

Abstract
Purpose
Due to the 4–6-month interval between a diagnostic amniocentesis and birth, clinical application of amniotic mesenchymal stem cell (AMSC)-based therapies demands a 3-stage cell manufacturing process, including isolation/primary expansion, cryopreservation, and thawing/secondary expansion. We sought to determine the feasibility and cell yield of such a staged cell manufacturing process, within regulatory guidelines.
Methods
Human AMSCs isolated from diagnostic amniocentesis samples (n=11) were processed under FDA-accredited Good Manufacturing Practice. Expanded cells were characterized by flow cytometry and cryopreserved for 3–5 months. Cell release criteria included: >90% CD29+, CD73+, and CD44+; <5% CD34+ and CD45+; negative mycoplasma QPCR and endotoxin assay; and ≥70% viability.
Results
Isolation and ample expansion of AMSCs was achieved in 54.5% (6/11) of the samples. Early processing and at least a 2mL sample were necessary for reliable cell manufacturing. Cell yield before cryopreservation was 223.2±65.4×106 cells (44.6-fold expansion), plus a 14.7×106-cell backup, after 36.3±7.8 days. Cell viability post-thaw was 88%. Expanded cells maintained a multipotent mesenchymal progenitor profile.
Conclusions
Human amniotic mesenchymal stem cells can be manufactured in large numbers from diagnostic amniocentesis, by an accredited staged processing, under definite procurement guidelines. These data further support the viability of clinical trials of amniotic mesenchymal stem cell-based therapies.
Keywords: amniotic fluid, mesenchymal amniocytes, amniocytes, mesenchymal stem cells, fetal cells, tissue engineering, fetal tissue engineering, cell therapy, cryopreservation, fetus
Among the different progenitor cell lineages naturally occurring in the amniotic fluid, the mesenchymal population has proven valuable in pre-clinical, large animal models of cell-based surgical therapy, through amniotic fluid-based fetal tissue engineering[1–4]. The concept involves the diagnosis of a structural birth defect by prenatal imaging, followed by isolation and expansion of amniotic mesenchymal stem cells (AMSCs) from a routine amniocentesis sample, which are then used to engineer a variety of tissue constructs in parallel to the remainder of gestation, so that a newborn, or fetus, could benefit from the availability of an autologous, custom-made graft for surgical repair of a given congenital anomaly[1–7].
Due to the prolonged 4–6-month interval between a typical diagnostic amniocentesis and birth, clinical translation of this therapeutic concept will require some means of cell preservation and storage during that intervening time period. This is particularly pertinent in light of the fact that, compared to mesenchymal stem cells from other sources, such as bone marrow and umbilical cord blood, AMSCs proliferate the fastest[8]. More specifically, we anticipate the need for a 3-stage cell manufacturing process, to include: isolation/primary expansion; cryopreservation; and thawing/secondary expansion. In this study, we sought to determine the feasibility and cell yield of such a staged cell manufacturing process, within applicable regulatory guidelines.
1. Materials and Methods
This study was approved by Children’s Hospital Boston’s Institutional Review Board, under protocol #S04-12-149. Human amniotic fluid specimens (n = 11) were obtained from routine diagnostic amniocentesis performed between 17 and 24 weeks gestation (1.5 to 7 mL per sample), in a sterile fashion, from fetuses with normal karyotypes. All samples were stored at 4°C until processed at a local, FDA-accredited Good Manufacturing Practice (GMP) facility within 72 hours of procurement. An overview of the staged cell manufacturing process and release criteria can be found in table 1.
Table 1.
Overview of a 3-stage amniotic mesenchymal stem cell manufacturing process as it relates to a tissue engineering application and respective cell release criteria.
