

Abstract
Following trauma to the brain significant changes occur in both the astroglial and vascular components of the neuropil. Angiogenesis is required to re-establish metabolic support and astrocyte activation encompasses several functions including scar formation and the production of growth factors. VEGF has seminal involvement in the process of brain repair and is upregulated during many pathological events. VEGF signaling is regulated mainly through its two primary receptors: flk-1 (KDR/VEGF-R2) is expressed on vascular endothelium and some neurons and flt-1 (VEGF-R1) in the CNS, is expressed predominantly by activated astrocytes. Using an injury model of chronic minipump infusion of neutralizing antibodies (NA) to block VEGF receptor signaling, this study takes advantage of these differences in VEGF receptor distribution in order to understand the role the cytokine plays after brain injury. Infusion of NA to flk-1 caused a significant decrease in vascular proliferation and increased endothelial cell degeneration compared to control IgG infusions but had no effect on astrogliosis. By contrast infusion of NA to flt-1 significantly decreased astroglial mitogenicity and scar formation and caused some increase in endothelial degeneration. Neutralization of the flt-1 receptor function, but not flk-1, caused significant reduction in the astroglial expression of the growth factors, CNTF and FGF by seven days. These data suggest that after CNS injury, endogenous VEGF upregulation (by astrocytes) induces angiogenesis and, by autocrine signaling, increases both astrocyte proliferation and facilitates expression of growth factors. It is likely that VEGF plays an important role in aspects of astroglial scar formation.
Keywords: Astrocytes, gliosis, angiogenesis, VEGF, CNTF, bFGF, nestin
Introduction
The CNS responds immediately to trauma by triggering a series of cellular events that are designed to repair the vasculature and wall off the injured area (Fitch and Silver 1997; Fawcett and Asher 1999; McGraw et al. 2001). Revascularization and repair of the blood-brain barrier reestablish metabolic and trophic support to the injured tissue (de Paermentier et al. 1986; Krum and Rosenstein 1988; Orita et al. 1989). Activated astrocytes form a gliotic scar to provide structural support, and isolate the injury site by reestablishing the glia limitans, thus restricting the migration of inflammatory cells into healthy tissue (Stichel and Müller 1998; Sofroniew 2005). Such reactive gliosis is characterized by astroglial proliferation, hypertrophy, process extension, and increased synthesis of intermediate filaments, including GFAP and vimentin, as well as production of many bioactive molecules (Eddleston and Mucke 1993; Silver and Miller 2004). Astroglial reactivity is stimulated by a number of factors, including thrombin, bFGF, CNTF, TGFα, TGFβ1, TGFβ2, TNFα, and IL-1 (Fawcett 1997; Campbell 2001; Swartz et al. 2001), several of which are microglial-derived cytokines (Liberto et al. 2004), and by trauma-induced release of purines such as ATP (Neary and Kang 2005).
One of the cytokines with seminal involvement in the process of brain repair is vascular endothelial growth factor (VEGF), which is upregulated during many pathological events in the CNS, including ischemia (Kovacs et al. 1996; Cobbs et al. 1998; Lennmyr et al. 1998; Issa et al. 1999; Lee et al. 1999; Pichiule 1999; Plate et al. 1999; Jin et al. 2000), cold lesions (Nag et al. 1997; Papavassiliou et al. 1997), spinal cord injuries (Bartholdi et al. 1997; Tsao et al. 1999; Vaquero et al. 1999), brain contusion (Skold et al. 2005) and direct wounding (Krum and Rosenstein 1998, 1999). VEGF is a secreted dimeric protein that is fundamentally important for angiogenesis during development, tissue regeneration/repair and tumor growth throughout the body (Leung et al. 1989; Klagsbrun and D’Amore 1996; Thomas 1996; Ferrara et al. 2003). The major isoform present in most mammalian tissues is VEGF165, a 45 kDa heparin binding, secreted glycoprotein (Ferrara et al. 2003). VEGF binds to the tyrosine kinase receptors flt-1(VEGF-R1) and (predominantly) flk-1 (KDR/VEGF-R2) that are expressed by vascular endothelial cells and triggers the mitotic and migratory processes necessary for angiogenesis in the periphery (Shibuya and Claesson-Welsh 2006). VEGF is also a vascular maintenance factor that promotes endothelial cell survival by acting through the flk-1 receptor (Yang, W. and de Bono 1997; Darland et al. 2003; Huang et al. 2003). In the intact adult CNS, VEGF protein expression is limited to the choroid plexus, area postrema, and cerebellar granule cells (Monacci et al. 1993), and VEGF receptors are normally expressed at very low levels (Peters et al. 1993; Kremer et al. 1997; Soker et al. 1998).
In the injured CNS, VEGF is implicated in post-traumatic angiogenesis, which is dependent upon the upregulation of endothelial flk-1 (Krum and Rosenstein 1998; Rosenstein et al. 1998; Issa et al. 1999; Krum and Rosenstein 1999; Plate et al. 1999; Proescholdt et al. 1999; Silverman et al. 1999; Jin et al. 2000; Beck et al. 2002; Harrigan et al. 2002; Krum et al. 2002; Mani et al. 2003; Croll et al. 2004; Skold et al. 2005). Concomitantly, VEGF protein is also strongly upregulated in astroglia and inflammatory cells near the damaged area (Bartholdi et al. 1997; Nag et al. 1997; Papavassiliou et al. 1997; Krum and Rosenstein 1998, 1999; Tsao et al. 1999; Vaquero et al. 1999; Salhia et al. 2000; Chodobski et al. 2003; Skold et al. 2005); in ischemia models, neurons have also been reported to express VEGF (Kovacs et al. 1996; Cobbs et al. 1998; Lennmyr et al. 1998; Issa et al. 1999; Lee et al. 1999; Pichiule 1999; Jin et al. 2000).
After traumatic insults, the primary VEGF receptors present a particularly interesting dichotomy in their cellular distribution. The flk-1 receptor is upregulated in neurons, while flt-1 is upregulated almost exclusively in reactive astrocytes (Krum and Rosenstein 1998, 1999; Lennmyr et al. 1998; Krum and Khaibullina 2003; Krum and Rosenstein 2004). Although some studies, using different experimental paradigms, have indicated endothelial cells upregulate flt-1 as well (Widenfalk et al. 2003; Skold et al. 2000), we have not observed unequivocal flt-1 expression in vascular endothelial cells adjacent to the wound site in our model; however, we find strong flt-1 expression in the astroglial endfeet that are closely applied to the endothelium (Krum and Rosenstein 1999; Krum et al. 2002). Several recent studies have demonstrated VEGF’s trophic and protective effects on neurons both in vivo and in vitro (Silverman et al. 1999; Jin et al. 2000, 2001; Matsuzaki et al. 2001; Lambrechts et al. 2003; Rosenstein et al. 2003; Azzouz et al. 2004; Khaibullina et al. 2004). It is now apparent, however, that astrocytes also respond to applied VEGF in vivo and in vitro (Krum et al. 2002; Khaibullina et al. 2004), and it has been recently demonstrated that endogenous VEGF is an important factor for stimulation of astroglial mitosis after brain injury (Krum and Khaibullina 2003).
Using our injury model of osmotic minipump infusion (Krum and Khaibullina 2003), the present study addresses specifically whether endogenous VEGF, acting via the flt-1 receptor, regulates astroglial proliferation and survival. Since the flt-1 receptor is strongly up-regulated in reactive astroglia and is negligible in blood vessels after a penetrating injury to the cerebrum (Krum and Rosenstein 1998, 1999; Krum et al. 2002; Krum and Khaibullina 2003), would its blockade by a specific neutralizing antibody abrogate astrocytic activation, survival and mitogenic responses without decreasing the angiogenic response? Conversely, would inhibition of endothelial flk-1 receptor activity using the same methodology cause a specific decrease in angiogenesis and endothelial survival, while sparing the astroglial response? The experimental mitigation of such responses in these experiments may provide important clues in understanding the relative contributions of reactive gliosis and revascularization to the overall process of CNS wound repair. This investigation takes advantage of the difference in VEGF receptor expression between astroglia and vascular endothelial cells in order to understand how VEGF regulates wound repair with regard to the relative mitotic activities and survival of these cell types.
The results show that inhibition of the flk-1 receptor caused significant decreases in vascular endothelial proliferation around the wound site with concomitant increases in vascular degeneration compared to IgG control infusions, yet there were no significant effects on astroglial maintenance or growth factor expression. In contrast, inhibition of the flt-1 signaling decreased astroglial participation at the injury site with respect to mitogenicity and scar formation. These results confirm that the differential VEGF receptor expression observed between astroglia and endothelial cells has functional importance. These data strongly suggest that, following a penetrating injury, endogenous VEGF acts through flt-1 receptors on reactive astrocytes to increase their proliferation, by autocrine means and serves to facilitate expression of the trophic factors bFGF and CNTF. Endogenous VEGF may thus play an important role in the formation and maintenance of the astroglial scar.
