

Abstract
Time-resolved visible pump/mid-infrared (mid-IR) probe spectroscopy in the region between 1600 and 1800 cm−1 was used to investigate electron transfer, radical pair relaxation, and protein relaxation at room temperature in the Rhodobacter sphaeroides reaction center (RC). Wild-type RCs both with and without the quinone electron acceptor QA, were excited at 600 nm (nonselective excitation), 800 nm (direct excitation of the monomeric bacteriochlorophyll (BChl) cofactors), and 860 nm (direct excitation of the dimer of primary donor (P) BChls (PL/PM)). The region between 1600 and 1800 cm−1 encompasses absorption changes associated with carbonyl (C=O) stretch vibrational modes of the cofactors and protein. After photoexcitation of the RC the primary electron donor P excited singlet state (P*) decayed on a timescale of 3.7 ps to the state 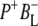 (where BL is the accessory BChl electron acceptor). This is the first report of the mid-IR absorption spectrum of
(where BL is the accessory BChl electron acceptor). This is the first report of the mid-IR absorption spectrum of 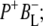 the difference spectrum indicates that the 9-keto C=O stretch of BL is located around 1670–1680 cm−1. After subsequent electron transfer to the bacteriopheophytin HL in ∼1 ps, the state
the difference spectrum indicates that the 9-keto C=O stretch of BL is located around 1670–1680 cm−1. After subsequent electron transfer to the bacteriopheophytin HL in ∼1 ps, the state 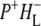 was formed. A sequential analysis and simultaneous target analysis of the data showed a relaxation of the
was formed. A sequential analysis and simultaneous target analysis of the data showed a relaxation of the 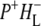 radical pair on the ∼20 ps timescale, accompanied by a change in the relative ratio of the
radical pair on the ∼20 ps timescale, accompanied by a change in the relative ratio of the 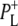 and
and 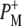 bands and by a minor change in the band amplitude at 1640 cm−1 that may be tentatively ascribed to the response of an amide C=O to the radical pair formation. We conclude that the drop in free energy associated with the relaxation of
bands and by a minor change in the band amplitude at 1640 cm−1 that may be tentatively ascribed to the response of an amide C=O to the radical pair formation. We conclude that the drop in free energy associated with the relaxation of 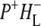 is due to an increased localization of the electron hole on the PL half of the dimer and a further consequence is a reduction in the electrical field causing the Stark shift of one or more amide C=O oscillators.
is due to an increased localization of the electron hole on the PL half of the dimer and a further consequence is a reduction in the electrical field causing the Stark shift of one or more amide C=O oscillators.
INTRODUCTION
The reaction center (RC) is a transmembrane pigment-protein complex responsible for light-driven charge separation in photosynthetic bacteria. The RC from the purple photosynthetic bacterium Rhodobacter (Rb.) sphaeroides was one of the first membrane proteins to be structurally characterized to atomic resolution by x-ray crystallography (1–3). An axis of pseudo-twofold symmetry relates the L and M subunits that, along with a third H subunit, form the basic core structure. The cofactors comprise two molecules of bacteriochlorophyll a (BChl, denoted PL and PM) that form the dimeric primary electron donor (P), two monomeric BChls (BL and BM), two bacteriopheophytins (BPhe, denoted HL and HM), two quinones (QA and QB), one nonheme iron (Fe), and one carotenoid. The BChl, BPhe, and quinone cofactors are arranged in two approximately symmetrical branches, termed L and M after the protein subunits.
The purple bacterial RC has been extensively investigated using ultrafast spectroscopic techniques, and it has been established that excitation of the P BChls results in the stepwise transfer of an electron along the L-branch of cofactors with an ∼100% efficiency. Electron transfer from P to the L-branch BPhe (HL) occurs in ∼4 ps at room temperature, whereas subsequent electron transfer to the ubiquinone QA occurs in ∼220 ps (4–7). The cation formed is shared between the PL and PM BChls that make up the P dimer. The role of the accessory BL molecule (located between the P and HL) in the electron transfer process, and in particular whether it acts as a real or a virtual intermediate, has been a matter of considerable debate. Much of this debate was caused by the difficulty in observing spectral changes related to the state 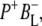 particularly in early studies (8,9). However, it is now generally accepted that the first step of electron transfer involves reduction of BL, and formation of the
particularly in early studies (8,9). However, it is now generally accepted that the first step of electron transfer involves reduction of BL, and formation of the 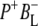 radical pair, with the rate of subsequent electron transfer from
radical pair, with the rate of subsequent electron transfer from 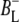 to HL being three to four times faster than the initial electron transfer from P* to BL (7,10,11). In the classical picture, excitation of the BPhes or accessory BChls of the RC results in ultrafast energy transfer to P, forming a P* state that initiates L-branch electron transfer.
to HL being three to four times faster than the initial electron transfer from P* to BL (7,10,11). In the classical picture, excitation of the BPhes or accessory BChls of the RC results in ultrafast energy transfer to P, forming a P* state that initiates L-branch electron transfer.
In this stepwise model of charge separation the free energy of the 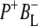 radical pair is described as being just below that of P*. It has been concluded that the free energy of the
radical pair is described as being just below that of P*. It has been concluded that the free energy of the 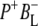 state lies ∼450 cm−1 below that of the initially excited primary donor P*, with a further (180 cm−1) drop in free energy on formation of
state lies ∼450 cm−1 below that of the initially excited primary donor P*, with a further (180 cm−1) drop in free energy on formation of 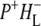 (12).
(12).
A number of experiments have challenged the classical picture in which excitation of the accessory BChl or BPhe cofactors leads uniquely to ultrafast energy transfer to P, forming P*. It was shown that after excitation of the monomeric BChls at 800 nm, direct formation of the radical pairs 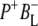 and
and 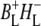 occurs without the involvement of P*, this very fast charge separation competing with energy transfer from these pigments to P. The work of van Brederode et al. (13–15) showed that this P*-independent charge separation occurs mainly after excitation of the blue side of the 800 nm absorbance band of the Rb. sphaeroides RC, and therefore involves the accessory BChl on the active L-branch (excitation of the BPhe on the L-branch also leads to direct charge separation).
occurs without the involvement of P*, this very fast charge separation competing with energy transfer from these pigments to P. The work of van Brederode et al. (13–15) showed that this P*-independent charge separation occurs mainly after excitation of the blue side of the 800 nm absorbance band of the Rb. sphaeroides RC, and therefore involves the accessory BChl on the active L-branch (excitation of the BPhe on the L-branch also leads to direct charge separation).
The picture of light-driven electron transfer in the RC is further complicated by relaxation events that occur on the timescale of the electron transfer process. For example, the multi-exponential decay of the recombination fluorescence from P* observed in RCs where electron transfer to QA is blocked has been interpreted in terms of relaxation of the originally formed radical pair states (9,16,17). In this model the multi-exponential decay of the fluorescence is thought to originate from a decrease in the free energy of the 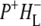 radical pair with time. Similarly, from a target analysis of data obtained in pump-probe measurements the radical pair
radical pair with time. Similarly, from a target analysis of data obtained in pump-probe measurements the radical pair 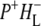 was observed to relax with a time constant in the tens of ps range (18). Such relaxations result from slow nuclear motions in the protein-cofactor system, and the role(s) played by the protein component of the RC during the initial energy and electron transfer events remains poorly understood.
was observed to relax with a time constant in the tens of ps range (18). Such relaxations result from slow nuclear motions in the protein-cofactor system, and the role(s) played by the protein component of the RC during the initial energy and electron transfer events remains poorly understood.
The purpose of the work described in this study was to examine photochemical charge separation in the Rb. sphaeroides RC through the application of femtosecond timescale infrared (IR) spectroscopy. In principle, identification of the rate of P+ formation and acceptor reduction should be straightforward when analyzing the large manifold of vibrational bands afforded by mid-IR spectroscopy (although at the cost of much lower extinction coefficients), because the spectra of the BChl and BPhe molecules in their neutral, excited, or radical state display very specific signatures in the mid-IR. Time-resolved mid-IR spectroscopy also opens up the unique possibility of investigating the protein response that follows electron transfer, and therefore the relaxation processes that occur in the vicinity of the cofactors on the timescale of electron transfer. For the bacterial RC, several studies of the initial electron transfer dynamics and protein response to this process have been reported (19–25). Hamm et al. (19) carried out measurements on Rb. sphaeroides RCs in the mid-IR region between 1000 and 1800 cm−1, with a time resolution of 400 fs. Two components were found in this work, one increasing with a time constant of 3.8 ps ascribed to the formation of 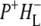 and one decaying with a time constant of 240 ps ascribed to electron transfer to the final
and one decaying with a time constant of 240 ps ascribed to electron transfer to the final 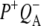 state. However, a fast kinetic component attributable to electron transfer from
state. However, a fast kinetic component attributable to electron transfer from 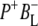 to
to 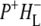 was not found in this study. A strong 200 fs component observed in a second IR experiment by Hamm et al. (20) was assigned to an ultrafast internal charge separation within the primary donor BChl dimer (i.e., formation of P+P−). In ps transient absorption studies of Rb. sphaeroides RCs in the range 1550–1800 cm−1 by Hochstrasser et al. (21), transitions of amide I were observed at a number of frequencies on a timescale shorter than 50 ps that suggested coupling of the electron transfer process with a specific part of protein backbone. Later in mid-IR experiments with 400 fs time-resolution (22) features related to the states P, P*, and P+ were identified in the difference spectra. The 9-keto modes of the P ground state were found at 1682 cm−1, and those of
was not found in this study. A strong 200 fs component observed in a second IR experiment by Hamm et al. (20) was assigned to an ultrafast internal charge separation within the primary donor BChl dimer (i.e., formation of P+P−). In ps transient absorption studies of Rb. sphaeroides RCs in the range 1550–1800 cm−1 by Hochstrasser et al. (21), transitions of amide I were observed at a number of frequencies on a timescale shorter than 50 ps that suggested coupling of the electron transfer process with a specific part of protein backbone. Later in mid-IR experiments with 400 fs time-resolution (22) features related to the states P, P*, and P+ were identified in the difference spectra. The 9-keto modes of the P ground state were found at 1682 cm−1, and those of 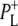 and
and 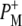 at 1714 cm−1 and 1702 cm−1, respectively. A fast transition at 1665 cm−1 was further assigned to either P* or
at 1714 cm−1 and 1702 cm−1, respectively. A fast transition at 1665 cm−1 was further assigned to either P* or 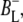 whereas a signal appearing concomitant with charge separation at 1665 cm−1 was proposed to be due to small frequency shifts and intensity changes of a large number of amide oscillators, in response to the charge separation. In Walker et al. (23) the electronic transitions of P* and P+ were measured in the 1700–2000 cm−1 region, and the P* transition was found to be present 300 fs after light absorption and decay to the P+ state in 3.4 ps. A further exploration of the near-IR region (24,25) led to the observation of four transitions assigned to the charge transfer states
whereas a signal appearing concomitant with charge separation at 1665 cm−1 was proposed to be due to small frequency shifts and intensity changes of a large number of amide oscillators, in response to the charge separation. In Walker et al. (23) the electronic transitions of P* and P+ were measured in the 1700–2000 cm−1 region, and the P* transition was found to be present 300 fs after light absorption and decay to the P+ state in 3.4 ps. A further exploration of the near-IR region (24,25) led to the observation of four transitions assigned to the charge transfer states 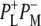 or
or 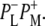 Two bands were observed in the P* state, at 5300 cm−1 and 2710 cm−1, and two bands in the
Two bands were observed in the P* state, at 5300 cm−1 and 2710 cm−1, and two bands in the 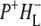 spectrum, at 8000 cm−1, and a hole transfer band at 2600 cm−1.
spectrum, at 8000 cm−1, and a hole transfer band at 2600 cm−1.
Recently, near-IR pump/UV probe spectroscopy was used to examine the initial electron transfer in wild-type RCs and 14 mutant complexes containing engineered tryptophan residues (26). Although the kinetics of charge separation were very different for the studied mutants, the protein relaxation kinetics measured as the decay of an initial absorption change of tryptophan after P* formation showed the same pattern for all the RCs. The conclusion drawn from these findings was that the initial charge separation is limited by the protein dynamics, which were initiated by the absorption of light rather than by charge separation.
