

Abstract
Ca2+-binding to calmodulin (CaM) causes facilitation and/or inactivation of recombinant Ca2+ channels. At the rat calyx of Held, before hearing onset, presynaptic Ca2+ currents (IpCa) undergo Ca2+/CaM-dependent inactivation during repetitive activation at around 1 Hz, implying that this may be a major cause of short-term synaptic depression. However, it remains open whether the Ca2+/CaM-dependent inactivation of IpCa persists in more mature animals. To address this question, we tested the effect of CaM inhibitors on the activity-dependent modulation of IpCa in calyces, before (postnatal day (P) 7–9) and after (P13–15) hearing onset. Our results indicate that the CaM-dependent IpCa inactivation during low-frequency stimulation, and the ensuing synaptic depression, occur only at calyces in the prehearing period. However, CaM immunoreactivity in P8 and P14 calyces was equally strong. Even at P13–15, high frequency stimulation (200–500 Hz) could induce IpCa inactivation, which was attenuated by EGTA (10 mm) or a CaM inhibitor peptide loaded into the terminal. Furthermore, the CaM inhibitor peptide attenuated a transient facilitation of IpCa preceding inactivation observed at 500 Hz stimulation, whereas it had no effect on sustained IpCa facilitations during trains of 50–200 Hz stimulation. These results suggest that the Ca2+/CaM-dependent IpCa modulation requires a high intraterminal Ca2+ concentration, which can be attained at immature calyces during low frequency stimulation, but only during unusually high frequency stimulation at calyceal terminals in the posthearing period.
At the calyx of Held synapse in the rodent auditory brainstem, repetitive activation at high frequencies (> 50 Hz) causes presynaptic Ca2+ currents (IpCa) to undergo Ca2+-dependent facilitation (Borst & Sakmann, 1998; Cuttle et al. 1998; Taschenberger et al. 2002; Tsujimoto et al. 2002) followed by divalent charge-carrier-dependent inactivation (Forsythe et al. 1998). This activity-dependent modulation of IpCa contributes to short-term synaptic plasticity (Forsythe et al. 1998; Inchauspe et al. 2004; Ishikawa et al. 2005; for a review, see Takahashi, 2005). When activated at low frequencies (< 10 Hz), IpCa remains unchanged in P14–17 rats (Takahashi et al. 2000). Recently, however, Xu & Wu (2005) reported that IpCa undergoes strong inactivation during low frequency activation, such as 2 Hz, at P8–13 rat calyces. This IpCa inactivation is Ca2+/CaM dependent, and could be a major cause of short-term synaptic depression.
During the second postnatal week, during which hearing onset occurs (P10–12, Jewett & Romano, 1972; Futai et al. 2001), calyces of Held undergo dramatic morphological, molecular and functional changes (Kandler & Friauf, 1993; Taschenberger & von Gersdorff, 2000; Futai et al. 2001; Iwasaki & Takahashi, 2001; Taschenberger et al. 2002; Fedchyshyn & Wang, 2005). During this period, Ca2+ channel subtypes in the nerve terminal, which mediate transmitter release, switch from mixed N-, P/Q- and R-types to predominantly P/Q-type (Iwasaki & Takahashi, 1998; Iwasaki et al. 2000), mobile Ca2+ buffer proteins, such as parvalbumin and calretinin, increase (Felmy & Schneggenburger, 2004), and Ca2+-dependent transmitter release modality changes (Fedchyshyn & Wang, 2005). As these developmental changes may potentially affect Ca2+/CaM-dependent IpCa inactivation, we examined IpCa inactivation more closely, and compared calyces in prehearing (P7–9) and posthearing (P13–15) periods. Our results indicate that at the calyx of Held, the occurrence of Ca2+/CaM-dependent inactivation of IpCa during low frequency activation and its contribution to synaptic depression are developmental phenomena restricted to the prehearing period, and that after the second postnatal week, the CaM-dependent IpCa inactivation can be induced only by the maximal frequency of stimulation.
Methods
Slice preparation and solutions
All experiments were performed in accordance with the guidelines of the Physiological Society of Japan. Brainstem slices were prepared from Wistar rats (P7–9 or P13–15) as previously described (Yamashita et al. 2003). Briefly, the rat was decapitated under halothane anaesthesia and the brain was quickly removed. Transverse slices (150–300 μm thick) containing the medial nucleus of the trapezoid body (MNTB) were cut using a tissue slicer (Linearslicer PRO-7; Dosaka, Japan).
