

Abstract
Glycosyl hydrolases hydrolyze the glycosidic bond either in carbohydrates or between carbohydrate and non-carbohydrate moiety. The β-glucuronidase (beta D-glucuronoside glucuronosohydrolase; EC 3.2.1.31) enzyme belongs to the family-2 glycosyl hydrolase. The E. coli borne β-glucuronidase gene (uidA) was devised as a gene fusion marker in plant genetic transformation experiments. Recent plant transformation vectors contain a novel β-glucuronidase (gusA) derived from Staphylococcus sp. RLH1 for E. coli uidA. It is known to have a ten fold higher sensitivity compared to E. coli β-glucuronidase. The functional superiority of Staphylococcus (gusA) over E. coli (uidA) activity is not fully known. The comparison of secondary structural elements among them revealed an increased percentage of random coils in Staphylococcus β-glucuronidase. The 3D model of gusA shows catalytic site residues 396Glu, 508Glu and 471Tyr of gusA in loop regions. Accessible surface area (ASA) calculations on the 3D model showed increased ASA for active site residues in Staphylococcus β-glucuronidase. Increased random coil, the presence of catalytic residues in loops, greater solvent accessibility of active residues and increased charged residues in gusA of Staphylococcus might facilitate interaction with the solvent. This hypothesizes the enhanced catalytic activity of β-glucuronidase in Staphylococcus sp. RLH1 compared to that in E. coli.
Keywords: β-glucuronidase, structure-function relationships, uidA, GUSPlus, catalytic activity
Background
Glycosyl hydrolases are a widespread group of enzymes hydrolyzing the glycosidic bond in carbohydrates or its derivatives. β-glucuronidase (EC 3.2.1.31) is a glycosyl hydrolase which hydrolyses β-glucuronic acid residues from the non-reducing termini of glycosaminoglycans (GAGs). Protein sequences of β-glucuronidase (GUS) from prokaryotes and eukaryotes are available at NCBI GenPept. In prokaryotes, the GUS of Escherichia coli is a well investigated glycosyl hydrolase. The E. coli β-glucuronidase gene (uidA) has been sequenced, and it is known to encode a stable enzyme [1]. The E. coli GUS is 603 amino acid residues long and it shares about 50% sequence identity with the human GUS with same substrate specificity. The human GUS contains three structural domains, namely, the sugar binding domain, immunoglobulin like beta-sandwich domain and TIM barrel domain [2]. It is also present in E. coli GUS. Jefferson [3] developed a gene fusion system using E. coli GUS gene (uidA) as a marker for analyzing gene expression in plant transformation experiments. The wide use of GUS as a tool in plant gene expression was primarily due to the stability and sensitivity of the reporter assay by providing quantitative and qualitative data on transgene expression. Transgenic plants expressing GUS show histochemical staining with the substrate 5-Bromo-4-chloro-3-indolyl-beta-D-glucuronide (X-Gluc) for GUS assay at pH 7.0.
The enzyme GUS acts by cleaving the β-glucuronic acid from the substrate X-Gluc and it produces blue color in transformed tissues. The recent version of CAMBIA plant transformation vectors (pCAMBIA 1305.1, pCAMBIA 1305.2, pCAMBIA 1105.1, pCAMBIA 1105.1R, pCAMBIA 0105.1R and pCAMBIA 0305.2) carry a novel GUS gene derived from Staphylococcus sp. RLH1. GUSPlus is a synthetic gene (gusA) from Staphylococcus encoding GUS protein detectable at ten-fold higher sensitivity to E. coli GUS [4,5]. The benefits of GUSPlus over E. coli GUS are: (1) greater sensitivity; (2) fast colour development with X-Gluc substrate; and (3) higher thermal stability. However, the molecular basis for the functional difference between GUS from Staphylococcus (GUSPlus) and E. coli is not fully understood. Here, we probe their functional difference using sequence and structural data analysis.
Methodology
GUS sequence data
Amino acid sequences of family-2 β-glucuronidase (beta-D-glucuronoside glucuronosohydrolase; EC 3.2.1.31) for Homo sapiens (NP_000172), Escherichia coli K12 (AAC74689) and Staphylococcus sp. RLH1 (AAK29422) were downloaded from GenPept at NCBI [6].
Sequence comparison
The percent identity shared between β-glucuronidase sequences was found by pairwise alignment using BLOSUM62 matrix [7]. The conserved catalytic site residues in β-glucuronidase were identified based on multiple sequence alignment (Figure 1) using ClustalW [8].
Figure 1.

Multiple sequence alignment of β-glucuronidase from Homo sapiens, E. coli and Staphylococcus sp. RLH1 showing the conserved active site residues: Glutamic acid (E: 451, 394, 396), Tyrosine (Y: 504, 468, 471) and Glutamic acid (E: 540, 504, 508) at corresponding positions as shown in boxes. The identity shared between the β-glucuronidase of Homo sapiens and E. coli is 42.6% and that of Homo sapiens and Staphylococcus sp. RLH1 is 41.8%.