In this work, we describe the mid-IR response of Rb. sphaeroides RCs after nonselective excitation in the visible region at 600 nm, and selective excitation in the near-IR at 805 nm and 860 nm. RCs both with and without the QA quinone were compared to examine the fate of the 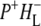 state. We conclude that characteristic absorption features of all the states involved in the primary events in the RC can be identified, including the poorly-characterized state
state. We conclude that characteristic absorption features of all the states involved in the primary events in the RC can be identified, including the poorly-characterized state 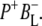 It is concluded that BL is strongly H-bonded, most likely to water molecule. A detailed kinetic scheme devised to fit the data is in good agreement with earlier reaction models proposed on the basis of visible pump-probe and fluorescence data. We identify signals that rise with the kinetics of charge separation at 1666/1656 cm−1 reflecting the protein response to charge separation. Finally, we find that a relaxation of the equilibrium between P* and the
It is concluded that BL is strongly H-bonded, most likely to water molecule. A detailed kinetic scheme devised to fit the data is in good agreement with earlier reaction models proposed on the basis of visible pump-probe and fluorescence data. We identify signals that rise with the kinetics of charge separation at 1666/1656 cm−1 reflecting the protein response to charge separation. Finally, we find that a relaxation of the equilibrium between P* and the 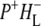 radical pair on the ∼20 ps timescale is accompanied by a change in the relative ratio of the
radical pair on the ∼20 ps timescale is accompanied by a change in the relative ratio of the 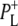 and
and 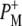 bands. We conclude that the drop in free energy associated with the relaxation of
bands. We conclude that the drop in free energy associated with the relaxation of 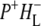 is due to an increased localization of the electron hole on the PL half of the dimer.
is due to an increased localization of the electron hole on the PL half of the dimer.
MATERIALS AND METHODS
All experiments were carried out with a femtosecond visible-pump/mid-IR-probe laser setup that has been described previously (27,28). The femtosecond pulse generator included an integrated Ti:sapphire oscillator and regenerative amplifier laser system (Hurricane, Spectra Physics, Mountain View, CA), and produced 800 nm, 85 fs pulses at a repetition rate of 1 kHz, and an energy of 0.6 mJ. Part of the light was passed through a noncollinear optical parametric amplifier, where white light and the second harmonic were generated. The 860 nm excitation light was produced using an 860-nm interference filter placed in the path of the white light seed. The pump pulses were directed through an optical delay line and a chopper operating at 500 Hz. The polarization of the excitation pulses was set to the magic angle (54.7°) with respect to the IR probe pulse using a polarization rotator placed behind the delay line. The second part of the 800 nm light (energy 360 μJ) was used to pump an optical parametric generator and amplifier with a difference frequency generator (TOPAS, Light Conversion, Vilnius, Lithuania) to produce the mid-IR probe pulses. The visible pump and mid-IR probe pulses were attenuated and focused onto the sample with 20 cm and 6 cm lenses, respectively, the size of the pump beam being ∼125 μm. After passing the sample the mid-IR probe pulses were dispersed using a spectrograph and imaged onto a 32-element HgCdT array detector operating at 77 K that provided a spectral window of ∼100 cm−1 or 200 cm−1 with a corresponding spectral resolution of 3 cm−1 or 6 cm−1, respectively. Signals from the detector array (Infrared Systems Development, Winter Park, Florida) were amplified and fed into 32 home-built integrate-and-holds, which were read out on every shot with a National Instruments (Austin, Texas) acquisition card (PCI6031E). A phase-locked chopper operating at 500 Hz was used to ensure that the sample was excited on every other shot and so the change in transmission and hence optical density could be measured. An overlap of the pump and probe pulses was found in GaAs sample and the instrument response function (IRF) was of the order of ∼100 fs. The sample was moved in a home-built Lissajous scanner, to ensure the excitation of a fresh part of the sample at every shot. Rb. sphaeroides RCs were isolated according to standard procedures (29). The construction, spectroscopic analysis and x-ray crystal structure of the mutant AM260W have been described in detail (30–32). The QA ubiquinone was removed chemically from R26 RCs using a procedure described previously (33) as modified by Woodbury et al. (34). For the vis/mid-IR experiments RCs were suspended in a D2O buffer of 100 mM Tris (pH 7.6) and the optical density of the sample was adjusted to between 0.2 and 0.3 at 800 nm for a 20-μm path length. A closed cell consisting of two CaF2 windows separated by a 20-μm Teflon spacer was filled with ∼40 μl of the RC solution and placed in a housing that was purged with a flow of N2 to reduce the effect of water vapor on the mid-IR pulses. The RCs were excited with three different wavelengths: at 600 nm (nonselective excitation of all BChls, fwhm = 9 nm), 805 nm (excitation mainly of the accessory BChls, fwhm = 8 nm), and 860 nm (selective excitation of P, fwhm = 10 nm), with excitation power of 300 nJ for 600 nm and 805 nm excitation and 250 nJ for 860 nm excitation. For each excitation wavelength experiments were repeated twice using fresh samples, with spectra being recorded at 80 different time points. In a single experiment a spectral probe window of ∼200 cm−1 was covered, so four partly overlapping regions were measured between 1580 and 2300 cm−1. All data were subjected to global and target analysis (18,35).
RESULTS
Reaction centers with QA removed: nonselective excitation at 600 nm
R26 RCs in which the QA ubiquinone had been removed (see Materials and Methods) were nonselectively excited at 600 nm, corresponding to a largely unstructured absorbance band arising from the Qx transitions of all four BChl cofactors. The resulting absorption changes in the mid-IR were measured in the frequency window 1780–1600 cm−1, covering the area corresponding to stretching modes of the carbonyl (C=O) groups of the cofactors and protein. In the case of the BChl and BPhe cofactors three such C=O groups, referred to as the 2a-acetyl, 9-keto, and 10a-ester carbonyls, are expected to contribute to the spectrum.
Fig. 1 shows two selected time traces detected at 1685 cm−1 (open squares) and 1715 cm−1 (solid squares). The fit through the data is the result of a global analysis of the whole data set using a sequential model with increasing lifetimes. The resulting evolution associated difference spectra (EADS; Fig. 2) reflect the spectral evolution of the system in time, but do not necessarily represent ‘pure’ states, as discussed in detail elsewhere (18,35). The global analysis yielded five kinetic components with lifetimes of ∼100 fs, 3.8 ps, 16 ps, 4 ns, and a nondecaying component (Table 1). The observation of the ‘pure’ ultrafast process of energy transfer is perturbed in fs mid-IR spectroscopy by a component that follows the IRF and describes a coherent interaction of the laser pulses with the sample. For this reason the resolved ∼100 fs components were not shown in any of EADS and SADS presented in this work. Also the negative signal before time zero visible in the 1685 cm−1 and 1715 cm−1 traces shown in Fig. 1 originated from perturbed free induction decay (36,37), and was not included in the fit. Because no reference probe pulse was used, the noise in the measured spectra (not shown) consisted mainly of so-called baseline noise, i.e., a flat, structureless offset in the spectra that was easily recognized by a singular vector decomposition of the residual matrix. For target analysis (and the time traces in Fig. 1), the outer product of the first two singular vector pairs of the residual matrix (being structureless in the time domain) was subtracted from the data, leading to reduction in the noise by a factor of 2.
FIGURE 1.

Two representative time traces measured using Rb. sphaeroides R26 RCs lacking QA, excited at 600 nm, and probed at 1685 cm−1 and 1715 cm−1. The solid lines through the data points are the result of a global fit using a sequential model with time constants of: τ1 = 120 fs, τ2 = 3.8 ps, τ3 = 16 ps, τ4 = 4 ns, and τ5= infinite. IRF width = 150 fs FWHM. The timescale is linear up to 3 ps and logarithmic thereafter.
FIGURE 2.

EADS of R26 RCs lacking QA, excited at 600 nm, resulting from a global analysis using a sequential model with increasing lifetimes. The measurements were carried out over a 1780–1600 cm−1 window with a spectral resolution of 6 cm−1.
TABLE 1.
Summary of the lifetimes resulting from global analysis of the data obtained in individual experiments
| Sample | Excitation wavelength (nm) | IR region/resolution (cm−1) | Lifetimes from global analysis |
|---|---|---|---|
| R26 - QA removed | 600 | 1780–1600/6 | ∼100 fs, 3.8 ps, 16 ps, 4.0 ns, infinite |
| R26 - QA removed | 805 | 1775–1590/6 | ∼100 fs, 3.0 ps, 12 ps, 4.0 ns, infinite |
| R26 - QA removed | 860 | 1775–1590/6 | ∼100 fs, 3.0 ps, 17 ps, 4.7 ns, infinite |
| R26 - QA removed | 805 | 1783–1693/3 | 2.6 ps, 7.2 ps, 1.7 ns, infinite |
| AM260W | 860 | 1775–1590/6 | ∼100 fs, 3.0 ps, 17 ps, 430 ps, infinite |
| R26 - QA removed | 600 | 1908–1736/6 | ∼100 fs, 3.8 ps, infinite |
| 2160–1930/6 | |||
| 2254–2060/6 | |||
| 2280–2111/3 | |||
| R26 - QA intact | 860 | 1780–1590/6 | ∼100 fs, 3.6 ps, 280 ps, infinite |
| R26 - QA intact | 600 | 1775–1590/6 | ∼100 fs, 4.4 ps, 222 ps, infinite |
In the EADS shown in Fig. 2 negative bands refer to ground state vibrational modes and positive bands to the shifted vibrational modes in the excited or charge separated state. Ultrafast Qx to Qy relaxation of the excited states of P and energy transfer from BL* and BM* led to the formation of the state assigned to the excited state of the primary electron donor P* (Fig. 2, red spectrum), which decayed in 3.8 ps into the state represented by the green spectrum. The 16 ps (green) and 4 ns (blue) spectra were very similar, but nevertheless some dynamics could be seen. In seeking to assign these spectra it should be noted that measurements of delayed fluorescence in isolated RCs in which electron transfer to QA was blocked (9) or QA was replaced (34), and transient absorption experiments on isolated RCs containing QA (38) and on membrane-bound RCs (18), have shown a multi-exponential decay of P* plus an increase in the amount of 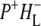 on a timescale of ∼20 ps. These observations have led to the conclusion that the equilibrium between P* and the
on a timescale of ∼20 ps. These observations have led to the conclusion that the equilibrium between P* and the 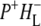 radical pair shifts toward the latter due to a drop in its free energy. A kinetic model taking these observations (9,34,38) into account was developed by van Stokkum et al. (18). Guided by this, in this study the 16 ps spectrum was assigned to the initially-formed
radical pair shifts toward the latter due to a drop in its free energy. A kinetic model taking these observations (9,34,38) into account was developed by van Stokkum et al. (18). Guided by this, in this study the 16 ps spectrum was assigned to the initially-formed 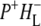 state (denoted (
state (denoted (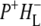 )1), whereas the 4 ns spectrum was assigned to a relaxed form of this state (denoted
)1), whereas the 4 ns spectrum was assigned to a relaxed form of this state (denoted  These spectra are clearly rich in features and display a number of negative bands, several of which can be assigned with reference to steady-state FTIR difference spectra. The negative band centered at 1687 cm−1, which is more intense in the 16 ps and 4 ns spectra than in the 3.8 ps spectrum, is assigned to the 9-keto C=O modes of PL and PM in the ground state. In FTIR experiments, where the state
These spectra are clearly rich in features and display a number of negative bands, several of which can be assigned with reference to steady-state FTIR difference spectra. The negative band centered at 1687 cm−1, which is more intense in the 16 ps and 4 ns spectra than in the 3.8 ps spectrum, is assigned to the 9-keto C=O modes of PL and PM in the ground state. In FTIR experiments, where the state 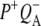 was studied on a slow (ms) timescale, two bands corresponding to the 9-keto mode of P in the ground state were found, at 1692 cm−1 for PL and 1683 cm−1 for PM (39–41). The two positive bands at 1715 cm−1 and 1705 cm−1 are ascribed to the 9-keto modes of PL and PM, respectively, in the cation state P+; for convenience the cation state of these BChls is henceforth referred to as
was studied on a slow (ms) timescale, two bands corresponding to the 9-keto mode of P in the ground state were found, at 1692 cm−1 for PL and 1683 cm−1 for PM (39–41). The two positive bands at 1715 cm−1 and 1705 cm−1 are ascribed to the 9-keto modes of PL and PM, respectively, in the cation state P+; for convenience the cation state of these BChls is henceforth referred to as 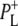 and
and 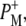 although it should be noted that the single positive charge is shared between the two BChls of the dimer. These attributions are summarized in Table 2.
although it should be noted that the single positive charge is shared between the two BChls of the dimer. These attributions are summarized in Table 2.
TABLE 2.
