

Abstract
Paired-pulse facilitation (PPF) and depression (PPD) are forms of short-term plasticity that are generally thought to reflect changes in transmitter release probability. However, desensitization of postsynaptic AMPA receptors (AMPARs) significantly contributes to PPD at many glutamatergic synapses. To clarify the involvement of AMPAR desensitization in synaptic PPD, we compared PPD with AMPAR desensitization, induced by paired-pulse glutamate application in patches excised from postsynaptic cells at the calyx of Held synapse of developing rats. We found that AMPAR desensitization contributed significantly to PPD before the onset of hearing (P10–12), but that its contribution became negligible after hearing onset. During postnatal development (P7–21) the recovery of AMPARs from desensitization became faster. Concomitantly, glutamate sensitivity of AMPAR desensitization declined. Single-cell reverse transcription-polymerase chain reaction (RT-PCR) analysis indicated a developmental decline of GluR1 expression that correlated with speeding of the recovery of AMPARs from desensitization. Transmitter release probability declined during the second postnatal week (P7–14). Manipulation of the extracellular Ca2+/Mg2+ ratio, to match release probability at P7–8 and P13–15 synapses, revealed that the release probability is also an important factor determining the involvement of AMPAR desensitization in PPD. We conclude that the extent of involvement of AMPAR desensitization in short-term synaptic depression is determined by both pre- and postsynaptic mechanisms.
Synaptic depression is widely observed at a variety of synapses, and contributes to dynamic neuronal gain-control at some synapses (Abbott et al. 1997), while limiting the speed of high-fidelity transmission at others (Brenowitz et al. 1998). As synaptic depression is stronger when transmitter release probability is high (Takeuchi, 1958), it has been proposed to result primarily from depletion of releasable synaptic vesicles (Betz, 1970). However, other mechanisms also contribute to synaptic depression (for review, see Zucker & Regehr, 2002). Notably, AMPAR desensitization underlies synaptic depression at a subset of glutamatergic synapses where cyclothiazide (CTZ) reduces synaptic depression (Otis et al. 1996; Neher & Sakaba, 2001; Rozov et al. 2001; Taschenberger et al. 2002, 2005; DiGregorio et al. 2007). At the calyx of Held, in the rodent auditory brainstem, CTZ reduces synaptic depression before P10 (Neher & Sakaba, 2001; Taschenberger et al. 2002), but its effect becomes much weaker after P12 (Joshi & Wang, 2002; Taschenberger et al. 2002, 2005).
At immature calyceal synapses, the significant involvement of AMPAR desensitization in synaptic depression may arise from slow clearance of glutamate from the synaptic cleft (Taschenberger et al. 2005), owing to the spoon-shaped structure of immature nerve terminals (Kandler & Friauf, 1993; Sätzler et al. 2002). Other mechanisms that can potentially increase involvement of AMPAR desensitization in synaptic depression include (i) high sensitivity of AMPARs to desensitization by glutamate, (ii) slow recovery of postsynaptic AMPARs from desensitization (Joshi et al. 2004), and (iii) high transmitter release probability (Taschenberger & von Gersdorff, 2000; Iwasaki & Takahashi, 2001). To determine whether these mechanisms underlie the developmental decrease of AMPAR desensitization involvement in PPD, we recorded EPSCs and AMPAR currents from the principal neurons in the medial nucleus of the trapezoid body (MNTB) in slices of developing rat brainstem, and compared PPD of EPSCs with AMPAR desensitization induced by paired-pulse glutamate application in patches excised from the MNTB neurons. To assess molecular mechanisms, we performed quantitative RT-PCR analysis for mRNAs harvested from individual MNTB neurons, where the recovery time of AMPAR from desensitization had been determined in excised patch recordings. To assess further how alterations of release probability affect the involvement of AMPAR desensitization in synaptic depression, we matched release probability between synapses before and after hearing onset by manipulating the extracellular Ca2+/Mg2+ concentration ratio. Our results suggest that all factors (i–iii) participate in the developmental decrease in the contribution of AMPAR desensitization to synaptic depression.
Methods
Preparation and solutions
All experiments were performed in accordance with the guidelines of the Physiological Society of Japan. Transverse brainstem slices (150–200 μm thick) containing the MNTB were prepared from Wistar rats (P7–21) killed by decapitation under halothane anaesthesia as previously described (Forsythe & Barnes-Davies, 1993). Slices were incubated at 36–37°C for 1 h, and then maintained at room temperature (22–26°C). MNTB principal neurons were visually identified with a 40× water immersion objective (Zeiss, Germany or Olympus, Japan) attached to an upright microscope (Axioskop, Zeiss or BX51WI, Olympus). The standard aCSF for perfusion contained (in mm): 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 10 glucose, 3 myo-inositol, 2 sodium pyruvate and 0.5 ascorbic acid (pH 7.4 when bubbled with 5% CO2 and 95% O2). The aCSF routinely contained bicuculline methiodide (10 μm; Sigma, USA or Fluka, Switzerland) and strychnine hydrochloride (0.5 μm, Sigma) to block inhibitory synaptic responses. Recording patch pipettes were filled with a solution containing (in mm): 110 CsF, 30 CsCl, 10 Hepes, 5 EGTA, 1 MgCl2 and 5 N-(2,6-diethylphenylcarbamoylmethyl)-triethylammonium chloride (QX-314, Alomone Laboratories, Israel) (pH adjusted to 7.3 with CsOH).
Recordings
Whole-cell and outside-out patch clamp recordings were made from the MNTB principal neurons using a patch-clamp amplifier (Axopatch 200B, Axon Instruments, USA) at a holding potential of −70 mV, unless otherwise noted. Patch pipettes were pulled from standard-wall borosilicate glass (GC150F-7.5, Harvard Apparatus, UK) to give a resistance of 2–3 MΩ. The access resistance was 4–10 MΩ, which was routinely compensated by 70–80%. EPSCs were evoked by extracellular stimulation using a bipolar tungsten electrode positioned halfway between the midline and the MNTB. EPSCs derived from the calyx of Held synapse were identified as those evoked in an all-or-none manner for a graded stimulus intensity and those having amplitude > 1 nA at −70 mV (Forsythe & Barnes-Davies, 1993). All recordings were performed at room temperature (25–27°C).
Fast L-glutamate application
Double-barrelled application pipettes were fabricated from theta glass tubes (outer diameter, 2.0 mm; TG200-15, Harvard Apparatus, UK) pulled to a tip diameter of 200–300 μm (Koike et al. 2000). Control aCSF and test solution (containing 10 mm l-glutamate) were continuously passed through each barrel of the application pipette using a gravity-fed pressure. The application pipette was rapidly moved by a square voltage pulse (15–25 V, 0.7–1 ms) applied to a piezoelectric device (Burleigh Instruments, USA). The time required for solution exchange was measured from a change in junction potential when the solution was switched from the standard aCSF to one diluted 10-fold. The 10–90% rise time and decay time of the solution exchange ranged from 130 to 170 μs and from 90 to 150 μs, respectively (Koike-Tani et al. 2005). To block NMDA receptors (NMDARs) d-APV (50 μm, Tocris, UK) was included in all solutions.
