

Abstract
We describe the comprehensive characterization of homeodomain DNA-binding specificities from a metazoan genome. The analysis of all 84 independent homeodomains from D. melanogaster reveals the breadth of DNA sequences that can be specified by this recognition motif. The majority of these factors can be organized into 11 different specificity groups, where the preferred recognition sequence between these groups can differ at up to 4 of the 6 core recognition positions. Analysis of the recognition motifs within these groups led to a catalog of common specificity determinants that may cooperate or compete to define the binding site preference. Using these recognition principles, a homeodomain can be reengineered to create factors where its specificity is altered at the majority of recognition positions. This resource also allows prediction of homeodomain specificities from other organisms, which is demonstrated by the prediction and analysis of human homeodomain specificities.
Introduction
In humans, as well as many other metazoans, homeodomains comprise the second largest class of sequence-specific transcription factors (TFs) (Tupler et al., 2001). Homeotic genes were first identified in D. melanogaster because their altered activity resulted in dramatic phenotypes such as the formation of an additional pair of wings (Lewis, 1978). Cloning of these genes led to the landmark observation that they contain a common sequence motif that encodes a DNA-binding domain (Gehring et al., 1994a). Subsequent studies have identified a large number of additional homeodomain proteins in Drosophila that regulate diverse developmental processes. A remarkable number of these genes have mammalian homologs with conserved developmental functions and biochemical properties (Banerjee-Basu and Baxevanis, 2001; Mukherjee and Burglin, 2007).
Insights into the mechanisms of sequence-specific DNA binding by homeodomains have been provided by the three-dimensional structures of individual protein-DNA complexes coupled with directed mutagenesis and biochemical analysis (Ades and Sauer, 1995; Gehring et al., 1994b; Wolberger, 1996). The homeodomain consists of approximately 60 amino acids that fold into a stable 3-helix bundle preceded by a flexible N-terminal arm. Interactions with a 5 to 7 base pair DNA binding site are formed by positioning a single “recognition” helix in the major groove and the N-terminal arm in the minor groove (Figure 1A and B). Despite a common DNA-binding architecture, there is significant variation in the sequence composition within the homeodomain family; for example the two superclasses of homeodomains, denoted as typical and atypical (Banerjee-Basu and Baxevanis, 2001; Mukherjee and Burglin, 2007), share low sequence identity and recognize substantially different DNA sequences, yet their docking with the DNA is nearly identical (Kissinger et al., 1990; Wolberger et al., 1991). This conserved binding geometry allows differences in amino acid sequence and DNA-binding specificity for various homeodomains to be interpreted within a common structural framework. Residues at positions 2, 3 and 5–8 on the N-terminal arm, as well as residues at positions 47, 50, 51, 54 and 55 on the recognition helix, can all contribute to DNA-binding specificity (Ades and Sauer, 1995; Damante et al., 1996; Ekker et al., 1994; Fraenkel et al., 1998; Passner et al., 1999; Piper et al., 1999; Wolberger et al., 1991) (Figure 1B and C).
Figure 1.
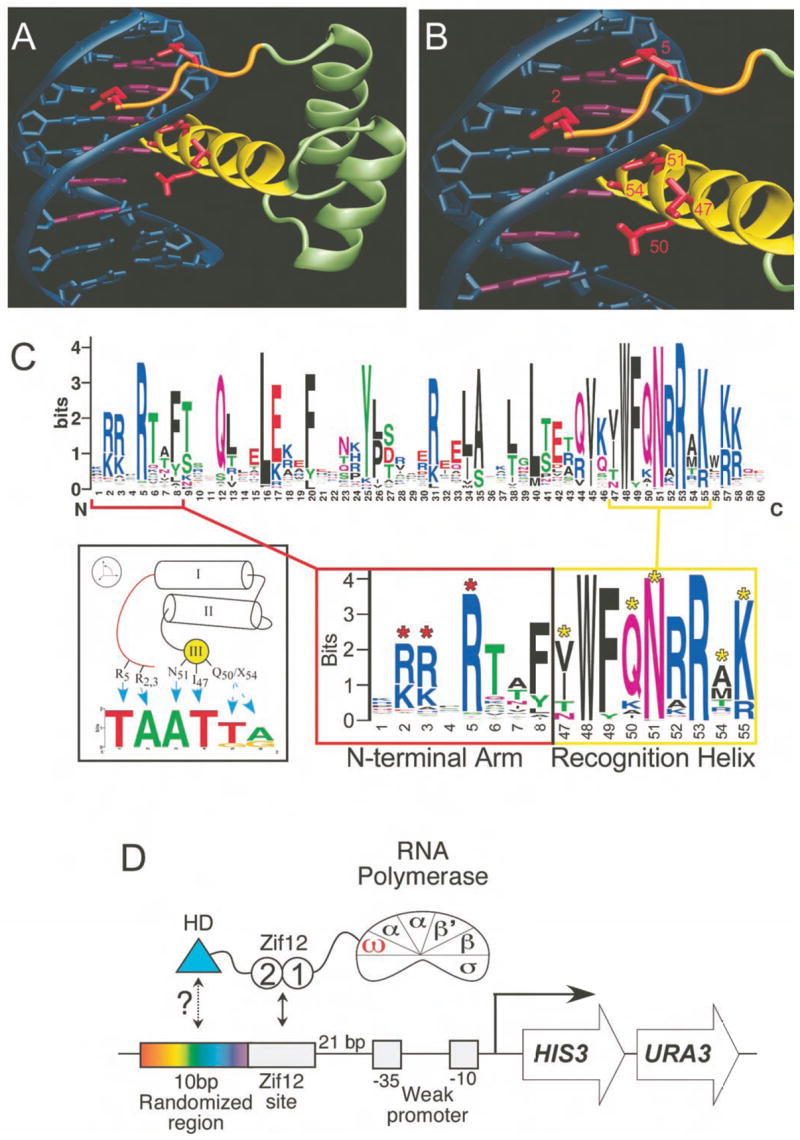
DNA recognition by the homeodomain family. A) The structure of Msx-1 bound to DNA is representative of homeodomain-DNA interactions (Hovde et al., 2001). B) Detailed view of the recognition contacts (red), where residues at positions 2 and 5 of the N-terminal arm (orange) interact with bases in the minor groove and residues at positions 47, 50, 51 and 54 of the recognition helix (yellow) are positioned to make contacts in the major groove. C) (Top) Sequence logo representation of the diversity in our set of 84 homeodomains. (Bottom) Windows highlighting the diversity in the DNA-recognition regions - the N-terminal arm (red) and recognition helix (yellow). The key recognition positions are indicated with asterisks. D) Cartoon depicting recruitment of omega-Zif12-HD (homeodomain) fusions to the weak promoter driving the HIS3 and URA3 reporters used in the B1H system (Meng et al., 2005; Meng and Wolfe, 2006).
How specific sequence variations between homeodomains lead to different recognition preferences has been defined in several cases. Seminal experiments demonstrated that Lys50 promotes recognition of TAATCC by the Bicoid class of homeodomains instead of the TAAT(T/G)(A/G) recognized by the Gln50-containing Antp and En classes (Hanes and Brent, 1989; Percival-Smith et al., 1990; Treisman et al., 1989). Beachy and colleagues mapped differences in binding site position 2 specificity for the posterior HOX protein Abd-B (TTATGG) and more anterior HOX family members (TAATGG) to amino acids at positions 3, 6 and 7 in the N-terminal arm (Ekker et al., 1994). Interestingly, substitutions at amino acids that overlap with these positions (6–8) are sufficient to switch the specificity of an NK-2 type homeodomain (CAAGTG) to the specificity of an Antp-type homeodomain (TAAGTG) at the neighboring base, binding site position 1 (Damante et al., 1996). This complexity is not limited to the N-terminal arm, as residues at different amino acid positions, such as 47 and 54, can potentially contact the same base pair (Fraenkel et al., 1998; Gruschus et al., 1997; Wolberger et al., 1991). This diversity in potential recognition contacts has hindered efforts to globally reengineer homeodomain specificity (Mathias et al., 2001). Consequently, a comprehensive description of the determinants of homeodomain DNA-binding specificity remains an important goal.