Assignments of positive and negative bands observed in time-resolved IR difference spectra and light-minus-dark FTIR difference spectra
| Bands | Assignments |
|---|---|
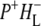 EADS without QA EADS without QA
|
|
| P/P+ modes | |
| 1736(−)/1751(+) | 10a-ester C=O of PL/PM and 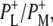 respectively respectively |
| 1715(+), 1705 (+) | 9-keto modes of 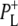 and and 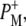 respectively respectively |
| 1687(−) | 9-keto modes of PL and PM |
| 1624(−)/1635(+) | 2a-acetyl modes of 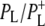
|
| 1642(−)/1647(+) | 2a-acetyl modes of 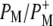
|
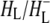 and protein modes and protein modes |
|
| 1747(−), 1732(−) | Free and H-bonded (to Trp L100) 10a-ester C=O of HL |
| 1675(−)/1591(+) | 9-keto C=O of HL hydrogen bonded to Glu L104 |
| 1656(−)/1663(+) | Upshift of an amide I C=O |
| 1656(−)/1646(+) | Conformational changes in the protein backbone due to HL transition from the neutral to the anion state and/or further 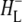 relaxation relaxation |
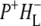 EADS without QA (3 cm−1 resolution) EADS without QA (3 cm−1 resolution) |
|
| 1739(−)/1751(+) | 10a-ester C=O of PL/PM and 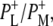 respectively respectively |
| 1746(−), 1733(−) | Free and H-bonded (to Trp L100)10a-ester C=O HL |
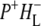 EADS with QA EADS with QA
|
|
| 1656(−)/1646(+) | Conformational changes in the protein backbone due to HL transition from the neutral to the anion state and/or further 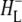 relaxation relaxation |
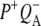 EADS EADS |
|
| P/P+ modes | |
| 1739(−)/1750(+) | 10a-ester C=O of PL/PM and 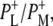 respectively respectively |
| 1715(+), 1705(+) | 9-keto modes of 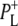 and and 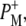 respectively respectively |
| 1687(−) | 9-keto modes of PL and PM |
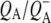 and protein modes and protein modes |
|
| 1728(+) | Transition of 10a-ester C=O of HL in electric field of 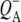
|
| 1666(−)/1656(+) | Protein amide I transition on 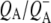
|
| 1650(−)/1640(+) | Protein backbone C=O H-bonded to QA transition on 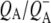
|
| 1630(−) | QA mode |
| 1600(−) | QA mode |
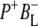 SADS SADS |
|
| 1687(−) | 9-keto C=O of PL and PM |
| 1739(−)/1750(+) | 10a-ester C=O of PL/PM and 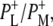 respectively respectively |
| 1715(+), 1705(+) | 9-keto modes of  and and 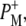 respectively respectively |
| 1650(−)/1640(+) | Response of protein C=O group |
| 1680–1670(−) | The ground state 9-keto C=O of BL |
| 1657(+) | 9-keto C=O of 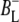 - strong H-bond with protein - strong H-bond with protein |
Note that the discussed  and
and 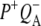 spectra are EADS, resulting from the global analysis, whereas only the
spectra are EADS, resulting from the global analysis, whereas only the 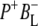 spectrum is SADS as resolved in the target analysis.
spectrum is SADS as resolved in the target analysis.
Whereas the C=O modes of BChl and BPhe pigments upshift in the cation state, they downshift in both the excited (42) and anion states (43). The negative shoulder at 1675 cm−1 in the 16 ps and 4 ns spectra in Fig. 2 can be ascribed to the 9-keto C=O of HL in the neutral state, which is hydrogen bonded to Glu L104 (44). In FTIR experiments this mode has been reported to undergo a very strong downshift to 1591 cm−1 upon charge separation (43), and evidence for this can be seen in the strongly positive signal appearing at the edge of the spectral window around 1600 cm−1 in the spectra of the 16 ps (Fig. 2, green) and 4 ns (Fig. 2, blue) components.
The C=O modes of the protein are expected to absorb in the range 1665–1620 cm−1 and partly overlap with signals due to BChl a 9-keto and 2a-acetyl C=O modes. The negative band at 1656 cm−1 in the 16 ps and 4 ns spectra in Fig. 2 can be ascribed to an upshift of one or more amide C=O groups to 1663 cm−1 in response to the formation of 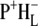 (21,22,43,45,46). Note however, that a contradictory assignment of this negative band at 1656 cm−1 has been proposed (44). In this assignment the 1656 cm−1 mode downshifts to 1646 cm−1 due to conformational changes in the protein backbone in response to electron transfer to HL (39), and/or further relaxation of
(21,22,43,45,46). Note however, that a contradictory assignment of this negative band at 1656 cm−1 has been proposed (44). In this assignment the 1656 cm−1 mode downshifts to 1646 cm−1 due to conformational changes in the protein backbone in response to electron transfer to HL (39), and/or further relaxation of 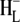 (44). Due to the low extinction coefficient of the BChl a 2a-acetyl modes and the presence of partially overlapping protein C=O modes an unequivocal assignment of the bands observed in the region between 1620 and 1650 cm−1 was not possible. The negative bands at 1642 and 1624 cm−1 in the 16 ps and 4 ns spectra in Fig. 2 are probably attributable to the 2a-acetyl modes of PM and PL, respectively (39–41,45,47–49) and in the cation state these modes are expected to shift to higher frequency. The positive band at 1635 cm−1 could be due to the 2a-acetyl C=O of
(44). Due to the low extinction coefficient of the BChl a 2a-acetyl modes and the presence of partially overlapping protein C=O modes an unequivocal assignment of the bands observed in the region between 1620 and 1650 cm−1 was not possible. The negative bands at 1642 and 1624 cm−1 in the 16 ps and 4 ns spectra in Fig. 2 are probably attributable to the 2a-acetyl modes of PM and PL, respectively (39–41,45,47–49) and in the cation state these modes are expected to shift to higher frequency. The positive band at 1635 cm−1 could be due to the 2a-acetyl C=O of 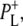 upshifted from 1624 cm−1. However, it was not possible to assign the positive band at 1646 cm−1 to the upshifted 2-acetyl C=O of
upshifted from 1624 cm−1. However, it was not possible to assign the positive band at 1646 cm−1 to the upshifted 2-acetyl C=O of 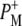 because it might also be due to a perturbation of an amide I oscillator in the 1646/1656 cm−1 region (21,22,39) or further relaxation of the HL anion state (44).
because it might also be due to a perturbation of an amide I oscillator in the 1646/1656 cm−1 region (21,22,39) or further relaxation of the HL anion state (44).
It has been reported in the literature that in the 10a-ester region between 1760 and 1710 cm−1 a negative band at 1740 cm−1 and positive band 1750 cm−1 arise from the neutral and cation state, respectively of both PL and PM (39–41,45). This was indeed observed in the spectra of the 16 ps, 4 ns, and infinite components as a bleaching at ∼1736 cm−1 and an absorption increase at 1751 cm−1. It is possible that negative bands related to free and H-bonded modes of the 10a-ester group of HL at 1747 cm−1 and 1732 cm−1, respectively (44), could also contribute to this region of the EADS, but were not resolved due to the relatively low spectral resolution (6 cm−1). These attributions are returned to below, in the discussion of data recorded at a higher spectral resolution. Finally, the spectrum of the nondecaying component in Fig. 2 (cyan) shows a decrease in intensity for all bands. The evolution observed from the 4 ns spectrum (blue) to the nondecaying spectrum (cyan) mainly reflects the decay of the radical pair 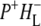 in the absence of electron transfer to QA.
in the absence of electron transfer to QA.
Reaction centers with QA removed: selective excitation at 805 and 860 nm
R26 RCs lacking QA were also excited selectively at 805 nm and 860 nm. At 805 nm the laser pulse mainly excited the accessory BChls, although there was also the possibility of a small amount of excitation of P via the high energy exciton component of the Qy transition of the dimer. At 860 nm the laser pulse selectively excited the P BChls. A global analysis of each of the data sets using a sequential model with increasing lifetimes showed five EADS representing the time-evolution of the system (data not shown). The resulting lifetimes were very similar in the data sets collected with 805, 860 nm, and 600-nm excitation (Table 1). For the four slower components the EADS resulting from a global analysis of the data sets obtained with 805 and 860 nm excitation (data not shown) were very similar to the spectra presented in Fig. 2.
Reaction centers assembled without QA: selective excitation at 860 nm
Charge separation in RCs with an alanine (Ala) to tryptophan (Trp) mutation of residue M260 in the binding pocket of the QA ubiquinone was also investigated using 860 nm excitation. A combination of kinetic spectroscopy, x-ray crystallography, and FTIR spectroscopy has shown that the introduction of a bulky tryptophan residue in the QA binding pocket causes this AM260W RC to assemble without a QA quinone (30–32,50). Despite this, the structure of the RC outside the immediate vicinity of the QA pocket is not altered and the kinetics of electron transfer from P to HL are not affected (30). In agreement with this the data obtained for AM260W mutant RCs upon excitation with 860 nm light were very similar to those obtained with the QA-removed R26 RCs, a global analysis of the data yielding five EADS with lifetimes of 0.1 ps, 3 ps, 17 ps, 430 ps, and a nondecaying component (Table 1). The longest-lived component (430 ps) was ascribed to the (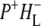 )2 state. The nature of the nondecaying spectrum is not totally clear. Recently we have carried out a more detailed analysis of the spectral evolution in AM260W RCs and concluded that a further relaxation of (
)2 state. The nature of the nondecaying spectrum is not totally clear. Recently we have carried out a more detailed analysis of the spectral evolution in AM260W RCs and concluded that a further relaxation of (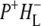 )2 occurs on a timescale of ∼500 ps to form (
)2 occurs on a timescale of ∼500 ps to form (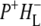 )3, and this will be described in more detail in a future publication (N. P. Pawlowicz , I. H. M. van Stokkum, J. Breton, M. L. Groot , R. van Grondelle, M. R. Jones, unpublished). The lineshapes of these spectra (data not shown) were similar to those obtained with 860 nm excitation of QA removed RCs.
)3, and this will be described in more detail in a future publication (N. P. Pawlowicz , I. H. M. van Stokkum, J. Breton, M. L. Groot , R. van Grondelle, M. R. Jones, unpublished). The lineshapes of these spectra (data not shown) were similar to those obtained with 860 nm excitation of QA removed RCs.
The summary of the attribution of the various states in the data sets obtained on excitation of the QA-deficient RCs at 805 nm and 860 nm is shown in Table 1. The first picosecond timescale EADS was attributed to P*. The next state represents a mixture of mainly (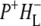 )1 with some contribution from
)1 with some contribution from 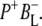 The longest-lived component was ascribed to the
The longest-lived component was ascribed to the 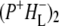 state, whereas the nondecaying components still contained mainly contributions from the relaxed form
state, whereas the nondecaying components still contained mainly contributions from the relaxed form 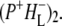 Due to the limited time range of the experiment of 3 ns, no evidence of 3P state formation was observed in these EADS.
Due to the limited time range of the experiment of 3 ns, no evidence of 3P state formation was observed in these EADS.
Reaction centers with QA removed: high spectral resolution with 805 nm excitation
The absorption changes after 805 nm excitation of R26 RCs lacking QA were also measured in 1783–1693 cm−1 range with a spectral resolution of 3 cm−1, as opposed to the 6 cm−1 resolution applicable to the spectra discussed in the last three sections. This was done to provide more detailed information on the two forms of 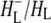 observed in steady-state FTIR measurements (44). The attributions are summarized in Table 2. As can be seen in Fig. 3, in the mid-IR spectra of both the
observed in steady-state FTIR measurements (44). The attributions are summarized in Table 2. As can be seen in Fig. 3, in the mid-IR spectra of both the 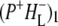 and
and 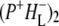 states (green and blue spectra, respectively) three negative bands were resolved in the C=O region of 10a-ester modes of
states (green and blue spectra, respectively) three negative bands were resolved in the C=O region of 10a-ester modes of 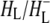 at 1733, 1739, and 1746 cm−1. Two of these wavelengths were similar to those obtained by steady-state FTIR spectroscopy with 4 cm−1 spectral resolution (44). The light-induced FTIR difference spectrum of
at 1733, 1739, and 1746 cm−1. Two of these wavelengths were similar to those obtained by steady-state FTIR spectroscopy with 4 cm−1 spectral resolution (44). The light-induced FTIR difference spectrum of 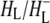 shows two bands related to free and H-bonded modes in the 10a-ester region of HL, at 1732 and 1747 cm−1, respectively (44), whereas only one band at 1743 cm−1 is found in the FTIR difference spectrum of isolated BPhe in tetrahydrofuran (THF) solvent (44). Following the same arguments, the 1746 and 1733 cm−1 negative bands observed in the green and blue spectra in Fig. 3 could correspond to a free and H-bonded conformer of the 10a ester C=O group of HL, respectively, with Trp L100 acting as the H-bond donor (43,50).
shows two bands related to free and H-bonded modes in the 10a-ester region of HL, at 1732 and 1747 cm−1, respectively (44), whereas only one band at 1743 cm−1 is found in the FTIR difference spectrum of isolated BPhe in tetrahydrofuran (THF) solvent (44). Following the same arguments, the 1746 and 1733 cm−1 negative bands observed in the green and blue spectra in Fig. 3 could correspond to a free and H-bonded conformer of the 10a ester C=O group of HL, respectively, with Trp L100 acting as the H-bond donor (43,50).