Data analysis
All records were low pass filtered at 5 kHz and digitized at 50 kHz using a Digidata 1322A analog–digital converter with pCLAMP8 software (Axon Instruments). All data are expressed as means ±s.e.m. Statistical analysis was made using ANOVA followed by Student's paired t test unless otherwise noted. Differences were considered significant as P < 0.05.
Single cell quantitative RT-PCR
Single cell quantitative RT-PCR analysis was carried out as previously described (Koike-Tani et al. 2005). After performing rapid l-glutamate application experiments on outside-out patches excised from an MNTB principal neuron, the cytoplasm of the neuron was harvested into a patch pipette, and submitted to real-time PCR-based quantification.
Immunocytochemistry
Immunocytochemical examinations were performed as previously described (Ishikawa et al. 2003). Immuno-affinity-purified rabbit anti-GluR1 antibody (Upstate, Lake Placid, NY, USA; diluted 1 : 100) was used for detecting GluR1. Calyces were labelled using mouse monoclonal anti-synaptophysin antibody (Chemicon, Temecula, CA, USA; diluted 1 : 200). The immunofluorescence intensities of GluR1 were densitometrically evaluated.
Immunoblotting
Immunoblotting procedures were as previously described (Kimura et al. 2003). GluR1 was detected using ECL-plus chemiluminescence (GE Healthcare) combined with anti-GluR1 antibody (diluted 1 : 1000).
Variance–mean analysis of EPSCs
Variance–mean analysis was used to estimate the mean release probability per site (p). EPSCs, evoked at 0.05 Hz, were recorded in the presence of the low-affinity glutamate receptor antagonist kynurenic acid (2 mm, Tocris) to prevent AMPAR saturation. Under various conditions for altering p, 15 successive EPSCs were collected for constructing a variance–mean plot. At P13–15 synapses, the K+ channel blocker 4-aminopyridine (4-AP, 5–10 μm) (Wako, Japan) or tetraethylammomium chloride (TEA-Cl, 50–100 μm) (Nacalai, Japan) was added to the aCSF for acquiring data at high p. Variance–mean plots were analysed by fitting the simple parabola equation:
where I and σ2 represent the mean amplitude and variance of EPSCs, respectively, q denotes mean quantal size and N denotes the number of release sites (Clements & Silver, 2000). In this method, assuming that σ2 arises entirely from stochastic changes in p, q can be estimated from the initial slope of the parabola, Nq from the larger X intercept of the parabola, and p can be estimated as I/Nq.
Results
Developmental changes in the effect of CTZ on the paired-pulse depression of EPSCs
Using paired-pulse stimulation protocols, we evoked EPSCs at the calyx of Held in slices taken from rats at different postnatal periods (P7, P14 and P21). At P7, EPSCs showed PPD at a wide range of interstimulus intervals (ISIs, from 5 ms to 10 s, Fig. 1). The magnitude of depression became much smaller at P14 (Fig. 1), as reported previously (Taschenberger & von Gersdorff, 2000; Iwasaki & Takahashi, 2001). At P21, PPD at short ISIs was reduced, revealing a peak at around 30 ms ISI. This peak may be produced by a combination of synaptic depression and facilitation, although the latter is masked by the former (Barnes-Davies & Forsythe, 1995; Borst et al. 1995). As previously reported for synaptic depression during high frequency stimulation (50 or 100 Hz; Joshi & Wang, 2002; Taschenberger et al. 2002), bath-application of CTZ (100 μm) significantly attenuated PPD at the ISIs between 10 ms and 1 s at P7, but had no effect at P14 and P21.
Figure 1. Developmental reductions in the magnitude of PPD and in the effect of CTZ on PPD.
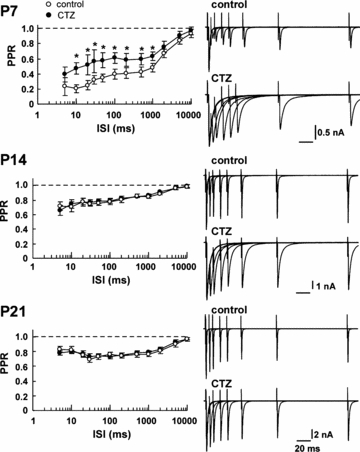
EPSCs were evoked in MNTB neurons using a paired-pulse stimulation protocol (ISI, 5 ms to 10 s) in the absence (control, ○) or presence of CTZ (100 μm, •) at P7, P14 and P21. Ordinates in left panels indicate paired-pulse ratios (PPRs) of EPSCs (the second EPSC amplitude relative to the first one) plotted at each ISI (abscissae, logarithmic coordinates). Right panels show EPSCs evoked by paired-pulse stimulations at different ISIs (5–200 ms, superimposed) before (upper traces) and after (lower traces) CTZ applications. Asterisks indicate significant difference by paired t test (P < 0.05). In this and the following figures, error bars represent the s.e.m.
Developmental speeding in the recovery of AMPARs from desensitization
To assess the involvement of AMPAR desensitization in PPD, we examined the recovery time of AMPARs from desensitization, by applying l-glutamate (10 mm) to outside-out patches excised from postsynaptic MNTB neurons by a pair of brief (0.7–1.0 ms) pulses at various interpulse intervals (IPIs, Fig. 2A). At P7, desensitization was observed for IPIs of up to 1 s, and the recovery time of AMPAR currents in patches (AMPAR patch currents) could be fitted with a sum of two exponential functions, with fast and slow time constants of 46.7 ms (73.2%) and 435 ms, respectively (weighted mean, 151 ms, data derived from 4 to 7 cells). As animals matured, the recovery time course became faster and mono-exponential, with a time constant of 26.5 ms at P14 (4–7 cells) and 16.1 ms at P21 (6–9 cells). These results agree with those reported for mice by Joshi et al. (2004), except that their time constant in P6–8 mice (62.5 ms, single exponential fit to data up to 300 ms IPI) is much shorter than our weighted mean time constant in P7 rats (151 ms).
Figure 2. Developmental speeding in the recovery of AMPARs from desensitization.

A, developmental decrease in the recovery time from desensitization of AMPA patch currents induced by paired-pulse glutamate applications to outside-out patches excised from MNTB neurons. Data derived from P7, P14 and P21 rats. Insets show AMPA patch currents induced by the paired pulse protocol (IPI, 10–100 ms, superimposed, averaged from 3 traces). Graphs indicate the amplitude of the second AMPA patch current relative to the first one (I2/I1, ordinates) at various IPIs (abscissae, 5–200 ms). IPIs up to 1 s are shown in the top inset at P7. The fast (τf) and slow (τs) time constants and their weighted means (τm) are indicated at P7, and time constants from single exponential fits (τ) are indicated at P14 and P21. B, paired-pulse AMPA patch currents (left traces superimposed) in the presence of CTZ (100 μm) at P7, P14 and P21. Bar graphs show I2/I1 (IPI, 10 ms) in the absence (open columns) or presence (filled columns) of CTZ.