Materials and Methods
Minipump Implantation
Osmotic minipumps (Model 2001, Alza Corp, Palo Alto, CA; delivery rate 1 μl/hr) were filled with either 1) 20 μg/ml (delivery of 0.48 μg/day) goat anti-mouse flt-1 neutralizing antibody (flt-1-NA; R & D Systems) in artificial CSF (aCSF; Harvard Apparatus); 2) 1 μg/ml goat anti-mouse flk-1 neutralizing antibody (flk-1-NA; R & D Systems) in aCSF (delivery of 0.024 μg/day); or 3) with 20 μg/ml normal goat IgG (R & D Systems) in aCSF to provide isotypic control infusions. The antibodies are directed toward the extracellular domain of each receptor. Although there is no specific information about which of the seven immunoglobulin-like extracellular domains are targeted, the antibodies are specific in that they have very limited cross-reactivity with other VEGF receptors (e.g., flt-1-NA has 0.2% cross-reactivity with flk-1; flk-1-NA has <2% cross-reactivity with flt-1). The dosages employed for the present study are over twice what the Neutralization Dose50 (ND50) is for each neutralizing antibody, according to the manufacturer’s specifications. Neutralizing antibodies to VEGF receptors have been used previously to successfully block endogenous rodent flt-1 and flk-1 (Witte et al. 1998; Dias et al. 2001; Kunkel et al. 2001). Young adult Wistar rats were anaesthetized with ketamine/xylazine (60 mg/kg/5 mg/kg) and each received a single infusion cannula that was placed in the right striatum, as described previously (Rosenstein et al. 1998; Krum et al. 2002; Krum and Khaibullina 2003) Briefly, after a skin incision over the right side of the skull, a pocket was formed over the neck and shoulder blades to hold the minipump. A 2 mm hole was drilled in the skull 1 mm posterior to the coronal suture and 5 mm lateral to the sagittal suture. Using a stereotaxic apparatus, the cannula was inserted through the cortex and into the striatum to a final depth of 3 mm. The cannula was cemented in place and the incision sutured. Survival times were 3 and 7 days, with 6–7 animals/group used for each time period. After the appropriate time interval the animals were anaesthetized as described above and were perfused intracardially with 4% paraformaldehyde in 0.1 M cacodylate buffer with 3% sucrose. The cannulae were removed and the brains blocked for vibratome sectioning followed by processing for routine paraffin embedment. All surgical procedures were performed within the guidelines provided by and with the approval of the GWUMC Intitutional Animal Care and Use Committee.
Immunohistochemistry
Paraffin or vibratome sections were immunostained for the following antigens: cell proliferation marker, proliferating cell nuclear antigen (PCNA; monoclonal, 1:500, SIGMA); an endothelial basement membrane marker, laminin (polyclonal, 1:200, SIGMA); an astrocytic marker, glial fibrillary acidic protein (GFAP; monoclonal, 1:500, SIGMA); nestin (1:200); basic fibroblast growth factor (bFGF; 1:100; Santa Cruz); ciliary neurotrophic factor (CNTF; 1:500); TUNEL (Promega, as per kit instructions). Some of the antibodies were used to co-localize antigens to a particular cell phenotype, such as PCNA/laminin or PCNA/GFAP for proliferating endothelial cells and astroglia respectively. TUNEL was co-localized with either GFAP or laminin. TUNEL and PCNA were visualized using nickel enhancement. Controls included omission of primary antibodies and replacement of the primary antibody with normal serum from the appropriate species. None of the immunohistochemical controls exhibited specific immunoreactivity.
While most of the immunoreactions were carried out using 7 mm paraffin sections, free-floating vibratome sections (60 μm) were used for nestin, and VEGF receptor (flt-1 and flk-1) immunostaining. Paraffin sections were deparaffinized and dehydrated prior to incubation with antisera. All sections were incubated overnight at 4°C with the primary antibodies, which were diluted in 0.05 M TBS containing 1% normal serum. After washing in TBS, the sections were incubated in the appropriate dilutions of secondary antibody for 30 min, followed by exposure to peroxidase-anti-peroxidase. Visualization of the reaction product was done by using either diaminobenzidine (DAB) alone or by nickel intensification of the DAB reaction product if the sections were to be double-labeled.
For confocal fluorescence microscopy, sections incubated with primary antibodies were exposed to a solution of the appropriate fluorochrome(s) for 1 hr. at room temperature and viewed with a Bio-Rad 1024 laser confocal microscope.
Calculation of proliferation indices for vessels and astrocytes
PCNA is a reliable marker for proliferating cells particularly when only the granular, darkly stained reaction product present within nuclei is counted as an indication of proliferative activity. Results obtained with PCNA immunostaining are very similar to those obtained by 3[H]-thymidine autoradiography (Galand and Degraef 1989; Thaete et al. 1989; Hall et al. 1990; Yamada et al. 1992; Amat et al. 1996). Bromodeoxyuridine (BrdU) is also widely used as a proliferation marker, but its use was precluded in our immunohistochemical protocol. Double immunostaining for BrdU with laminin, GFAP or the other markers used was unsatisfactory due to the rigorous processing necessary for BrdU immunolocalization.
Following double-immunolabeling of sections for laminin and PCNA, the proliferation index (P.I.) for vascular profiles was calculated for controls and for each VEGF concentration, as described previously (Krum et al. 2002; Krum and Khaibullina 2003). Because the concentration of infused agents falls off as an exponential function of the distance from the infusion site (Kasamatsu et al. 1981; Lum et al. 1984), the tissue at a distance of 600 μm around the perimeter of the entire infusion track were assessed for the presence of PCNA (in the endothelial nuclei) and laminin-positive vessels. Two observers blinded to the treatments counted darkly stained PCNA(+) nuclei. It was not possible to visualize unlabeled (PCNA-negative) endothelial nuclei with these immunolabeling methods, so a true endothelial proliferation index could not be calculated. Instead, the values were averaged and expressed as P.I.s for vascular profiles (P.I.=[#PCNA(+) vessel profiles] ÷ [total # vessel profiles] × 100). Astroglia that were double-labeled for PCNA/GFAP, as well as GFAP(+) astrocytes with visible nuclei that were not immunoreactive for PCNA, were counted in six areas within 600 μm of the infusion track. Astroglial proliferation indices were calculated using P.I. = [#PCNA(+)/GFAP(+) cells] ÷ [total # GFAP(+) cells] × 100.
Quantitation of vascular and astroglial TUNEL immunoreactivity
Quantification of TUNEL-positive nuclei was performed using sections that were double-labeled for laminin/TUNEL (endothelial cells) and GFAP/TUNEL (astrocytes), as described previously (Krum and Khaibullina 2003). Similar to the measurement of vascular proliferation index, TUNEL(+) endothelial cell nuclei and laminin(+) vessels were counted at a distance of 600 μm around the perimeter of the entire infusion track by two observers blinded to the treatments. Similar to vascular PCNA counting, it was not possible to visualize unlabeled (TUNEL-negative) endothelial nuclei with these immunolabeling methods to obtain a strict measurement of endothelial degeneration. Instead, the values were averaged and expressed as a percentage ([#TUNEL(+) vessel profiles] ÷ [total # vessel profiles] × 100). GFAP(+) astroglia were also counted within 600 μm of the infusion site. Since it was possible to visualize unlabeled astroglial nuclei as well as TUNEL(+) astroglia, a true measure of astroglial degeneration was obtained and expressed as a percentage.
Quantitation of reactive astroglia
The total number of astrocytes adjacent to the wound site (within 600 μm; derived from the PCNA/GFAP and TUNEL/GFAP counts) were added together for each animal and averaged in order to determine the relative abundance of reactive astroglia resulting from each treatment.
Quantitation of bFGF(+) and CNTF(+) astrocytes
To determine the percentage of reactive astroglia that express bFGF or CNTF in response to VEGF receptor neutralization, paraffin sections were fluorescently immunostained for bFGF/GFAP or CNTF/GFAP and viewed with a Biorad confocal microscope. Using the confocal images, astroglia that were double-labeled, as well as astrocytes that were immunoreactive for GFAP alone, were counted within 600 – 700 μm of the infusion site. Two sections of the wound area from each animal, taken 100 μm apart, were counted and the percentage of double-labeled astrocytes was determined for each animal. Percentages were then averaged for each experimental and control group (n = 6 each).
Measurement of wound areas
For each animal, five sections were taken at approximately 100 μm intervals through the central portion of the infusion site. The length (from cortex to striatum) and width of the wound cavities were measured using a 4X objective in conjunction with a stage micrometer. Length and width measurements were multiplied to obtain the areas of the wounds. Wound cavity areas were averaged separately for each time point in both VEGF receptor-NA infusions and controls.
Statistics
Data were reported as mean ± standard error of mean (S.E.M.). The Students t-test for paired data and ANOVA tests with Tukey correction were used for statistical analysis. Statistical significance was set at P < 0.05.