FIGURE 3.

R26 RCs lacking QA excited at 805 nm and probed using a spectral resolution of 3 cm−1. The EADS are the result of a global analysis of the data using a sequential model with increasing lifetimes of 2.6 ps, 7.2 ps, 1.7 ns, and 4 ns.
As discussed above, the negative band at 1739 cm−1 in the spectra of 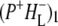 and
and 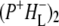 in Fig. 3 is attributable to the 10a ester C=O of PL/PM, with the positive band at 1751 cm−1 corresponding to the 10a ester C=O of
in Fig. 3 is attributable to the 10a ester C=O of PL/PM, with the positive band at 1751 cm−1 corresponding to the 10a ester C=O of 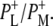 These frequencies match closely values from steady-state FTIR spectroscopy of 1740 cm−1 (negative) and 1750 cm−1 (positive) (39–41,45).
These frequencies match closely values from steady-state FTIR spectroscopy of 1740 cm−1 (negative) and 1750 cm−1 (positive) (39–41,45).
Reaction centers with QA removed: spectral changes in four other frequency ranges on 600 nm excitation
Experiments were also carried out on R26 RCs lacking QA in four other partly overlapping frequency regions. These were 2254–2060 cm−1, 2160–1930 cm−1, and 1908–1736 cm−1, each with a spectral resolution of 6 cm−1, and 2280–2111 cm−1 with a higher resolution of 3 cm−1. The excitation wavelength was 600 nm in each case. Each frequency region was analyzed separately, resulting in two EADS that are shown in Fig. 4. Also shown on the same scale are spectra covering the 1780–1600 cm−1 region (these are the EADS shown on an expanded scale in Fig. 2). The region between 2153 and 2075 cm−1 is not shown due to the high IR absorption by D2O. This D2O feature aside, no sharp spectral features were found in the frequency regions between 2280 and 1780 cm−1, but nevertheless the fitted lifetimes associated with these EADS were consistent with those found in the 1780–1600 cm−1 region.
FIGURE 4.

EADS of R26 RCs lacking QA calculated from measurements carried out over a broad spectral range between 2280–1600 cm−1. The spectra between 2153–2075 cm−1 are not shown due to the high IR absorption by D2O in this region. The lifetimes shown are the result of global analysis of the data obtained in 1780–1600 cm−1 region (Fig. 2). Excitation was at 600 nm in each case.
In the region above 1780 cm−1 only two components were seen in the analysis, a component with a lifetime of 3.8 ps attributed to the P* state (red) and a nondecaying component attributed to the final 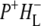 state (green). The positive feature between 1880 and 2247 cm−1 in the spectrum of this
state (green). The positive feature between 1880 and 2247 cm−1 in the spectrum of this 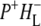 state is the shoulder of the broad positive band that has been attributed to an electronic transition (hole transfer) within the P+ dimer (51), and its position measures the resonance interaction between the two BChl molecules when P is oxidized. This attribution was made on the basis of the absence of this band in spectra of monomeric BChl a in THF (44) and its absence in the spectra of mutants in which either PL or PM was replaced by BPhe a (23,44,51). The measured transition energy and dipole strength of the band yields the resonance interaction matrix element that causes the remaining unpaired electron of P+ to move back and forth between PL and PM, and the energy difference between the two states in which a positive charge is fully localized on either PL or PM (51). From FTIR experiments that yielded a light-minus-dark
state is the shoulder of the broad positive band that has been attributed to an electronic transition (hole transfer) within the P+ dimer (51), and its position measures the resonance interaction between the two BChl molecules when P is oxidized. This attribution was made on the basis of the absence of this band in spectra of monomeric BChl a in THF (44) and its absence in the spectra of mutants in which either PL or PM was replaced by BPhe a (23,44,51). The measured transition energy and dipole strength of the band yields the resonance interaction matrix element that causes the remaining unpaired electron of P+ to move back and forth between PL and PM, and the energy difference between the two states in which a positive charge is fully localized on either PL or PM (51). From FTIR experiments that yielded a light-minus-dark 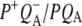 difference spectrum it was found that this hole transfer band is in fact centered at 2600 ± 100 cm−1 (51–53) and is not shifted in D2O (51). In previous experiments where a pure
difference spectrum it was found that this hole transfer band is in fact centered at 2600 ± 100 cm−1 (51–53) and is not shifted in D2O (51). In previous experiments where a pure 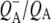 difference spectrum was generated in D2O no contribution to the 2600 cm−1 feature was found, showing it to arise from the P+ component (41,51,54). This 2600 cm−1 band is also absent in the difference spectrum of monomeric BChl a+/BChl a in solvent (44) and in the
difference spectrum was generated in D2O no contribution to the 2600 cm−1 feature was found, showing it to arise from the P+ component (41,51,54). This 2600 cm−1 band is also absent in the difference spectrum of monomeric BChl a+/BChl a in solvent (44) and in the 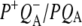 FTIR difference spectra of mutants with a heterodimeric BChl:BPhe primary donor, so it has been assigned to the dimeric nature of the oxidized primary donor P+ (23,44,51). In the work of Walker et al. (23) the time evolution of the 2600 cm−1 band was studied with a subpicosecond resolution and compared with measurements of the electron transfer rate in the RC. The P+ transition appeared in 3.4 ps, coincident with decay of the P* state, and was assigned to a transition between the symmetric and antisymmetric combination of the localized hole states of the dimer (23). The spectral kinetics and measured anisotropy of the transition permitted a direct verification of the nature of the eigenstates of P+ (23). The fs IR experiments reported here showed the significant charge transfer character of P* in agreement with for example findings of Lockhart and Boxer (55,56) based on Stark spectroscopy. The broad featureless band appears within the time resolution of the experiment and the signal approximately doubled in size with a time constant of 3.8 ps, in contrast to the findings of Walker et al. (23), where this signal was observed to rise concomitant with the appearance of P+ in 3.4 ps, indicating no charge transfer character of P*.
FTIR difference spectra of mutants with a heterodimeric BChl:BPhe primary donor, so it has been assigned to the dimeric nature of the oxidized primary donor P+ (23,44,51). In the work of Walker et al. (23) the time evolution of the 2600 cm−1 band was studied with a subpicosecond resolution and compared with measurements of the electron transfer rate in the RC. The P+ transition appeared in 3.4 ps, coincident with decay of the P* state, and was assigned to a transition between the symmetric and antisymmetric combination of the localized hole states of the dimer (23). The spectral kinetics and measured anisotropy of the transition permitted a direct verification of the nature of the eigenstates of P+ (23). The fs IR experiments reported here showed the significant charge transfer character of P* in agreement with for example findings of Lockhart and Boxer (55,56) based on Stark spectroscopy. The broad featureless band appears within the time resolution of the experiment and the signal approximately doubled in size with a time constant of 3.8 ps, in contrast to the findings of Walker et al. (23), where this signal was observed to rise concomitant with the appearance of P+ in 3.4 ps, indicating no charge transfer character of P*.
Reaction centers containing QA: nonselective excitation at 600 nm
To characterize the normal end product of membrane-spanning electron transfer, the state 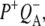 R26 RCs containing QA were nonselectively excited at 600 nm, and data recorded in the frequency range 1590–1775 cm−1. The EADS generated by global analysis of the data set are shown in Fig. 5 (note that the ∼100 fs component is not shown). The obtained lifetimes are reported in Table 1 and band attributions for
R26 RCs containing QA were nonselectively excited at 600 nm, and data recorded in the frequency range 1590–1775 cm−1. The EADS generated by global analysis of the data set are shown in Fig. 5 (note that the ∼100 fs component is not shown). The obtained lifetimes are reported in Table 1 and band attributions for 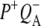 are summarized in Table 2. Electron transfer from
are summarized in Table 2. Electron transfer from 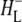 to QA was observed to occur with a lifetime of 222 ps, forming
to QA was observed to occur with a lifetime of 222 ps, forming 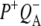 (blue spectrum). This final nondecaying EADS (blue spectrum) showed clear differences from the 222 ps component (green) in the 1675–1610 cm−1 region, where the absorption changes are attributable to a variety of contributions that include the protein responses to charge separation.
(blue spectrum). This final nondecaying EADS (blue spectrum) showed clear differences from the 222 ps component (green) in the 1675–1610 cm−1 region, where the absorption changes are attributable to a variety of contributions that include the protein responses to charge separation.
FIGURE 5.

R26 RCs with QA excited at 600 nm and probed in the region 1775–1590 cm−1. Displayed are EADS resulting from a global analysis using a sequential model with increasing lifetimes of 0.1 ps, 4.4 ps, 222 ps, and a nondecaying component.
The spectrum of the 222 ps component was similar to those obtained for the 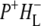 state in the experiments described above with RCs lacking QA (Fig. 2). The nondecaying spectrum (Fig. 5, blue) was attributable to the state
state in the experiments described above with RCs lacking QA (Fig. 2). The nondecaying spectrum (Fig. 5, blue) was attributable to the state 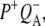 with the 1675–1610 cm−1 region showing a pattern that is characteristic for contributions from
with the 1675–1610 cm−1 region showing a pattern that is characteristic for contributions from 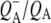 (39,41,57,58), with bands that were negative at 1666 cm−1, positive at 1656 cm−1, negative at 1650 cm−1, positive at 1640 cm−1, and negative around 1630 and 1600 cm−1. The 1666(−)/1656(+) signal has been ascribed to a change in a protein amide I transition (21), whereas the 1650(−)/1640(+) signal has been attributed to the response of a protein backbone C=O that is connected to QA via an H-bond that is perturbed upon reduction of QA (39,45,59). An H-bond between the carbonyl of QA and the peptide N-H of Ala M260 residue is inferred from the x-ray crystal structure of the Rb. sphaeroides RC (40,45). In agreement with this, the band at 1650 cm−1 is present in the
(39,41,57,58), with bands that were negative at 1666 cm−1, positive at 1656 cm−1, negative at 1650 cm−1, positive at 1640 cm−1, and negative around 1630 and 1600 cm−1. The 1666(−)/1656(+) signal has been ascribed to a change in a protein amide I transition (21), whereas the 1650(−)/1640(+) signal has been attributed to the response of a protein backbone C=O that is connected to QA via an H-bond that is perturbed upon reduction of QA (39,45,59). An H-bond between the carbonyl of QA and the peptide N-H of Ala M260 residue is inferred from the x-ray crystal structure of the Rb. sphaeroides RC (40,45). In agreement with this, the band at 1650 cm−1 is present in the 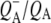 FTIR difference spectra of Rb. sphaeroides and Rhodopseudomonas viridis RCs (41).
FTIR difference spectra of Rb. sphaeroides and Rhodopseudomonas viridis RCs (41).
In the 10a-ester region a set of bands were observed in the 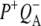 EADS (Fig. 5, blue) that were somewhat different from those seen in preceding
EADS (Fig. 5, blue) that were somewhat different from those seen in preceding 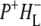 EADS (Fig. 5, green). Both spectra showed bands attributable to the 9-keto C=O of PLPM,
EADS (Fig. 5, green). Both spectra showed bands attributable to the 9-keto C=O of PLPM, 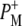 and
and 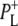 at 1687(−), 1705(+), and 1715(+) cm−1, respectively, and a differential feature at 1739(−)/1750(+) attributed to the 10a-ester C=O of PL/PM and
at 1687(−), 1705(+), and 1715(+) cm−1, respectively, and a differential feature at 1739(−)/1750(+) attributed to the 10a-ester C=O of PL/PM and 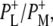 respectively. However an additional small positive band was seen at 1728 cm−1 in the
respectively. However an additional small positive band was seen at 1728 cm−1 in the 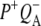 EADS (Fig. 5, blue) that has been ascribed in
EADS (Fig. 5, blue) that has been ascribed in 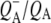 FTIR difference spectra to a shift of the 10a-ester C=O of HL in response to the electric field created by
FTIR difference spectra to a shift of the 10a-ester C=O of HL in response to the electric field created by 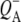 (60). As a general point, complex signals appear in this region due to heterogeneity in the conformation and hydrogen bonding of the 10a-ester C=O of HL, and this heterogeneity may be related to the functional heterogeneity observed in electron transfer kinetics.