CTZ blocks desensitization of recombinant AMPARs composed of GluR flip splice variants, whereas desensitization of AMPARs composed of flop variants is relatively resistant to CTZ (Partin et al. 1994, 1996), raising the possibility that the lack of effect of CTZ on synaptic depression at more mature calyces might arise from AMPARs composed of GluR flop variants (Taschenberger et al. 2002). Despite the fact that GluR flop variants increase in MNTB neurons during postnatal development, CTZ blocks desensitization of AMPAR patch currents, induced by a sustained application of l-glutamate, throughout development (P7–21) to a similar extent (Koike-Tani et al. 2005). To make a more direct comparison between the effect of CTZ on AMPAR patch currents and that on PPD, we used a paired-pulse protocol for testing the desensitization of AMPAR patch currents induced by rapid l-glutamate application. As shown in Fig. 2B, CTZ (100 μm) abolished desensitization of AMPAR patch currents throughout the ages examined (P7–P21). These results, together with those of PPD (Fig. 1), strongly suggest that at the calyces of Held, AMPAR desensitization is significantly involved in PPD at P7, whereas its involvement becomes much less at P14 or later. In agreement with this conclusion, low-affinity AMPAR antagonists reduce synaptic depression during 100 Hz stimulation trains at P5–10, but not at P12–18, rat calyces (Taschenberger et al. 2002; Renden et al. 2005; but see Wong et al. 2003).
Developmental changes in the AMPAR subunit transcripts
We next explored what molecular mechanisms underlie the developmental speeding in the recovery of AMPARs from desensitization, by performing quantitative single-cell RT-PCR analysis. After establishing a whole-cell recording in MNTB neurons, the intracellular contents were harvested into the patch pipette and submitted to quantitative RT-PCR analysis (Lambolez et al. 1992; Geiger et al. 1995; Koike-Tani et al. 2005). As we previously reported (Koike-Tani et al. 2005), the expression of GluR1–4 mRNAs in MNTB neurons is differentially regulated during development from P7 to P21. Thus GluR1 mRNAs are down-regulated, whereas GluR4 mRNAs are up-regulated (Fig. 3A). During development, the proportion of GluR flip splice variant mRNAs in total GluR mRNAs decreased from 16.8 ± 5.1% at P7 to 5.3 ± 2.0% at P14 and further down to 3.2 ± 1.5% at P21 (Fig. 3B).
Figure 3. GluR mRNAs measured using single cell RT-PCR in developing MNTB neurons.
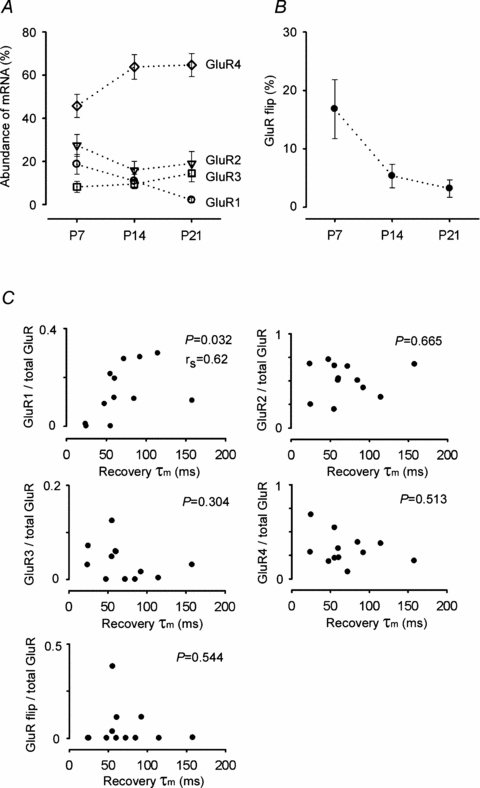
A, abundance of individual GluR (GluR1–4) mRNAs relative to total GluR mRNAs at different postnatal periods. Developmental changes in the relative abundance (%) of GluR1 (○), GluR2 (∇), GluR3 (□), GluR4 (⋄), derived from 15 to 19 MNTB neurons at each age, are plotted. B, developmental decrease in the relative abundance of the total GluR flip splice variants. C, the relationships between the abundance of GluR1–4 mRNAs or that of total flip mRNAs in individual MNTB neurons at P7–13 (ordinates), and the weighted mean time constant (τm) of recovery from desensitization of AMPA patch currents, recorded from outside-out patches excised from the same neuron (abscissae). Only GluR1 abundance displays a significant correlation with τm in Spearman correlation analysis (P= 0.032). rs indicates Spearman's rank correlation coefficient.
Recombinant AMPARs composed of edited (G) form GluRs (2–4) have a faster recovery time from desensitization compared with those composed of unedited (R) form of GluRs or GluR1, which lacks the R/G site (Lomeli et al. 1994; Krampfl et al. 2002). Of GluR (2–4) mRNAs, G-form mRNAs comprised 80% at P7 and > 99% at P14. Among recombinant homomeric AMPARs, GluR1 AMPARs have the slowest recovery time from desensitization (Grosskreutz et al. 2003). However, it is unknown whether GluR1 assembled into native AMPARs, possibly as heterotetramers (Rosenmund et al. 1998; Brorson et al. 2004), can slow the recovery time from desensitization. After recording AMPAR currents with the paired-pulse protocol in patches excised from an MNTB neuron, we made whole-cell recordings with a new pipette from the same neuron, and harvested the intracellular contents for quantitative RT-PCR analysis. We then examined whether there was a correlation between the abundance of individual GluR subunits in each MNTB neuron and the weighted mean time constant of recovery from desensitization. As illustrated in Fig. 3C, the time constant of recovery from desensitization was slower in patches from MNTB neurons having abundant GluR1 mRNA (rs = 0.62, P = 0.03, Spearman correlation analysis). By contrast, there was no correlation between the recovery time constant and the abundance of GluR2, GluR3, GluR4, or GluR1–4 flip mRNAs. These results suggest that GluR1 in native AMPARs slows their recovery from desensitization. Thus, the developmental down-regulation of GluR1 mRNAs is most likely to underlie the developmental speeding in the recovery of AMPARs from desensitization.