Slices were incubated for 1 h at 36–37°C in artificial cerebrospinal fluid (aCSF) containing (mm): 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 10 glucose, 3 myo-inositol, 2 sodium pyruvate, and 0.5 ascorbic acid (pH 7.4 when bubbled with 95% O2 and 5% CO2, 310–320 mosmol l−1), and maintained thereafter at room temperature (22–28°C). All recordings were made at room temperature. The aCSF routinely contained bicuculline methiodide (10 μm) and strychnine hydrochloride (0.5 μm) to block GABAA receptors and glycine receptors, respectively. For measuring IpCa, NaCl in the aCSF was replaced by equimolar TEA-Cl (10 mm). Furthermore, TTX (1 μm) and 4-AP (0.5 mm) were added to the aCSF to block sodium and potassium currents. The presynaptic pipette solutions contained (mm): 105 caesium gluconate, 30 CsCl, 10 Hepes, 0.5 EGTA, 1 MgCl2, 12 phosphocreatine (Na salt), 3 ATP (Mg salt), 0.3 GTP (Na salt) (pH 7.3–7.4 adjusted with CsOH, 290–300 mosmol l−1). In some experiments, myosin light chain kinase (MLCK) peptide (30 μm; Calbiochem, USA) was dissolved in the presynaptic pipette solution and loaded into the presynaptic terminal by diffusion. When 10 mm EGTA was added to the pipette solution, caesium gluconate was reduced to maintain constant osmolarity. The postsynaptic pipette solution contained (mm): 110 CsF, 30 CsCl, 10 Hepes, 5 EGTA, 1 MgCl2 and 5 QX-314 (pH adjusted to 7.3–7.4 with CsOH, 285–295 mosmol l−1). For measuring EPSCs, d(–)2-amino-5-phosphonovaleric acid (d-APV, 50 μm) was added to the aCSF to block NMDA receptor-mediated currents. At P7–9 (but not at P13–15) calyceal synapses, AMPA receptor desensitization and saturation are involved in synaptic depression during repetitive stimulation (Taschenberger et al. 2002). To minimize these effects, cyclothiazide (100 μm) and kynurenate (2 mm) were included in the aCSF for recording EPSCs in P7–9 rats.
Recordings and data analysis
Whole-cell recordings were made from the calyx of Held presynaptic terminals and postsynaptic MNTB principal neurons using a patch-clamp amplifier (EPC 7 or EPC 9/2, HEKA Electronik, Germany) as previously described (Yamashita et al. 2003). The presynaptic pipette was pulled to give a resistance of 5–7 MΩ and recordings had a series resistance of 9–25 MΩ, which was compensated by up to 75% for a final value of 6–7 MΩ. Presynaptic Ca2+ currents were evoked by 1 ms depolarizing command pulses to 0 mV under voltage-clamp at a holding potential of −80 mV unless otherwise noted. The resistance of the postsynaptic pipette was 2–4 MΩ, and series resistance was typically 7–15 MΩ, which was not compensated for.
EPSCs were evoked by afferent fibre stimulation with a bipolar tungsten electrode positioned halfway between the midline and the MNTB. Records were low-pass filtered at 5 kHz and digitized at 50 kHz. To examine the effect of calmidazolium on IpCa and EPSCs, we incubated slices with aCSF containing calmidazolium (20 μm; Sigma, USA) at least 30 min before and during recordings. Calmidazolium had no effect on the basal amplitude of IpCa and EPSCs (data not shown).
All values are given as means ±s.e.m. and significance of difference was evaluated by Student's unpaired t test or two-way ANOVA. P < 0.05 was taken as the level of significance.
Immunocytochemistry
Tissue fixation and immunocytochemistry of brainstem containing the MNTB region of Wistar rats (P8 and P14) were performed as previously described (Ishikawa et al. 2003). To visualize CaM and synaptophysin, we used anti-CaM antibody (mouse monoclonal, Upstate Biotechnology, USA; diluted 1 : 500) and anti-synaptophysin antibody (rabbit polyclonal, Zymed Laboratories, USA; diluted 1 : 200), together with goat secondary antibodies conjugated with Alexa fluor 488 and Alexa fluor 588 (Invitrogen, USA; diluted 1 : 200). In the primary antibody absorbing tests for evaluating the specificity of the CaM immuno-reactivity, both recombinant CaM (Upstate) and CaM from bovine brain (Calbiochem) were incubated with primary antibodies for 30 min at room temperature.
Results
Developmental changes in CaM-dependent inactivation of presynaptic Ca2+ currents
IpCa at P8–13 calyces undergoes inactivation during low frequency stimulation when recorded with a whole-cell pipette solution containing 50 μm BAPTA (Xu & Wu, 2005), whereas no such inactivation is observed for IpCa at P14–17 calyces with a pipette solution containing 0.5 mm EGTA (Takahashi et al. 2000). To investigate whether the discrepancy between these results reflected the different Ca2+ buffer strengths in the pipette solutions, we evoked IpCa at P7–9 calyces, under whole-cell voltage-clamp, with a pipette solution containing 0.5 mm EGTA, using a pair of brief (1 ms) depolarizing pulses (from −80 mV to 0 mV). In this protocol, IpCa showed paired-pulse inactivation (PPI) at interstimulus interval (ISIs) of 0.05–2 s (Fig. 1A and B). The maximal inhibition (15.6 ± 2.4%, n = 6) was observed at 0.5 s ISI. Preincubation of slices with the CaM inhibitor calmidazolium (20 μm) significantly attenuated the PPI of IpCa. Furthermore, intraterminal application of the myosin light chain kinase (MLCK) peptide (30 μm), a specific inhibitor of CaM (Török & Trentham, 1994), also attenuated the PPI of IpCa. These results are consistent with those reported by Xu & Wu (2005), indicating that the Ca2+/CaM-dependent inactivation of IpCa is reproducible at P7–9 calyces with a pipette solution containing 0.5 mm EGTA.