A complete survey of DNA-binding specificity on a large family of DNA-binding domains has not been previously attempted. We have recently described a bacterial one-hybrid (B1H) system that allows the specificities of a DNA-binding domain to be rapidly characterized with sufficient ease that multiple factors can be assayed in parallel (Meng et al., 2005; Meng and Wolfe, 2006). Using this system, we analyze the DNA-binding specificities for all 84 homeodomains in D. melanogaster that are not associated with an additional DNA-binding domain as well as 16 mutant homeodomains with changes in residues that contribute to DNA recognition. Our analysis reveals a diverse array of DNA-binding specificities with a minimum of seventeen unique specificities in D. melanogaster, of which the majority of homeodomains can be clustered into 11 specificity groups. Members of a given specificity group typically share common recognition residues. Combining this data with previous structural and biochemical work on the homeodomain family, we propose and evaluate a detailed set of recognition determinants for homeodomains and use this information to broadly and accurately predict the specificities of homeodomains in the human genome.
Results
Analysis of homeodomains using a modified bacterial one-hybrid (B1H) system
We have modified our B1H system to rapidly characterize the DNA-binding specificity of a homeodomain (Meng et al., 2005; Meng and Wolfe, 2006). Homedomains are expressed as fusions to both the omega subunit of RNA-polymerase (Dove and Hochschild, 1998), which provides better dynamic range than fusions to alpha (data not shown), and to zinc fingers 1 and 2 of the protein Zif268 (Zif12; Figure 1D). Because zinc finger-homeodomain chimeras exhibit increased affinity and specificity (Pomerantz et al., 1995), even homeodomains with relatively low DNA binding activity can be readily characterized, A library with 10 randomized base pairs adjacent to a Zif12 binding site (ZF10) was used to isolate recognition sequences that are complementary to the homeodomain in this selection system (Figure 1D and Supplementary Figure 1).
This system was used to determine DNA-binding specificities for all 84 of the homeodomains in the D. melanogaster genome that are not associated with an auxiliary DNA-binding domain (Supplementary Figure 2 and Supplementary Table 1). These homeodomains cluster into previously described families (Banerjee-Basu and Baxevanis, 2001; Mukherjee and Burglin, 2007) based on their amino acid similarity (Supplementary Figure 3), where approximately 85% of these homeodomains are in the “typical” superclass. Present in the collection of Drosophila homeodomains are diverse sets of amino acids at DNA-recognition positions, which suggests that a range of DNA-binding specificities is possible (Figure 1C). One notable exception is Asn at position 51 of the recognition helix, which is present in all but one of these homeodomains.
Comparisons to earlier studies confirm that the motifs obtained by the B1H method accurately reflect the DNA-binding specificities of homeodomains. For example, all of our specificities for the homeotic (HOX) gene family share a common consensus –T(A/T)AT(T/G)(A/G) (Supplementary Figure 4), consistent with previous studies (Pearson et al., 2005). Furthermore, subtle differences in the specificity of Ubx, Dfd and Abd-B that were previously observed in biochemical assays (Ekker et al., 1994; Ekker et al., 1992) are also present in our data, such as the preference of Abd-B for Thy over Ade at binding site position 2. Thus, even subtle differences in homeodomain specificity can be captured by the B1H analysis. The accuracy of our B1H-generated data was further validated by competition gel mobility shift assays performed for 9 factors that display different specificities (Supplementary Figure 5).
Global alignment and clustering of homeodomain binding sites
Remarkable diversity exists in the B1H-determined DNA-binding specificities for the entire set of homeodomains (Supplementary Figure 2). The conservation of Asn51, which specifies Ade at binding site position 3 (Fraenkel et al., 1998; Wolberger et al., 1991), in combination with our ability to infer the orientation of each homeodomain on its binding site (Supplementary Figure 6 and Supplementary Table 2) provides a basis for aligning all of these recognition sequences. Using this master alignment (Supplementary Table 3), hierarchical clustering of the D. melanogaster homeodomains was performed based on the similarity of their DNA-binding specificities (Figure 2A). The majority of these factors can be organized into eleven different specificity groups and the average specificity of these groups was determined for the purposes of comparison (Figure 2). In this analysis, we used only the core 6 base pair element recognized by these factors. Consistent with the idea that many homeodomain proteins prefer similar TAAT-related motifs, slightly more than half (43) of the homeodomains fall into the Antp or En specificity groups. There are also a number of specificity groups, such as the Abd-B and NK-1 group, which differ in sequence preference from the Antp or En groups at only one or two positions. However, other groups, such as the TGIF-Exd group, differ at four positions relative to the Antp or En groups. Outside of these specificity groupings are six factors that exhibit unique specificities. The observed diversity of specificities reveals the adaptability of the homeodomain architecture for the recognition of a variety of DNA sequences.
Figure 2.

Clustering of the 84 Drosophila homeodomains. (A) Clustering based on the similarity between the recognition motifs of these factors, which we have organized into eleven different specificity groups. (B) The typical and atypical homeodomains are distributed into separate groups. The average specificity of each group is indicated under the Group recognition motif, and to the right is the Sequence logo of the key recognition positions. (C) The specificity groups (colored rectangles) are mapped onto the homeodomain amino acid sequence similarity tree. In instances where neighbors have been assigned to different specificity groups (indicated by red brackets) any difference in residue type at a key recognition position (5, 47, 50, 54 or 55) is noted (ND = No difference).
Clustering the D. melanogaster homeodomains by specificity has revealed that homeodomains that share strong amino acid sequence similarity are not always found in the same specificity group (Figure 2C). In 10 examples, two factors share strong sequence similarity, but fall into different specificity groups. In eight of these comparisons, this difference can be explained by the presence of a different residue at one or more of the key DNA-recognition positions (5, 47, 50, 51, 54 and 55, see below). Pairs of factors with high overall sequence similarity, but different specificities, may represent recently diverged gene duplications where one factor has acquired new target genes.
Distinguishing features of homeodomain specificity groups
The contribution of specific residues toward binding site preference for one or more group members has been demonstrated in previous studies. Below, we use correlations between the average group recognition motifs and the amino acid distributions at key DNA recognition positions (Figure 2B) to systematically describe the characteristics of each group that lead to differences in binding specificity.
Typical superclass
Antp and En groups
The largest groups of homeodomains provide a reference point to describe how differences in amino acid sequence correlate with DNA-binding specificity. The Antp and En groups share similar recognition motifs and amino acid distributions at the key recognition positions. However, at binding site position 5, the En group prefers Thy, whereas the Antp group tolerates either Gua or Thy. There is a corresponding difference at amino acid position 54: Ala for the En group and Met for the Antp group. In the Antp-DNA structure, the side chain of Met54 is neighboring this base pair (Fraenkel and Pabo, 1998).
Bcd group
Typical homeodomains utilize Lys50 to specify Cyt at binding site positions 5 and 6 through the interaction of Lys50 with the complementary Gua at these positions (Tucker-Kellogg et al., 1997).