Developmental decline of GluR1 proteins
To ensure that GluR1 protein expression in MNTB neurons undergoes developmental decline in parallel with its transcript, we examined GluR1 immunoreactivity (IR) in developing MNTB neurons (Fig. 4A). As animals matured, the intensity of GluR1 IR decreased. Western blot analysis of the tissue derived from the MNTB region at different postnatal days indicated a developmental decline of GluR1 protein (Fig. 4B), in agreement with a previous report (Caicedo & Eybalin, 1999; but see Hermida et al. 2006). The parallel decrease in GluR1 mRNA and protein observed in MNTB neurons is consistent with the fact that GluR proteins undergo a rapid turnover, and are replaced by 50% in 24 h (Mammen et al. 1997; Huh & Wenthold, 1999).
Figure 4. Developmental changes in the GluR1 immunoreactivity in MNTB neurons.

A, GluR1-IR of MNTB principal neurons in developing rats. Confocal laser scanning microscope pictures showing GluR1-IR (left column, green), synaptophysin-IR (middle column, red) and merged views (right column). Graph in the right panel shows the developmental decline in GluR1-IR. Ordinate indicates relative IR normalized to that of the GluR1 at P7. B, Western blot analysis of GluR1 subunit. Samples from the MNTB region at each age (P7, P14–16 and P21) were separated by SDS-PAGE for immunoblot detection. Open arrowheads indicate the molecular weight, and a filled arrowhead indicates the position of GluR1. An asterisk indicates non-specific signals originated from the ABC detection method. The GluR1 signal intensity at each lane was normalized to the mean value at P7.
Developmental decrease in glutamate sensitivity of AMPAR desensitization
Transmitter glutamate remaining in the synaptic cleft after the initial EPSC can contribute to PPD by desensitizing AMAPRs (Otis et al. 1996). Might the l-glutamate concentration required to cause AMPAR desensitization change with development? To address this question, we applied l-glutamate twice, first for a sustained period (>16 s) at 0.1–100 μm from one barrel, followed by a 100 ms test pulse (10 mm) (Fig. 5A). The resting-state AMPAR desensitization curve thus produced showed higher sensitivity of AMPARs to be desensitized by l-glutamate at P7–8 than P13–15 (Fig. 5B). The IC50 for l-glutamate-induced desensitization was 5.7 μm in P7 patches, and 13.8 μm in P14 patches (Fig. 5B). Thus, the glutamate sensitivity of AMPAR desensitization decreases by 2.3 times during the second postnatal week.
Figure 5. Developmental change in the glutamate sensitivity of AMPAR desensitization.
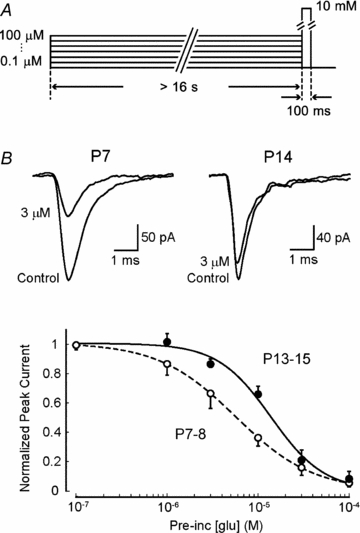
A, the protocol of glutamate application: AMPAR currents were induced by a 10 mm glutamate application with a 100 ms test pulse to outside-out patches excised from P7–8 or P13–15 MNTB neurons, after sustained applications (> 16 s) of l-glutamate at various concentrations (0.1–100 μm). B, sample records show AMPAR currents (averaged from 5 traces) in P7 and P14 patches, in the absence and presence of 3 μm l-glutamate (superimposed). The graph indicates the relative amplitude of AMPAR patch currents (ordinate, normalized to those without prior glutamate application) at P7–8 (○) and P13–15 (•) induced by the test pulse after sustained applications of l-glutamate at various concentrations (abscissa). The lines were drawn following least squares fitting of the Hill equation (I = Imax[glu]−n/{[glu]−n+ IC50−n}, where the Hill coefficient (n) was 1.0 for P7 and 1.5 for P13–15 AMPAR patch currents).
The effect of release probability on the involvement of AMPAR desensitization in synaptic depression
At synapses with relatively high release probability, postsynaptic AMPARs have a higher chance of being occupied by synaptically released glutamate. At such synapses, repetitive synaptic transmission results in a higher rate of AMPAR desensitization, thereby increasing synaptic depression. At the rat calyx of Held, release probability decreases during postnatal development (Iwasaki & Takahashi, 2001; Taschenberger et al. 2002). To assess the effect release probability has on the contribution of AMPAR desensitization to PPD, we estimated release probability using the variance–mean analysis (Clements & Silver, 2000; Scheuss et al. 2002), and determined the extracellular divalent cationic compositions that could match release probabilities between P7–8 and P13–15 (Fig. 6A). This method gives the number of independent release sites (N), the mean release probability per site (p), and the mean quantal amplitude (q), from the variance–mean plot (see Methods). Kynurenic acid (2 mm) was added to the aCSF to minimize an involvement of AMPAR saturation in the variance–mean relationship (Foster & Regehr, 2004). For the parabola fit, release probability was altered by simply changing [Ca2+]o/[Mg2+]o ratio in aCSF at P7, whereas at P14, addition of 4-AP (10 μm) or TEA (50–100 μm) was often required to attain high release probability (Fig. 6A, see Methods). The p-values estimated from the variance–mean analysis were 0.45 ± 0.04 at P7–8 (n = 6) and 0.19 ± 0.02 at P13–15 (n = 9) (Fig. 6B). The N-values were 358 ± 122 at P7–8 and 719 ± 194 at P13–15. These estimates are comparable to previous estimates at P8–10 rat calyces (500–900, Scheuss et al. 2002) and the number of morphologically identified active zones (Taschenberger et al. 2002; Sätzler et al. 2002; see Discussion). Although the absolute values for N and p can depend upon the methods used, the relative magnitudes of developmental changes in N and p, during the second postnatal week, are consistent with those previously estimated using the tetanic stimulation method at this synapse (Iwasaki & Takahashi, 2001; Taschenberger et al. 2002). At P7–8, reducing [Ca2+]o to 1.3 mm and increasing [Mg2+]o to 1.7 mm reduced p to 0.24 ± 0.05 (n = 6), which was similar to p at P13–15 (Fig. 6B). At P13–15, increasing [Ca2+]o to 4 mm and omitting [Mg2+]o from the aCSF increased p to 0.45 ± 0.04 (n = 6), which was similar to that at P7–8 in normal aCSF (Fig. 6B).
Figure 6. Compensations for differences in release probability by manipulation of extracellular divalent cation concentrations at different ages.