Figure 1. Developmental changes in IpCa inactivation at the calyx of Held.
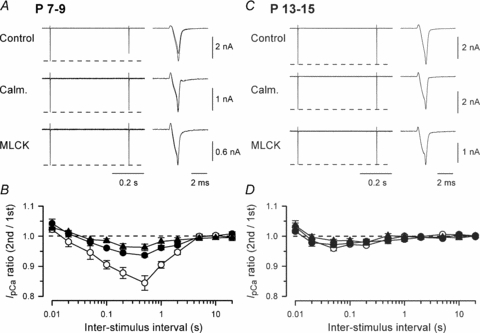
IpCa was induced by paired depolarizing command pulses (from −80 mV to 0 mV, 1 ms in duration) in P7–9 (A and B) or P13–15 (C and D) rat calyces. A and C, sample records of IpCa after application of calmidazolium (Calm. 20 μm by preincubation), or MLCK peptide (MLCK, 30 μm, by intraterminal application), or without application of CaM inhibitors (Control). The fist and second IpCa are superimposed on the right panel at faster time scales. Dashed lines indicate the first IpCa amplitudes. B and D, the ratio of the second IpCa amplitude relative to the first one (IpCa ratio on ordinate) at different ISIs (abscissa) after calmidazolium treatment (•), intraterminal application of MLCK peptide (▴) or with no inhibitor application (○). At P7–9 (B), maximal IpCa inactivation (at 0.5 s ISI) was 15.6 ± 2.4% (n = 6) in control, 6.4 ± 0.6% (n = 5) after calmidazolium treatment, and 3.7 ± 1.1% (n = 5) after intraterminal MLCK peptide application. At P13–15 (D), maximal IpCa inactivation (at 50 ms ISI) was 4.1 ± 0.7% (n = 7) in control, 2.8 ± 1.0% (n = 4) after calmidazolium treatment, and 1.5 ± 0.5% (n = 5) after intraterminal MLCK peptide application. Results were essentially the same when the charge instead of amplitude was measured for IpCa. In this and following figures, data points and bars indicate mean and s.e.m.
We next investigated whether the CaM-dependent IpCa inactivation persists after hearing onset (Jewett & Romano, 1972). At P13–15 calyces, IpCa exhibited much less PPI (at 0.01–20 s ISIs, Fig. 1C and D). Neither calmidazolium (20 μm) nor MLCK peptide (30 μm) affected the PPI of IpCa. These results indicate that the Ca2+/CaM-dependent IpCa inactivation at 0.1–2 s ISI is a developmental phenomenon, occurring only at immature calyces of Held.
Contribution of CaM-dependent IpCa inactivation to synaptic depression
In simultaneous pre- and postsynaptic whole-cell recording at P7–9 calyces, the Ca2+/CaM-dependent IpCa inactivation has been shown to contribute to synaptic depression during low frequency stimulation (Xu & Wu, 2005). However, under whole-cell recording, mobile endogenous Ca2+ buffers in the nerve terminal may be washed out and replaced by Ca2+ buffer in the pipette solution. To evaluate the contribution of Ca2+/CaM-dependent IpCa to synaptic depression at synapses with no presynaptic dialysis, we used calmidazolium, which inhibits Ca2+/CaM-dependent IpCa inactivation following extracellular application (Fig. 1). When EPSCs were evoked by afferent fibre stimulation at 0.5–5 Hz, they were depressed to a low steady level (Fig. 2A), which was lower at higher frequency of stimulation (Fig. 2B). Calmidazolium significantly attenuated the magnitude of synaptic depression, raising the steady level during repetitive stimulation, by 13–19% (Fig. 2B) and paired-pulse ratio of EPSCs by 13–21% (Fig. 2C). These results suggest that Ca2+/CaM-dependent IpCa inactivation does contribute to synaptic depression at P7–9 calyceal synapse, as postulated by Xu & Wu (2005). At P13–15, however, calmidazolium (20 μm) no longer attenuated synaptic depression (Fig. 2D–F), as expected from the lack of CaM-dependent IpCa inactivation (Fig. 1C and D). Thus, the occurrence of Ca2+/CaM-dependent IpCa inactivation during low frequency stimulation and the ensuing synaptic depression are restricted to the prehearing period.
Figure 2. Developmental changes in the contribution of Ca2+/CaM-dependent IpCa inactivation to synaptic depression at the calyx of Held.