Physical and chemical parameter estimation
The physical and chemical parameters were predicted using the ExPASy ProtParam tool available online as described elsewhere [9]. The computed parameters include the molecular weight, theoretical pI (isoelectric point), amino acid composition, atomic composition, instability index and aliphatic index (Table 1 in supplementary material).
Secondary structure analysis
The GUS sequence from E. coli and Staphylococcus were analyzed for percentage composition of alpha helix, beta strands and random coils (Table 2 in supplementary material) using the GORIV secondary structure prediction method available at NPS@: network protein sequence analysis [10].
3D model building
3D structure of GUS from E. coli and Staphylococcus sp. were predicted using the crystal structure of human GUS as template using Discovery Studio (Accelrys Software Inc, version 1.7). The coordinates of the template human GUS (PDB Id: 1BHG) was downloaded from PDB [11]. Model validation was done using Ramachandran Plot. The resulting structure was energy minimized using the CHARMM force field and the process was repeated till the structure with desired energy levels was obtained. Superposition of the modeled structures of E. coli and Staphylococcus GUS was carried out using the Combinatorial Extension (CE) method and the root mean square deviation (RMSD) was calculated [12].
Accessible surface area (ASA) calculations
The accessible molecular surface was calculated for human, E. coli and Staphylococcus GUS model structures using WHAT IF [13]. The Statistics about accessibilities (SHOACC) option was used to find out the accessibility of residues. The side chain accessibility for the catalytic site residues of human, E. coli and Staphylococcus GUS were plotted as a function of residue position (Figure 2).
Figure 2.
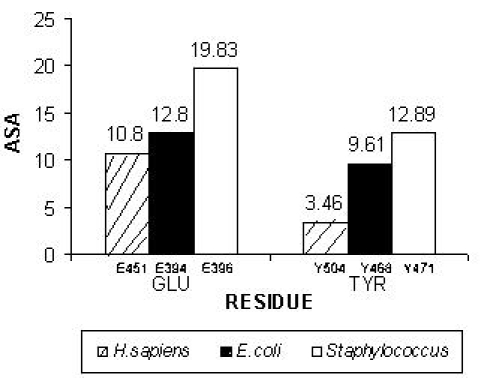
Plot of accessible surface area (ASA) as a function of residue position is shown for active residues. The catalytic sites: Glutamic acid (E: 451, 394, 396) and Tyrosine (Y: 504, 468, 471) corresponding to Homo sapiens, E. coli and Staphylococcus sp. RLH1 is shown. The Staphylococcus sp. RLH1 GUS was found to have high accessible surface area compared to others at the active site residues.
Discussion
The functional superiority of GUS from Staphylococcus over E. coli is intriguing. However, the molecular basis of functional advantage for Staphylococcus GUS is not known. Here, we probe GUS functional dominance in Staphylococcus over E. coli using sequence analysis and predicted 3D models. Pair-wise alignment of E. coli and Staphylococcus GUS revealed an identity of 45.7% and a similarity of 63.5%. Similar alignments of GUS from human and E. coli show 42.6% identity and that of human and Staphylococcus is 41.8% identity. Nevertheless, E. coli and Staphylococcus GUS showed conservation of catalytic site residues at positions corresponding to human GUS. Based on earlier reports for human β-glucuronidase, 451Glu was identified to be the proton donor and 540Glu as the nucleophile which stabilize the carbonium ion in the catalytic reaction [2] and [14]. 504Tyr is located in the active site of human GUS and its role in catalysis has not been defined. Results show conservation of 394Glu, 504Glu, 468Tyr in E. coli and 396Glu, 508Glu, 471Tyr in Staphylococcus GUS at positions corresponding to the catalytic site residues of human GUS (Figure 1). Subsequently, the amino acid sequence of GUS from E. coli and Staphylococcus were analyzed for its physicochemical properties (Table 1 in supplementary material).
The E. coli GUS is 603 residues long with a predicted molecular weight of 68.447 KDa. Similarly, the Staphylococcus GUS is 602 amino acid residues with a molecular weight of 68.701 KDa. Further the instability index predicted for E. coli and Staphylococcus GUS did not show difference between them (instability index provides an estimate of the stability of the enzyme in a test tube). It is known that a protein whose instability index is smaller to 40 is predicted as stable, a value above 40 predicts that the protein may be unstable [15]. However, the significant functional difference observed was with respect to the composition of the charged amino acid residues. The Staphylococcus GUS contains ten negatively charged residues and six positively charged residues more than that in E. coli GUS (Table 1 see supplementary material). The increased percentage of charged residues is likely to enhance hydrophilicity in Staphylococcus GUS favoring interactions with the solvent environment. Further, analysis of the predicted secondary structures of GUS from E. coli and Staphylococcus revealed that they predominantly contain random coils (Table 2 under supplementary material). The Staphylococcus GUS contains slightly more coils than the E. coli GUS. Subsequently, the 3D models GUS from E. coli and Staphylococcus was built with human GUS as template using Discovery Studio. The predicted model is validated using Ramachandran plot for disallowed contacts. Structural superposition (Cα) was carried out between E. coli and Staphylococcus GUS models and a RMSD of 4.4Å was observed.