Results
Wound area
All animals survived without any apparent complications from the infusions. Cavities were present around the site of cannula implantation, as described previously (Rosenstein et al. 1998; Krum et al. 2002; Krum and Khaibullina 2003). In general, the inflammatory cell influx into the wound areas appeared similar in all types of infusions, and by seven days a typical connective tissue down-growth from the pial surface had occurred (Logan and Berry 2002). A glia limitans, demarcated by GFAP/nestin(+) astrocytes at the edge of the cavity, was present in all infusion paradigms. Analysis of cavity sizes revealed no significant differences between control IgG infusions and either of the receptor antibody infusions (Fig. 1).
Fig. 1.
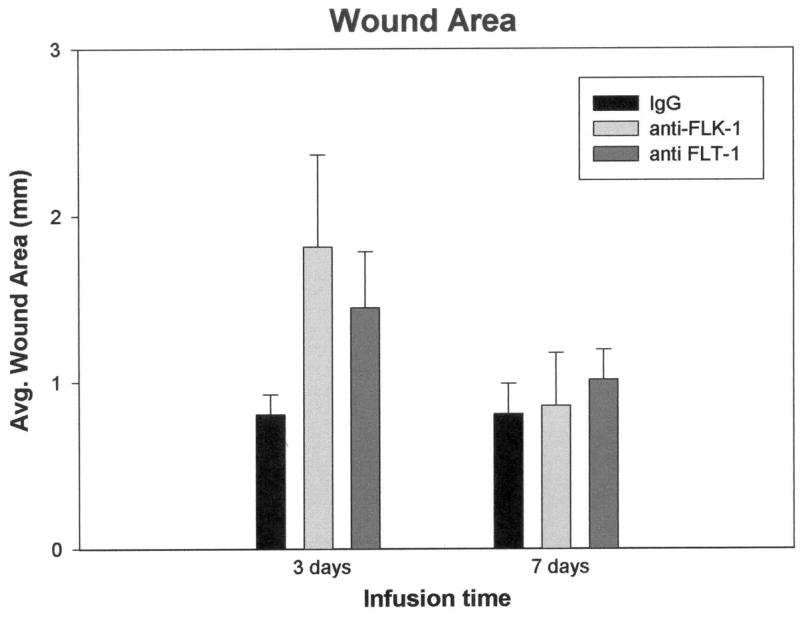
There was more variability in wound size for the experimental infusions vs. IgG controls, particularly at three days post-infusion, but the differences were not significant at either time point. Error bars represent SEM.
Vascular proliferation and degeneration
Vascular proliferation, as defined by PCNA labeling indices, was significantly decreased in both three and seven day anti-flk-1 infusions by 65% and 50%, respectively, compared to the IgG controls (Fig. 2A). In contrast, although infusion of flt-1-NA produced a trend toward diminished vascular proliferation compared to control infusions (particularly at 3 days postoperative), neutralization of flt-1 did not have a statistically significant effect on vascular PCNA labeling indices.
Fig. 2.

Vascular effects of VEGF receptor antibody infusions. A. At both time points, the vascular proliferation index quantified within 600 μm of the infusion sites was significantly decreased only after flk-1-NA infusions, compared to IgG controls. Vessels were double-labeled with laminin and PCNA. B. flk-1-NA administration resulted in significant increases in vascular TUNEL labeling at each time point, while flt-1-NA infusion caused significantly increased TUNEL labeling only at three days. *P < 0.05 (vs. control). Error bars represent SEM. C. A seven day IgG infusion immunostained for laminin shows profuse vessels filling the wound cavity (W). D. Numerous vessels lie adjacent to the wound cavity of a seven day flt-1-NA infusion. E. A seven day flk-1-NA infusion shows sparse vasculature around the wound cavity (W). Bar = 75 μm.
TUNEL/GFAP and TUNEL/laminin immunohistochemistry was used to determine the index of vascular and astroglial degeneration. TUNEL immunoreactivity is indicative of DNA fragmentation that occurs in both apoptosis and necrosis (Grasl-Kraupp et al. 1995; Newcomb et al. 1999; Fujikawa et al. 2000). Following flk-1-NA infusion, the TUNEL/laminin index was significantly increased by approximately 5-fold at both time points (Fig. 2B). Although control infusions produced an increased vascular density at the wound site (Fig. 2C), laminin immunostaining revealed a generally decreased density of vessels adjacent to the wounds in flk-1-NA infusions (Fig. 2 D), presumably because of the combined effects of reduced endothelial proliferation and increased vascular degeneration. The majority of the capillaries having TUNEL(+) endothelial cell nuclei appeared to be shrunken, collapsed, and somewhat fragmented. Interestingly, flt-1-NA infusion also produced a significant (almost two-fold) increase in TUNEL(+) vessels at the three day time point, but had no statistically significant effect on the vascular TUNEL labeling index at seven days.
Astroglial proliferation and degeneration
Infusion of neutralizing antibody to flt-1 significantly reduced astroglial proliferation by approximately 55% compared to controls at 3 days and by >80% at 7 days (Fig. 3A). In contrast, infusion of neutralizing antibody to flk-1 had no significant effect on astroglial proliferation, which was comparable to controls. Notably, the astroglial TUNEL index, indicative of degenerating cells, was not significantly different from controls in either the flt-1 or the flk-1 antibody infusions, although it trended higher for flt-1-NA at three days (Fig. 3B).
Fig. 3.
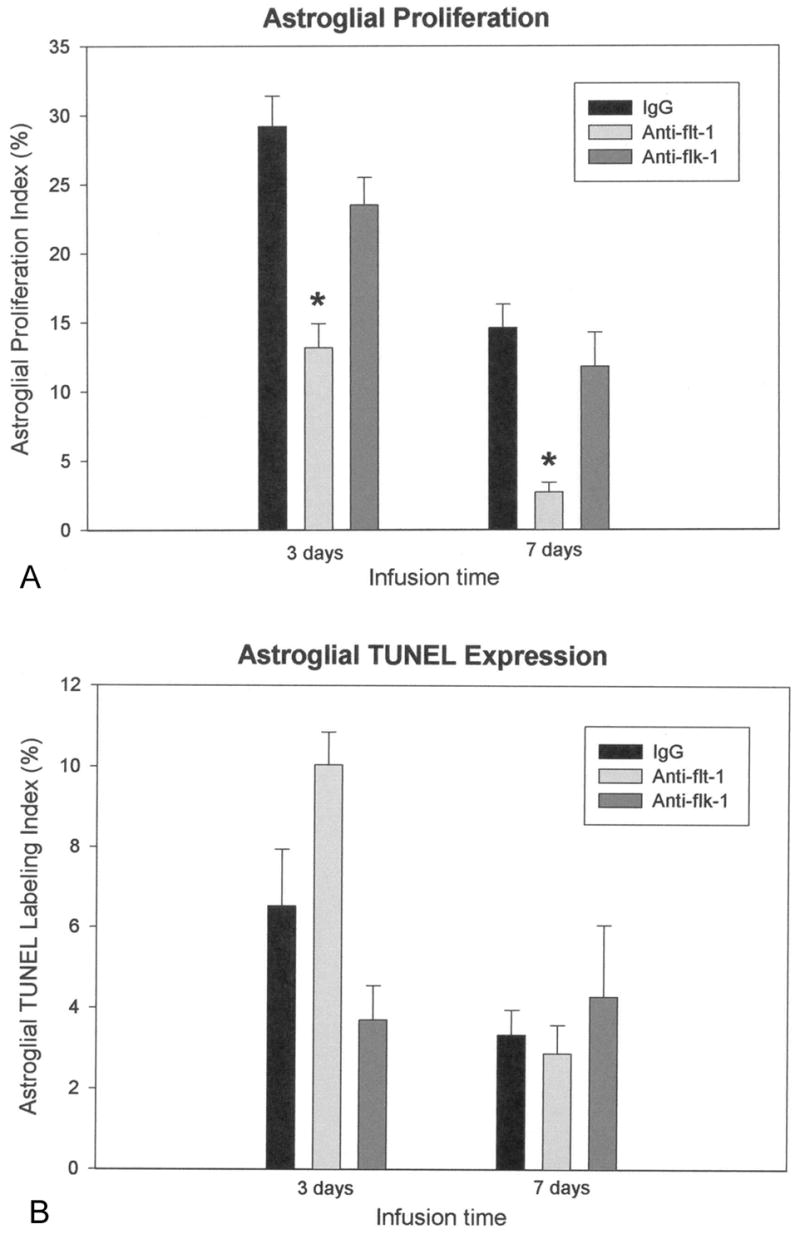
Astroglial effects of VEGF receptor antibody infusions. A. Quantification of the astroglial proliferation index within 600 μm of the infusion sites revealed significant decreases only after flt-1-NA infusion at both three and seven days, compared to IgG control infusions. B. Astroglial TUNEL expression was not significantly different from IgG control infusions after administration of either receptor antibody. *P < 0.05 (vs. control). Error bars represent SEM.