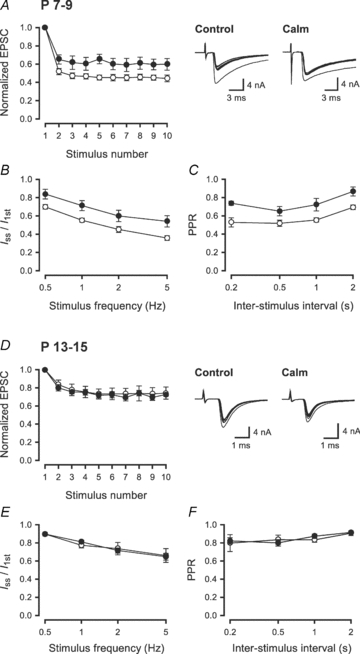
Nerve-evoked EPSCs were recorded from MNTB principal neurons in P7–9 (A–C) and P13–15 (D–F) rats. A and D, the EPSC amplitudes, during a 2 Hz train (10 stimuli), relative to the first one, with (•) or without (○) calmidazolium pretreatment. Sample records show the 1st–10th EPSCs (superimposed). In A, cyclothiazide (100 μm) and kynurenate (2 mm) were included in the aCSF to block AMPA receptor desensitization and saturation. B and E, magnitude of synaptic depression, at P7–9 (B) and P13–15 (E) synapses, during trains of stimulation at 0.5–5 Hz, expressed as the steady-state amplitude (Iss; mean of the 6th to 10th events) relative to the first EPSC amplitude (I1st) with (•) or without (○) calmidazolium pretreatment. Calmidazolium significantly reduced the magnitude of synaptic depression (P < 0.01, n = 5–9) at P7–9 synapses (B), but had no effect at P13–14 synapses (E, P > 0.9, n = 4–9). C and F, paired-pulse ratio (PPR) of EPSCs at different ISIs with (•) or without (○) calmidazolium pretreatment at P7–9 (C) and P13–15 (F) synapses. The PPR was measured as the ratio of the second EPSC amplitude relative to the first one during trains of stimulation at different frequencies.
Mechanisms underlying the developmental decline of CaM-dependent IpCa inactivation
What mechanisms underlie the developmental decline of Ca2+/CaM-dependent IpCa inactivation? Might CaM expression in the nerve terminal be down-regulated during development? Presynaptic terminals, identified with synaptophysin immunoreactivity, as well as postsynaptic cell bodies, showed strong CaM-immunoreactivity at both P8 and P14 (Fig. 3A). The CaM-immunofluorescence signals were observed practically in all calyces in the MNTB region. The CaM-specificity of the signals was verified by their disappearance after absorbing the primary antibody with CaM protein (P14 with CaM). The average intensity of CaM-immunofluorescence signals at P14 calyces was similar to that at P8 calyces (Fig. 3B), indicating that the level of CaM expression in the nerve terminal was unchanged during the second postnatal week.
Figure 3. CaM immunoreactivities at the calyces of Held terminal at P8 and P14.
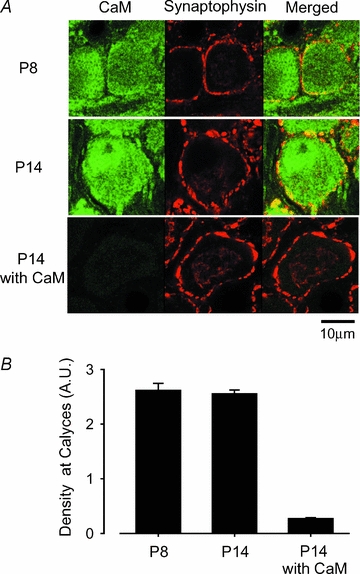
A, CaM immunoreactivity (left column, labelled green with Alexa fluor 488), synaptophysin immunoreactivity (middle column, labelled red with Alexa fluor 568), and their overlap (right column, yellow). Bottom panel in the left column (P14 with CaM) shows the background after absorbing the primary antibodies with CaM protein. B, densitometric measurements of CaM immunofluorescence intensity in the regions overlapped with synaptophysin immunofluorescence signals at P8 and P14, and the background intensity after antibody absorption at P14.
Ca2+ channel subtypes in calyceal terminals undergo a developmental switch from mixed N-, P/Q- and R-types to predominantly P/Q-type during the second postnatal week (Iwasaki & Takahashi, 1998; Iwasaki et al. 2000). We investigated whether this switch might be a cause for the reduction of CaM-dependent IpCa inactivation. At P7–9 calyces, after blocking N-type VGCCs using ω-conotoxin GVIA (2 μm), IpCa clearly showed PPI with its peak being observed at 0.5 s ISI (Fig. 4). The magnitude of inactivation was, however, slightly smaller than control, presumably because of less Ca2+ influx after blocking N-type VGCCs. These results indicate that the developmental reduction of N-type VGCCs cannot be a major cause for the developmental decline of CaM-dependent IpCa inactivation.
Figure 4. Effect of an N-type Ca2+ channel blocker on the Ca2+/CaM-dependent inactivation of IpCa.
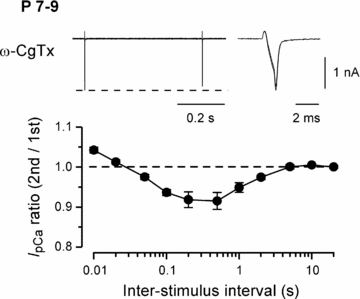
IpCa was induced by paired depolarizing pulses, as in Fig. 1, in the presence of ω-CgTX (2 μm) at P7–9 calyces (n = 5). Sample records, and the ratio of the second IpCa amplitude relative to the first one at different ISIs (abscissa), are shown as in Fig. 1.