A, release probability estimated using the variance–mean analysis at P7 and P13. Left panels, EPSC amplitudes in solutions containing 1 mm Ca2+ and 2 mm Mg2+ (i), 1.3 mm Ca2+ and 1.7 mm Mg2+ (ii), 2 mm Ca2+ and 1 mm Mg2+ (iii, standard aCSF) and 4 mm Ca2+ and 0 mm Mg2+ (iv) at P7. Solutions at P13 (lower panels) contain 1 mm Ca2+ and 2 mm Mg2+ (i), 2 mm Ca2+ and 1 mm Mg2+ (ii, standard aCSF), 4 mm Ca2+ and 0 mm Mg2+ (iii), and 4 mm Ca2+ and 0 mm Mg2+ with 10 μm 4-AP (iv). Averaged EPSCs from 15 events each are superimposed and shown in insets. Right panels show variance–mean plots and parabolic fits. In the presence of kynurenate the mean amplitude of EPSCs was 1.6 ± 0.5 nA (n = 6) at P7–8, and 1.0 ± 0.2 nA (n = 9) at P13–15 (no significant difference, P = 0.3, unpaired t test). B, release probabilities estimated for P7–8 and P13–15 synapses, in normal aCSF (open bars) and in aCSFs with modified extracellular [Ca2+]/[Mg2+] compositions (filled bars). The release probabilities were not significantly different between P7–8 synapses in normal aCSF (n = 6) and P13–15 synapses in modified aCSF (n = 6), as well as between P13–15 synapses in normal aCSF (n = 6) and P7–8 synapses in modified aCSF (n = 9).
Using the modified aCSF solutions we next examined the contribution of release probability to PPD. At P7–8 synapses, reducing p to the same level as at P13–15 markedly reduced PPD at ISIs shorter than 0.5 s, but it had no effect on PPRs at longer ISIs (0.5–10 s, Fig. 7A, online supplemental material, Supplemental Table 1). These PPD components correspond to those caused by Ca2+/calmodulin (CaM)-dependent inactivation of presynaptic Ca2+ currents (IpCa) (Xu & Wu, 2005; Nakamura et al. 2008). After matching p, the attenuating effect of CTZ on PPD became significantly less, being observed only at ISIs of 50–200 ms (Fig. 7C versus Fig. 1). At P13–15 synapses, matching p to the level seen at P7–8 increased PPD, but remained lower than that seen at P7–8 (Fig. 7B, Supplemental Table 1). In this condition, CTZ significantly attenuated PPD at the short ISIs (5–30 ms, Fig. 7D). The lack of CTZ effect on PPDs at longer intervals is consistent with the fast recovery of AMPARs from desensitization at P14 (Fig. 2). These results suggest that the release probability significantly, but not exclusively, contributes to the involvement of AMPAR desensitization in synaptic depression.
Figure 7. Effects of release probability on PPRs.

A, PPRs at P7–8 synapses in normal aCSF (○) and modified aCSF containing 1.3 mm Ca2+ and 1.7 mm Mg2+ ( , low Pr). Dashed lines in A and B (data in normal aCSF) were taken from Fig. 1. B, PPRs at P13–15 synapses in normal aCSF (○) and modified aCSF containing 4.0 mm Ca2+ and 0 mm Mg2+ (
, low Pr). Dashed lines in A and B (data in normal aCSF) were taken from Fig. 1. B, PPRs at P13–15 synapses in normal aCSF (○) and modified aCSF containing 4.0 mm Ca2+ and 0 mm Mg2+ ( , high Pr). C, PPRs at P7–8 synapses (n = 6) in the modified aCSF in the presence (•) and absence (
, high Pr). C, PPRs at P7–8 synapses (n = 6) in the modified aCSF in the presence (•) and absence ( ) of CTZ. D, PPRs at P13–15 synapses (n = 7) in the modified aCSF in the presence (•) and absence (
) of CTZ. D, PPRs at P13–15 synapses (n = 7) in the modified aCSF in the presence (•) and absence ( ) of CTZ. Asterisks in B and D indicate significant difference between values before and after CTZ application at a given ISI.
) of CTZ. Asterisks in B and D indicate significant difference between values before and after CTZ application at a given ISI.
An increase in transmitter release can secondarily cause synaptic depression via vesicle depletion. In the present study, involvement of AMPAR desensitization in PPD was assessed by the degree of PPD attenuated by CTZ. Since CTZ also increases transmitter release, via inhibiting presynaptic potassium conductance, at calyceal synapses of P13–16 (Ishikawa & Takahashi, 2001) and P7–8 (T. Yamashita and T. Takahashi, unpublished observation) rats, contribution of desensitization to PPD might be greater than those estimated here in both age groups.
Discussion
Contribution of transmitter release probability to the involvement of AMPAR desensitization in synaptic depression
Rodents start to detect sounds at P10–13 (Mikaelian & Ruben, 1964; Kikuchi & Hilding, 1965; Futai et al. 2001). Immature calyces of Held, before the hearing onset, show strong synaptic depression during repetitive stimulation (Taschenberger & von Gersdorff, 2000; Iwasaki & Takahashi, 2001) and compound ISI dependency (Fig. 1, see also Iwasaki & Takahashi, 2001) in PPD. At such synapses, at P7–8, CTZ attenuated PPD for a wide range of ISIs up to 1 s (Fig. 1), suggesting that AMPAR desensitization is involved in synaptic depression even during 1 Hz stimulation (cf. Renden et al. 2005). Reducing release probability down to the level of P13–15, by lowering the [Ca2+]o/[Mg2+]o ratio, attenuated PPD at ISIs shorter than 0.5 s (Fig. 7A), and reduced the CTZ-sensitive components of synaptic depression (Fig. 7CversusFig. 1), suggesting that high release probability at immature synapses contributes to involvement of AMPAR desensitization in synaptic depression. The number of morphologically defined active zone at the calyx of Held is estimated to be 300–500 at P5–10 and 700 at P14, and the total number of docked synaptic vesicles is 1700 at P5 and 2100 at P14 (Taschenberger et al. 2002; Sätzler et al. 2002). Thus, the average number of vesicles docked at each active zone can be estimated as 3.4–5.7 at P5 and 3 at P14. As the release probability is 0.45 at P7–8 and 0.19 at P13–15 (Fig. 6), a single presynaptic action potential can maximally induce the exocytosis of 2.6 vesicles at each active zone at P7, whereas the number is 0.6 at P14. Thus, the chance for postsynaptic AMPARs to be exposed twice to glutamate in the paired-pulse stimulation protocol is much higher at P7 than at P14. Furthermore, at immature calyces, glutamate simultaneously released from nearby release sites tends to summate in the synaptic cleft, thereby increasing involvement of desensitization in synaptic depression.
Conversely, at P13–15 calyces, raising release probability to the level of P7–8 synapses increased involvement of AMPAR desensitization in PPD (Fig. 7D). These results suggest that a developmental decline in release probability reduces the involvement of AMPAR desensitization in PPD. Thus, release probability is an important presynaptic determinant for the involvement of AMPAR desensitization in synaptic depression. At P13–15 synapses, release probability was 0.2 (Fig. 6), and the recovery time constant (τ) of AMPARs from desensitization was 27 ms (Fig. 2). This is comparable to an imaginary situation where they are exposed to transmitter glutamate every time of stimulation (p= 1.0) but recover five times more quickly from desensitization (τ= 5.4 ms). In such a situation, involvement of AMPAR desensitization in synaptic depression will be significant at frequencies higher than 185 Hz. In fact, at P14 calyces of Held, AMPAR desensitization is uninvolved in synaptic depression during 100 Hz stimulation (Taschenberger et al. 2002, 2005; but see Wong et al. 2003), but significantly involved during 300 Hz stimulation (Taschenberger et al. 2005).