(60). As a general point, complex signals appear in this region due to heterogeneity in the conformation and hydrogen bonding of the 10a-ester C=O of HL, and this heterogeneity may be related to the functional heterogeneity observed in electron transfer kinetics.
R26 RCs containing QA: selective excitation at 860 nm
R26 RCs with an intact QA were also excited at 860 nm and probed in the region 1780–1590 cm−1. On the basis of global analysis using a sequential model with increasing lifetimes three EADS were resolved, with associated lifetimes of 3.6 ps (P*), 280 ps 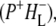 and a nondecaying component
and a nondecaying component 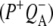 (Table 1). The lineshapes of the EADS (data not shown) were similar to those obtained for the equivalent states in the experiment with 600 nm excitation. Again, the nondecaying component showed bands related to a protein backbone C=O that is connected to QA at 1650(−)/1640(+), and a downshift of the 1666 cm−1 band to 1656 cm−1 in the amide I absorption range.
(Table 1). The lineshapes of the EADS (data not shown) were similar to those obtained for the equivalent states in the experiment with 600 nm excitation. Again, the nondecaying component showed bands related to a protein backbone C=O that is connected to QA at 1650(−)/1640(+), and a downshift of the 1666 cm−1 band to 1656 cm−1 in the amide I absorption range.
Summary of lifetimes obtained from global analysis
To summarize the results obtained in the individual experiments, all the lifetimes resulting from the global analysis of experimental data are listed in Table 1. As can be seen the lifetimes of the states resulting from independent global analysis were similar across the eight sets of data. The lifetimes associated with the EADS attributed to P* ranged between 2.6 ps and 4.4 ps. The lifetimes associated with the EADS attributed to the first charge separated state 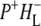 in RCs lacking QA were also in good agreement, ranging from 7.2 ps to 17 ps. Finally the lifetimes associated with the EADS attributed to the second charge separated state
in RCs lacking QA were also in good agreement, ranging from 7.2 ps to 17 ps. Finally the lifetimes associated with the EADS attributed to the second charge separated state 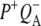 in RCs with an intact QA were 222 ps and 280 ps for 600 nm and 860 nm excitation, respectively.
in RCs with an intact QA were 222 ps and 280 ps for 600 nm and 860 nm excitation, respectively.
DISCUSSION
In the following we discuss several key points resulting from the analysis of this experimental data. The first major point of the discussion is the target analysis using the kinetic model that provided the best possible fit to the data. A difference spectrum for 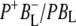 is extracted and its details are discussed. In addition consideration is given to the agreement between lifetimes resulting from the data and those extracted from similar kinetic models applied previously, including those that assume direct charge separation from BL*. The discussion then examines the agreement between the fs time-resolved mid-IR spectra of the
is extracted and its details are discussed. In addition consideration is given to the agreement between lifetimes resulting from the data and those extracted from similar kinetic models applied previously, including those that assume direct charge separation from BL*. The discussion then examines the agreement between the fs time-resolved mid-IR spectra of the 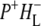 and
and 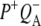 states and the sum of steady state FTIR spectra measured previously for
states and the sum of steady state FTIR spectra measured previously for 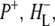 and
and 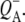 Features specific for the reduction of HL and QA observed in fs time-resolved spectra are assigned, as are changes in protein conformation due to the electron transfer. Finally the issue of charge delocalization on the primary electron donor is discussed.
Features specific for the reduction of HL and QA observed in fs time-resolved spectra are assigned, as are changes in protein conformation due to the electron transfer. Finally the issue of charge delocalization on the primary electron donor is discussed.
Target analysis of the seven data sets using a single model
The global analysis using a sequential model with increasing lifetimes described above gave very good fits to the seven sets of experimental data, but it has the disadvantage that it does not take into account the multi-exponential nature of the decay of the P* state or the documented involvement of 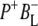 in electron transfer from P* to HL. To further disentangle the contributions of various states to the observed spectral evolution a simultaneous target analysis of all data sets was carried out. This analysis accounted for the multi-exponential decay of P* emission by including back reactions, and included a
in electron transfer from P* to HL. To further disentangle the contributions of various states to the observed spectral evolution a simultaneous target analysis of all data sets was carried out. This analysis accounted for the multi-exponential decay of P* emission by including back reactions, and included a 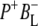 intermediate. On the basis of the sequential analysis presented above, and results from the literature obtained in visible region pump/probe experiments, rate constants for the forward and backward reactions were estimated and the full kinetic scheme depicted in Fig. 6 was obtained. According to this scheme the decay of the P* state is four-exponential, proceeding through the states
intermediate. On the basis of the sequential analysis presented above, and results from the literature obtained in visible region pump/probe experiments, rate constants for the forward and backward reactions were estimated and the full kinetic scheme depicted in Fig. 6 was obtained. According to this scheme the decay of the P* state is four-exponential, proceeding through the states 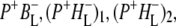 and
and 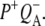 This kinetic scheme was similar to that used in previous reports to analyze visible-region pump-probe data (18) and the lifetimes obtained in this study were in good agreement with those obtained in a number of published studies (12,61–64).
This kinetic scheme was similar to that used in previous reports to analyze visible-region pump-probe data (18) and the lifetimes obtained in this study were in good agreement with those obtained in a number of published studies (12,61–64).
FIGURE 6.

Scheme for energy transfer and electron transfer in R26 and AM260W RCs. Green arrows (plus associated time constants) refer to forward processes, red arrows to back reactions. Forward electron transfer from the 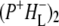 state in RCs with QA led to the
state in RCs with QA led to the 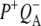 state, whereas in QA-deficient RCs it led to the triplet state of P (3P) and recombination to the ground state.
state, whereas in QA-deficient RCs it led to the triplet state of P (3P) and recombination to the ground state.
The kinetic model shown in Fig. 6 was applied simultaneously to all of the data sets described above, resulting in lifetimes of 150 fs, 3.7 ps, 0.7 ps, 23 ps, 290 ps, and two nondecaying components. The initially excited states Qx* and BL*/BM*, observed on excitation at 600 nm and 805 nm, respectively, are formed during the laser pulse. Energy transfer from BL*/BM* to P taking place in ∼150 fs after direct excitation of the accessory BChls was poorly resolved in our experiments (in addition there is the possibility of a small amount of direct charge separation from BL*, but for simplicity this was not included in the model). The P* state decays in 3.7 ps by charge separation to form 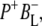 and this subsequently decays in 0.7 ps to form the
and this subsequently decays in 0.7 ps to form the 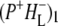 state. The next component, with a decay time of 23 ps, is relaxation of
state. The next component, with a decay time of 23 ps, is relaxation of 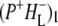 into
into 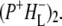 Decay of
Decay of 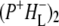 occurs in one of two ways, depending on whether or not the sample contains QA. In case of RCs with QA the
occurs in one of two ways, depending on whether or not the sample contains QA. In case of RCs with QA the 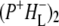 state decays in 290 ps to
state decays in 290 ps to  which is represented by a nondecaying component.
which is represented by a nondecaying component. 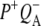 decays on a timescale of ms, which is assumed to be infinite taking into account the time range of our experiment. In case of RCs without QA the
decays on a timescale of ms, which is assumed to be infinite taking into account the time range of our experiment. In case of RCs without QA the 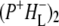 state decays in ∼20 ns, ∼30% into the triplet state of the primary electron donor (3P) and the other fraction recombines to the ground state of P in ∼6.7 ns.
state decays in ∼20 ns, ∼30% into the triplet state of the primary electron donor (3P) and the other fraction recombines to the ground state of P in ∼6.7 ns.
A representative set of species associated decay spectra (SADS) resulting from the target analysis are shown in Fig. 7 (again the SADS corresponding to the ultrafast ∼150 fs component was not shown). These SADS are derived from the data obtained with R26 RCs lacking QA that had been excited at 805 nm. The red spectrum, which is attributed to P* shows a negative band at 1687 cm−1 that is ascribed to the 9-keto modes of PL and PM in the ground state, and this downshifts to 1668 cm−1 in the excited state.
FIGURE 7.

SADS derived from data obtained with R26 RCs lacking QA that had been excited at 805 nm. The SADS were produced from a target analysis of all data sets using the kinetic scheme outlined in Fig. 6.
The green spectrum in Fig. 7 is that of the first intermediate in charge separation, 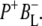 Due to its short lifetime and, as a consequence, low population it was difficult to detect the state
Due to its short lifetime and, as a consequence, low population it was difficult to detect the state 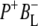 in the time-resolved difference spectra. For the same reasons the level of uncertainty in the
in the time-resolved difference spectra. For the same reasons the level of uncertainty in the 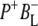 SADS in Fig. 7 is greater than in the remaining spectra, but nevertheless this spectrum was generally reproducible in the simultaneous target analysis of the seven data sets (see below). This spectrum contains the ground state bleaching of P around 1687 cm−1 plus an additional bleaching in the 1670–1680 cm−1 region that can be ascribed to BL. It is possible that the small positive band at 1657 cm−1 is attributable to the 9-keto C=O of the anion state of BL that would be consistent with the general picture that the stretching frequency of a keto C=O undergoes a downshift on reduction of a cofactor (44). In addition, it is likely that any shift of the stretching frequency of the 9-keto C=O of BL on formation of
SADS in Fig. 7 is greater than in the remaining spectra, but nevertheless this spectrum was generally reproducible in the simultaneous target analysis of the seven data sets (see below). This spectrum contains the ground state bleaching of P around 1687 cm−1 plus an additional bleaching in the 1670–1680 cm−1 region that can be ascribed to BL. It is possible that the small positive band at 1657 cm−1 is attributable to the 9-keto C=O of the anion state of BL that would be consistent with the general picture that the stretching frequency of a keto C=O undergoes a downshift on reduction of a cofactor (44). In addition, it is likely that any shift of the stretching frequency of the 9-keto C=O of BL on formation of 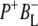 is complicated by changes arising from oxidation of the immediately adjacent P BChls, in addition to effects due to the reduction of BL. On the basis of resonance Raman studies (65), the frequency of the 9-keto C=O of BL in the ground state is thought to be in the region of 1687 cm−1 and downshifts to 1675 cm−1 in RCs where P is chemically oxidized as a result of the formation of a moderate strength H-bond, or strengthening of a weak H-bond. In Robert and Lutz (65) the H-bond donor was suggested to be a water molecule, and this was confirmed in a subsequent study that showed that this P+-induced downshift was lost in structurally-characterized mutant reaction centers in which this water molecule was absent (66). It is therefore possible that any shift in the stretching frequency of the 9-keto C=O of BL on formation of
is complicated by changes arising from oxidation of the immediately adjacent P BChls, in addition to effects due to the reduction of BL. On the basis of resonance Raman studies (65), the frequency of the 9-keto C=O of BL in the ground state is thought to be in the region of 1687 cm−1 and downshifts to 1675 cm−1 in RCs where P is chemically oxidized as a result of the formation of a moderate strength H-bond, or strengthening of a weak H-bond. In Robert and Lutz (65) the H-bond donor was suggested to be a water molecule, and this was confirmed in a subsequent study that showed that this P+-induced downshift was lost in structurally-characterized mutant reaction centers in which this water molecule was absent (66). It is therefore possible that any shift in the stretching frequency of the 9-keto C=O of BL on formation of 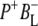 arises both from the reduction of BL, and also a change in the strength of any hydrogen bond interaction between this C=O group and the adjacent water molecule. On the other hand, on the basis of the FTIR difference spectrum of BChl a anion in THF (67) the 9-keto
arises both from the reduction of BL, and also a change in the strength of any hydrogen bond interaction between this C=O group and the adjacent water molecule. On the other hand, on the basis of the FTIR difference spectrum of BChl a anion in THF (67) the 9-keto 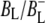 mode was assigned to be downshifted from 1683 cm−1 to 1620 cm−1. For this reason, it is also possible that the broad positive feature around 1600 cm−1 in fs mid-IR
mode was assigned to be downshifted from 1683 cm−1 to 1620 cm−1. For this reason, it is also possible that the broad positive feature around 1600 cm−1 in fs mid-IR 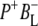 spectrum represents the downshifted BL 9-keto in the anion state similar to the frequency reported in (67). Furthermore, the differential signals in the region between 1650 and 1600 cm−1 could not be attributed with certainty, as multiple contributions are expected in this region from the acetyl C=O groups of P and B and protein C=O groups. These attributions of the 1670–1680(−)/1657(+) cm−1 and/or 1600(+) cm−1 modes to the 9-keto C=O of
spectrum represents the downshifted BL 9-keto in the anion state similar to the frequency reported in (67). Furthermore, the differential signals in the region between 1650 and 1600 cm−1 could not be attributed with certainty, as multiple contributions are expected in this region from the acetyl C=O groups of P and B and protein C=O groups. These attributions of the 1670–1680(−)/1657(+) cm−1 and/or 1600(+) cm−1 modes to the 9-keto C=O of 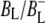 are only tentative at this stage and will require further investigation, possibly using the mutant reaction centers described above in which the water molecule adjacent to this group is excluded (66).
are only tentative at this stage and will require further investigation, possibly using the mutant reaction centers described above in which the water molecule adjacent to this group is excluded (66).