NK-1, Bar and Ladybird groups
Many of these homeodomains are members of the NK or DL homeodomain classes (Banerjee-Basu and Baxevanis, 2001) and generally have Thr at position 47 or 54. Compared to the Antp and En groups, the homeodomains with Thr47 have reduced specificity at binding site positions 4 and/or 5 (Supplementary Figure 7).
NK-2 group
The members of this group prefer Gua at position 4, due to an interaction between Tyr54 and the complementary Cyt (Gruschus et al., 1997). Their specificities vary at binding site position 1, which correlates with differences at residues 6 and 7 of the N-terminal arm (Damante et al., 1996) (Supplementary Figure 8).
Abd-B group
These factors prefer Thy over Ade at position 2. In Abd-B, this preference has been mapped to amino acid positions 3, 6 and 7 of the N-terminal arm (Ekker et al., 1994); however, the variability within the N-terminal arm precludes a simple correlation of binding preference and amino acid sequence.
Atypical homeodomains
The atypical groups generally prefer Gua at binding site position 2, and Cyt and Ade at positions 4 and 5 (Figures 2B and 3A). In CG11617, the Iroquois group and the TGIF group, the preference for Cyt and Ade at positions 4 and 5 correlates with the presence of Arg54, consistent with the structure of MATα2 (Wolberger et al., 1991) (Figure 3B). The single exception to this correlation, Onecut, contains a unique residue (Met50), which may contribute to its distinct binding preference. Likewise, with the exception of the Iroquois group, homeodomains that contain Arg55 prefer Gua at position 2, consistent with the Exd and Pbx structures (Passner et al., 1999; Piper et al., 1999).
Figure 3.
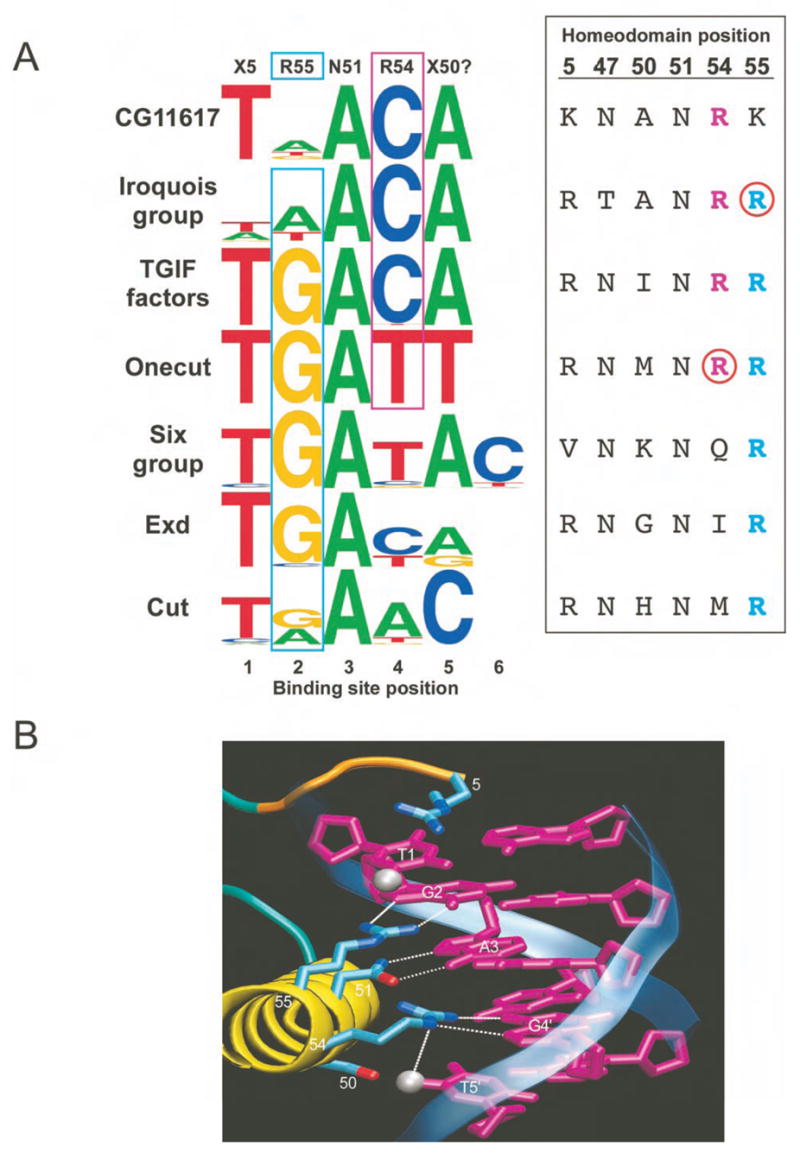
Atypical homeodomain specificity and correlations with positions 54 and 55. A) (Left) Sequence logos for types of atypical homeodomains (either groups or outliers). (Right) The corresponding amino acid sequences at the key DNA contact positions. Arg at position 54 (magenta) correlates with a preference for Cyt at binding site position 4. Arg at position 55 (cyan) correlates with a preference for Gua at binding site position 2. Notable exceptions are indicated by red circles. B) Structural model of DNA recognition for atypical family members constructed from a superposition of the contacts observed in the MATα2-DNA (Wolberger et al., 1991) and Exd-Ubx-DNA structures (Passner et al., 1999). The arginines potentially specify the contacted Gua and the 5′ Thy due to the favorable van der Waals interaction (~4 Å) with the T-methyl group (silver sphere).
TGIF-Exd group
Our data are consistent with previously described specificities for individual members of the TGIF - Exd group (TGA(C/t)A) (Bertolino et al., 1995; Chang et al., 1996).
Six group
All members of this group (So, Six4 and Optix) display a specificity that overlaps with the recognition motif TGATAC and share identical residues at the key DNA-recognition positions (47, 50, 51, 54 and 55). Our data are consistent with a known So motif ((T/C)GATAC) (Hazbun et al., 1997). A discrepancy between our data and a motif (TAAT) reported for an Optix homolog, Six3 (Zhu et al., 2002), is investigated in the analysis of human homeodomains described below.
Iroquois group
Our monomeric motif (ACA) reflects part of the palindromic, homodimer binding site (ACANNTGT) for a full-length Mirr protein (Bilioni et al., 2005). Homeodomains in this group have weak preferences at binding site positions 1 and 2, despite containing notable specificity determinants (Arg5 and Arg55). One striking feature of the Iroquois group is Ala at position 8 (Supplementary Figure 3). In other homeodomains, a large hydrophobic residue at this position binds in a cleft formed by the homeodomain helices and appears to position the N-terminal arm over the 5′ end of the binding site (Figure 4). To examine the effect of residue 8 on Iroquois specificity, an Ala8Phe mutation was introduced into Caup (Figure 4D). This mutation restores, albeit incompletely, the anticipated specificity at positions 1 and 2. The incomplete transformation suggests that additional determinants also contribute to specificity at the 5′ end of the binding site (Supplementary Figure 9).
Figure 4.