Analyses of the predicted models were performed to glean information for describing catalytic function. We observed that in E. coli GUS, the catalytic site residues expect for 394Glu were found in the secondary structural regions with 504Glu in the β strand and 468Tyr in the immediate environment of a β strand. However, in Staphylococcus GUS all the three catalytic site residues 396Glu, 508Glu and 471Tyr occur distinctly in the loop segments. The above has relevance to the observation we made based on the secondary structure analysis wherein there is an increased presence of coiled regions in Staphylococcus GUS. Interestingly, all the three catalytic site residues fall within the loop region of the 3D structure. The solvent accessibility of the protein structures as measured by the accessible surface area (ASA) calculations for human, E. coli and Staphylococcus GUS models for understanding of the functional relationship. 396Glu and 471Tyr of Staphylococcus GUS scored the highest ASA of 19.8301 and 12.8921 compared to E. coli (12.8080 and 9.6195) and human (10.8017 and 3.4625) for side-chains of corresponding residues. 396Glu corresponds to 451Glu of the human GUS was known to be the proton donor in the catalytic reaction. Thus, a high ASA for the side chains of Staphylococcus catalytic site residues (396Glu and 471Tyr) is expected to promote an increased interaction with the solvent environment favoring enhanced catalysis (Figure 2).
The increased random coils in Staphylococcus GUS are proposed to have effects on higher rates of catalysis. The studies of Agarwal [16] elucidate the importance of coiled regions in the process of catalysis. Thus, random coils and loop regions account for greater structural flexibility enzyme towards variation dynamics in solution. Solvent surrounding the enzyme play a role in the enzyme reaction. This provides energy for crossing the activation energy barrier. However, in other cases the required energy is provided by the thermodynamical fluctuations of the solvent. The fluctuations in the hydration shell and bulk-solvent surrounding the enzyme are correlated with the internal protein motions. The side-chains of several surface residues extend into the solvent and the motion of these residues is coupled to the motion of surrounding solvent molecules. Thus structural features aid in the internal motions at fast time-scales to control the chemical environment of the active-site favoring the catalytic step to proceed to the product state. Hence, dynamical coupling allows the transfer of energy from the solvent to the surface regions of the enzyme. This energy transfer changes the behavior of reaction trajectories through the network of protein vibrations that is eventually transferred to the active-site to promote catalysis.
Conclusion
Staphylococcus GUS with increased coiling contains all the three catalytic site residues 396Glu, 508Glu and 471Tyr in its loop regions. Moreover, the catalytic residues 396Glu and 471Tyr were found to have greater solvent accessible surface area compared to the corresponding residues in E. coli GUS. The presence of greater number of charged residues Staphylococcus GUS compared to E. coli GUS is an additional feature for explaining functional difference between them. These differential features provide insights to differentiate the observed catalysis rate in GUS from Staphylococcus and E. coli.
Supplementary material
Acknowledgments
This work is supported by Biotechnology Information System (BTIS) of the Department of Biotechnology, Government of India.
References
- 1.Jefferson RA, et al. Proc Natl Acad Sci USA. 1986;83:8447. doi: 10.1073/pnas.83.22.8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jain S, et al. Nat Struct Biol. 1996;3:375. doi: 10.1038/nsb0496-375. [DOI] [PubMed] [Google Scholar]
- 3.Jefferson RA. Pl Mol Biol Rep. 1987;5:387. [Google Scholar]
- 4.Jefferson RA, et al. US patent. 2003;6:391. [Google Scholar]
- 5.Broothaerts W, et al. Nature. 2005;433:629. doi: 10.1038/nature03309. [DOI] [PubMed] [Google Scholar]
- 6. http//ncbi.nlm.nih.gov.
- 7. http://www.ebi.ac.uk/emboss/align.
- 8. http://www.ebi.ac.uk/clustalw.
- 9. http://www.expasy.ch/tools/protparam.html.
- 10. http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_gor4.html.
- 11. http://www.rcsb.org/pdb.
- 12. http://cl.sdsc.edu/ce/ce_align.html.
- 13. http://swift.cmbi.ru.nl/servers/html/index.html.
- 14.Islam MR, et al. J Biol Chem. 1999;274:23451. doi: 10.1074/jbc.274.33.23451. [DOI] [PubMed] [Google Scholar]
- 15.Gasteiger E, et al. The Proteomics Protocols Handbook. New Jersey: Humana press Inc; 2005. [Google Scholar]
- 16.Agarwal PK, et al. Microbial Cell Factories. 2006;5:2. doi: 10.1186/1475-2859-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.