The negative band at 1739 cm−1 that upshifts to give a positive band at 1750 cm−1 in the SADS of 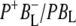 is attributable to the upshift of the mode of the 10a-ester C=O of P station formation of P+ (39). The positive band at 1715 cm−1 is attributable to the 9-keto C=O of
is attributable to the upshift of the mode of the 10a-ester C=O of P station formation of P+ (39). The positive band at 1715 cm−1 is attributable to the 9-keto C=O of 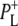 and, remarkably, the band of the 9-keto C=O of
and, remarkably, the band of the 9-keto C=O of 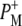 at 1705 cm−1 is significantly diminished. The most likely reason is a perturbed distribution of the unpaired electron within P+ due to the nearby negative charge localized on BL with the main contribution coming from
at 1705 cm−1 is significantly diminished. The most likely reason is a perturbed distribution of the unpaired electron within P+ due to the nearby negative charge localized on BL with the main contribution coming from 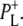 Upon further transfer of the electron to HL its influence on the electron distribution within P+ is reduced, leading to an increase of the 9-keto band of
Upon further transfer of the electron to HL its influence on the electron distribution within P+ is reduced, leading to an increase of the 9-keto band of 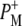 at 1705 cm−1 in the
at 1705 cm−1 in the 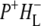 states (Fig. 7, blue and cyan spectra).
states (Fig. 7, blue and cyan spectra).
The blue and cyan spectra in Fig. 7 represent the product of the second step of charge separation, 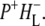 These SADS were similar to the corresponding EADS presented in the Results, and the attribution of the various bands is summarized in Table 2 (see Results for details). After electron transfer to HL in ∼0.7 ps, the
These SADS were similar to the corresponding EADS presented in the Results, and the attribution of the various bands is summarized in Table 2 (see Results for details). After electron transfer to HL in ∼0.7 ps, the 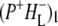 state was formed (Fig. 7, blue), which then relaxed with a lifetime of 23 ps to the
state was formed (Fig. 7, blue), which then relaxed with a lifetime of 23 ps to the 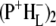 state (Fig. 7, cyan). Minor differences are apparent between these two spectra, mainly in the intensity of the bands at 1663(+) and 1656(−) cm−1 that have been ascribed to the response of an amide C=O (39), and the bands assigned to 9-keto modes of
state (Fig. 7, cyan). Minor differences are apparent between these two spectra, mainly in the intensity of the bands at 1663(+) and 1656(−) cm−1 that have been ascribed to the response of an amide C=O (39), and the bands assigned to 9-keto modes of 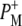 and
and 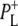 at 1705 and 1715 cm−1, respectively. Minor changes are also seen in the relative intensities of the product state bands at 1647 and 1635 cm−1. As the 2a-acetyl C=O modes of P upshift in the cation state, these positive bands are ascribed to the 2a-acetyl modes of
at 1705 and 1715 cm−1, respectively. Minor changes are also seen in the relative intensities of the product state bands at 1647 and 1635 cm−1. As the 2a-acetyl C=O modes of P upshift in the cation state, these positive bands are ascribed to the 2a-acetyl modes of 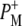 and
and 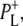 respectively, and the negative bands at 1642 cm−1 and 1624 cm−1 could correspond to the equivalent ground state modes of PM and PL (39). However, as discussed above, these attributions of the observed 2a-acetyl C=O modes are not certain due to other contributions that affect the region of this spectrum (21,22,39,44).
respectively, and the negative bands at 1642 cm−1 and 1624 cm−1 could correspond to the equivalent ground state modes of PM and PL (39). However, as discussed above, these attributions of the observed 2a-acetyl C=O modes are not certain due to other contributions that affect the region of this spectrum (21,22,39,44).
For RCs lacking QA, 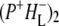 is the last charge separated state, and the triplet state of P is formed in ∼7 ns (Fig. 7, magenta spectrum). The triplet state spectrum shows a negative band at 1687 cm−1 from a mode that downshifts to give a positive band at 1672 cm−1. This positive band at 1672 cm−1 is the most prominent in the 3P spectrum, and was also observed in an FTIR study conducted on RCs at 85 K (66). The presence of the negative band at 1687 cm−1 strongly supports the assignment of this 1672 cm−1 band to the 9-keto C=O groups of the P BChls.
is the last charge separated state, and the triplet state of P is formed in ∼7 ns (Fig. 7, magenta spectrum). The triplet state spectrum shows a negative band at 1687 cm−1 from a mode that downshifts to give a positive band at 1672 cm−1. This positive band at 1672 cm−1 is the most prominent in the 3P spectrum, and was also observed in an FTIR study conducted on RCs at 85 K (66). The presence of the negative band at 1687 cm−1 strongly supports the assignment of this 1672 cm−1 band to the 9-keto C=O groups of the P BChls.
The final step of charge separation in RCs containing QA leads to formation of the 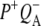 state in 290 ps. Two SADS of this state obtained with either 600 nm or 860 nm excitation are shown in Fig. 8 B. The 1675–1610 cm−1 region shows a pattern that is characteristic for contributions from
state in 290 ps. Two SADS of this state obtained with either 600 nm or 860 nm excitation are shown in Fig. 8 B. The 1675–1610 cm−1 region shows a pattern that is characteristic for contributions from 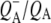 (39,41,57,58). The strong band shift signal at 1666(−)/1656(+) cm−1 has been ascribed to a protein response to the reduction of QA (58). The shoulder at 1728 cm−1 is assigned to the anion state of QA and has been observed previously in FTIR
(39,41,57,58). The strong band shift signal at 1666(−)/1656(+) cm−1 has been ascribed to a protein response to the reduction of QA (58). The shoulder at 1728 cm−1 is assigned to the anion state of QA and has been observed previously in FTIR 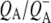 difference spectra together with a negative band at 1735 cm−1 (39,41,60) that is shifted to 1739 cm−1 in fs mid-IR spectrum, probably due to an overlap with 10a-ester modes of P.
difference spectra together with a negative band at 1735 cm−1 (39,41,60) that is shifted to 1739 cm−1 in fs mid-IR spectrum, probably due to an overlap with 10a-ester modes of P.
FIGURE 8.

Comparison of the P* and 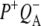 spectra resulting from target analysis of R26 RCs. (A) P* state spectra resulting from excitation at 805 nm (black) and 860 nm (red). (B)
spectra resulting from target analysis of R26 RCs. (A) P* state spectra resulting from excitation at 805 nm (black) and 860 nm (red). (B) 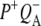 spectra resulting from excitation at 600 nm (black) and 860 nm (red).
spectra resulting from excitation at 600 nm (black) and 860 nm (red).
In summary, the SADS obtained for the various states formed during charge separation contained features that correlated well with previous data from steady-state FTIR spectroscopy, and most spectral features could be assigned with reasonable confidence. The majority of features arose from frequency shifts associated with oxidation of P or reduction of BL, HL, or QA, with some additional components arising from the response of the protein. In the next section the reproducibility of the various spectra is considered by comparing SADS obtained from individual data sets.
Comparison of the P*, P+HL−, and P+QA− SADS resulting from a simultaneous target analysis of R26 and AM260W RCs
To show the reproducibility of the fs mid-IR experiments the P*, 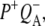 and
and 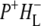 spectra resulting from individual experiments on different samples were compared. These comparisons are shown in Fig. 8 A (P*), Fig. 8 B (
spectra resulting from individual experiments on different samples were compared. These comparisons are shown in Fig. 8 A (P*), Fig. 8 B (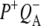 ) and Supplementary Material, Fig. S1 and Data S1
) and Supplementary Material, Fig. S1 and Data S1 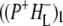 and
and 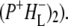
Fig. 8 A shows a comparison of the SADS corresponding to the P* state for R26 RCs excited at 805 nm (black) and 860 nm (red). The spectra are very similar with the main transition centered at 1668 cm−1. The SADS corresponding to the P* state derived from the remaining five data sets had similar line-shapes (not shown), and it was concluded that the spectral signature of this state was very reproducible, with relatively little distortion, apart from presence of some baseline noise.
Fig. 8 B shows SADS corresponding to the 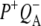 state derived from data on R26 RCs excited at 600 nm (black) and 860 nm (red). The most noticeable difference between the two is the smaller amplitude of the signals observed at 1675(−) cm−1 and 1667(+)/1656(−) cm−1 in the red spectrum. Other than this, the two spectra show good correspondence.
state derived from data on R26 RCs excited at 600 nm (black) and 860 nm (red). The most noticeable difference between the two is the smaller amplitude of the signals observed at 1675(−) cm−1 and 1667(+)/1656(−) cm−1 in the red spectrum. Other than this, the two spectra show good correspondence.
Fig. S1 and Data S1 shows the spectra of 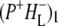 (black) and
(black) and 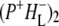 (red) states resulting from four separate experiments. The first, second, and third plot show data for RCs depleted of QA that were excited at 600 nm, 805 nm, and 860 nm, respectively, whereas the fourth plot shows data for the QA-deficient AM260W mutant excited at 860 nm. Once again a high degree of similarity was observed between the SADS from different experiments. This figure also allows the differences between the SADS corresponding to the
(red) states resulting from four separate experiments. The first, second, and third plot show data for RCs depleted of QA that were excited at 600 nm, 805 nm, and 860 nm, respectively, whereas the fourth plot shows data for the QA-deficient AM260W mutant excited at 860 nm. Once again a high degree of similarity was observed between the SADS from different experiments. This figure also allows the differences between the SADS corresponding to the 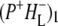 (black) and
(black) and 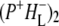 (red) states to be compared. A consistent difference was an increase in the relative amplitude of the
(red) states to be compared. A consistent difference was an increase in the relative amplitude of the 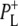 band at 1715 cm−1 relative to that of
band at 1715 cm−1 relative to that of 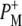 at 1705 cm−1 in the
at 1705 cm−1 in the 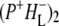 state, and this is also apparent on comparing the blue
state, and this is also apparent on comparing the blue 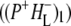 and cyan
and cyan 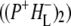 spectra shown in Fig. 7. This point is returned to below.
spectra shown in Fig. 7. This point is returned to below.
Comparison of the P+BL− SADS resulting from a simultaneous target analysis of R26 and AM260W RCs
Fig. 9 shows SADS for the 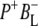 state. The green spectrum is that also shown in Fig. 7 (green), whereas the black spectrum is the average of the
state. The green spectrum is that also shown in Fig. 7 (green), whereas the black spectrum is the average of the 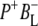 SADS from the five remaining data sets. Despite the relatively small amplitude of the signal corresponding to the
SADS from the five remaining data sets. Despite the relatively small amplitude of the signal corresponding to the 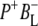 state, and the larger noise therefore present in the
state, and the larger noise therefore present in the 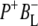 spectra, there is a good level of agreement between the two spectra shown in Fig. 9. The pattern of negative and positive bands typical for PL, PM, and
spectra, there is a good level of agreement between the two spectra shown in Fig. 9. The pattern of negative and positive bands typical for PL, PM, and 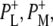 respectively, was reproduced reasonably well in the single (Fig. 9, green) and averaged (Fig. 9, black)
respectively, was reproduced reasonably well in the single (Fig. 9, green) and averaged (Fig. 9, black) 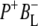 SADS. These encompass the transitions of the 10a-ester C=O of PL/PM and
SADS. These encompass the transitions of the 10a-ester C=O of PL/PM and 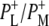 at 1739(−)/1750(+) cm−1, respectively, and the 9-keto C=O modes of PL/PM at 1687(−) cm−1,
at 1739(−)/1750(+) cm−1, respectively, and the 9-keto C=O modes of PL/PM at 1687(−) cm−1, 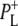 at 1715(+) cm−1, and
at 1715(+) cm−1, and 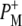 at 1705(+) cm−1. The broad negative feature between 1690 and 1670 cm−1 represents the ground states of the 9-keto C=O groups of PLPM and BL. As discussed above, signals attributable to PLPM are expected to appear between 1690 and 1680 cm−1 (40), so it is possible that the negative signal between 1680 and 1670 cm−1 can be tentatively assigned to BL. Note that the similarities between
at 1705(+) cm−1. The broad negative feature between 1690 and 1670 cm−1 represents the ground states of the 9-keto C=O groups of PLPM and BL. As discussed above, signals attributable to PLPM are expected to appear between 1690 and 1680 cm−1 (40), so it is possible that the negative signal between 1680 and 1670 cm−1 can be tentatively assigned to BL. Note that the similarities between 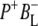 and
and 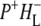 spectra are due to P/P+ transitions, that play a dominant role in those two spectra on top of
spectra are due to P/P+ transitions, that play a dominant role in those two spectra on top of 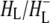 and
and 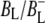 signals. Nevertheless, the transition observed at 1670–1680(−)/1657(+) cm−1 tentatively assigned to
signals. Nevertheless, the transition observed at 1670–1680(−)/1657(+) cm−1 tentatively assigned to 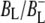 is evidently absent in the
is evidently absent in the 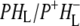 difference spectrum.
difference spectrum.