| Samples | Tests | Release Criteria |
|---|---|---|
|
Phase I Primary Expansion
Pre-Freeze |
Sterility | No growth for 14 days |
|
Phase II Secondary Expansion
Post-Harvest and prior to scaffold seeding |
Immunophenotyping | <5% CD34+, CD45+ cells
>90% CD29+, CD73+, CD44+ cells |
| Mycoplasma PCR | Negative | |
| Cell Count | >6 ×108 cells | |
| Cell Viability | ≥ 70% | |
| Sterility | No Growth until time of product release | |
| Phase III Pre-Surgical Preparation | Endotoxin | < 5EU/ml |
| Gram Stain | No organism seen |
Isolation and Primary Expansion
The mesenchymal cell population was isolated from the amniotic samples based on methods as previously described by our group[1, 5]. Briefly, the sample was centrifuged at 400 g for 15 minutes. The pellet was resuspended in growth medium (2mL medium per 10mL amniotic fluid) consisting of high-glucose Dulbeco Modified Eagle Medium with L-glutamine (DMEM; Lonza, Walkersville, MD), 20 % fetal bovine serum (FBS; Hyclone, Logan, UT), Gentamicin (Lonza) and 5 ng/ml of basic Fibroblast Growth Factor (Promega, Madison, WI) and plated into 1 well of a BD Bioacoat™ Collagen I-coated 24 well plate (BD Biosciences, San Jose, CA) in a 5% carbon dioxide incubator at 37°C. Non-adherent cells were removed 48 hours later and cultures were fed as needed until they reached 70–80% confluence. At harvest, cells were washed once with PBS (Invitrogen, Carlsbad, CA) and detached with a trypsin-like solution (TrypLE Express; Invitrogen) for 3–5 minutes at 37°C. Cells were passaged in T75 flasks without counting using growth medium. Before being frozen down, cell viability was assessed by Trypan blue exclusion. Viable cells were characterized by flow cytometric analysis of specific surface antigens, 14-day sterility tests, mycoplasma QPCR, and endotoxin assays (details below).
Cryopreservation
Cells were then seeded at 3×103 cells per cm2 in 10xT175 flasks and expanded through one more passage prior to being frozen in Plasmalyte-A (Baxter Healthcare, Charlotte, NC) containing 2.5% human serum albumin (Baxter) and 10% DMSO (Cryoserv; Edwards Lifesciences, Irvine, CA) using a control rate freezer (Cryo, Rockville, MD) and stored in the vapor phase of a liquid nitrogen tank. Cryopreservation was for 3–5 months.
Secondary Expansion
Frozen cells were thawed and diluted 10 times in the same growth medium as described for isolation and primary expansion and immediately plated at 3×103 cells per cm2. The medium was changed the following day to remove dead cells and residual DMSO. Secondary expansion was up to at least 6×108 cells, after which cell viability was assessed by Trypan blue exclusion and viable cells were again characterized by flow cytometry, 14-day sterility tests, mycoplasma QPCR, and endotoxin assays (details below).
Flow Cytometry
At both expansion stages, cells were stained following standard protocol with a panel of 15 antibodies: CD90 FITC, HLA ABC FITC, CD9 FITC (BD Biosciences), CD73 PE, CD106 PE, CD166 PE (BD Biosciences, San Jose, CA), CD45 PerCP-Cy5.5, HLA-DR PerCP, CD117 PerCP-Cy5.5 (BD Biosciences), CD34 PE-Cy7, CD10 PE-Cy7 (BD Bioscience), CD44 PE-Cy7 (eBioscience, San Diego, CA), CD29 APC, CD13 APC (BD Biosciences) and CD105 APC (eBioscience). Nonspecific cell staining was excluded using mouse isotype immunoglobulin controls. The data was acquired using the 6-color BD FACSCanto system (BD Biosciences) and analyzed with FlowJo (Treestar Inc., Ashland, OR).
Sterility Testing
Fourteen-day sterility cultures were performed in accordance with criteria set forth in the FDA’s GMP regulations, Title 21 of CFR, section 610.12 for sterility testing of pharmaceutical products. This method is in compliance with federal guidelines for final product testing. The cultures were prepared using Millipore’s Steritest Filtration System (Millipore, Billerica, MA) and were incubated in appropriate media as per the manufacturer’s instructions for 14 days. The validation of this system, procedural controls, test organisms, and products has demonstrated that the Millipore Steritest system is valid for the isolation of microorganism contamination of cellular products and/or supplies as low as 10 CFU/ml for test organisms used. For cellular products cultured in the presence of antibiotics such as Gentamicin, Millipore TTHVAB210 canisters (Millipore) were used for the test samples. These canisters contain a low absorption Durapore membrane filter (0.45 μm) that is efficient in rinsing away any residual antimicrobial agents from a test sample.
One canister of each set was filled with Fluid Thioglycolate Medium (FTM); the other was filled with Soy Casein Media (SCM). FTM media and test samples were incubated at 30–35° C for 14 days. The SCM canisters were incubated at room temperature for the same period. The canisters were examined for turbidity and evidence of growth on the third, fourth, or fifth day, and on the seventh and fourteenth day of testing. Turbidity is equivalent to identification of positive cultures. Positive cultures undergo hospital-based gram stain, organism identification and sensitivity testing.
Mycoplasma Assay
Mycoplasma PCR testing was performed using MycoSensor™ QPCR Assay Kit (Stratagene, La Jolla, CA), per manufacturer’s instructions.