Immunostaining for GFAP revealed both decreased density of reactive astrocytes adjacent to the wounds and decreased hypertrophy in the majority of astrocytes at both 3 and 7 days in the anti-flt-1 infusions compared to controls (Fig. 4A – D). Consistent with the decreased proliferation index observed in the flt-1-NA infusions, the overall number of astrocytes within the 600 μm region adjacent to the wounds trended lower at 3 days and was significantly decreased at 7 days compared to both flk-1-NA and control infusions (Fig. 4E).
Fig. 4.

Quantification of astroglia within 600 μm of the infusion site; GFAP/TUNEL immunolabeling. Dark nuclei are TUNEL (+). A. 3 day IgG infusion and B. 3 day flk-1-NA have comparable numbers of astrocytes adjacent to the wound sites (W). C. 3 day flt-1-NA infusion shows a slightly decreased astroglial population compared to A and B. D, E. After 7 days, IgG and flk-1-NA infusions show large numbers of GFAP(+) reactive astrocytes near the wound area, while in F, flt-1-NA administration exhibited markedly reduced numbers of reactive astrocytes. Bar = 100 μm. G. Counts of astrocyte numbers at infusion sites. A statistically significant decrease in astrocytes was detected at 7 days only with flt-1-NA infusions compared to IgG controls. *P < 0.05 (vs. control). Error bars represent SEM.
Nestin expression
There were no qualitative or quantitative differences in nestin(+) astroglial distribution between IgG and flk-1-NA infusions at either time point, but the flt-1-NA infusions had fewer astrocytes, and therefore, fewer nestin(+) astroglia (Fig. 5 A–C). Nestin(+) astrocytes were confined to an area that extended approximately 1 mm from the wound edges in every case. Nestin was consistently co-localized with GFAP, such that portions of the astroglial processes were nestin(+), although a few GFAP(+) cells did not express nestin protein (not shown). Astrocytes in the contralateral cortex and striatum did not express nestin in any of the experimental paradigms.
Fig. 5.

Nestin (red)/GFAP (green) fluorescent immunolabeling reveals a reduced presence of double-labeled astrocytes (yellow) after flt-1-NA infusion directly adjacent to the wounds (W) at the 7 day time point . A, IgG infusion. B, flk-1-NA infusion. C, flt-1-NA infusion. Bar = 75 μm.
CNTF and bFGF immunoreactivity
Labeling indices were derived by counting astrocytes that were fluorescently double labeled with bFGF/GFAP or CNTF/GFAP to determine if inhibition of endogenous VEGF-astrocyte interactions would affect astroglial expression of either trophic factor. There were no significant differences between control and receptor antibody infusions at 3 days, but by 7 days, only flt-1-NA administration had significantly decreased the percentage of astrocytes that were immunoreactive for bFGF or CNTF (Fig. 6 A – H). Flt-1 blockade produced astrocytic bFGF and CNTF labeling indices that were only 50% of control infusions, while flk-1 blockade had no effect on astrocytic expression of these growth factors.
Fig. 6.
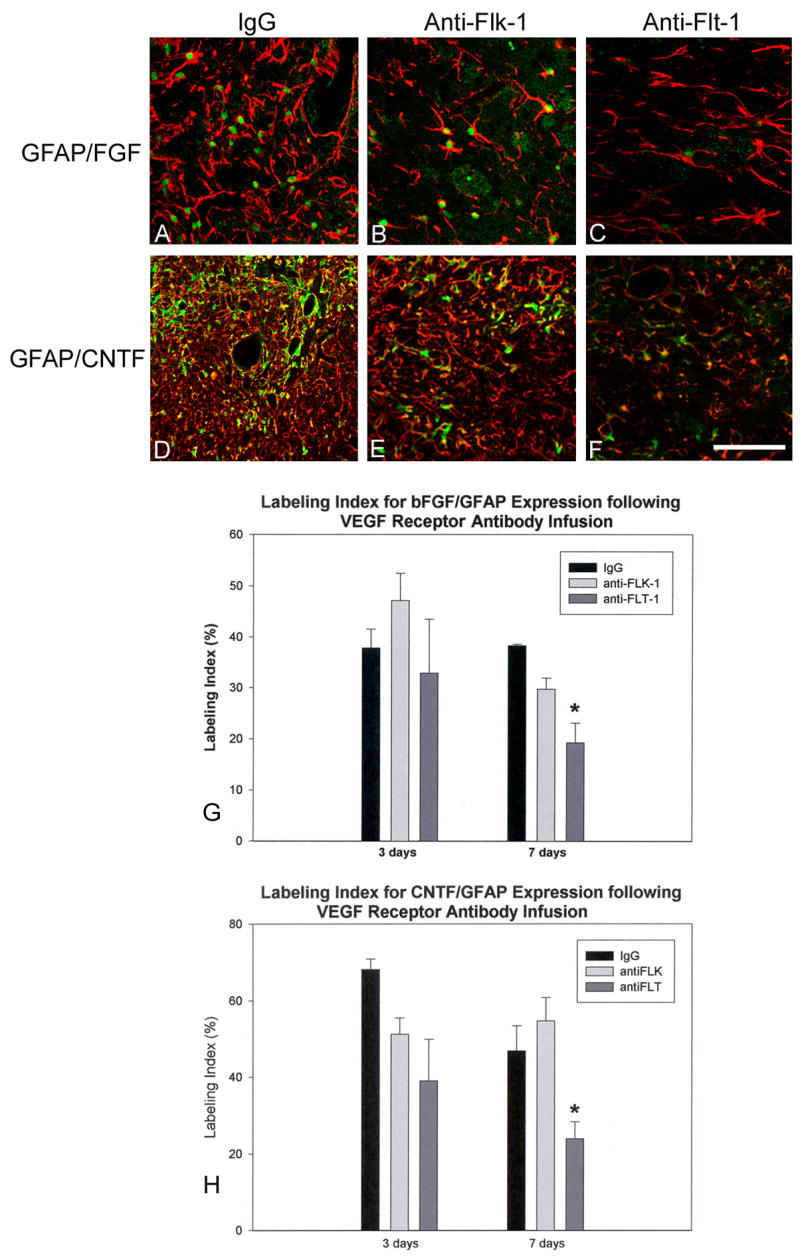
A – C, GFAP (red)/bFGF (green) fluorescent immunostaining adjacent to infusion sites (7 day time point). bFGF(+) cells have green nuclei. Numerous astrocytes are double labeled in the IgG infusion (A) and the flk-1-NA infusion (B) compared to the few observed in the flt-1-NA infusions (C). D – F, GFAP (red)/CNTF (green) fluorescent immunolabeling near infusion area (7 day time point) show a similar pattern of double labeling. IgG (D) and flk-1-NA infusions (E) show more CNTF(+)/GFAP(+) astrocytes (yellow) than the flt-1-NA infusions (F). Bar = 100 μm. G and H show that only the 7 day flt-1-NA infusions have significantly decreased numbers of astrocytes that are double stained for bFGF (G) and CNTF (H). *P < 0.05 (vs. control). Error bars represent SEM.
Discussion
Endogenous VEGF is induced by various types of CNS injuries and plays a role in neuronal survival and brain repair (Widenfalk et al. 2003; Krum and Rosenstein 2004; Yasuhara et al. 2005; Skold et al. 2006). It is also now apparent that a reduction in VEGF levels in mice can cause neurodegeneration, while increased VEGF levels delay neurodegeneration in models of amyotrophic lateral sclerosis (Lambrechts et al. 2003; Wang et al. 2007). Exogenous VEGF delivered to a wound site results in increased angiogenesis and astroglial proliferation and smaller wound areas when compared to controls (Krum et al. 2002) and can afford neuroprotection in stroke models (Manoonkitiwongsa et al 2004; Kaya et al. 2005). We have previously investigated the role of endogenously-derived VEGF in wound healing by inhibiting it with a specific neutralizing antibody using the same infusion model. The permanent insertion of the infusion cannula creates an injury site similar to a stab wound, and the injured tissue is directly and continuously exposed to the infusate (Krum et al. 2002; Krum and Khaibullina 2003). It was clear in that study that VEGF is an important component of the healing response, since its inhibition significantly decreases both astroglial and vascular proliferation, with a concomitant increase in wound size. In the present study, we took advantage of the differential expression of VEGF receptors by CNS cells, where flk-1 is expressed primarily by endothelial cells and neurons and reactive astroglia predominantly express flt-1 in order to determine if the reduction in astroglial proliferation was the result of VEGF blockade alone, or if it was because of the decrement in vascular growth. The specific blockade of each receptor in situ over periods of three to seven days allowed us to conclude that VEGF directly stimulates astroglia to proliferate and upregulate growth factor production via their expression of the flt-1 receptor, since blockade of the flk-1 receptor resulted only in decreased vascular proliferation and survival and had no effect on the measured astroglial parameters.