FIGURE 9.

Overlaid 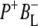 SADS of R26 RCs. The green spectrum is that shown in Fig. 7 (green), obtained after excitation of RCs not containing QA with 805 nm. The black spectrum is the average of the remaining five
SADS of R26 RCs. The green spectrum is that shown in Fig. 7 (green), obtained after excitation of RCs not containing QA with 805 nm. The black spectrum is the average of the remaining five 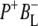 SADS.
SADS.
Fig. S2 and Data S1 show the decomposition of eight representative time traces into the time-dependent contributions of each of the species that participate in the spectral evolution. The selected time traces were determined for R26 RCs with QA removed excited at 805 nm (Fig. S2 A and Data S1) and for R-26 RCs containing QA excited at 860 nm (Fig. S2 B and Data S1). As can be seen in these concentration profiles at each single frequency the contributions from most of the intermediates can be identified. We selected time traces where mainly one or only a few species can be selectively observed. For example, the time trace measured at 1668 cm−1 represents the P* state; at 1686 cm−1 the signals are negative due to the bleaching of the 9-keto C=O of PL and PM in the ground state; at 1698 cm−1 and 1717 cm−1 the transient signal has become positive due to formation of the first and second radical pairs: 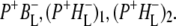 The time traces measured at 1666 cm−1 and 1654 cm−1 are related to the amide I transition in
The time traces measured at 1666 cm−1 and 1654 cm−1 are related to the amide I transition in 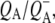 note that in the trace at 1666 cm−1 the signals from P* (see trace at 1668 cm−1), amide I and QA are observed; in the trace at 1654 cm−1 the signals from amide I,
note that in the trace at 1666 cm−1 the signals from P* (see trace at 1668 cm−1), amide I and QA are observed; in the trace at 1654 cm−1 the signals from amide I, 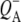 and the 9-keto C=O of
and the 9-keto C=O of 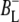 are observed, whereas in the trace at 1678 cm−1 we mainly detect the ground state contributions of BL and HL to the signal. The trace at 1728 cm−1 shows the shift of the 10a-ester transition of HL due to the electric field of
are observed, whereas in the trace at 1678 cm−1 we mainly detect the ground state contributions of BL and HL to the signal. The trace at 1728 cm−1 shows the shift of the 10a-ester transition of HL due to the electric field of 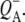 An instructive aspect of these traces is that they emphasize the low amplitude of the contribution of
An instructive aspect of these traces is that they emphasize the low amplitude of the contribution of 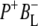 (blue traces), due to the transient nature of this species.
(blue traces), due to the transient nature of this species.
Comparison of the mid-IR spectral evolution with visible region pump-probe data
The target analysis described in Figs. 6 and 7 involved a kinetic scheme similar to that used in previous reports to analyze visible-region pump-probe data (18) and the lifetimes obtained in this study are in good agreement with those previously obtained. In (18) membrane-bound wild-type Rb. sphaeroides RCs with QA reduced were measured using fs pump/probe spectroscopy. The target analysis of the data based on a 5-compartment reversible kinetic model involving relaxation of the second radical pair showed lifetimes of 0.18 ps for B* decay, 5.1 ps for P* decay, 1.9 ps for 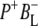 decay, 22 ps for
decay, 22 ps for 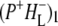 decay, and a long-lived component representing relaxation of
decay, and a long-lived component representing relaxation of 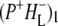 into
into 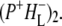 The lifetimes of this model with the given time constants are approximately the same as those obtained from global analysis by using the 5-component forward kinetic model in this study. In Holzwarth and Muller (38), fs pump/probe experiments were carried out on RCs with QA. Global analysis of the data with a 5-component kinetic model resulted in lifetimes of 3.1 ps related to P* decay, 1.5 ps to
The lifetimes of this model with the given time constants are approximately the same as those obtained from global analysis by using the 5-component forward kinetic model in this study. In Holzwarth and Muller (38), fs pump/probe experiments were carried out on RCs with QA. Global analysis of the data with a 5-component kinetic model resulted in lifetimes of 3.1 ps related to P* decay, 1.5 ps to 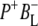 state decay, 148 ps to
state decay, 148 ps to 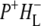 state decay and a long-lived component due to
state decay and a long-lived component due to 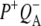 decay. A 10.8 ps component resulting from this analysis was not attributed to any state. These lifetimes obtained on the basis of a forward kinetic model are in good agreement with the lifetimes obtained in van Stokkum et al. (18). In Schmidt et al. (12) and Hamm et al. (61) RCs containing QA were studied by ps pump/probe spectroscopy. Again five lifetimes were resolved: 2.3 ps related to
decay. A 10.8 ps component resulting from this analysis was not attributed to any state. These lifetimes obtained on the basis of a forward kinetic model are in good agreement with the lifetimes obtained in van Stokkum et al. (18). In Schmidt et al. (12) and Hamm et al. (61) RCs containing QA were studied by ps pump/probe spectroscopy. Again five lifetimes were resolved: 2.3 ps related to 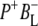 state formation, 0.9 ps to electron transfer to HL, 200 ps attributed to
state formation, 0.9 ps to electron transfer to HL, 200 ps attributed to 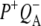 formation and long-lived component related to decay of this state. An additional 7 ps component was related to P* decay and ascribed to the intrinsic multi-exponential character of P* state decay (12,61–64).
formation and long-lived component related to decay of this state. An additional 7 ps component was related to P* decay and ascribed to the intrinsic multi-exponential character of P* state decay (12,61–64).
We conclude that the kinetic scheme obtained in this study is in a good agreement with that proposed in van Stokkum et al. (18), where vis-NIR pump-probe data were analyzed on the basis of a target analysis using a similar reversible model, as well as to the forward sequential model applied to the results in Hamm et al. (61) and Arlt et al. (62).
Comparison of fs mid-IR spectra and FTIR spectra of radical pair states
A good test of the quality and veracity of the fs time-resolved IR difference spectra described in this report is their correspondence with spectra obtained through steady-state FTIR difference spectroscopy. To compare the two types of spectra, 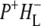 and
and 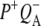 difference spectra were simulated using published results obtained from FTIR spectroscopy carried out on photochemically generated radical states P+/P (39,47–49),
difference spectra were simulated using published results obtained from FTIR spectroscopy carried out on photochemically generated radical states P+/P (39,47–49), 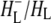 (43,44) and
(43,44) and 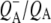 (39–41,45,47–49). Because the FTIR difference spectra were not normalized, the spectra of these radical states were summed with weights that produced the best agreement (in terms of amplitude) to the time-resolved mid-IR spectra. The weights were 0.5 and 1.5 for P+/P and
(39–41,45,47–49). Because the FTIR difference spectra were not normalized, the spectra of these radical states were summed with weights that produced the best agreement (in terms of amplitude) to the time-resolved mid-IR spectra. The weights were 0.5 and 1.5 for P+/P and 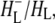 respectively, and 1.5 and 0.5 for P+/P and
respectively, and 1.5 and 0.5 for P+/P and 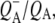 respectively. Fig. 10 A compares the FTIR difference spectrum of the
respectively. Fig. 10 A compares the FTIR difference spectrum of the 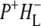 state (blue) and the EADS attributed to the
state (blue) and the EADS attributed to the 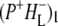 state as obtained by fs mid-IR spectroscopy after 600 nm excitation of R26 RCs with QA (black) and without QA (red). Variations in baseline aside, given the large difference in the way they were obtained there is a good degree of similarity between the steady state and time resolved spectra. The most conspicuous difference is in the spectral range of HL, where two negative bands at 1743 cm−1 and 1733 cm−1 are present in the FTIR difference spectrum of
state as obtained by fs mid-IR spectroscopy after 600 nm excitation of R26 RCs with QA (black) and without QA (red). Variations in baseline aside, given the large difference in the way they were obtained there is a good degree of similarity between the steady state and time resolved spectra. The most conspicuous difference is in the spectral range of HL, where two negative bands at 1743 cm−1 and 1733 cm−1 are present in the FTIR difference spectrum of 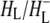 in the 10a-ester region (measured with spectral resolution of 4 cm−1) (43,44). These have been attributed to the ground state of two different forms of HL with a free or hydrogen-bonded 10a-ester carbonyl group (43,44). In our experiments using a 6 cm−1 spectral resolution only one negative band at 1736 cm−1 was resolved. This difference was probably due to differences in spectral resolution, as more structure was seen in this region of the
in the 10a-ester region (measured with spectral resolution of 4 cm−1) (43,44). These have been attributed to the ground state of two different forms of HL with a free or hydrogen-bonded 10a-ester carbonyl group (43,44). In our experiments using a 6 cm−1 spectral resolution only one negative band at 1736 cm−1 was resolved. This difference was probably due to differences in spectral resolution, as more structure was seen in this region of the 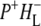 spectrum in time-resolved experiments carried out with a 3 cm−1 spectral resolution (Fig. 3, green and blue). In the three spectra presented in Fig. 10A this mode upshifted to 1751 cm−1 in the cation state of 10a-ester of
spectrum in time-resolved experiments carried out with a 3 cm−1 spectral resolution (Fig. 3, green and blue). In the three spectra presented in Fig. 10A this mode upshifted to 1751 cm−1 in the cation state of 10a-ester of 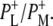 These frequencies match closely values from steady-state FTIR spectroscopy of 1740 cm−1 (negative) and 1750 cm−1 (positive) (39–41,45).
These frequencies match closely values from steady-state FTIR spectroscopy of 1740 cm−1 (negative) and 1750 cm−1 (positive) (39–41,45).
FIGURE 10.

Comparison of fs mid-IR spectra and FTIR spectra of radical pair states. (A) Comparison of EADS of the 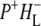 state measured for the samples with (black) and without quinone QA (red) excited with 600 nm light and summed FTIR spectra of P+ (39,47–49) and
state measured for the samples with (black) and without quinone QA (red) excited with 600 nm light and summed FTIR spectra of P+ (39,47–49) and 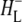 (blue) (43,44) (note that the FTIR spectra were not normalized). (B) Comparison of the EADS of the
(blue) (43,44) (note that the FTIR spectra were not normalized). (B) Comparison of the EADS of the 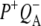 state (black line) formed after 600 nm excitation and summed FTIR spectra of P+ (39,47–49) and
state (black line) formed after 600 nm excitation and summed FTIR spectra of P+ (39,47–49) and 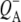 (blue line) (39–41,45,47–49).
(blue line) (39–41,45,47–49).
A second difference between the time-resolved and steady state IR spectra in Fig. 10 A concerns the bands attributable to the 9-keto C=O groups of the P BChls. The FTIR difference spectrum has positive bands at 1705 cm−1 and 1715 cm−1 arising from 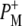 and
and 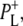 respectively, together with negative bands at 1683 cm−1 and 1692 cm−1 arising from the neutral states of PL and PM, respectively. In the case of the negative bands, the ground state mode of PL is much more prominent in the FTIR spectrum (blue) than in the fs spectra (red and black) where it appears only as a shoulder of the 1692 cm−1 band. Similarly the positive band attributable to
respectively, together with negative bands at 1683 cm−1 and 1692 cm−1 arising from the neutral states of PL and PM, respectively. In the case of the negative bands, the ground state mode of PL is much more prominent in the FTIR spectrum (blue) than in the fs spectra (red and black) where it appears only as a shoulder of the 1692 cm−1 band. Similarly the positive band attributable to 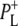 at 1715 cm−1 is more prominent in the fs spectra (red and black) than in the FTIR spectrum (blue). The origin of these differences remains to be determined, but could reflect different extents of relaxation of the cation state of the P dimer in the different types of sample and different experimental conditions. These small differences aside, the agreement between the fs mid-IR and FTIR spectra is very good.
at 1715 cm−1 is more prominent in the fs spectra (red and black) than in the FTIR spectrum (blue). The origin of these differences remains to be determined, but could reflect different extents of relaxation of the cation state of the P dimer in the different types of sample and different experimental conditions. These small differences aside, the agreement between the fs mid-IR and FTIR spectra is very good.
