]
- Ohkubo K, Suzuki K, Otorii T. [Pressor and depressor responses to intracerebroventricularly administered prostaglandins and arachidonic acid in anaesthetized rabbits] Nippon Yakurigaku Zasshi. 1984;84:327–335. [PubMed] [Google Scholar]
- Okuyama S, Aihara H. Hyperalgesic action in rats of intracerebroventricularly administered arachidonic acid, PG E2 and PG F2 alpha: effects of analgesic drugs on hyperalgesia. Arch Int Pharmacodyn Ther. 1985;278:13–22. [PubMed] [Google Scholar]
- Oomagari K, Buisson B, Dumuis A, Bockaert J, Pin JP. Effect of Glutamate and Ionomycin on the Release of Arachidonic Acid, Prostaglandins and HETEs from Cultured Neurons and Astrocytes. Eur J Neurosci. 1991;3:928–939. doi: 10.1111/j.1460-9568.1991.tb00028.x. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, Malan TP, Jr, Porreca F. Spinal and supraspinal mechanisms of neuropathic pain. Ann N Y Acad Sci. 2000;909:12–24. doi: 10.1111/j.1749-6632.2000.tb06673.x. [DOI] [PubMed] [Google Scholar]
- Pinardi G, Sierralta F, Miranda HF. Atropine reverses the antinociception of nonsteroidal anti-inflammatory drugs in the tail-flick test of mice. Pharmacol Biochem Behav. 2003;74(3):603–608. doi: 10.1016/s0091-3057(02)01046-8. [DOI] [PubMed] [Google Scholar]
- Pompeia C, Lima T, Curi R. Arachidonic acid cytotoxicity: can arachidonic acid be a physiological mediator of cell death? Cell Biochem Funct. 2003;21:97–104. doi: 10.1002/cbf.1012. [DOI] [PubMed] [Google Scholar]
- Pound EM, Kang JX, Leaf A. Partitioning of polyunsaturated fatty acids, which prevent cardiac arrhythmias, into phospholipid cell membranes. J Lipid Res. 2001;42:346–351. [PubMed] [Google Scholar]
- Raja SN, Meyer RA, Campbell JN. Peripheral mechanisms of somatic pain. Anesthesiology. 1988;68:571–590. doi: 10.1097/00000542-198804000-00016. [DOI] [PubMed] [Google Scholar]
- Robinson MB, Dowd LA. Heterogeneity and functional properties of subtypes of sodium-dependent glutamate transporters in the mammalian central nervous system. Adv Pharmacol. 1997;37:69–115. doi: 10.1016/s1054-3589(08)60948-5. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes HM, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Rychkov GY, Litjens T, Roberts ML, Barritt GJ. Arachidonic acid inhibits the store-operated Ca2+ current in rat liver cells. Biochem J. 2005;385:551–556. doi: 10.1042/BJ20041604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- Sawin PD, Traynelis VC, Rich G, Smith BA, Maves TJ, Follett KA, Moore SA. Chymopapain-induced reduction of proinflammatory phospholipase A2 activity and amelioration of neuropathic behavioral changes in an in vivo model of acute sciatica. J Neurosurg. 1997;86:998–1006. doi: 10.3171/jns.1997.86.6.0998. [DOI] [PubMed] [Google Scholar]
- Schafers M, Marziniak M, Sorkin LS, Yaksh TL, Sommer C. Cyclooxygenase inhibition in nerve-injury- and TNF-induced hyperalgesia in the rat. Exp Neurol. 2004;185:160–168. doi: 10.1016/j.expneurol.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Scholer DW, Ku EC, Boettcher I, Schweizer A. Pharmacology of diclofenac sodium. Am J Med. 1986;80:34–38. doi: 10.1016/0002-9343(86)90077-x. [DOI] [PubMed] [Google Scholar]
- Semba J, Wakuta MS. Regional differences in the effects of glutamate uptake inhibitor L-trans-pyrrolidine-2,4-dicarboxylic acid on extracellular amino acids and dopamine in rat brain: an in vivo microdialysis study. Gen Pharmacol. 1998;31:399–404. doi: 10.1016/s0306-3623(98)00047-0. [DOI] [PubMed] [Google Scholar]
- Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci. 2003;23:2899–2910. doi: 10.1523/JNEUROSCI.23-07-02899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syriatowicz JP, Hu D, Walker JS, Tracey DJ. Hyperalgesia due to nerve injury: role of prostaglandins. Neuroscience. 1999;94:587–594. doi: 10.1016/s0306-4522(99)00365-6. [DOI] [PubMed] [Google Scholar]