Using either antibody, we could not detect any differences in wound size when compared to IgG controls, suggesting that the simultaneous inhibition of endogenously VEGF-induced astroglial and vascular proliferation is necessary to augment wound size during the repair process (Krum and Khaibullina 2003). The relative loss of vasculature at the wound site due to flk-1-NA infusion would, logically, promote hypoxic degeneration of adjacent cells. It is possible, however, that tissue damage could be abrogated to a degree by the presence of unchecked astroglial activation and subsequent scarring, which may have many beneficial functions that include clearance of glutamate and potassium ions or production of cytokines and growth factors, among other considerations (Eddleston and Mucke 1993; Stichel and Müller 1998; Krum et al. 2002; Faulkner et al. 2004; Sofroniew 2005). After infusion of flt-1-NA, on the other hand, the relative loss of reactive astroglia could be compensated for by a relatively intact and modestly angiogenic vasculature that might offset the production of a larger wound area.
Vascular effects of VEGF receptor blockade
Infusion of flk-1-NA caused a significant decrease in vascular proliferation at three and seven days, and also produced a significant increase in vascular TUNEL staining at both time points, indicative of vascular degeneration adjacent to the infusion/wound site. The decreased density of vessels, and their shrunken, fragmented appearance (compared to control infusions) were also indicative of ongoing vascular degeneration. We have observed similar effects with blockade of VEGF itself in our previous study (Krum and Khaibullina 2003). Controls exhibited an increased vascular density at the wound sites compared to adjacent uninjured brain areas, which presumably was induced by secreted endogenous VEGF (Krum and Rosenstein 1998, 1999).
In contrast, infusion of flt-1-NA had no statistically significant effect on vascular proliferation, although there was a marked trend toward decreased vascular proliferation after flt-1 blockade, and there was only a partial effect on vessel degeneration. Consistent with the results of the present study, others have shown that flk-1 is the primary receptor that mediates VEGF-induced endothelial proliferation and survival in peripheral blood vessels (Gerber et al. 1998; Zachary and Gliki 2001). It is thought that flt-1 only weakly transduces signals for these functions (Zachary and Gliki 2001), but at three days there was a significant increase in vascular TUNEL labeling in our model. Notably, flt-1 signaling is involved the prevention of oxygen-induced vessel loss in the retina (Shih et al. 2003a, b). It appears that CNS endothelial cells share proliferative (flk-1-dependent) VEGF receptor characteristics with peripheral vascular endothelium, but may be more closely related to retinal endothelium in terms of their survival mechanisms.
While it has been shown that flt-1 is a decoy receptor for VEGF that acts as a sink to regulate the amount of VEGF available to bind to endothelial flk-1, thus influencing angiogenic activity, there is also circumstantial evidence that flt-1 can be phosphorylated by VEGF which stimulates direct effects on growth and migration of non-endothelial cells (see Meyer and Rahimi 2003). If blockade of flt-1 in our experiments thwarts such a decoy effect, we would expect that the vascular proliferation index would be increased during flt-1 blockade, since theoretically more VEGF would be available to bind with endothelial flk-1 receptors, but this was not observed. Since we have not detected flt-1 expression in vascular endothelial cells in our model, the relative decrease in activated astrocytes that occurs after flt-1 blockade (see below) may result in decreased amounts of endogenous VEGF (since astrocytes are a primary source of secreted VEGF), which would likely lead to decreased angiogenic activity.
Astroglial effects of VEGF receptor blockade
Flk-1-NA infusion had no significant effect on astrocyte proliferation, degeneration, number, or the expression of nestin, VEGF, CNTF, or bFGF at the wound sites at either time point. All of these parameters were comparable to those of the IgG control infusions, strongly suggesting that flk-1 is not a primary functional VEGF receptor for activated astrocytes, at least within the experimental parameters of the present study.
In sharp contrast to the results from flk-1 blockade, infusion with flt-1-NA produced significant decrements in astroglial proliferation at both time points, and a significant decrease in astrocyte number was observed after seven days of infusion. Likewise, seven days of flt-1-NA administration decreased astrocytic expression of both CNTF and bFGF by 50% compared to control infusions. These growth factors are produced by activated astroglia. bFGF is trophic for glia, blood vessels and neurons (Finklestein et al. 1988; Gomez-Pinilla et al. 1992; Yang and Cui 1998), while CNTF is neurotrophic (Sleeman et al. 2000). The results suggest that VEGF may have a regulatory influence over astroglial production of these trophic factors, which could positively influence wound repair and neurite growth. There appears to be a delayed effect of flt-1 receptor blockade since both the decrease in number of astrocytes and the decrease in percentage of them that are labeled for both FGF and CNTF are only significant after 7 days of flt-1-NA infusion, and not at 3 days. Thus, VEGF, via its interaction with flt-1, appears to be involved in several aspects of the astroglial activation cascade, including mitotic activity and upregulation of bFGF and CNTF. This observation fits with the recent finding that flt-1 signaling is also involved in inflammatory cytokine/chemokine production by monocytes (Selvaraj et al. 2003).
In our previous work using VEGF-NA infusions (Krum and Khaibullina 2003), there was a very large increase in TUNEL(+) astrocytes at each time point. Interestingly, blockade of flt-1 activity trended toward an increase in the number of TUNEL(+) astrocytes but the change was not statistically significant. Thus, although flt-1 signaling is necessary for astroglial mitogenesis, it remains unclear if VEGF/flt-1 interactions have a major role in astroglial survival after injury. The role of flt-1 in promoting VEGF’s survival effects on various cell types is only now beginning to be defined. Flt-1 signaling is responsible for preventing oxygen-induced vessel loss in the retina (Shih et al. 2003a, b) and for promoting the survival of chronic lymphocytic leukemia B cells (Lee, Y. K. et al. 2005). It is unknown if there are survival factors for astrocytes after injury; it is possible that there may be only limited astroglial survival, and the prevention of mitogenesis brings about the observation of fewer astrocytes at the wound site.
Nestin is strongly expressed in mature reactive astroglia, and may be a part of the activation cascade that represents a less mature state that is responsive to trophic factors and is capable of migratory activity (Krum and Rosenstein 1999). It is possible that nestin(+) cells are newly arrived to the infusion site from the subventricular zone (SVZ), since evidence suggests that progenitor cells migrate to a wound site and stroke penumbra (Arvidsson et al. 2002; Jin et al. 2003; Zhang et al. 2004). There were no differences in nestin(+) astroglial distribution between flk-1-NA infusions and the controls at either time point, but there was an observable reduction in nestin expression after flt-1-NA. This reduction was not as pronounced as what we observed previously, in which nestin immunoreactivity was almost completely abrogated ipsilateral to the wound/infusion site after VEGF-NA application (Krum and Khaibullina 2003). The profound loss of nestin expression around the wound site after VEGF-NA infusion may reflect a combined sensitivity of astroglia to the loss of both VEGF-supported vascularity and a lack of direct astroglial stimulation via the flt-1 receptor. In the present study, nestin immunoreactivity (upregulation) was not observed in the contralateral cortex/striatum in either controls or in antibody-infused brains.
The experiments in this study afforded us the ability to document the segregation of VEGF receptor function between astrocytes and the vasculature in vivo. The results shed new light on the function of flt-1 in astrocytes, indicating that it is the receptor most responsible for binding endogenous VEGF and transmitting the proliferation signal for these cells. Based on our TUNEL results, flt-1 activation may not be a strong promoter of astroglial survival, but our results support its role in transmitting survival signals in CNS endothelial cells, similar to what occurs in hypoxic retinal vasculature (Shih et al. 2003a, b). We have also largely confirmed the role of flk-1 as an important effector of endothelial survival and proliferation in the CNS, as it is in peripheral blood vessels. Thus, inhibition of astrocyte proliferation and growth factor production, without decreasing angiogenesis which is essential to wound repair, can be accomplished by the administration of flt-1-NA. Future studies could examine if other astrocyte-derived scar components, e.g. extracellular matrix molecules, could be modified by inhibition of VEGF’s specific proliferation and stimulatory effects on astroglia. Such information could be highly useful for strategies designed to promote neurite outgrowth at sites of astroglial scarring (Rhodes and Fawcett 2004; Silver and Miller 2004).