Developmental changes in GluR subunits underlying decreased involvement of AMPAR desensitization in synaptic depression
Matching release probability between P7–8 and P13–15 revealed that there must be other factors that contribute to the developmental decline in the involvement of AMPAR desensitization in PPD (Fig. 7C and D). In the cerebral cortex, recovery of AMPARs from desensitization in bipolar cells is slow, whereas that in multipolar cells is fast (Rozov et al. 2001). CTZ attenuates synaptic depression at the pyramidal–bipolar cell synapse, but not at the pyramidal–multipolar cell synapse. Thus, both at brainstem calyceal synapses and cerebral cortical synapses, slow recovery of AMPARs from desensitization underlies the involvement of desensitization in synaptic depression.
Our single cell RT-PCR analysis indicated that receptors in patches from cells containing abundant GluR1 transcripts had a slower recovery from desensitization (Fig. 3). Recombinant homomeric GluR1 AMPARs have slow recovery kinetics from desensitization, whereas GluR4 homomeric AMPARs have very fast (millisecond order) recovery kinetics (Grosskreutz et al. 2003). It has been postulated that AMPARs having a fast desensitization on-rate may have slow recovery kinetics from desensitization (Quirk et al. 2004). However, in native AMPARs, recovery kinetics from desensitization was not correlated with the abundance of GluR4, or the flip/flop ratio of GluR splice variants (Fig. 3C). A close correlation between GluR1 abundance and slow recovery of AMPARs from desensitization suggests that GluR1 abundance determines the kinetics of recovery from desensitization in native AMPARs.
Our results indicate that the sensitivity of AMPAR desensitization to l-glutamate decreases during development from P7 to P14 (Fig. 5). Given that homomeric GluR1 receptors have a relatively high sensitivity to desensitization by glutamate (Robert & Howe, 2003), the developmental decline in the GluR1 subunit might also underlie the developmental reduction in glutamate sensitivity of AMPAR desensitization. In this respect, the level of GluR1 expression in the postsynaptic neuron is an important postsynaptic determinant for the involvement of AMPAR desensitization in synaptic depression. This conclusion is based on an assumption that subsynaptic and extrasynaptic receptors are identical. Although this assumption remains to be established, the fact that AMPA receptors shuttle between subsynaptic and extrasynaptic regions by lateral diffusion (Borgdorff & Choquet, 2002; Ashby et al. 2004) tends to support this assumption.
Developmental speeding in transmitter clearance contributing to a decrease in the involvement of AMPAR desensitization in paired-pulse synaptic depression
Using the non-desensitizing AMPAR agonist kainate, Taschenberger et al. (2005) estimated the recovery time of subsynaptic AMPARs from desensitization as 484 ms at P5–7 and 21 ms at P12–14. The value at P12–14 is comparable to the recovery time constant of AMPAR patch currents from AMPAR desensitization at P14 (27 ms, Fig. 2A), whereas the value at P5–7 is more than three times longer than that of AMPAR patch currents at P7 (151 ms), suggesting that glutamate clearance from the synaptic cleft is retarded at P7, but no longer at P14. The developmental acceleration of glutamate clearance may arise from morphological reformation of calyx terminal from a spoon-shape to finger-like structure (Kandler & Friauf, 1993; Sätzler et al. 2002; Wimmer et al. 2006). At P7 calyces, at an ISI of 1 s, CTZ significantly attenuated PPD (Fig. 1), whereas AMPAR patch currents fully recovered from desensitization after a 1 s interval (Fig. 2A). These results together suggest that slow clearance of glutamate from the synaptic cleft contributes to involvement of AMPAR desensitization in synaptic depression, as previously suggested at immature calyces of Held (Koike-Tani et al. 2005; Renden et al. 2005; Taschenberger et al. 2005) and at the chick magnocellular synapses (Otis et al. 1996). Whilst our experiments were performed at room temperature, the speed of AMPAR desensitization and glutamate clearance from the synaptic cleft are faster at physiological temperature, although release probability is unchanged (Kushmerick et al. 2006; Postlethwaite et al. 2007; but see Yang & Wang, 2006). In this respect, the relative contribution of pre- and postsynaptic mechanisms to the involvement of AMPAR desensitization in PPD may be different at physiological temperature.
Developmental acquisition of high-fidelity synaptic transmission
Before the onset of hearing, presynaptic action potentials at the calyx of Held are broad because of slow inactivation of Na+ channels (Leão et al. 2005), and the slow activation kinetics and low density of K+ channels (Nakamura & Takahashi, 2007). This limits the maximal frequency of reliable presynaptic firing to 200 Hz (Nakamura & Takahashi, 2007). Postsynaptically NMDARs broaden action potentials, thereby limiting reliable synaptic transmission below 10 Hz (Futai et al. 2001). In addition, the high release probability, high glutamate sensitivity of AMPAR desensitization, slow recovery of AMPARs from desensitization, and slow glutamate clearance from the synaptic cleft together promote postsynaptic AMPAR desensitization, thereby depressing postsynaptic potentials below firing threshold during high frequency synaptic transmission. During postnatal development, all these desensitizing factors are reduced, thereby establishing high-fidelity high frequency synaptic transmission for binaural sound source localization at this auditory relay synapse.
Acknowledgments
We thank Seiji Ozawa and Keisuke Tsuzuki for advise on the single-cell RT-PCR technique and Stephen Heinemann for providing us cDNAs for GluR. We also thank David Digregorio and Mark Farrant for helpful comments and Shizuka Seino for her excellent technical assistance. This study was supported by Grant-in-Aid for Specially Promoted Research from the Ministry of Education, Culture, Sports, Science and Technology.