Binding of Ca2+ to the C-terminal lobe of CaM facilitates VGCCs (Ca2+-dependent facilitation, CDF), whereas Ca2+ binding to its N-terminal lobe inactivates VGCCs (Ca2+-dependent inactivation, CDI) (DeMaria et al. 2001; Liang et al. 2003). In cells expressed with recombinant P/Q-type channels, intracellular EGTA has no effect on C-lobe-dependent CDF but blocks N-lobe-dependent CDI (Liang et al. 2003; see also Lee et al. 2000). Whilst it remains open whether such a differential mechanism operates for native VGCCs, we examined whether EGTA affects Ca2+/CaM-dependent inactivation of IpCa at P7–9 calyces. As shown in Fig. 5A, EGTA (10 mm) markedly reduced Ca2+/CaM-dependent inactivation of IpCa, suggesting that CaM N-lobe might be involved in IpCa inactivation. Because EGTA has a slow binding on-rate (Smith et al. 1984) it can reduce Ca2+ mainly in the region distant from the site of Ca2+ entry. Might it be that even in mature calyces, if Ca2+ accumulates during high frequency stimulation, IpCa undergoes Ca2+/CaM-dependent inactivation? When we repetitively activated IpCa by 1 ms depolarizing square pulses at 50–200 Hz for 2 s, IpCa underwent facilitation, which was followed by inactivation at 200 Hz. EGTA (10 mm) loaded into calyces attenuated both facilitation and inactivation of IpCa (Fig. 5B). Thus, the Ca2+-dependent inactivation of IpCa can be induced in mature calyces even after hearing onset.
Figure 5. Inhibitory effects of EGTA on the activity-dependent inactivation and facilitation of IpCa.
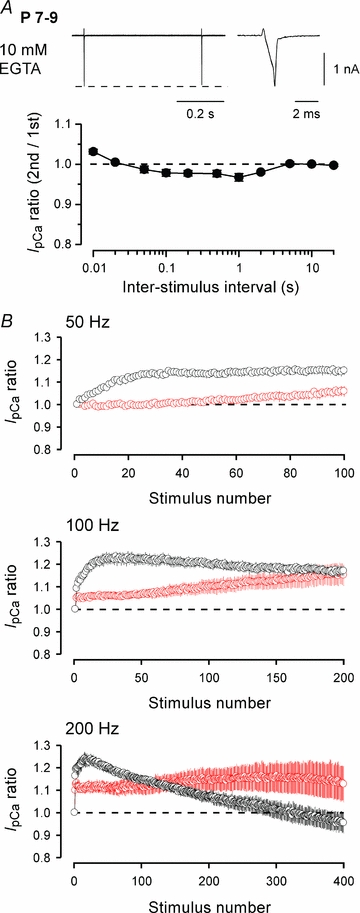
IpCa was evoked by 1 ms depolarizing command pulses after dialysing calyces with a patch pipette solution containing 10 mm EGTA. A, at P7–9 calyces, EGTA markedly reduced IpCa inactivation induced by the paired-pulse protocol. The maximal IpCa inactivation (at 1 s ISI) was 3.3 ± 0.8% (n = 5), which was significantly less than control in Fig. 1 (P < 0.01). B, at P13–15 calyces, IpCa was evoked by 2 s trains of 1 ms command pulses at 50 Hz, 100 Hz and 200 Hz, with 0.5 mm (black, n = 4) or 10 mm (red, n = 4) EGTA included in the patch pipette solution. On the ordinate, the IpCa amplitude is normalized to the initial one.
We next investigated whether the IpCa modulation observed during trains of repetitive stimulation at mature calyces was CaM dependent. In these experiments we evoked IpCa by trains of action potential (AP)-waveform pulses (Fig. 6A). With this protocol IpCa showed predominant facilitation during stimulation at 50–200 Hz (Fig. 6B–D), but a sustained inactivation followed a transient facilitation when stimulated at 500 Hz (Fig. 6E). MLCK peptide (30 μm), loaded into calyces, had no effect on IpCa facilitation at 50–200 Hz, but attenuated both facilitation and inactivation of IpCa at 500 Hz.
Figure 6. Involvement of CaM in activity-dependent inactivation and facilitation of IpCa at P13–15 calyces.
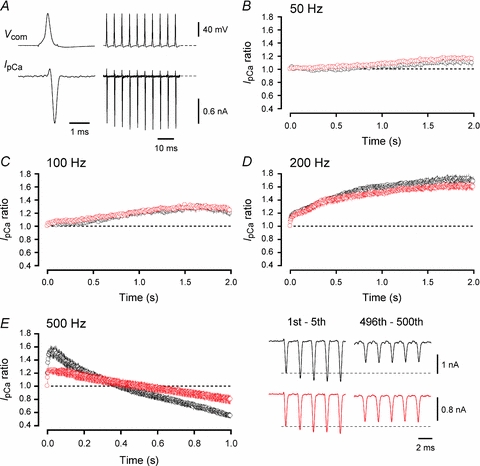
A, sample records of IpCa evoked by 200 Hz trains of AP-waveform command pulses (Vcom). The AP-waveforms were obtained from another calyceal terminal at the resting potential of −71 mV. The initial AP (shown on a fast time scale) had an overshoot of 22 mV. B–E, the IpCa amplitude, normalized to the first one, during 50 Hz (B), 100 Hz (C), 200 Hz (D) and 500 Hz (E) trains, in the presence (red) and absence (black) of MLCK peptide (30 μm) in the calyceal terminal. Averaged data are from 4 to 5 calyceal terminals. Sample records in E show the 1st–5th and 496th–500th IpCa during 500 Hz trains, in the absence (black traces) and presence (red traces) of MLCK peptide. The maximal IpCa facilitation and inactivation was 55.2 ± 5.3% and 45.9 ± 4.9%, respectively, in control (n = 5), and 27 ± 4.2% (P < 0.03) and 21.3 ± 6.2% (P < 0.02), respectively, in the presence of MLCK peptide (n = 4).