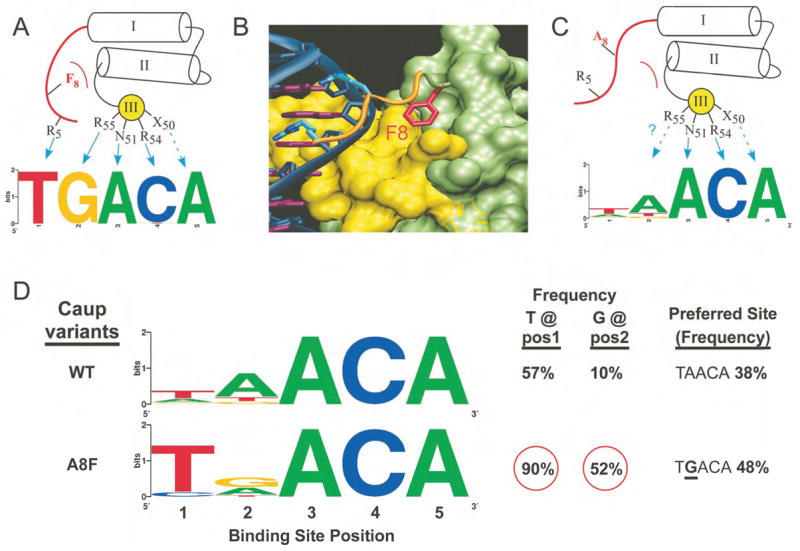
The role of position 8 in organizing the N-terminal arm. A) A large hydrophobic residue at position 8 docks into a pocket formed by the three-helix bundle of the homeodomain fold anchoring the N-terminal arm over the minor groove. B) Surface rendering of the homeodomain (residues 9–60, recognition helix shown in yellow; Msx-1 structure (Hovde et al., 2001)). Phe8 (red) sits in a structural pocket. C) Iroquois family members contain Ala at position 8, allowing the N-terminal arm to sample other conformations that reduce the specificity of the factor. D) Reintroduction of the Phe at position 8 in Caup (A8F) dramatically alters the specificity of the protein at positions 1 and 2 of the binding site.
Our assessment of the typical and atypical superclasses suggests two overlapping, but distinct sets of protein-DNA interactions (Figure 2B and 3B). Both classes generally share Arg5 and Asn51, which typically specify Thy and Ade at binding site positions 1 and 3, as well as common set of phosphate contacting residues (Supplementary Figure 3), which should result in a similar docking arrangement of all of these homeodomains with the DNA. Thus, specificity differences between these homeodomains primarily arise from distinct combinations of residues that directly interact with DNA or that influence these contact residues, rather than changes in the overall conformation of the homeodomain-DNA complex.
Common specificity determinants for homeodomain proteins
Computational and qualitative approaches were used to decipher how variations in homeodomain amino acid sequences across all specificity groups lead to differences in the preferred bases at each binding site position. Mutual information (MI) analysis was used to identify potential specificity determinants by evaluating homeodomain residues that co-vary with changes in binding site preferences (Gutell et al., 1992; Mahony et al., 2007). A simple MI analysis identified some expected correlations at the protein/DNA interface (Supplementary Table 4), but was complicated by the limited variability at some individual positions (Supplementary Figure 10A). To compensate for differences in variability, the MI matrix was transformed into a joint rank product matrix (Supplementary Figure 10B). This plot identifies many known homeodomain-DNA interactions; for example, strong MI is observed between recognition helix positions 50 and 54 and binding site positions 6 and 4, respectively. However, a strong correlation between residue 47 and binding site position 2 is likely due to evolutionary linkage; the residue present at position 47 correlates to the superclass of the homeodomain (atypical or typical) and each superclass typically prefers different bases at this position. Although evolutionary history complicates MI analysis, novel positions are identified that may be new hallmarks for predicting binding specificity.
To identify which amino acids lead to different binding site preferences, we examined the correlations between amino acid sequence and recognition preference in the context of homeodomain structures and existing or new mutagenesis experiments. The keystone for this analysis is recognition of Ade at position 3 by Asn51. Inferences about specificity determinants may not be valid in the absence of this interaction. Below, residues that most frequently contribute to specificity are summarized for each position in the binding site (Figure 5) and a more detailed analysis is available in the supplementary discussion.
Figure 5.
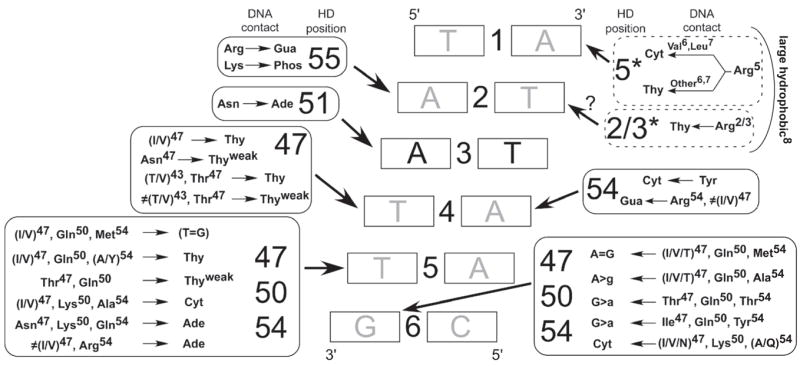
Catalog of common specificity determinants for Asn51-containing homeodomains. Amino acid positions that are most likely to influence the sequence preference at a particular position are indicated in boxes (solid line – major groove, dotted line – minor groove) surrounding the core 6 bp binding element. An arrow points from the box of potential interactions to the base within each base pair that it describes. For simplicity some interactions, such as Lys50 with binding site positions 5 and 6, are described as influencing specificity on the primary strand of the DNA when in reality direct contacts are made to the complementary strand. DNA recognition by residues in the N-terminal arm is also dependent on the type of residue at position 8 as observed for the Iroquois group.
Binding Site (BS) Position 1
89% of the aligned recognition sequences have Thy at this position. Consistent with this preference, the majority of homeodomains (94%) have Arg5 in the N-terminal arm, which specifies Thy (Ades and Sauer, 1995).
BS Position 2
Preferences for Ade, Gua or Thy are observed among the different homeodomains. 83% of the aligned recognition sequences have Ade at this position. Most typical homeodomains contain Arg2 or Arg3, which help specify Ade (Ades and Sauer, 1995; Hovde et al., 2001). Most atypical homeodomains contain Arg55, which can specify Gua.
BS Position 3
Asn51 specifies Ade at this position.
BS Position 4
Any base can be specified at this position. Thy is the most common base (80%) and is strongly correlated with the presence of Ile or Val at position 47.
BS Position 5
Preferences for Ade, Thy and Cyt are observed among different homeodomains. For many specificity groups, correlations exist between combinations of residues at positions 47, 50 and 54 and certain base preferences.
BS Position 6
Preferences for Ade, Gua and Cyt are observed among the different homeodomains. Like binding site position 5, residues at positions 47, 50 and 54 appear to be the primary determinants of specificity.
These results imply that there is rarely a simple one-to-one correlation between a specific residue and the preferred base at a binding site position. This complexity precludes the construction of a basic “recognition code” that defines specificity based on a subset of residues at key recognition positions; however, this analysis reveals some general principles regarding how certain combinations of residues influence specificity. Multiple homeodomain positions can contact a single base pair (e.g. residues 47 and 54 at base position 4 and residues 3 and 55 at base position 2), and when more than one determinant is present for a single base pair, these residues can be in competition (see next section). In addition, other residues can indirectly contribute to specificity by influencing the conformation of potential contact residues. For example Ala8 affects specificity in the N-terminal arm (Figure 4). Similarly, Lys50 displays distinct base preferences in the Bcd and Six groups, likely due to different neighboring residues at positions 47 and 54. These examples support the general conclusion that the contribution of individual specificity determinants to DNA recognition is modulated by additional residues at the protein-DNA interface.
Bcd uses competing contact residues
We have used Bcd to explore the role of competition in determining specificity, as it contains Ile47 and Arg54, which can specify Thy and Cyt, respectively, at binding site position 4. At this position, Bcd displays a strong preference for Thy, a weak preference for Gua and no evidence of tolerance for Cyt (Figure 6A and Supplementary Figure 11). The weak preference for Gua at position 4 has been previously demonstrated (Dave et al., 2000), and is likely due to Lys50, as this residue can interact simultaneously with the carbonyls of the base at position 4 on the primary strand and position 5 on the complementary strand in the context of the consensus binding site, TAATCC (Tucker-Kellogg et al., 1997).