Acknowledgments
We thank Newton More for his technical expertise in immunofluorescence methods. This work was supported by National Institutes of Health Grants NS 38128 and 45189 (J.M.K. and J.M.R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2–3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- Amat JA, Ishiguro H, Nakamura K, Norton WT. Phenotypic diversity and kinetics of proliferating microglia and astrocytes following cortical stab wounds. GLIA. 1996;16:368–382. doi: 10.1002/(SICI)1098-1136(199604)16:4<368::AID-GLIA9>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nature Med. 2002;8:963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- Azzouz M, Ralph GS, Storkebaum ELEW, Mitrophanous KA, Kingsman SM, Carmeliet P, Mazarakis ND. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature. 2004;429:413–417. doi: 10.1038/nature02544. [DOI] [PubMed] [Google Scholar]
- Bartholdi D, Rubin BP, Schwab ME. VEGF mRNA induction correlates with changes in the vascular architecture upon spinal cord damage in the rat. Eur J Neurosci. 1997;9:2549–2560. doi: 10.1111/j.1460-9568.1997.tb01684.x. [DOI] [PubMed] [Google Scholar]
- Beck H, Acker T, Puschel AW, Fujisawa H, Carmeliet P, Plate KH. Cell type-specific expression of neuropilins in an MCA-occlusion model in mice suggests a potential role in post-ischemic brain remodeling. J Neuropathol Exp Neurol. 2002;61:339–350. doi: 10.1093/jnen/61.4.339. [DOI] [PubMed] [Google Scholar]
- Campbell IL. Cytokine-mediated inflammation and signaling in the intact central nervous system. Prog Brain Res. 2001;132:481–498. doi: 10.1016/S0079-6123(01)32097-6. [DOI] [PubMed] [Google Scholar]
- Chodobski A, Chung I, Kozniewska E, Ivanenko T, Chang W, Harrington JF, Duncan JA, Szmydynger-Chodobska J. Early neutrophilic expression of vascular endothelial growth factor after traumatic brain injury. Neuroscience. 2003;122:853–867. doi: 10.1016/j.neuroscience.2003.08.055. [DOI] [PubMed] [Google Scholar]
- Cobbs CS, Chen J, Greenberg DA, Graham SH. Vascular endothelial growth factor expression in transient focal cerebral ischemia in the rat. Neurosci Lett. 1998;249:79–82. doi: 10.1016/s0304-3940(98)00377-2. [DOI] [PubMed] [Google Scholar]
- Croll SD, Ransohoff RM, Cai N, Zhang Q, Martin FJ, Wei T, Kasselman LJ, Kintner J, Murphy AJ, Yancopoulos GD, Weigand SJ. VEGF-mediated inflammation precedes angiogenesis in adult brain. Exp Neurol. 2004;187:388–402. doi: 10.1016/j.expneurol.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Darland DC, Massingham LJ, Smith SR, Piek E, Saint-Geniez M, D’Amore PA. Pericyte production of cell-associated VEGF is differentiation-dependent and is associated with endothelial survival. Dev Biol. 2003;264:275–288. doi: 10.1016/j.ydbio.2003.08.015. [DOI] [PubMed] [Google Scholar]
- de Paermentier F, Heuschling P, Knoops B, Janssens de Varebeke P, van den Bosch de Aguilar P. A new model for quantification of microvascular regeneration after a lesion of the rat cerebral cortex. Brain Res. 1986;398:419–424. doi: 10.1016/0006-8993(86)91508-8. [DOI] [PubMed] [Google Scholar]
- Dias S, Hattori K, Heissig B, Zhu Z, Wu Y, Witte L, Hicklin DJ, Tateno M, Bohlen P, Moore MA, Rafii S. Inhibition of both paracrine and autocrine VEGF/VEGFR-2 signaling pathways is essential to induce long-term remission of xenotransplanted human leukemias. Proc Nat Acad Sci (USA) 2001;98:10857–10862. doi: 10.1073/pnas.191117498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddleston M, Mucke L. Molecular profile of reactive astrocytes--implications for their role in neurologic disease. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JW. Astrocytic and neuronal factors affecting axon regeneration in the damaged central nervous system. Cell Tissue Res. 1997;290:371–377. doi: 10.1007/s004410050943. [DOI] [PubMed] [Google Scholar]
- Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res Bull. 1999;49:377–391. doi: 10.1016/s0361-9230(99)00072-6. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nature Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Finklestein SP, Apostolides PJ, Caday CG, Prosser J, Philips MF, Klagsbrun M. Increased basic fibroblast growth factor (bFGF) immunoreactivity at the site of focal brain wounds. Brain Res. 1988;460:253–259. doi: 10.1016/0006-8993(88)90370-8. [DOI] [PubMed] [Google Scholar]
- Fitch MT, Silver J. Activated macrophages and the blood-brain barrier: inflammation after CNS injury leads to increases in putative inhibitory molecules. Exp Neurol. 1997;148:587–603. doi: 10.1006/exnr.1997.6701. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Shinmei SS, Cai B. Kainic acid-induced seizures produce necrotic, not apoptotic, neurons with internucleosomal DNA cleavage: implications for programmed cell death mechanisms. Neuroscience. 2000;98:41–53. doi: 10.1016/s0306-4522(00)00085-3. [DOI] [PubMed] [Google Scholar]
- Galand P, Degraef C. Cyclin/PCNA immunostaining as an alternative to tritiated thymidine pulse labelling for marking S phase cells in paraffin sections from animal and human tissues. Cell Tissue Kinet. 1989;22:383–392. doi: 10.1111/j.1365-2184.1989.tb00223.x. [DOI] [PubMed] [Google Scholar]
- Gerber HP, Dixit V, Ferrara N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem. 1998;273:13313–13316. doi: 10.1074/jbc.273.21.13313. [DOI] [PubMed] [Google Scholar]
- Gomez-Pinilla F, Lee JW, Cotman CW. Basic FGF in adult rat brain: cellular distribution and response to entorhinal lesion and fimbria-fornix transection. J Neuroscience. 1992;12:345–355. doi: 10.1523/JNEUROSCI.12-01-00345.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995;21:1465–1468. doi: 10.1002/hep.1840210534. [DOI] [PubMed] [Google Scholar]
- Hall PA, Levison DA, Woods AL, Yu CC, Kellock DB, Watkins JA, Barnes DM, Gillett CE, Camplejohn R, Dover R. Proliferating cell nuclear antigen (PCNA) immunolocalization in paraffin sections: an index of cell proliferation with evidence of deregulated expression in some neoplasms. J Pathol. 1990;162:285–294. doi: 10.1002/path.1711620403. [DOI] [PubMed] [Google Scholar]
- Harrigan MR, Ennis SR, Masada T, Keep RF. Intraventricular infusion of vascular endothelial growth factor promotes cerebral angiogenesis with minimal brain edema. Neurosurgery. 2002;50:589–598. doi: 10.1097/00006123-200203000-00030. [DOI] [PubMed] [Google Scholar]
- Huang J, Frischer JS, Serur A, Kadenhe A, Yokoi A, McCrudden KW, New T, O’Toole K, Zabski S, Rudge JS, Holash J, Yancopoulos GD, Yamashiro DJ, Kandel JJ. Regression of established tumors and metastases by potent vascular endothelial growth factor blockade. Proc Nat Acad Sci (USA) 2003;100:7785–7790. doi: 10.1073/pnas.1432908100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa R, Krupinski J, Bujny T, Kumar S, Kaluza J, Kumar P. Vascular endothelial growth factor and its receptor, KDR, in human brain tissue after ischemic stroke. Lab Invest. 1999;79:417–425. [PubMed] [Google Scholar]
- Jin K, Mao XO, Batteur SP, McEachron E, Leahy A, Greenberg DA. Caspase-3 and the regulation of hypoxic neuronal death by vascular endothelial growth factor. Neuroscience. 2001;108:351–358. doi: 10.1016/s0306-4522(01)00154-3. [DOI] [PubMed] [Google Scholar]
- Jin K, Sun Y, Xie L, Peel A, Mao XO, Batteur S, Greenberg DA. Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol Cell Neurosci. 2003;24:171–189. doi: 10.1016/s1044-7431(03)00159-3. [DOI] [PubMed] [Google Scholar]
- Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor rescues HN33 neural cells from death induced by serum withdrawal. J Mol Neurosci. 2000;14:197–203. doi: 10.1385/JMN:14:3:197. [DOI] [PubMed] [Google Scholar]
- Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Nat Acad Sci (USA) 2000;97:10242–10247. doi: 10.1073/pnas.97.18.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin KL, Mao XO, Nagayama T, Goldsmith PC, Greenberg DA. Induction of vascular endothelial growth factor and hypoxia-inducible factor-1alpha by global ischemia in rat brain. Neuroscience. 2000;99:577–585. doi: 10.1016/s0306-4522(00)00207-4. [DOI] [PubMed] [Google Scholar]
- Kasamatsu T, Itakura T, Jonsson G. Intracortical spread of exogenous catecholamines: effective concentration for modifying cortical plasticity. J Pharm Exp Therap. 1981;217:841–850. [PubMed] [Google Scholar]