Discussion
Developmental decline of Ca2+/CaM-dependent IpCa inactivation
At immature calyces of Held (P7–9), we confirmed the findings by Xu & Wu (2005) that IpCa evoked by a pair of brief depolarizing pulses undergoes prominent PPI at long ISIs (0.05–2 s), and that these PPI can be attenuated by the CaM inhibitors calmidazolium or MLCK peptide. In contrast, at P13–15 calyces, the Ca2+/CaM-dependent PPI of IpCa was absent, despite the fact that CaM expression in calyceal terminals was similar at P8 and P14. Even at P13–15, however, repetitive activation of IpCa at high frequencies showed IpCa inactivation, which could be attenuated by 10 mm EGTA or MLCK peptide. These results suggest that the developmental decline of the Ca2+/CaM-dependent inactivation of IpCa results from a developmental change in the intraterminal Ca2+ dynamics, rather than the Ca2+/CaM-dependent mechanism.
CaM has four Ca2+ binding sites with Kd ranging from 1.7 μm to 8.9 μm (Shifman et al. 2006). At P8–10 calyces, the volume-averaged intraterminal Ca2+ concentration reaches 400 nm during an AP (Helmchen et al. 1997). After hearing onset, local Ca2+ transients, evoked by an AP and assessed by confocal spot measurements, become sparser and smaller (Nakamura et al. 2007). Developmental changes in intraterminal Ca2+ dynamics have been suggested by the finding that 10 mm EGTA loaded into calyces reduces EPSCs by more than 50% in the prehearing period (Borst & Sakmann, 1996), but that the reduction becomes marginal after hearing onset, despite the fact that BAPTA attenuates EPSCs to a similar extent throughout the developmental period (Fedchyshyn & Wang, 2005). As the Ca2+-binding rate of EGTA is two orders of magnitude slower than that of BAPTA (Smith et al. 1984), it has been proposed that the number of VGCCs comprising the Ca2+ domain may decrease during development (Fedchyshyn & Wang, 2005). The intraterminal Ca2+ dynamics may also change as a result of developmental increase in endogenous Ca2+ buffers (Lohmann & Friauf, 1996; Felmy & Schneggenburger, 2004), or more restricted Ca2+ diffusion owing to the developmental restructuring of calyces (Kandler & Friauf, 1993; Taschenberger et al. 2002).
Ca2+-dependent Ca2+ current facilitation
At P13–15 calyces, during repetitive stimulation (50–500 Hz), IpCa showed facilitation as reported previously (Cuttle et al. 1998; Forsythe et al. 1998; Taschenberger et al. 2002; Tsujimoto et al. 2002). This facilitation could be attenuated by 10 mm EGTA (Fig. 5B), but the CaM inhibitor MLCK peptide had no effect on the facilitation evoked by AP-waveform command pulses at 50–200 Hz. Given that neuronal calcium sensor 1 (NCS-1) mimics and partially occludes IpCa facilitation produced by a pair of depolarizing pulses at 5–20 ms intervals (Tsujimoto et al. 2002), Ca2+-dependent IpCa facilitations observed at 50–200 Hz is likely to be mediated by NCS-1. During 500 Hz stimulation, however, MLCK peptide attenuated IpCa facilitation, suggesting an additional involvement of CaM at this maximal frequency. These observations are in agreement with the fact that NCS-1 has more than 10 times higher Ca2+-binding affinity than CaM (Burgoyne, 2007). Among EF-hand splice variants of VGCC α1A subunit, EFa channels are sensitive to global changes in intracellular Ca2+ concentration, whereas EFb channels are insensitive, being spared even in the presence of 10 mm BAPTA (Chaudhuri et al. 2004). Thus, EFa channels are more likely to be involved in the facilitation of IpCa at the calyx of Held.