- Tal M, Bennett GJ. Extra-territorial pain in rats with a peripheral mononeuropathy: mechano-hyperalgesia and mechano-allodynia in the territory of an uninjured nerve. Pain. 1994;57:375–382. doi: 10.1016/0304-3959(94)90013-2. [DOI] [PubMed] [Google Scholar]
- Trotti D, Aoki M, Pasinelli P, Berger UV, Danbolt NC, Brown RH, Jr, Hediger MA. Amyotrophic lateral sclerosis-linked glutamate transporter mutant has impaired glutamate clearance capacity. J Biol Chem. 2001;276:576–582. doi: 10.1074/jbc.M003779200. [DOI] [PubMed] [Google Scholar]
- Trotti D, Volterra A, Lehre KP, Rossi D, Gjesdal O, Racagni G, Danbolt NC. Arachidonic acid inhibits a purified and reconstituted glutamate transporter directly from the water phase and not via the phospholipid membrane. J Biol Chem. 1995;270:9890–9895. doi: 10.1074/jbc.270.17.9890. [DOI] [PubMed] [Google Scholar]
- Urban MO, Hama AT, Bradbury M, Anderson J, Varney MA, Bristow L. Role of metabotropic glutamate receptor subtype 5 (mGluR5) in the maintenance of cold hypersensitivity following a peripheral mononeuropathy in the rat. Neuropharmacology. 2003;44:983–993. doi: 10.1016/s0028-3908(03)00118-7. [DOI] [PubMed] [Google Scholar]
- Volterra A, Trotti D, Cassutti P, Tromba C, Galimberti R, Lecchi P, Racagni G. A role for the arachidonic acid cascade in fast synaptic modulation: ion channels and transmitter uptake systems as target proteins. Adv Exp Med Biol. 1992;318:147–158. doi: 10.1007/978-1-4615-3426-6_13. [DOI] [PubMed] [Google Scholar]
- Volterra A, Trotti D, Racagni G. Glutamate uptake is inhibited by arachidonic acid and oxygen radicals via two distinct and additive mechanisms. Mol Pharmacol. 1994;46:986–992. [PubMed] [Google Scholar]
- Wall PD, Waxman S, Basbaum AI. Ongoing activity in peripheral nerve: injury discharge. Exp Neurol. 1974;45:576–589. doi: 10.1016/0014-4886(74)90163-0. [DOI] [PubMed] [Google Scholar]
- White BC, Sullivan JM, DeGracia DJ, O’Neil BJ, Neumar RW, Grossman LI, Rafols JA, Krause GS. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci. 2000;179:1–33. doi: 10.1016/s0022-510x(00)00386-5. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999;353:1959–1964. doi: 10.1016/S0140-6736(99)01307-0. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Yaksh TL. Spinal pharmacology of thermal hyperesthesia induced by constriction injury of sciatic nerve. Excitatory amino acid antagonists. Pain. 1992;49:121–128. doi: 10.1016/0304-3959(92)90198-K. [DOI] [PubMed] [Google Scholar]
- Yeo JF, Ong WY, Ling SF, Farooqui AA. Intracerebroventricular injection of phospholipases A2 inhibitors modulates allodynia after facial carrageenan injection in mice. Pain. 2004;112:148–155. doi: 10.1016/j.pain.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Yue HY, Fujita T, Kumamoto E. Phospholipase A2 activation by melittin enhances spontaneous glutamatergic excitatory transmission in rat substantia gelatinosa neurons. Neuroscience. 2005;135:485–495. doi: 10.1016/j.neuroscience.2005.05.040. [DOI] [PubMed] [Google Scholar]
- Zerangue N, Arriza JL, Amara SG, Kavanaugh MP. Differential modulation of human glutamate transporter subtypes by arachidonic acid. J Biol Chem. 1995;270:6433–6435. doi: 10.1074/jbc.270.12.6433. [DOI] [PubMed] [Google Scholar]
- Zhu X, Eisenach JC. Cyclooxygenase-1 in the spinal cord is altered after peripheral nerve injury. Anesthesiology. 2003;99:1175–1179. doi: 10.1097/00000542-200311000-00026. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol. 2001;429:23–37. doi: 10.1016/s0014-2999(01)01303-6. [DOI] [PubMed] [Google Scholar]