- Kaya D, Gürsoy-Özdemir Y, Yemisci M, Tuncer N, Aktan S, Dalkara T. VEGF protects against focal ischemia without increasing blood-brain barrier permeability when administered intracerebroventricularly. J Cereb Blood Flow Metab. 2005;25:1111–1118. doi: 10.1038/sj.jcbfm.9600109. [DOI] [PubMed] [Google Scholar]
- Khaibullina AA, Rosenstein JM, Krum JM. Vascular endothelial growth factor promotes neurite maturation in primary CNS neuronal cultures. Brain Res Dev Brain Res. 2004;148:59–68. doi: 10.1016/j.devbrainres.2003.09.022. [DOI] [PubMed] [Google Scholar]
- Klagsbrun M, D’Amore PA. Vascular endothelial growth factor and its receptors. Cytokine Growth Factor Rev. 1996;7:259–270. doi: 10.1016/s1359-6101(96)00027-5. [DOI] [PubMed] [Google Scholar]
- Kovacs Z, Ikezaki K, Samoto K, Inamura T, Fukui M. VEGF and flt. Expression time kinetics in rat brain infarct. Stroke. 1996;27:1865–1872. doi: 10.1161/01.str.27.10.1865. [DOI] [PubMed] [Google Scholar]
- Kremer C, Breier G, Risau W, Plate KH. Up-regulation of flk-1/vascular endothelial growth factor receptor 2 by its ligand in a cerebral slice culture system. Cancer Res. 1997;57:3852–3859. [PubMed] [Google Scholar]
- Krum JM, Khaibullina A. Inhibition of endogenous VEGF impedes revascularization and astroglial proliferation: roles for VEGF in brain repair. Exp Neurol. 2003;181:241–257. doi: 10.1016/s0014-4886(03)00039-6. [DOI] [PubMed] [Google Scholar]
- Krum JM, Mani N, Rosenstein JM. Angiogenic and astroglial responses to vascular endothelial growth factor administration in adult rat brain. Neuroscience. 2002;110:589–604. doi: 10.1016/s0306-4522(01)00615-7. [DOI] [PubMed] [Google Scholar]
- Krum JM, Rosenstein JM. Patterns of angiogenesis in neural transplant models: II Fetal neocortical transplants. J Comp Neurol. 1988;271:331–345. doi: 10.1002/cne.902710304. [DOI] [PubMed] [Google Scholar]
- Krum JM, Rosenstein JM. VEGF mRNA and its receptor flt-1 are expressed in reactive astrocytes following neural grafting and tumor cell implantation in the adult CNS. Exp Neurol. 1998;154:57–65. doi: 10.1006/exnr.1998.6930. [DOI] [PubMed] [Google Scholar]
- Krum JM, Rosenstein JM. Transient coexpression of nestin, GFAP, and vascular endothelial growth factor in mature reactive astroglia following neural grafting or brain wounds. Exp Neurol. 1999;160:348–360. doi: 10.1006/exnr.1999.7222. [DOI] [PubMed] [Google Scholar]
- Kunkel P, Ulbricht U, Bohlen P, Brockmann MA, Fillbrandt R, Stavrou D, Westphal M, Lamszus K. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res. 2001;61:6624–6628. [PubMed] [Google Scholar]
- Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, van Marion I, Al-Chalabi A, Bornes S, Musson R, Hansen V, Beckman L, Adolfsson R, Pall HS, Prats H, Vermeire S, Rutgeerts P, Katayama S, Awata T, Leigh N, Lang-Lazdunski L, Dewerchin M, Shaw C, Moons L, Vlietinck R, Morrison KE, Robberecht W, Van Broeckhoven C, Collen D, Andersen PM, Carmeliet P. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. [see comment] Nature Gen. 2003;34:383–394. doi: 10.1038/ng1211. [DOI] [PubMed] [Google Scholar]
- Lee MY, Ju WK, Cha JH, Son BC, Chun MH, Kang JK, Park CK. Expression of vascular endothelial growth factor mRNA following transient forebrain ischemia in rats. Neurosci Lett. 1999;265:107–110. doi: 10.1016/s0304-3940(99)00219-0. [DOI] [PubMed] [Google Scholar]
- Lee YK, Shanafelt TD, Bone ND, Strege AK, Jelinek DF, Kay NE. VEGF receptors on chronic lymphocytic leukemia (CLL) B cells interact with STAT 1 and 3: implication for apoptosis resistance. Leukemia. 2005;19:513–523. doi: 10.1038/sj.leu.2403667. [DOI] [PubMed] [Google Scholar]
- Lennmyr F, Ata KA, Funa K, Olsson Y, Terent A. Expression of vascular endothelial growth factor (VEGF) and its receptors (Flt-1 and Flk-1) following permanent and transient occlusion of the middle cerebral artery in the rat. J Neuropathol Exp Neurol. 1998;57:874–882. doi: 10.1097/00005072-199809000-00009. [DOI] [PubMed] [Google Scholar]
- Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- Liberto CM, Albrecht PJ, Herx LM, Yong VW, Levison SW. Pro-regenerative properties of cytokine-activated astrocytes. Journal of Neurochemistry. 2004;89:1092–1100. doi: 10.1111/j.1471-4159.2004.02420.x. [DOI] [PubMed] [Google Scholar]
- Logan A, Berry M. Cellular and molecular determinants of glial scar formation. Adv Exp Med Biol. 2002;513:115–158. doi: 10.1007/978-1-4615-0123-7_4. [DOI] [PubMed] [Google Scholar]
- Lum JT, Nguyen T, Felpel LP. Drug distribution in solid tissue of the brain following chronic local perfusion utilizing implanted osmotic minipumps. J Pharm Meth. 1984;12:141–147. doi: 10.1016/0160-5402(84)90031-7. [DOI] [PubMed] [Google Scholar]
- Mani N, Khaibullina A, Krum JM, Rosenstein JM. Activation of receptor-mediated angiogenesis and signaling pathways after VEGF administration in fetal rat CNS explants. J Cereb Blood Flow Metab. 2003;23:1420–1429. doi: 10.1097/01.WCB.0000090620.86921.9C. [DOI] [PubMed] [Google Scholar]
- Manoonkitiwongsa P, Schultz R, McCreery D, Whitter E, Lyden P. Neuroprotection of ischemic brain by vascular endothelial growth factor is critically dependent on proper dosage and may be compromised by angiogenesis. J Cereb Blood Flow Metab. 2004;24:693–702. doi: 10.1097/01.WCB.0000126236.54306.21. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Tamatani M, Yamaguchi A, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M. Vascular endothelial growth factor rescues hippocampal neurons from glutamate-induced toxicity: signal transduction cascades. FASEB J. 2001;15:1218–1220. [PubMed] [Google Scholar]
- McGraw J, Hiebert GW, Steeves JD. Modulating astrogliosis after neurotrauma. J Neurosci Res. 2001;63:109–115. doi: 10.1002/1097-4547(20010115)63:2<109::AID-JNR1002>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Meyer RD, Rahimi N. Comparative structure-function analysis of VEGFR-1 and VEGFR-2: What have we learned from chimeric systems? Ann NY Acad Sci. 2003;995:200–207. doi: 10.1111/j.1749-6632.2003.tb03223.x. [DOI] [PubMed] [Google Scholar]
- Monacci WT, Merrill MJ, Oldfield EH. Expression of vascular permeability factor/vascular endothelial growth factor in normal rat tissues. Am J Physiol. 1993;264:995–1002. doi: 10.1152/ajpcell.1993.264.4.C995. [DOI] [PubMed] [Google Scholar]
- Nag S, Takahashi JL, Kilty DW. Role of vascular endothelial growth factor in blood-brain barrier breakdown and angiogenesis in brain trauma. J Neuropathol Exp Neurol. 1997;56:912–921. doi: 10.1097/00005072-199708000-00009. [DOI] [PubMed] [Google Scholar]
- Neary JT, Kang Y. Signaling from P2 nucleotide receptors to protein kinase cascades induced by CNS injury: implications for reactive gliosis and neurodegeneration. Mol Neurobiol. 2005;31:95–103. doi: 10.1385/MN:31:1-3:095. [DOI] [PubMed] [Google Scholar]
- Newcomb JK, Zhao X, Pike BR, Hayes RL. Temporal profile of apoptotic-like changes in neurons and astrocytes following controlled cortical impact injury in the rat. Exp Neurol. 1999;158:76–88. doi: 10.1006/exnr.1999.7071. [DOI] [PubMed] [Google Scholar]
- Orita T, Akimura T, Kamiryo T, Nishizaki T, Furutani Y, Harada K, Ikeyama Y, Aoki H. Cerebral endothelial regeneration following experimental brain injury. Variation in the regeneration process according to the severity of injury. Acta Neuropathol. 1989;77:397–401. doi: 10.1007/BF00687374. [DOI] [PubMed] [Google Scholar]
- Papavassiliou E, Gogate N, Proescholdt M, Heiss JD, Walbridge S, Edwards NA, Oldfield EH, Merrill MJ. Vascular endothelial growth factor (vascular permeability factor) expression in injured rat brain. J Neurosci Res. 1997;49:451–460. doi: 10.1002/(sici)1097-4547(19970815)49:4<451::aid-jnr6>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Peters KG, De Vries C, Williams LT. Vascular endothelial growth factor receptor expression during embryogenesis and tissue repair suggests a role in endothelial differentiation and blood vessel growth. Proc Natl Acad Sci U S A. 1993;90:8915–8919. doi: 10.1073/pnas.90.19.8915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichiule P, Chavez JC, Xu K, LaManna JC. Vascular endothelial growth factor upregulation in transient global ischemia by cardiac arrest and resuscitation in rat brain. Mol Brain Res. 1999;74:83–90. doi: 10.1016/s0169-328x(99)00261-2. [DOI] [PubMed] [Google Scholar]