Physiological roles of CaM-dependent IpCa modulations
At calyceal synapses in the prehearing period, with non-dialysed presynaptic terminals, synaptic depression induced by repetitive stimulation of afferent fibres was attenuated by calmidazolium. Ca2+/CaM is thought to accelerate vesicle replenishment at calyceal synapses (Sakaba & Neher, 2001). If this mechanism predominates, the CaM blocker should have increased the magnitude of depression. Thus, Ca2+/CaM-dependent IpCa inactivation seems to be a more important mechanism underlying synaptic depression during low frequency stimulation (< 30 Hz) at the immature calyx of Held, as postulated by Xu & Wu (2005). However, at posthearing calyces Ca2+/CaM-dependent IpCa inactivation became evident only after prolonged high frequency stimulation. At calyceal synapses of P15 mice, at room temperature, transmission starts to fail at 20 Hz and failures become prominent at 50 Hz, unless NMDA receptors are pharmacologically blocked (Futai et al. 2001). In this respect, Ca2+/CaM-dependent IpCa inactivation during prolonged transmission at 500 Hz is unlikely to play a physiological role. It remains to be seen, however, whether this mechanism operates for synaptic depression in physiological conditions. At physiological temperature, calyceal synapses can follow short trains of afferent fibre stimulations at 600–800 Hz (Taschenberger & von Gersdorff, 2000), but the build-up of intraterminal Ca2+ may be reduced by faster activation/inactivation kinetics of IpCa (Kushmerick et al. 2006).
Short-term synaptic depression at immature calyces, before hearing onset, involves multiple mechanisms, in addition to classically documented depletion of releasable synaptic vesicles (Betz, 1970). These mechanisms include desensitization of postsynaptic AMPA receptors (Taschenberger et al. 2002, 2005), autoreceptor inhibition via G protein-coupled receptors (Iwasaki & Takahashi, 2001; Kimura et al. 2003) and Ca2+/CaM-dependent IpCa inactivation (Xu & Wu, 2005; present study). As animals mature, most of these mechanisms become less significant, with vesicle depletion remaining as the most important mechanism underlying short-term depression at the calyx of Held.
Acknowledgments
This study was supported by a Grant-in-Aid for Specially Promoted Research from the Ministry of Education, Culture, Sports, Science and Technology to T.T. and Research Fellowship from the Japanese Society for the Promotion of Science to T.Y. We thank Mark Farrant for helpful comments and Masahiro Kaneko for providing action-potential waveform command templates.
References
- Betz WJ. Depression of transmitter release at the neuromuscular junction of the frog. J Physiol. 1970;206:629–644. doi: 10.1113/jphysiol.1970.sp009034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JGG, Sakmann B. Calcium influx and transmitter release in a fast CNS synapse. Nature. 1996;383:431–434. doi: 10.1038/383431a0. [DOI] [PubMed] [Google Scholar]
- Borst JGG, Sakmann B. Facilitation of presynaptic calcium currents in the rat brainstem. J Physiol. 1998;513:149–155. doi: 10.1111/j.1469-7793.1998.149by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne RD. Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat Rev Neurosci. 2007;8:182–193. doi: 10.1038/nrn2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Chang S-Y, DeMaria CD, Alvania RS, Soong TW, Yue DT. Alternative splicing as a molecular switch for Ca2+/calmodulin-dependent facilitation of P/Q-type Ca2+ channels. J Neurosci. 2004;24:6334–6342. doi: 10.1523/JNEUROSCI.1712-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuttle MF, Tsujimoto T, Forsythe ID, Takahashi T. Facilitation of the presynaptic calcium current at an auditory synapse in rat brainstem. J Physiol. 1998;512:723–729. doi: 10.1111/j.1469-7793.1998.723bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- Fedchyshyn MJ, Wang LY. Developmental transformation of the release modality at the calyx of Held synapse. J Neurosci. 2005;25:4131–4140. doi: 10.1523/JNEUROSCI.0350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felmy F, Schneggenburger R. Developmental expression of the Ca2+-binding proteins calretinin and parvalbumin at the calyx of Held of rats and mice. Eur J Neurosci. 2004;20:1473–1482. doi: 10.1111/j.1460-9568.2004.03604.x. [DOI] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- Futai K, Okada M, Matsuyama K, Takahashi T. High-fidelity transmission acquired via a developmental decrease in NMDA receptor expression at an auditory synapse. J Neurosci. 2001;21:3342–3349. doi: 10.1523/JNEUROSCI.21-10-03342.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F, Borst JGG, Sakmann B. Calcium dynamics associated with a single action potential in a CNS presynaptic terminal. Biophys J. 1997;72:1458–1471. doi: 10.1016/S0006-3495(97)78792-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inchauspe CG, Martini FJ, Forsythe ID, Uchitel OD. Functional compensation of P/Q by N-type channels blocks short-term plasticity at the calyx of Held presynaptic terminal. J Neurosci. 2004;24:10379–10383. doi: 10.1523/JNEUROSCI.2104-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Kaneko M, Shin H-S, Takahashi T. Presynaptic N-type and P/Q-type Ca2+ channels mediating synaptic transmission at the calyx of Held of mice. J Physiol. 2005;568:199–209. doi: 10.1113/jphysiol.2005.089912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Nakamura Y, Saitoh N, Li W-B, Iwasaki S, Takahashi T. Distinct roles of Kv1 and Kv3 potassium channels at the calyx of Held presynaptic terminal. J Neurosci. 