Endotoxin Assay
Endotoxin levels were determined by the gel-clot limulus amebocyte lysate (LAL) test method in compliance with the FDA’s GMP regulations, Title 21 of CFR, Section 211. Acceptable release criteria are endotoxin level of is 5.0 EU/mL or less.
2. Results
Isolation and expansion of AMSCs in sufficient numbers so as to meet the cell release criteria was achieved in 54.5% (6/11) of the samples. Both the size of the amniotic fluid sample and the time between procurement and initial processing seemed to have an impact on the feasibility of the cell manufacturing process. At least a 2mL sample was shown to be necessary and cell isolation could only be achieved within 48 hours of procurement.
As expected, AMSCs were found to proliferate quite rapidly. Average cell yield during primary expansion was 223.2±65.4×106 cells (44.6-fold expansion), plus a 14.7×106-cell backup, after 36.3±7.8 days. Average cell viability post-thaw was 88%. At both stages in which immunophenotyping was performed, all viable cells expressed markers compatible with a multipotent mesenchymal progenitor lineage, including CD73 (SH3), CD105 (SH2), CD44, CD29, CD90, and CD13. As expected, these cells also stained positive for HLA-A,B,C but were negative for CD45 and CD34. A representative phenotypic profile of the AMSCs post-secondary expansion is shown in Figure 1. Our protocol consistently generated cells with the profile shown therein, yet only 5 markers were necessary for the release criteria as indicated in Table 1. In order to pass such criteria, cells had to be negative (criteria set at <5% positive) for both the panleukocyte marker CD45 and the hematopoietic stem cell marker CD34 and >90% positive for CD29, CD44 and CD73. At all cell manufacturing stages, cultured AMSCs displayed the typical “fibroblast-like” morphology under light microscopy, as shown in Figure 2.
Figure 1.
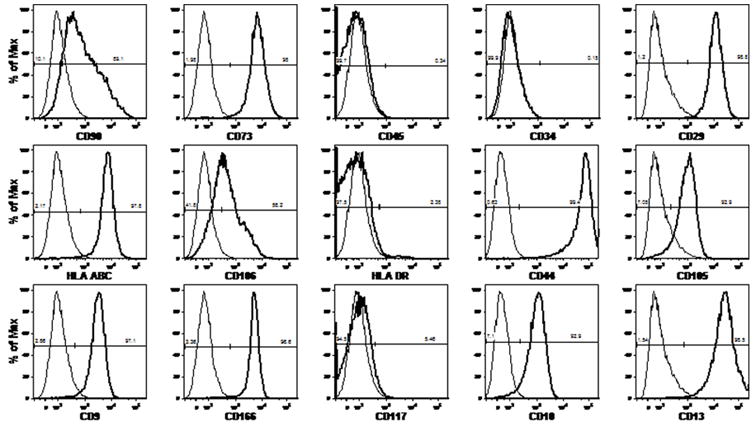
Representative phenotypic signature of amniotic mesenchymal stem cells after secondary expansion. The numbers indicated on the plots represent the percentage of positive (number on the right) or negative (number on the left) cells for each surface marker.
Figure 2.
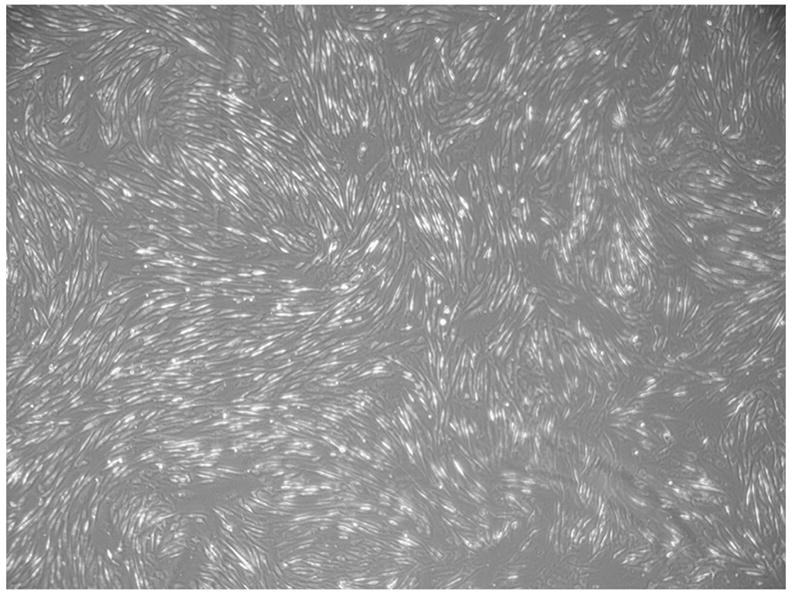
Representative phase micrograph of cultured of amniotic mesenchymal stem cells. At all cell manufacturing stages they displayed the typical “fibroblast-like” morphology shown here.