Figure 6.
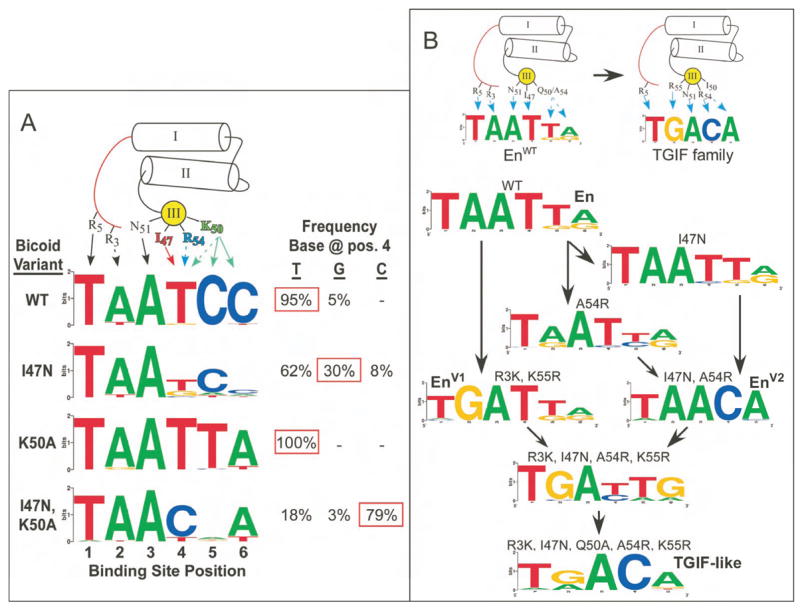
Exploring DNA-binding specificity through mutagenesis. A) Mutational analysis of binding site position 4 in Bcd. Three different mutants (I47N, K50A and I47N with K50A) were characterized to determine the alteration in base preference at this position. The frequency that each base was recovered at position 4 is indicated to the right of the Sequence logo for each factor. B) Conversion of Engrailed (En) into a homeodomain with TGIF-like specificity. (Top) Schematic representation of the critical base contacts responsible for specificity in En and TGIF family members. (Bottom) Flow diagram of the mutations required to complete the specificity conversion. Two intermediate specificity conversions (EnV1 and EnV2) were obtained first, and these mutations were combined along with Q50A to produce TGIF-like specificity.
The absence of Cyt in the recognition motif at position 4 suggests that Ile47 or Lys50 may prevent Arg54 from contributing to the base preference. When Ile47 is mutated to Asn, a residue commonly found in atypical homeodomains that contain Arg54, a slight tolerance for Cyt is observed, indicating the influence of Arg54 (Figure 6A). When Lys50 is mutated to Ala, a complete shift to an En-like specificity (TAATTA) is observed. In the double mutant Ile47Asn and Lys50Ala, a preference for Cyt at position 4 - the base specified by Arg54 in most atypical homeodomains - is revealed. Thus, three different potential specificities are embedded within Bcd. Lys50 and Arg54 are less influential, likely because they are more flexible and are able to make other favorable interactions: Lys50 with bases at positions 5 and 6, and Arg54 with the phosphodiester backbone.
Engineering the DNA-binding specificity of En
We used our catalog of specificity determinants to shift the specificity of En from a typical homeodomain (TAATTA) to a TGIF-type atypical homeodomain (TGACA). En and TGIF differ in binding site preference at four out of six positions (Figure 6B) and share only 28% amino acid sequence identity overall. While homeodomain specificities have been previously altered at one or two binding site positions, attempts to produce more dramatic changes have failed (Mathias et al., 2001).
Two partial conversions were performed in parallel to assess the flexibility of the En-scaffold for each end of the binding site (Figure 6B): two mutations (R3K and K55R) were sufficient to alter specificity at position 2 (TGATTA) and two other mutations (I47N and A54R) altered specificity at positions 4–6 (TAACA). The combination of both pairs of mutations (R3K, I47N, A54R and K55R) resulted in the desired 5′ specificity, but an intermediate 3′ specificity (TGA(T/C)(T/A)(G/A); Figure 6B), which suggests additional competing specificity determinants. Gln50, although passive in the I47N, A54R mutant, might influence specificity in the quadruple mutant context. Indeed, addition of the Q50A mutation creates an almost complete conversion to the desired TGACA specificity, as demonstrated by motif clustering analysis (Supplementary Figure 12). The intermediate and final transformations of binding specificity demonstrate that En is a robust scaffold for engineering novel DNA-binding specificities (Supplementary Figure 13). In addition, these results highlight how the impact of an individual specificity determinant (i.e. Gln50) can be influenced by its context at the homeodomain-DNA interface.
Predicting the specificity of the human homeodomains
We used our analysis of Drosophila homeodomain specificities to predict the specificity of most human homeodomain proteins. Pairs of homeodomains with the highest overall sequence similarity can have different specificities, likely due to differences at their key recognition positions (Figure 2C). Therefore, three criteria were employed in making predictions for the independent human homeodomains: 1) the presence of Asn51, 2) the overall sequence similarity of each human homeodomain to each fly homeodomain, and 3) the number of identical residues at five recognition positions (5, 47, 50, 54 and 55). The recognition motifs for 153 of 193 human homeodomains (79%) were constructed from the selected binding sites of up to three fly factors that share the highest overall sequence homology and the most similar recognition residues (Supplementary Figure 14). A cross-validation test with the fly homeodomain set was used to assess the accuracy of these predictions (Supplementary Table 5). The human predictions were binned into four confidence levels based on the cross-validation analysis (Supplementary Table 6) from highest (1) to lowest (4). 113 (74%) of the predictions fall in the top two confidence levels. These predictions were confirmed for six human homeodomains (BarHL1, Nkx3-2, PitX2, Six3, TGIF2, Vsx1) by determining their specificities using the B1H system (Figure 7). The determined and predicted specificities are very similar (all p-values < 2×10−6), indicating that this approach should be applicable to homeodomains from a broad range of species. This conclusion is supported by an independent comparison with the specificities for non-fly homeodomains in TRANSFAC (Matys et al., 2003) with our predicted specificities for these factors (Supplementary Table 7). Predictions of homeodomain specificities from other species can be made through our web-page where a user enters the homeodomain amino acid sequence and a recognition motif is generated if homeodomains are present in our dataset that meet the user-defined criteria (Supplementary Figure 15). Our specificity predictions for the human homeodomain set, their corresponding PWMs, and the interactive prediction tool are available at http://ural.wustl.edu/flyhd.
Figure 7.

Comparison of the predicted and determined recognition motifs for 6 human homeodomains. The specificities of the human factors were determined using the B1H system. In each case the “Determined” compares favorably with the “Predicted” motif generated using our algorithm. The p-value for each comparison was calculated from the weight matrices for each motif as described in the Methods with additional metrics of these comparisons in Supplementary Table 9. Of particular note, the specificity of Six3 is consistent with other Six family members; it does not specify TAAT as previously described (Zhu et al., 2002).
Discussion
A major limitation for understanding transcriptional regulation in animal cells is the paucity of defined specificities for the majority of encoded transcription factors. The B1H system offers many potential advantages for the analysis of transcription factor specificity. First, selected binding sites are assayed for the ability to activate a biological response in the context of competition from a pool of potential sites in the E. coli genome. More importantly, the ability to determine the orientation of the homeodomain on each selected binding site allows even partially symmetric sites to be properly aligned when constructing recognition motifs (Supplementary Figure 6). Correct alignment of selected sites is not only important for ranking predicted recognition sequences in genomic DNA sequences, but it is also required to understand the structural basis for variations in DNA binding specificity.