- Plate KH, Beck H, Danner S, Allegrini PR, Wiessner C. Cell type specific upregulation of vascular endothelial growth factor in an MCA-occlusion model of cerebral infarct. J Neuropathol Exp Neurol. 1999;58:654–666. doi: 10.1097/00005072-199906000-00010. [DOI] [PubMed] [Google Scholar]
- Proescholdt MA, Heiss JD, Walbridge S, Muhlhauser J, Capogrossi MC, Oldfield EH, Merrill MJ. Vascular endothelial growth factor (VEGF) modulates vascular permeability and inflammation in rat brain. J Neuropathol Exp Neurol. 1999;58:613–627. doi: 10.1097/00005072-199906000-00006. [DOI] [PubMed] [Google Scholar]
- Rhodes KE, Fawcett JW. Chondroitin sulphate proteoglycans: preventing plasticity or protecting the CNS? J Anat. 2004;204:33–48. doi: 10.1111/j.1469-7580.2004.00261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstein JM, Krum JM. New roles for VEGF in nervous tissue - beyond blood vessels. Exp Neurol. 2004;187:246–253. doi: 10.1016/j.expneurol.2004.01.022. [DOI] [PubMed] [Google Scholar]
- Rosenstein JM, Mani N, Khaibullina A, Krum JM. Neurotrophic effects of vascular endothelial growth factor on organotypic cortical explants and primary cortical neurons. J Neurosci. 2003;23:11036–11044. doi: 10.1523/JNEUROSCI.23-35-11036.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstein JM, Mani N, Silverman WF, Krum JM. Patterns of brain angiogenesis after vascular endothelial growth factor administration in vitro and in vivo. Proc Nat Acad Sci (USA) 1998;95:7086–7091. doi: 10.1073/pnas.95.12.7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salhia B, Angelov L, Roncari L, Wu X, Shannon P, Guha A. Expression of vascular endothelial growth factor by reactive astrocytes and associated neoangiogenesis. Brain Res. 2000;883:87–97. doi: 10.1016/s0006-8993(00)02825-0. [DOI] [PubMed] [Google Scholar]
- Selvaraj SK, Giri RK, Perelman N, Johnson C, Malik P, Kalra VK. Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood. 2003;102:1515–1524. doi: 10.1182/blood-2002-11-3423. [DOI] [PubMed] [Google Scholar]
- Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Experimental Cell Research. 2006;312:549–560. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Shih SC, Ju M, Liu N, Mo JR, Ney JJ, Smith LE. Transforming growth factor beta1 induction of vascular endothelial growth factor receptor 1: mechanism of pericyte-induced vascular survival in vivo. Proc Nat Acad Sci (USA) 2003;100:15859–15864. doi: 10.1073/pnas.2136855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih SC, Ju M, Liu N, Smith LE. Selective stimulation of VEGFR-1 prevents oxygen-induced retinal vascular degeneration in retinopathy of prematurity. [see comment] J Clin Invest. 2003;112:50–57. doi: 10.1172/JCI17808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nature Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Silverman WF, Krum JM, Mani N, Rosenstein JM. Vascular, glial and neuronal effects of vascular endothelial growth factor in mesencephalic explant cultures. Neuroscience. 1999;90:1529–1541. doi: 10.1016/s0306-4522(98)00540-5. [DOI] [PubMed] [Google Scholar]
- Skold MK, Cullheim S, Hammarberg H, Piehl F, Suneson A, Lake S, Sjogren A, Walum E, Risling M. Induction of VEGF and VEGF receptors in the spinal cord after mechanical spinal injury and prostaglandin administration. Eur J Neurosci. 2000;12:3675–3686. doi: 10.1046/j.1460-9568.2000.00263.x. [DOI] [PubMed] [Google Scholar]
- Skold MK, Risling M, Holmin S. Inhibition of vascular endothelial growth factor receptor 2 activity in experimental brain contusions aggravates injury outcome and leads to early increased neuronal and glial degeneration. Eur J Neurosci. 2006;23:21–34. doi: 10.1111/j.1460-9568.2005.04527.x. [DOI] [PubMed] [Google Scholar]
- Skold MK, von Gertten C, Sandberg-Nordqvist AC, Mathiesen T, Holmin S. VEGF and VEGF receptor expression after experimental brain contusion in rat. J Neurotrauma. 2005;22:353–367. doi: 10.1089/neu.2005.22.353. [DOI] [PubMed] [Google Scholar]
- Sleeman MW, Anderson KD, Lambert PD, Yancopoulos GD, Wiegand SJ. The ciliary neurotrophic factor and its receptor, CNTFR alpha. Pharm Acta Helvet. 2000;74:265–272. doi: 10.1016/s0031-6865(99)00050-3. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- Stichel CC, Müller HW. The CNS lesion scar: new vistas on an old regeneration barrier. Cell Tissue Res. 1998;294:1–9. doi: 10.1007/s004410051151. [DOI] [PubMed] [Google Scholar]
- Swartz KR, Liu F, Sewell D, Schochet T, Campbell I, Sandor M, Fabry Z. Interleukin-6 promotes post-traumatic healing in the central nervous system. Brain Res. 2001;896:86–95. doi: 10.1016/s0006-8993(01)02013-3. [DOI] [PubMed] [Google Scholar]
- Thaete LG, Ahnen DJ, Malkinson AM. Proliferating cell nuclear antigen (PCNA/cyclin) immunocytochemistry as a labeling index in mouse lung tissues. Cell Tissue Res. 1989;256:167–173. doi: 10.1007/BF00224731. [DOI] [PubMed] [Google Scholar]
- Thomas KA. Vascular endothelial growth factor, a potent and selective angiogenic agent. J Biol Chem. 1996;271:603–606. doi: 10.1074/jbc.271.2.603. [DOI] [PubMed] [Google Scholar]
- Tsao MN, Li YQ, Lu G, Xu Y, Wong CS. Upregulation of vascular endothelial growth factor is associated with radiation-induced blood-spinal cord barrier breakdown. J Neuropathol Exp Neurol. 1999;58:1051–1060. doi: 10.1097/00005072-199910000-00003. [DOI] [PubMed] [Google Scholar]
- Vaquero J, Zurita M, de Oya S, Coca S. Vascular endothelial growth/permeability factor in spinal cord injury. J Neurosurg. 1999;90:220–223. doi: 10.3171/spi.1999.90.2.0220. [DOI] [PubMed] [Google Scholar]
- Wang Y, Mao XO, Xie L, Banwait S, Marti HH, Greenberg DA, Jin K. Vascular endothelial growth factor overexpression delays neurodegeneration and prolongs survival in amyotrophic lateral sclerosis mice. J Neurosci. 2007;27:304–307. doi: 10.1523/JNEUROSCI.4433-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widenfalk J, Lipson A, Jubran M, Hofstetter C, Ebendal T, Cao Y, Olson L. Vascular endothelial growth factor improves functional outcome and decreases secondary degeneration in experimental spinal cord contusion injury. Neuroscience. 2003;120:951–960. doi: 10.1016/s0306-4522(03)00399-3. [DOI] [PubMed] [Google Scholar]
- Witte L, Hicklin DJ, Zhu Z, Pytowski B, Kotanides H, Rockwell P, Bohlen P. Monoclonal antibodies targeting the VEGF receptor-2 (Flk1/KDR) as an anti-angiogenic therapeutic strategy. Cancer Met Rev. 1998;17:155–161. doi: 10.1023/a:1006094117427. [DOI] [PubMed] [Google Scholar]
- Yamada K, Yoshitake K, Sato M, Ahnen DJ. Proliferating cell nuclear antigen expression in normal, preneoplastic, and neoplastic colonic epithelium of the rat. Gastroenterology. 1992;103:160–167. doi: 10.1016/0016-5085(92)91109-h. [DOI] [PubMed] [Google Scholar]
- Yang SY, Cui JZ. Expression of the basic fibroblast growth factor gene in mild and more severe head injury in the rat. J Neurosurg. 1998;89:297–302. doi: 10.3171/jns.1998.89.2.0297. [DOI] [PubMed] [Google Scholar]
- Yang W, de Bono DP. A new role for vascular endothelial growth factor and fibroblast growth factors: increasing endothelial resistance to oxidative stress. FEBS Lett. 1997;403:139–142. doi: 10.1016/s0014-5793(96)01486-x. [DOI] [PubMed] [Google Scholar]
- Yasuhara T, Shingo T, Muraoka K, wen Ji Y, Kameda M, Takeuchi A, Yano A, Nishio S, Matsui T, Miyoshi Y, Hamada H, Date I. The differences between high and low-dose administration of VEGF to dopaminergic neurons of in vitro and in vivo Parkinson’s disease model. Brain Res. 2005;1038:1–10. doi: 10.1016/j.brainres.2004.12.055. [DOI] [PubMed] [Google Scholar]
- Zachary I, Gliki G. Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc Res. 2001;49:568–581. doi: 10.1016/s0008-6363(00)00268-6. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zhang Z, Wang L, Wang Y, Gousev A, Zhang L, Ho KL, Morshead C, Chopp M. Activated neural stem cells contribute to stroke-induced neurogenesis and neuroblast migration toward the infarct boundary in adult rats. J Cereb Blood Flow Metab. 2004;24:441–448. doi: 10.1097/00004647-200404000-00009. [DOI] [PubMed] [Google Scholar]