All samples that met the expansion numbers set forth in the cell release criteria passed the 14-day sterility tests and had negative mycoplasma QPCR and endotoxin assays at the end of both primary and secondary expansions.
3. Discussion
Fetal cells are known to survive at lower oxygen tensions than those tolerated by mature cells and also to commonly lack long extensions and strong intercellular adhesions. Probably because of those characteristics, fetal cells display better survival after refrigeration and cryopreservation protocols when compared with adult cells. This enhanced cryopreservation endurance, however, is tissue-specific. Data from primates and humans have shown that fetal hematopoietic stem cells, as well as fetal lung, kidney, intestine, thyroid and brain tissues can be well preserved at low temperatures, whereas non-hematopoietic liver and spleen tissues can also be cryopreserved, but not as easily[9]. The present data suggests that AMSCs are among those that can withstand protracted cryopreservation while maintaining their identity and still proliferating in large numbers, so as to render viable clinical trials of AMSC-based therapies.
Understandably, clinical trials of cell-based therapies fall under the jurisdiction of the FDA and, as such, must overcome numerous regulatory hurdles before they can be brought to fruition. This study is part of an extensive development aimed at paving the way for a number of foreseeable, and also possibly as yet unsuspected, trials of AMSC-based therapies. One might wonder why we used fetal bovine serum, rather than human serum, in light of the fact that the FDA typically defers the approval of novel cell-based therapies that include cell exposure to xenogeneic materials. Indeed, in a previous study from our group, we have shown that a commercially available serum derived from allogeneic, pooled human donors can be used to reliably isolate and expand amniotic fluid-derived MSCs ex vivo, regardless of gestational age, at rates comparable to that of cells cultured in FBS[10]. While the use of animal-derived products for clinical-grade cell manufacturing is not ideal, it is not absolutely forbidden. Select, controlled animal-based products, including FBS, can be safely used and have been previously approved by the FDA for clinical trials of cell-based therapies. In the case of AMSC-based methods, the need for a somewhat extended, staged cell manufacturing course actually affords the possibility of using select/approved animal products at the early stages of the process, which can then be washed out and replaced by human-derived or synthetic products in the latter stages of the process. For example, one could use FBS for the isolation and primary expansion of AMSCs as described above, but human serum during secondary expansion.
In addition to the deliberately simple and easily reproducible methodology for isolation of amniotic and placental MSCs described here and in our previous work, other methods that rely on oxygen tension, two-stage cultures, or alternative media formulations have been reported[11, 12]. It remains to be seen whether these alternatives would lead to any advantage, or disadvantage, in clinical-grade processing. Also, it remains to be determined whether amniotic stem cells recently obtained by somewhat different methods and described as more primitive than purely mesenchymal are actually the same cells that we and others refer to as AMSCs, which have been shown to be able to give rise to cells from more than one germ layer[13, 14].
Among the experimentally proven alternatives for clinical application of AMSC-based fetal tissue engineering, its application in the repair of congenital diaphragmatic hernia seems closest to clinical trials[2, 3]. Table 2 offers an illustrative overview of a 3-stage AMSC manufacturing process, as described above, only adapted for use in the engineering of clinical-grade diaphragmatic tendons for the repair of congenital diaphragmatic hernia, as we have previousuly described experimentally[2, 3]. This study indicates that such a process would certainly be viable.
Table 2.
Overview of a 3-stage amniotic mesenchymal stem cell manufacturing process as it relates to the engineering of diaphragmatic constructs for implantation in the neonate.
|
Fertile experimental work from multiple groups has recently introduced a number of promising novel therapeutic concepts utilizing these cells, not only in tissue engineering, but also cell transplantation, gene therapy, and others[15]. In addition to our ongoing efforts to overcome yet a few more regulatory requirements before the first clinical trial of AMSC-based therapy can become a reality, the present data lend concrete support to the prospect of AMSC banking for uses later in life, thus potentially expanding the reach of this therapeutic notion.