This study provides a complete analysis of homeodomain specificities in a metazoan and it dramatically increases the number of characterized homeodomains in this organism, as only 18 of 84 had any binding site information in the FlyREG database (Bergman et al., 2005). We find that the homeodomain family displays an extensive range of specificities in which a wide variety of bases can be preferred at most positions within the core 6 bp binding site. Overall, the majority of homeodomains (93%) in our dataset can be clustered into 11 different specificity groups with an additional 6 homeodomains that display unique specificities. This clustering strategy allowed us to describe how common variations in residues at a given position in the homeodomain contribute to differences in specificity. However, even within these groups there are homeodomains that display differences in binding site preference. For example, members of the NK-2 group differ in their base preference at the 5′-most position and Exd specificity clearly differs from other members of the TGIF group (Supplementary Figure 8, Figure 3A). In addition, differences outside the core 6 base pair binding site motifs lead to further diversity among homeodomain specificities (Supplementary Figure 2). Thus, the 17 specificities described by the 11 groups and 6 unique homeodomains represent the minimum number of different specificities recognized by Drosophila homeodomains.
Our analysis demonstrates that the overall sequence similarity between two homeodomains is a useful, but sometimes misleading indicator of the degree of similarity in their DNA-binding specificities. Once factors are clustered into specificity groups, it is possible to compare binding specificity with their degree of sequence homology (Figure 2C). As expected, a substantial correlation between sequence similarity and preferred recognition motif is observed. However, we find multiple examples where pairs of closely related homeodomains cluster into different specificity groups. In both naturally-occurring and engineered homeodomains, single amino acid changes at putative DNA recognition positions are sufficient to alter specificity. These observations illustrate the importance of defining the amino acid positions that contribute to variations in binding site specificity in order to make accurate specificity predictions.
In addition to providing a better understanding of DNA-recognition for this family, this dataset provides a resource for the prediction and interpretation of homeodomain binding sites in regulatory targets within the D. melanogaster genome. The specificity of individual homeodomains has proven instrumental in the identification of functional regulatory sites utilized by these factors in vivo (a subset of examples in D. melanogaster are listed in Supplementary Table 8) and in the computational identification of target genes with evolutionarily conserved binding sites (Berman et al., 2004; Kheradpour et al., 2007; Schroeder et al., 2004; Sinha et al., 2003). Comparisons with chromatin immunoprecipitation (ChIP) data confirm that Bicoid monomer binding sites are enriched at sites that are occupied in vivo (Li et al., 2008) and that the combination of ChIP data and analysis of conserved transcription factor binding sites generally provides significant improvement in the prediction of functional targets over either method alone (Kheradpour et al., 2007). The complete analysis of D. melanogaster specificities also highlights the importance of identifying factors with overlapping specificities, as conserved binding sites may reflect recognition sequences for a number of potential factors.
Homeodomains can bind DNA as monomers, homodimers, heterodimers or higher order complexes; in several examples, the preferred recognition sequence of monomers in these complexes may even be modified (Pearson et al., 2005; Ryoo and Mann, 1999; Wilson and Desplan, 1999). Both structural data and our analysis suggest that a likely site for modified specificities is in the flexible N-terminal arm (Figures 1, 2 and 4). The recently described structures of Scr-Exd heterodimers bound to DNA reveal how complex formation can alter the interaction of residues within and beyond the N-terminal arm with DNA (Joshi et al., 2007). Thus, while the primary sequence determinants within the N-terminal arm help define sequence preferences, intramolecular (e.g. Ala8 in Caup; Figure 4) or intermolecular (e.g. Scr-Exd) interactions can also influence recognition. It is currently unclear how frequently monomeric specificities are modified by protein-protein interactions, but our systematic characterization of monomeric specificities provides a foundation to explore this question.
The analysis of homeodomain specificities in D. melanogaster also provides the basis to predict most homeodomains specificities in other organisms. We predicted the DNA-binding specificities of 79% of the independent homeodomains in the human genome with moderate to high confidence (Supplementary Figure 14). This prediction scheme can be applied to homeodomains from any species, providing a resource to help identify binding sites in cis-regulatory regions. In the future, incorporation of a probabilistic recognition code to approximate the specificities of factors that do not have good homologs in our database should allow more comprehensive specificity predictions based on homeodomain amino acid sequence (Benos et al., 2002; Liu and Stormo, 2005).
Continued analysis of homeodomain specificity will lead to more detailed understanding of recognition by this family. Our current experiments have led to a catalogue of specificity determinants that can be used to rationally engineer the DNA-binding specificity of homeodomains. The throughput of the B1H system will facilitate the synthesis of a more comprehensive recognition model as more naturally-occurring and mutant homeodomains are characterized. The B1H system can also be used to perform selections on pools of mutagenized homeodomains to assess the range of residues that are compatible with recognition of a given motif. Given the high success rate of the B1H method, a systematic characterization of other classes of DNA-binding domains can be used to produce a complete map of transcription factor specificities in a genome.
Experimental Procedures
Homeodomain binding site selections
A detailed description of the general B1H selection protocol has been previously described (Meng et al., 2005; Meng and Wolfe, 2006), modifications to this procedure and a detailed description of the construction of the ZF10 randomized library are presented in the Supplementary Methods. The 84 independent D. melanogaster homeodomains were identified as described in Supplementary Methods. The sequences of the homeodomains used in the B1H selection and the raw selected binding sites are found in Supplementary Table 1.
Construction of the master alignment of sites for clustering and MI analysis
The master alignment contains 1860 binding sites for 83 of the 84 Drosophila homeodomain proteins as well as Oct1 (Lag1 was excluded because it lacks Asn51). These alignments were constructed from overrepresented motifs identified for each factor using CONSENSUS (Hertz and Stormo, 1999). Details on the alignment construction, motif clustering and MI analysis can be found in the Supplementary Methods. All Sequence logos (Schneider and Stephens, 1990) for these factors were generated using WebLogo (Crooks et al., 2004). Because the number of selected binding sites that comprise a particular logo is modest (22 on average), the significance of bases that are absent or occur infrequently in a motif cannot be fully assessed.
Specificity Predictions for the human homeodomain set
193 homeodomains containing proteins were annotated in the SMART human genome database and 175 of these were independent homeodomains containing Asn51. To predict the DNA-binding specificity of this set we used the DNA-binding specificity of up to 3 of the fly homeodomains with the highest BLOSUM45 similarity scores (calculated from a sequence-to-profile multiple sequence alignment (Edgar, 2004) between the query sequence and the 84 fly homeodomain profiles) provided that: 1) they contained Asn51; 2) they contained identical residues at the other 5 key recognition positions (5, 47, 50, 54 and 55); and 3) they passed a BLOSUM45 similarity score threshold. The similarity score threshold was set to 200, based on a cross validation analysis of the fly homeodomain set (data not shown). Additionally, once a reference protein passed all of our filters, additional reference proteins were only added to the predictive set if their similarity score was within 40 similarity score units of the most similar reference protein. If no reference homeodomain passed these three criteria, we considered up to 3 homeodomains within the set that contained identical residues at 4 of the 5 key recognition positions, as long as they also passed the similarity score threshold. Specificity predictions comprise all of the selected binding sites for all of the reference homeodomains that passed the filters. In some cases no fly homeodomains met these criteria and consequently no prediction was made.