Supplementary material
Online supplemental material for this paper can be accessed at: http://jp.physoc.org/cgi/content/full/jphysiol.2007.142547/DC1 and http://www.blackwell-synergy.com/doi/suppl/10.1113/jphysiol.2007.142547
References
- Abbott LF, Varela JA, Sen K, Nelson SB. Synaptic depression and cortical gain control. Science. 1997;275:220–224. doi: 10.1126/science.275.5297.221. [DOI] [PubMed] [Google Scholar]
- Ashby MC, De La Rue SA, Ralph GS, Uney J, Collingridge GL, Henley JM. Removal of AMPA receptors (AMPARs) from synapses is preceded by transient endocytosis of extrasynaptic AMPARs. J Neurosci. 2004;24:5172–5176. doi: 10.1523/JNEUROSCI.1042-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes-Davies M, Forsythe ID. Pre- and postsynaptic glutamate receptors at a giant excitatory synapse in rat auditory brain stem slices. J Physiol. 1995;488:387–406. doi: 10.1113/jphysiol.1995.sp020974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ. Depression of transmitter release at the neuromuscular junction of the frog. J Physiol. 1970;206:629–644. doi: 10.1113/jphysiol.1970.sp009034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgdorff AJ, Choquet D. Regulation of AMPA receptor lateral movements. Nature. 2002;417:649–653. doi: 10.1038/nature00780. [DOI] [PubMed] [Google Scholar]
- Borst JGG, Helmchen F, Sakmann B. Pre- and postsynaptic whole-cell recordings in the medial nucleus of the trapezoid body of the rat. J Physiol. 1995;489:825–840. doi: 10.1113/jphysiol.1995.sp021095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz S, David J, Trussell L. Enhancement of synaptic efficacy by presynaptic GABAB receptors. Neuron. 1998;20:135–141. doi: 10.1016/s0896-6273(00)80441-9. [DOI] [PubMed] [Google Scholar]
- Brorson JR, Li D, Suzuki T. Selective expression of heteromeric AMPA receptors driven by flip-flop differences. J Neurosci. 2004;24:3461–3470. doi: 10.1523/JNEUROSCI.5023-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caicedo A, Eybalin M. Glutamate receptor phenotypes in the auditory brainstem and mid-brain of the developing rat. Eur J Neurosci. 1999;11:51–74. doi: 10.1046/j.1460-9568.1999.00410.x. [DOI] [PubMed] [Google Scholar]
- Clements JD, Silver RA. Unveiling synaptic plasticity: a new graphical and analytical approach. Trends Neurosci. 2000;23:105–113. doi: 10.1016/s0166-2236(99)01520-9. [DOI] [PubMed] [Google Scholar]
- DiGregorio DA, Rothman JS, Nielsen TA, Silver RA. Desensitization properties of AMPA receptors at the cerebellar mossy fiber granule cell synapses. J Neurosci. 2007;27:8344–8357. doi: 10.1523/JNEUROSCI.2399-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ID, Barnes-Davies M. The binaural auditory pathway: membrane currents limiting multiple action potential generation in the rat medial nucleus of the trapezoid body. Proc R Soc Lond Biol Sci. 1993;251:143–150. doi: 10.1098/rspb.1993.0021. [DOI] [PubMed] [Google Scholar]
- Foster KA, Regehr WG. Variance-mean analysis in the presence of a rapid antagonist indicates vesicle depletion underlies depression at the climbing fiber synapse. Neuron. 2004;43:119–131. doi: 10.1016/j.neuron.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Futai K, Okada M, Matsuyama K, Takahashi T. High-fidelity transmission acquired via a developmental decrease in NMDA receptor expression at an auditory synapse. J Neurosci. 2001;21:3342–3349. doi: 10.1523/JNEUROSCI.21-10-03342.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- Grosskreutz J, Zoerner A, Schlesinger F, Krampfl K, Dengler R, Bufler J. Kinetic properties of human AMPA-type glutamate receptors expressed in HEK293 cells. Eur J Neurosci. 2003;17:1173–1178. doi: 10.1046/j.1460-9568.2003.02531.x. [DOI] [PubMed] [Google Scholar]
- Hermida D, Elezgarai I, Puente N, Alonso V, Anabitarte N, Bilbao A, Doñate-Oliver F, Grandes P. Developmental increase in postsynaptic α-amino-3-hydroxy-5-methyl-4 isoxazolepropionic acid receptor compartmentalization at the calyx of Held synapse. J Comp Neurol. 2006;495:624–634. doi: 10.1002/cne.20911. [DOI] [PubMed] [Google Scholar]
- Huh KH, Wenthold RJ. Turnover analysis of glutamate receptors identifies a rapidly degraded pool of the N-methyl-d-aspartate receptor subunit, NR1, in cultured cerebellar granule cells. J Biol Chem. 1999;274:151–157. doi: 10.1074/jbc.274.1.151. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Nakamura Y, Saitoh N, Li WB, Iwasaki S, Takahashi T. Distinct roles of Kv1 and Kv3 potassium channels at the calyx of Held presynaptic terminal. J Neurosci. 2003;23:10445–10453. doi: 10.1523/JNEUROSCI.23-32-10445.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Takahashi T. Mechanisms underlying presynaptic facilitatory effect of cyclothiazide at the calyx of Held of juvenile rats. J Physiol. 2001;533:423–431. doi: 10.1111/j.1469-7793.2001.0423a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Takahashi T. Developmental regulation of transmitter release at the calyx of Held in rat auditory brainstem. J Physiol. 2001;534:861–871. doi: 10.1111/j.1469-7793.2001.00861.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi I, Shokralla S, Titis P, Wang LY. The role of AMPA receptor gating in the development of high-fidelity neurotransmission at the calyx of Held synapse. J Neurosci. 2004;24:183–196. doi: 10.1523/JNEUROSCI.1074-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi I, Wang LY. Developmental profiles of glutamate receptors and synaptic transmission at a single synapse in the mouse auditory brainstem. J Physiol. 2002;540:861–873. doi: 10.1113/jphysiol.2001.013506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandler K, Friauf E. Pre- and postnatal development of efferent connections of the cochlear nucleus in the rat. J Comp Neurol. 1993;328:161–184. doi: 10.1002/cne.903280202. [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Hilding D. The development of the organ of Corti in the mouse. Acta Otolaryngol. 1965;60:207–222. doi: 10.3109/00016486509127003. [DOI] [PubMed] [Google Scholar]
- Kimura M, Saitoh N, Takahashi T. Adenosine A1 receptor-mediated presynaptic inhibition at the calyx of Held of immature rats. J Physiol. 2003;553:415–426. doi: 10.1113/jphysiol.2003.048371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, Tsukada S, Tsuzuki K, Kijima H, Ozawa S. Regulation of kinetic properties of GluR2 AMPA receptor channels by alternative splicing. J Neurosci. 2000;20:2166–2174. doi: 10.1523/JNEUROSCI.20-06-02166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike-Tani M, Saitoh N, Takahashi T. Mechanisms underlying developmental speeding in AMPA-EPSC decay time at the calyx of Held. J Neurosci. 2005;25:199–207. doi: 10.1523/JNEUROSCI.3861-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krampfl K, Schlesinger F, Zörner A, Kappler M, Dengler R, Bufler J. Control of kinetic properties of GluR2 flop AMPA-type channels: impact of R/G nuclear editing. Eur J Neurosci. 2002;15:51–62. doi: 10.1046/j.0953-816x.2001.01841.x. [DOI] [PubMed] [Google Scholar]