Acknowledgments
This work was supported by the Center for Human Cell Therapy, as a sub-award of the NIH/NHLBI grant # U24 HL074355-01A1. S.A.S. was supported by the Joshua Ryan Rappaport Fellowship, at the Dept. of Surgery, Children’s Hospital Boston.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaviani A, Perry TE, Dzakovic A, et al. The amniotic fluid as a source of cells for fetal tissue engineering. J Pediatr Surg. 2001;36:1662–5. doi: 10.1053/jpsu.2001.27945. [DOI] [PubMed] [Google Scholar]
- 2.Fuchs JR, Kaviani A, Oh JT, et al. Diaphragmatic reconstruction with autologous tendon engineered from mesenchymal amniocytes. J Pediatr Surg. 2004;39:834–8. doi: 10.1016/j.jpedsurg.2004.02.014. discussion 834–8. [DOI] [PubMed] [Google Scholar]
- 3.Kunisaki SM, Fuchs JR, Kaviani A, et al. Diaphragmatic repair through fetal tissue engineering: a comparison between mesenchymal amniocyte- and myoblast-based constructs. J Pediatr Surg. 2006;41:34–9. doi: 10.1016/j.jpedsurg.2005.10.011. discussion 34–9. [DOI] [PubMed] [Google Scholar]
- 4.Kunisaki SM, Freedman DA, Fauza DO. Fetal tracheal reconstruction with cartilaginous grafts engineered from mesenchymal amniocytes. J Pediatr Surg. 2006;41:675–82. doi: 10.1016/j.jpedsurg.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 5.Kaviani A, Guleserian K, Perry TE, et al. Fetal tissue engineering from amniotic fluid. J Am Coll Surg. 2003;196:592–7. doi: 10.1016/s1072-7515(02)01834-3. [DOI] [PubMed] [Google Scholar]
- 6.Kunisaki SM, Jennings RW, Fauza DO. Fetal cartilage engineering from amniotic mesenchymal progenitor cells. Stem Cells Dev. 2006;15:245–53. doi: 10.1089/scd.2006.15.245. [DOI] [PubMed] [Google Scholar]
- 7.Shanti RM, Steigman SA, Li WJ, et al. Human fetal bone engineering with electrospun nanofibers and amniotic mesenchymal stem cells. Section on Surgery of the American Academy of Pediatrics 2007 Annual Meeting; San Francisco, CA. 2007. [Google Scholar]
- 8.Kunisaki SM, Fuchs JR, Steigman SA, et al. A Comparative Analysis of Cartilage Engineered from Different Perinatal Mesenchymal Progenitor Cells. Tissue Eng. 2007 doi: 10.1089/ten.2006.0407. in press. [DOI] [PubMed] [Google Scholar]
- 9.Fauza DO. Tissue engineering and transplantation in the fetus. In: Lanza RLRVJP, editor. Principles of tissue engineering. San Diego: Academic Press; 2007. [Google Scholar]
- 10.Kunisaki SM, Armant M, Kao GS, et al. Tissue engineering from human mesenchymal amniocytes: a prelude to clinical trials. J Pediatr Surg. 2007 doi: 10.1016/j.jpedsurg.2007.01.031. in press. [DOI] [PubMed] [Google Scholar]
- 11.Tsai MS, Lee JL, Chang YJ, et al. Isolation of human multipotent mesenchymal stem cells from second-trimester amniotic fluid using a novel two-stage culture protocol. Hum Reprod. 2004;19:1450–6. doi: 10.1093/humrep/deh279. [DOI] [PubMed] [Google Scholar]
- 12.In’t Anker PS, Scherjon SA, Kleijburg-van der Keur C, et al. Isolation of mesenchymal stem cells of fetal or maternal origin from human placenta. Stem Cells. 2004;22:1338–45. doi: 10.1634/stemcells.2004-0058. [DOI] [PubMed] [Google Scholar]
- 13.Tsai MS, Hwang SM, Tsai YL, et al. Clonal amniotic fluid-derived stem cells express characteristics of both mesenchymal and neural stem cells. Biol Reprod. 2006;74:545–51. doi: 10.1095/biolreprod.105.046029. [DOI] [PubMed] [Google Scholar]
- 14.De Coppi P, Bartsch G, Jr, Siddiqui MM, et al. Isolation of amniotic stem cell lines with potential for therapy. Nat Biotechnol. 2007;25:100–6. doi: 10.1038/nbt1274. [DOI] [PubMed] [Google Scholar]
- 15.Fauza D. Amniotic fluid and placental stem cells. Best Pract Res Clin Obstet Gynaecol. 2004;18:877–91. doi: 10.1016/j.bpobgyn.2004.07.001. [DOI] [PubMed] [Google Scholar]