Cross-validation analysis and comparison of predicted and determined motifs
To assess the accuracy of the specificity predictions we performed a cross-validation analysis where the binding specificity of each fly homeodomain was predicted based on the information of all of the other homeodomain proteins. All TRANSFAC 10.2 datasets associated with proteins classified as homeodomains (TRANSFAC classes C0006, C0027, C0047, C0053) and that contain at least 20 binding sites were extracted from the database (Matys et al., 2006). The 47 groups of binding sites that met these requirements were reanalyzed with CONSENSUS to generate new motifs. 27 of these 47 transcription factors were sufficiently similar to a D. melanogaster homeodomain to make a prediction based on our criteria (described in the text). In some cases (8), multiple homeodomains were associated with one dataset in TRANSFAC and vice versa (5). In these cases, we compared the predicted matrix for a factor to each of the CONSENSUS matrices associated with it. We used the Average Log Likelihood Ratio (ALLR) score to determine the best local alignment (Matalign-v2a, Wang, T & Stormo, G. D. unpublished) between the predicted and CONSENSUS matrices. Based on these alignments, we assessed the degree of similarity using the ALLR similarity score, the ALLR based distance and the e-value computed by Matalign.
Supplementary Material
Acknowledgments
We would like to thank Xiangdong Meng for his valuable advice and technical support. We would like to thank the Berkeley Drosophila Genome Project (BDGP) for producing the cDNA clones used in this study, the Drosophila Genomics Resource Center (DGRC) for distributing the clones, and Mark Stapleton and Susan Celniker for sharing unpublished results. Some of these ORFs were obtained from clones produced by BDGP under National Institutes of Health grant (HG002673 to S. E. Celniker). We would like to thank Adam Richards for technical support. S.A.W. and M.B.N. were supported by NIH grant 1R21HG003721 from NHGRI. A.W. was supported in part by NIH grant 1R21HG003721 from NHGRI. M.H.B. and A.W. were supported in part by a New Scholar in Aging Award from the Ellison Medical Foundation and American Cancer Society grant RSG-05-026-01-CCG. R.G.C. was supported by training grant T32 GM08802. G.D.S. was supported by NIH grant HG00249 from NHGRI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ades SE, Sauer RT. Specificity of minor-groove and major-groove interactions in a homeodomain-DNA complex. Biochemistry. 1995;34:14601–14608. doi: 10.1021/bi00044a040. [DOI] [PubMed] [Google Scholar]
- Banerjee-Basu S, Baxevanis AD. Molecular evolution of the homeodomain family of transcription factors. Nucl Acids Res. 2001;29:3258–3269. doi: 10.1093/nar/29.15.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benos PV, Lapedes AS, Stormo GD. Probabilistic code for DNA recognition by proteins of the EGR family. Journal of molecular biology. 2002;323:701–727. doi: 10.1016/s0022-2836(02)00917-8. [DOI] [PubMed] [Google Scholar]
- Bergman CM, Carlson JW, Celniker SE. Drosophila DNase I footprint database: a systematic genome annotation of transcription factor binding sites in the fruitfly, Drosophila melanogaster. Bioinformatics (Oxford, England) 2005;21:1747–1749. doi: 10.1093/bioinformatics/bti173. [DOI] [PubMed] [Google Scholar]
- Berman BP, Pfeiffer BD, Laverty TR, Salzberg SL, Rubin GM, Eisen MB, Celniker SE. Computational identification of developmental enhancers: conservation and function of transcription factor binding-site clusters in Drosophila melanogaster and Drosophila pseudoobscura. Genome Biol. 2004;5:R61. doi: 10.1186/gb-2004-5-9-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolino E, Reimund B, Wildt-Perinic D, Clerc RG. A novel homeobox protein which recognizes a TGT core and functionally interferes with a retinoid-responsive motif. J Biol Chem. 1995;270:31178–31188. doi: 10.1074/jbc.270.52.31178. [DOI] [PubMed] [Google Scholar]
- Bilioni A, Craig G, Hill C, McNeill H. Iroquois transcription factors recognize a unique motif to mediate transcriptional repression in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:14671–14676. doi: 10.1073/pnas.0502480102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CP, Brocchieri L, Shen WF, Largman C, Cleary ML. Pbx modulation of Hox homeodomain amino-terminal arms establishes different DNA-binding specificities across the Hox locus. Mol Cell Biol. 1996;16:1734–1745. doi: 10.1128/mcb.16.4.1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome research. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damante G, Pellizzari L, Esposito G, Fogolari F, Viglino P, Fabbro D, Tell G, Formisano S, Di Lauro R. A molecular code dictates sequence-specific DNA recognition by homeodomains. Embo J. 1996;15:4992–5000. [PMC free article] [PubMed] [Google Scholar]
- Dave V, Zhao C, Yang F, Tung CS, Ma J. Reprogrammable recognition codes in bicoid homeodomain-DNA interaction. Mol Cell Biol. 2000;20:7673–7684. doi: 10.1128/mcb.20.20.7673-7684.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove SL, Hochschild A. Conversion of the omega subunit of Escherichia coli RNA polymerase into a transcriptional activator or an activation target. Genes Dev. 1998;12:745–754. doi: 10.1101/gad.12.5.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekker SC, Jackson DG, von Kessler DP, Sun BI, Young KE, Beachy PA. The degree of variation in DNA sequence recognition among four Drosophila homeotic proteins. EMBO J. 1994;13:3551–3560. doi: 10.1002/j.1460-2075.1994.tb06662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekker SC, von Kessler DP, Beachy PA. Differential DNA sequence recognition is a determinant of specificity in homeotic gene action. EMBO J. 1992;11:4059–4072. doi: 10.1002/j.1460-2075.1992.tb05499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraenkel E, Pabo CO. Comparison of X-ray and NMR structures for the Antennapedia homeodomain-DNA complex. Nat Struct Biol. 1998;5:692–697. doi: 10.1038/1382. [DOI] [PubMed] [Google Scholar]
- Fraenkel E, Rould MA, Chambers KA, Pabo CO. Engrailed homeodomain-DNA complex at 2.2 A resolution: a detailed view of the interface and comparison with other engrailed structures. Journal of molecular biology. 1998;284:351–361. doi: 10.1006/jmbi.1998.2147. [DOI] [PubMed] [Google Scholar]
- Gehring WJ, Affolter M, Burglin T. Homeodomain proteins. Annu Rev Biochem. 1994a;63:487–526. doi: 10.1146/annurev.bi.63.070194.002415. [DOI] [PubMed] [Google Scholar]
- Gehring WJ, Qian YQ, Billeter M, Furukubo-Tokunaga K, Schier AF, Resendez-Perez D, Affolter M, Otting G, Wuthrich K. Homeodomain-DNA recognition. Cell. 1994b;78:211–223. doi: 10.1016/0092-8674(94)90292-5. [DOI] [PubMed] [Google Scholar]
- Gruschus JM, Tsao DH, Wang LH, Nirenberg M, Ferretti JA. Interactions of the vnd/NK-2 homeodomain with DNA by nuclear magnetic resonance spectroscopy: basis of binding specificity. Biochemistry. 1997;36:5372–5380. doi: 10.1021/bi9620060. [DOI] [PubMed] [Google Scholar]