- Kushmerick C, Renden R, von Gersdorff H. Physiological temperatures reduce the rate of vesicle pool depletion and short-term depression via an acceleration of vesicle recruitment. J Neurosci. 2006;26:1366–1377. doi: 10.1523/JNEUROSCI.3889-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambolez B, Audinat E, Bochet P, Crépel F, Rossier J. AMPA receptor subunits expressed by single Purkinje cells. Neuron. 1992;9:247–258. doi: 10.1016/0896-6273(92)90164-9. [DOI] [PubMed] [Google Scholar]
- Leão RM, Kushmerick C, Pinaud R, Renden R, Li GL, Taschenberger H, Spirou G, Levinson SR, von Gersdorff H. Presynaptic Na+ channels: locus, development, and recovery from inactivation at a high-fidelity synapse. J Neurosci. 2005;25:3724–3738. doi: 10.1523/JNEUROSCI.3983-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomeli H, Mosbacher J, Melcher T, Höger T, Geiger JR, Kuner T, Monyer H, Higuchi M, Bach A, Seeburg PH. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science. 1994;266:1709–1713. doi: 10.1126/science.7992055. [DOI] [PubMed] [Google Scholar]
- Mammen AL, Huganir RL, O'Brien RJ. Redistribution and stabilization of cell surface glutamate receptors during synapse formation. J Neurosci. 1997;17:7351–7358. doi: 10.1523/JNEUROSCI.17-19-07351.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikaelian D, Ruben RJ. Development of hearing in the normal CBA-J mouse. Acta Otolaryngol. 1964;59:409–422. [Google Scholar]
- Nakamura Y, Takahashi T. Developmental changes in potassium currents at the rat calyx of Held presynaptic terminal. J Physiol. 2007;581:1101–1112. doi: 10.1113/jphysiol.2007.128702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Yamashita T, Saitoh N, Takahashi T. Developmental changes in calcium/calmodulin-dependent inactivation of calcium currents at the calyx of Held. J Physiol. 2008 doi: 10.1113/jphysiol.2007.142521. doi 10.1113/jphysiol.2007.142521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Combining deconvolution and noise analysis for the estimation of transmitter release rates at the calyx of Held. J Neurosci. 2001;21:444–461. doi: 10.1523/JNEUROSCI.21-02-00444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis T, Zhang S, Trussell LO. Direct measurement of AMPA receptor desensitization induced by glutamatergic synaptic transmission. J Neurosci. 1996;16:7496–7504. doi: 10.1523/JNEUROSCI.16-23-07496.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partin KM, Fleck MW, Mayer ML. AMPA receptor flip/flop mutants affecting deactivation, desensitization, and modulation by cyclothiazide, aniracetam, and thiocyanate. J Neurosci. 1996;16:6634–6647. doi: 10.1523/JNEUROSCI.16-21-06634.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partin KM, Patneau DK, Mayer ML. Cyclothiazide differentially modulates desensitization of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor splice variants. Mol Pharmacol. 1994;46:129–138. [PubMed] [Google Scholar]
- Postlethwaite M, Hennig MH, Steinert JR, Graham BP, Forsythe ID. Acceleration of AMPA receptor kinetics underlies temperature-dependent changes in synaptic strength at the rat calyx of Held. J Physiol. 2007;579:69–84. doi: 10.1113/jphysiol.2006.123612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk JC, Siuda ER, Nisenbaum ES. Molecular determinants responsible for differences in desensitization kinetics of AMPA receptor splice variants. J Neurosci. 2004;24:11416–11420. doi: 10.1523/JNEUROSCI.2464-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renden R, Taschenberger H, Puente N, Rusakov DA, Duvoisin R, Wang LY, Lehre KP, von Gersdorff H. Glutamate transporter studies reveal the pruning of metabotropic glutamate receptors and absence of AMPA receptor desensitization at mature calyx of Held synapses. J Neurosci. 2005;25:8482–8497. doi: 10.1523/JNEUROSCI.1848-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert A, Howe JR. How AMPA receptor desensitization depends on receptor occupancy. J Neurosci. 2003;23:847–858. doi: 10.1523/JNEUROSCI.23-03-00847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stern-Bach Y, Stevens CF. The tetrameric structure of a glutamate receptor channel. Science. 1998;280:1596–1599. doi: 10.1126/science.280.5369.1596. [DOI] [PubMed] [Google Scholar]
- Rozov A, Jerecic J, Sakmann B, Burnashev N. AMPA receptor channels with long-lasting desensitization in bipolar interneurons contribute to synaptic depression in a novel feedback circuit in layer 2/3 of rat neocortex. J Neurosci. 2001;21:8062–8071. doi: 10.1523/JNEUROSCI.21-20-08062.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sätzler K, Söhl LF, Bollmann JH, Borst JG, Frotscher M, Sakmann B, Lübke JH. Three-dimensional reconstruction of a calyx of Held and its postsynaptic principal neuron in the medial nucleus of the trapezoid body. J Neurosci. 2002;22:10567–10579. doi: 10.1523/JNEUROSCI.22-24-10567.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuss V, Schneggenburger R, Neher E. Separation of presynaptic and postsynaptic contributions to depression by covariance analysis of successive EPSCs at the calyx of Held synapse. J Neurosci. 2002;22:728–739. doi: 10.1523/JNEUROSCI.22-03-00728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi N. The effect of temperature on the neuromuscular junction of the frog. Jpn J Physiol. 1958;8:391–404. doi: 10.2170/jjphysiol.8.391. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, Leão RM, Rowland KC, Spirou GA, von Gersdorff H. Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron. 2002;36:1127–1143. doi: 10.1016/s0896-6273(02)01137-6. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, Scheuss V, Neher E. Release kinetics, quantal parameters and their modulation during short-term depression at a developing synapse in the rat CNS. J Physiol. 2005;568:513–537. doi: 10.1113/jphysiol.2005.093468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, von Gersdorff H. Fine-tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J Neurosci. 2000;20:9162–9173. doi: 10.1523/JNEUROSCI.20-24-09162.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer VC, Horstmann H, Groh A, Kuner T. Donut-like topology of synaptic vesicles with a central cluster of mitochondria wrapped into membrane protrusions: a novel structure-function module of the adult calyx of Held. J Neurosci. 2006;109:109–116. doi: 10.1523/JNEUROSCI.3268-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AYC, Graharm BP, Billups B, Forsythe ID. Distinguishing between presynaptic and postsynaptic mechanisms of short-term depression during action potential trains. J Neurosci. 2003;23:4868–4877. doi: 10.1523/JNEUROSCI.23-12-04868.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wu LG. The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron. 2005;46:633–645. doi: 10.1016/j.neuron.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Yang YM, Wang LY. Amplitude and kinetics of action potential-evoked Ca2+ current and its efficacy in triggering transmitter release at the developing calyx of Held synapse. J Neurosci. 2006;26:5698–5708. doi: 10.1523/JNEUROSCI.4889-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.