2003;23:10445–10453. doi: 10.1523/JNEUROSCI.23-32-10445.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Momiyama A, Uchitel OD, Takahashi T. Developmental changes in calcium channel types mediating central synaptic transmission. J Neurosci. 2000;20:59–65. doi: 10.1523/JNEUROSCI.20-01-00059.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Takahashi T. Developmental changes in calcium channel types mediating synaptic transmission in rat auditory brainstem. J Physiol. 1998;509:419–423. doi: 10.1111/j.1469-7793.1998.419bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Takahashi T. Developmental regulation of transmitter release at the calyx of Held in rat auditory brainstem. J Physiol. 2001;534:861–871. doi: 10.1111/j.1469-7793.2001.00861.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett DL, Romano MN. Neonatal development of auditory system potentials averaged from the scalp of rat and cat. Brain Res. 1972;36:101–115. doi: 10.1016/0006-8993(72)90769-x. [DOI] [PubMed] [Google Scholar]
- Kandler K, Friauf E. Pre- and postnatal development of efferent connections of the cochlear nucleus in the rat. J Comp Neurol. 1993;328:161–184. doi: 10.1002/cne.903280202. [DOI] [PubMed] [Google Scholar]
- Kimura M, Saitoh N, Takahashi T. Adenosine A1 receptor-mediated presynaptic inhibition at the calyx of Held of immature rats. J Physiol. 2003;553:415–426. doi: 10.1113/jphysiol.2003.048371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushmerick C, Renden R, von Gersdorff H. Physiological temperatures reduce the rate of vesicle pool depletion and short-term depression via an acceleration of vesicle recruitment. J Neurosci. 2006;26:1366–1377. doi: 10.1523/JNEUROSCI.3889-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Scheuer T, Catterall WA. Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J Neurosci. 2000;20:6830–6838. doi: 10.1523/JNEUROSCI.20-18-06830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA, Yue DT. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39:951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- Lohmann C, Friauf E. Distribution of the calcium-binding proteins parvalbumin and calretinin in the auditory brainstem of adult and developing rats. J Comp Neurol. 1996;367:90–109. doi: 10.1002/(SICI)1096-9861(19960325)367:1<90::AID-CNE7>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, DiGregorio D, Takahashi T. Single action potential-evoked Ca2+ transients at the calyx of Held presynaptic terminal. Neurosci Res. 2007;58S:S71. [Google Scholar]
- Sakaba T, Neher E. Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron. 2001;32:1119–1131. doi: 10.1016/s0896-6273(01)00543-8. [DOI] [PubMed] [Google Scholar]
- Shifman JM, Choi MH, Mihalas S, Mayo SL, Kennedy MB. Ca2+/calmodulin-dependent protein kinase II (CaMKII) is activated by calmodulin with two bound calciums. Proc Natl Acad Sci U S A. 2006;103:13968–13973. doi: 10.1073/pnas.0606433103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PD, Liesegang GW, Berger RL, Czerlinski G, Podolsky RJ. A stopped-flow investigation of calcium ion binding by ethylene glycol bis (b-aminoethyl ether)-N,N-tetraacetic acid. Anal Biochem. 1984;143:188–195. doi: 10.1016/0003-2697(84)90575-x. [DOI] [PubMed] [Google Scholar]
- Takahashi T. Dynamic aspects of presynaptic calcium currents mediating synaptic transmission. Cell Calcium. 2005;37:507–511. doi: 10.1016/j.ceca.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Hori T, Kajikawa Y, Tsujimoto T. The role of GTP-binding protein activity in fast central synaptic transmission. Science. 2000;289:460–463. doi: 10.1126/science.289.5478.460. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, Leão RM, Rowland KC, Spirou GA, von Gersdorff H. Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron. 2002;36:1127–1143. doi: 10.1016/s0896-6273(02)01137-6. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, Scheuss V, Neher E. Release kinetics, quantal parameters and their modulation during short-term depression at a developing synapse in the rat CNS. J Physiol. 2005;568:513–537. doi: 10.1113/jphysiol.2005.093468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, von Gersdorff H. Fine-tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J Neurosci. 2000;20:9162–9173. doi: 10.1523/JNEUROSCI.20-24-09162.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Török K, Trentham DR. Mechanism of 2-chloro(ɛ-amino-Lys75)-[6-[4-(N,N-diethylamino)phenyl]-1,3,5-triazin-4-yl]calmodulin interactions with smooth muscle myosin light chain kinase and derived peptides. Biochemistry. 1994;33:12807–12820. doi: 10.1021/bi00209a012. [DOI] [PubMed] [Google Scholar]
- Tsujimoto T, Jeromin A, Saitoh N, Roder JC, Takahashi T. Neuronal calcium sensor 1 and activity-dependent facilitation of P/Q-type calcium currents at presynaptic nerve terminals. Science. 2002;295:2276–2279. doi: 10.1126/science.1068278. [DOI] [PubMed] [Google Scholar]
- Xu J, Wu L-G. The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron. 2005;46:633–645. doi: 10.1016/j.neuron.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Ishikawa T, Takahashi T. Developmental increase in vesicular glutamate content does not cause saturation of AMPA receptors at the calyx of Held synapse. J Neurosci. 2003;23:3633–3638. doi: 10.1523/JNEUROSCI.23-09-03633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]