- Gutell RR, Power A, Hertz GZ, Putz EJ, Stormo GD. Identifying constraints on the higher-order structure of RNA: continued development and application of comparative sequence analysis methods. Nucl Acids Res. 1992;20:5785–5795. doi: 10.1093/nar/20.21.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanes SD, Brent R. DNA specificity of the bicoid activator protein is determined by homeodomain recognition helix residue 9. Cell. 1989;57:1275–1283. doi: 10.1016/0092-8674(89)90063-9. [DOI] [PubMed] [Google Scholar]
- Hazbun TR, Stahura FL, Mossing MC. Site-specific recognition by an isolated DNA-binding domain of the sine oculis protein. Biochemistry. 1997;36:3680–3686. doi: 10.1021/bi9625206. [DOI] [PubMed] [Google Scholar]
- Hertz GZ, Stormo GD. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics (Oxford, England) 1999;15:563–577. doi: 10.1093/bioinformatics/15.7.563. [DOI] [PubMed] [Google Scholar]
- Hovde S, Abate-Shen C, Geiger JH. Crystal structure of the Msx-1 homeodomain/DNA complex. Biochemistry. 2001;40:12013–12021. doi: 10.1021/bi0108148. [DOI] [PubMed] [Google Scholar]
- Joshi R, Passner JM, Rohs R, Jain R, Sosinsky A, Crickmore MA, Jacob V, Aggarwal AK, Honig B, Mann RS. Functional specificity of a Hox protein mediated by the recognition of minor groove structure. Cell. 2007;131:530–543. doi: 10.1016/j.cell.2007.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradpour P, Stark A, Roy S, Kellis M. Reliable prediction of regulator targets using 12 Drosophila genomes. Genome research. 2007;17:1919–1931. doi: 10.1101/gr.7090407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissinger CR, Liu B, Martin-Blanco E, Kornberg TB, Pabo CO. Crystal structure of an engrailed homeodomain-DNA complex at 2.8 A resolution: A framework for understanding homeodomain-DNA interactions. Cell. 1990;63:579–590. doi: 10.1016/0092-8674(90)90453-l. [DOI] [PubMed] [Google Scholar]
- Lewis EB. A gene complex controlling segmentation in Drosophila. Nature. 1978;276:565–570. doi: 10.1038/276565a0. [DOI] [PubMed] [Google Scholar]
- Li XY, Macarthur S, Bourgon R, Nix D, Pollard DA, Iyer VN, Hechmer A, Simirenko L, Stapleton M, Hendriks CL, et al. Transcription Factors Bind Thousands of Active and Inactive Regions in the Drosophila Blastoderm. PLoS biology. 2008;6:e27. doi: 10.1371/journal.pbio.0060027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Stormo GD. Combining SELEX with quantitative assays to rapidly obtain accurate models of protein-DNA interactions. Nucleic acids research. 2005;33:e141. doi: 10.1093/nar/gni139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahony S, Auron PE, Benos PV. Inferring protein-DNA dependencies using motif alignments and mutual information. Bioinformatics (Oxford, England) 2007;23:i297–304. doi: 10.1093/bioinformatics/btm215. [DOI] [PubMed] [Google Scholar]
- Mathias JR, Zhong H, Jin Y, Vershon AK. Altering the DNA-binding specificity of the yeast Matalpha 2 homeodomain protein. J Biol Chem. 2001;276:32696–32703. doi: 10.1074/jbc.M103097200. [DOI] [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, et al. TRANSFAC(R): transcriptional regulation, from patterns to profiles. Nucl Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Brodsky MH, Wolfe SA. A bacterial one-hybrid system for determining the DNA-binding specificity of transcription factors. Nat Biotechnol. 2005;23:988–994. doi: 10.1038/nbt1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Wolfe SA. Identifying DNA sequences recognized by a transcription factor using a bacterial one-hybrid system. Nat protocols. 2006;1:30–45. doi: 10.1038/nprot.2006.6. [DOI] [PubMed] [Google Scholar]
- Mukherjee K, Burglin TR. Comprehensive Analysis of Animal TALE Homeobox Genes: New Conserved Motifs and Cases of Accelerated Evolution. J Mol Evol. 2007;65:137–153. doi: 10.1007/s00239-006-0023-0. [DOI] [PubMed] [Google Scholar]
- Passner JM, Ryoo HD, Shen L, Mann RS, Aggarwal AK. Structure of a DNA-bound Ultrabithorax-Extradenticle homeodomain complex. Nature. 1999;397:714–719. doi: 10.1038/17833. [DOI] [PubMed] [Google Scholar]
- Pearson JC, Lemons D, McGinnis W. Modulating Hox gene functions during animal body patterning. Nat Rev Genet. 2005;6:893–904. doi: 10.1038/nrg1726. [DOI] [PubMed] [Google Scholar]
- Percival-Smith A, Müller M, Affolter M, Gehring WJ. The interaction with DNA of wild-type and mutant fushi tarazu homeodomains. EMBO J. 1990;9:3967–3974. doi: 10.1002/j.1460-2075.1990.tb07617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper DE, Batchelor AH, Chang CP, Cleary ML, Wolberger C. Structure of a HoxB1-Pbx1 heterodimer bound to DNA: role of the hexapeptide and a fourth homeodomain helix in complex formation. Cell. 1999;96:587–597. doi: 10.1016/s0092-8674(00)80662-5. [DOI] [PubMed] [Google Scholar]
- Pomerantz JL, Sharp PA, Pabo CO. Structure-based design of transcription factors. Science. 1995;267:93–96. doi: 10.1126/science.7809612. [DOI] [PubMed] [Google Scholar]
- Ryoo HD, Mann RS. The control of trunk Hox specificity and activity by Extradenticle. Genes Dev. 1999;13:1704–1716. doi: 10.1101/gad.13.13.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucl Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder MD, Pearce M, Fak J, Fan H, Unnerstall U, Emberly E, Rajewsky N, Siggia ED, Gaul U. Transcriptional control in the segmentation gene network of Drosophila. PLoS biology. 2004;2:E271. doi: 10.1371/journal.pbio.0020271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, van Nimwegen E, Siggia ED. A probabilistic method to detect regulatory modules. Bioinformatics (Oxford, England) 2003;(19 Suppl 1):i292–301. doi: 10.1093/bioinformatics/btg1040. [DOI] [PubMed] [Google Scholar]
- Treisman J, Gönczy P, Vashishtha M, Harris E, Desplan C. A single amino acid can determine the DNA binding specificity of homeodomain proteins. Cell. 1989;59:553–562. doi: 10.1016/0092-8674(89)90038-x. [DOI] [PubMed] [Google Scholar]
- Tucker-Kellogg L, Rould MA, Chambers KA, Ades SE, Sauer RT, Pabo CO. Engrailed (Gln50-->Lys) homeodomain-DNA complex at 1.9 A resolution: structural basis for enhanced affinity and altered specificity. Structure. 1997;5:1047–1054. doi: 10.1016/s0969-2126(97)00256-6. [DOI] [PubMed] [Google Scholar]
- Tupler R, Perini G, Green MR. Expressing the human genome. Nature. 2001;409:832–833. doi: 10.1038/35057011. [DOI] [PubMed] [Google Scholar]
- Wilson DS, Desplan C. Structural basis of Hox specificity. Nat Struct Biol. 1999;6:297–300. doi: 10.1038/7524. [DOI] [PubMed] [Google Scholar]
- Wolberger C. Homeodomain interactions. Curr Opin Struct Biol. 1996;6:62–68. doi: 10.1016/s0959-440x(96)80096-0. [DOI] [PubMed] [Google Scholar]
- Wolberger C, Vershon AK, Liu B, Johnson AD, Pabo CO. Crystal structure of a MATalpha2 homeodomain-operator complex suggests a general model for homeodomain-DNA interactions. Cell. 1991;67:517–536. doi: 10.1016/0092-8674(91)90526-5. [DOI] [PubMed] [Google Scholar]
- Zhu CC, Dyer MA, Uchikawa M, Kondoh H, Lagutin OV, Oliver G. Six3-mediated auto repression and eye development requires its interaction with members of the Groucho-related family of co-repressors. Development (Cambridge, England) 2002;129:2835–2849. doi: 10.1242/dev.129.12.2835. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.