Fig. 10 B shows a comparison of the EADS for the 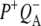 state measured by mid-IR femtosecond spectroscopy after 600 nm excitation (black line), and the FTIR difference spectrum that was calculated from the difference spectra of the individual P+ (39,47–49) and
state measured by mid-IR femtosecond spectroscopy after 600 nm excitation (black line), and the FTIR difference spectrum that was calculated from the difference spectra of the individual P+ (39,47–49) and 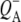 components (39–41,45,47–49) (blue line). Once again there is good correspondence in terms of the general line-shape despite the lower spectral resolution of the time-resolved experiment. The calculated spectrum of
components (39–41,45,47–49) (blue line). Once again there is good correspondence in terms of the general line-shape despite the lower spectral resolution of the time-resolved experiment. The calculated spectrum of 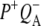 state shows positive bands at 1750, 1705, 1655 cm−1, and negative bands at 1740 and 1725 cm−1 and in the 1683–1690 cm−1 region. The EADS of
state shows positive bands at 1750, 1705, 1655 cm−1, and negative bands at 1740 and 1725 cm−1 and in the 1683–1690 cm−1 region. The EADS of 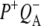 has a single negative band at 1687 cm−1, whereas the FTIR spectrum has a more complex line-shape with a main negative band at 1682 cm−1 and a shoulder at 1687 cm−1. Likewise the single negative band at 1666 cm−1 in the EADS corresponds to two bands in the FTIR spectrum, at 1670 cm−1 and 1664 cm−1. The positive band at 1730 cm−1 and negative band at 1725 cm−1 observed in the FTIR spectrum were not resolved by the fs mid-IR technique, but nevertheless the shoulder at 1730 cm−1 is present in time-resolved spectrum.
has a single negative band at 1687 cm−1, whereas the FTIR spectrum has a more complex line-shape with a main negative band at 1682 cm−1 and a shoulder at 1687 cm−1. Likewise the single negative band at 1666 cm−1 in the EADS corresponds to two bands in the FTIR spectrum, at 1670 cm−1 and 1664 cm−1. The positive band at 1730 cm−1 and negative band at 1725 cm−1 observed in the FTIR spectrum were not resolved by the fs mid-IR technique, but nevertheless the shoulder at 1730 cm−1 is present in time-resolved spectrum.
The response of the protein to photochemical charge separation
One motivation for studying RC charge separation in the IR region is that, unlike the case for vis/near-IR spectroscopy, the signals obtained report on the protein environment in addition to the cofactors participating in the reaction. In principle, signals from the protein arise from two different effects. First, hydrogen bonding between a cofactor and its protein binding site is likely to be perturbed after oxidation or reduction of the cofactor. Second, each step in electron transfer brings about a new charge distribution on the cofactors that may induce a local perturbation of polarizable groups of the protein, leading to localized structural changes (which in turn could affect hydrogen bonding to an adjacent cofactor). These electrostatic interactions are maximal close to the cofactors, but because of dipolar coupling they could also propagate on a larger scale. Such electrostatic effects could induce large rearrangements of the protein backbone and will be observed as changes of coupling constants within polarizable groups of the protein backbone and side chains.
As discussed above, the results obtained in this report through fs mid-IR spectroscopy are in very good agreement with data measured using the steady-state FTIR technique by Bagley et al. (40), Mantele et al. (44,49), Nabedryk et al. (45), and Breton et al. (57,58,68). This implies that there are no large scale structural rearrangements that occur on the timescale of electron transfer but that subsequently relax after formation of the final charge separated state. Such transient rearrangements would be expected to affect the line-shape of fs IR difference spectra, but not contribute to steady-state FTIR spectra. A small number of the spectral features in the fs IR spectra could be correlated to features in FTIR spectra that have been attributed to protein responses. For example, spectra of the 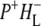 state (Fig. 2, green and blue; Fig. 5, green; Fig. 7, blue and cyan; and Fig. 9) showed the upshift of a band from 1656(−) to 1663(+) cm−1 that has been attributed to a response of the amide I transition (21,22). The presence of this feature in the ps spectra of the
state (Fig. 2, green and blue; Fig. 5, green; Fig. 7, blue and cyan; and Fig. 9) showed the upshift of a band from 1656(−) to 1663(+) cm−1 that has been attributed to a response of the amide I transition (21,22). The presence of this feature in the ps spectra of the 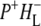 states shows that this response occurs on the timescale of electron transfer to the pheophytin. Likewise, in spectra of the
states shows that this response occurs on the timescale of electron transfer to the pheophytin. Likewise, in spectra of the 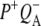 state (Fig. 5, blue, and Fig. 8 B) the downshift of a mode from 1666(−) to 1656(+) cm−1 has been attributed to a change in a protein amide I transition (39–41,57,58), whereas the downshift of a mode from 1650(−) to 1640(+) cm−1 has been attributed to a response of the protein backbone connected to QA via an H-bond (44) that becomes perturbed after electron transfer to QA (39). Again, the presence of these features in the fs IR spectra indicates that these protein responses occur on the timescale of electron transfer, rather than being slower timescale responses to formation of the charge separated state. This conclusion is in general agreement with findings from previous studies (21,22).
state (Fig. 5, blue, and Fig. 8 B) the downshift of a mode from 1666(−) to 1656(+) cm−1 has been attributed to a change in a protein amide I transition (39–41,57,58), whereas the downshift of a mode from 1650(−) to 1640(+) cm−1 has been attributed to a response of the protein backbone connected to QA via an H-bond (44) that becomes perturbed after electron transfer to QA (39). Again, the presence of these features in the fs IR spectra indicates that these protein responses occur on the timescale of electron transfer, rather than being slower timescale responses to formation of the charge separated state. This conclusion is in general agreement with findings from previous studies (21,22).
In the fs mid-IR spectra presented we observe an amide (protein) response during all the steps of the electron transfer. This observation supports the conclusion drawn by Adolphs et al. (69) that the electronic states of RC chromophore are strongly coupled to a protein dipole.
In this study the relaxation of the radical pair 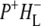 was resolved, presenting an opportunity to determine whether the processes that cause a drop in free energy of ∼60 meV are associated with a specific spectral signature. In Fig. 11 the average of all collected
was resolved, presenting an opportunity to determine whether the processes that cause a drop in free energy of ∼60 meV are associated with a specific spectral signature. In Fig. 11 the average of all collected 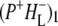 (black) and
(black) and 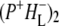 (red) SADS is plotted, together with the difference between the two spectra (green). The main features in the green spectrum are the increase of the
(red) SADS is plotted, together with the difference between the two spectra (green). The main features in the green spectrum are the increase of the 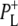 band at 1715 cm−1 (and a corresponding decrease of the
band at 1715 cm−1 (and a corresponding decrease of the 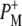 band at 1705 cm−1) and a decrease of the band-shift signal at 1666(+)/1656(−), which has been attributed to an amide response (21,22,39). This would imply that on the timescale of ∼20 ps the hole becomes more localized on PL and the unpaired electron on PM, and that this localization process is concurrent with a decrease in the strength of the electric field that causes the Stark shift of nearby amide oscillators. Interestingly, the timescale of this process coincides partially with the time dependence of protein dynamics as recently measured by the Stark shift of tryptophan absorption, which was 10 ps and 190 ps (26). There, for 14 mutants in which the initial electron transfer varied from 2 ps to tens of ps, it was shown that the protein relaxation kinetics on the ps timescale are independent of electron transfer kinetics, and protein conformational changes are initiated by the absorption of light and not by charge separation. This conclusion supports the idea that protein dynamics play a key role in the kinetics of the primary electron transfer process, what may suggest that the electron transfer kinetics are actually driven by protein dynamics, rather than protein responding to charge separation. Possibly an ongoing conformational change is the cause of the increased localization of the electron hole in the P dimer. We stress that the amide response is only observed in the cation of P and not in the P excited state, and therefore seems to be a local electric field probe.
band at 1705 cm−1) and a decrease of the band-shift signal at 1666(+)/1656(−), which has been attributed to an amide response (21,22,39). This would imply that on the timescale of ∼20 ps the hole becomes more localized on PL and the unpaired electron on PM, and that this localization process is concurrent with a decrease in the strength of the electric field that causes the Stark shift of nearby amide oscillators. Interestingly, the timescale of this process coincides partially with the time dependence of protein dynamics as recently measured by the Stark shift of tryptophan absorption, which was 10 ps and 190 ps (26). There, for 14 mutants in which the initial electron transfer varied from 2 ps to tens of ps, it was shown that the protein relaxation kinetics on the ps timescale are independent of electron transfer kinetics, and protein conformational changes are initiated by the absorption of light and not by charge separation. This conclusion supports the idea that protein dynamics play a key role in the kinetics of the primary electron transfer process, what may suggest that the electron transfer kinetics are actually driven by protein dynamics, rather than protein responding to charge separation. Possibly an ongoing conformational change is the cause of the increased localization of the electron hole in the P dimer. We stress that the amide response is only observed in the cation of P and not in the P excited state, and therefore seems to be a local electric field probe.
FIGURE 11.

The average of all the 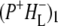 (black) and
(black) and 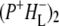 (red) spectra, and difference between them (green). An increase or decrease of an intensity of the indicated bands was marked with arrows directed up or down, respectively.
(red) spectra, and difference between them (green). An increase or decrease of an intensity of the indicated bands was marked with arrows directed up or down, respectively.
Charge delocalization within primary electron donor and relaxation of P+HL−
A feature of charge separation in the bacterial RC is that the hole created by membrane-spanning electron transfer is shared between the two BChls that constitute the primary electron donor. This P+ dimer gives rise to a number of distinctive spectroscopic signals. In particular, a number of studies on Rb. sphaeroides RCs have shown the presence of a broad positive band centered at ∼2600 cm−1 (±100 cm−1) in the light-minus-dark difference 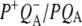 spectrum (20,23,44,51–53), which has been attributed to an electronic transition of the dimeric P+. In this study the fs mid-IR experiments carried out in the region above 2000 cm−1 showed the shoulder of this broad positive band around 2200 cm−1 (Fig. 4). The fact that half the signal is already present at time zero showed the P* charge transfer character, in agreement with earlier Stark measurements (55,56).
spectrum (20,23,44,51–53), which has been attributed to an electronic transition of the dimeric P+. In this study the fs mid-IR experiments carried out in the region above 2000 cm−1 showed the shoulder of this broad positive band around 2200 cm−1 (Fig. 4). The fact that half the signal is already present at time zero showed the P* charge transfer character, in agreement with earlier Stark measurements (55,56).
The details of the distribution of spin density between PL and PM after oxidation of P may also be relevant to the finding that fitting of the experimental data required two 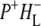 states in those samples where electron transfer to QA was blocked by quinone removal or exclusion through mutagenesis. In all such cases, the initially-formed
states in those samples where electron transfer to QA was blocked by quinone removal or exclusion through mutagenesis. In all such cases, the initially-formed 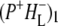 state was found to decay to a spectrally-distinct
state was found to decay to a spectrally-distinct 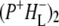 state with a lifetime that varied between 7.2 ps and 17 ps (Table 1). In Fig. 11 the SADS obtained for the states
state with a lifetime that varied between 7.2 ps and 17 ps (Table 1). In Fig. 11 the SADS obtained for the states 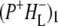 and
and 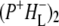 from the target analysis of four of the data sets are compared (black for
from the target analysis of four of the data sets are compared (black for 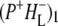 and red for
and red for 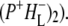 In all four data sets, the relative amplitude of the bands at 1715 and 1705 cm−1 underwent a small but consistent change during the time evolution of the system from the
In all four data sets, the relative amplitude of the bands at 1715 and 1705 cm−1 underwent a small but consistent change during the time evolution of the system from the 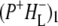 state to the relaxed state
state to the relaxed state 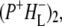 with the 1715 cm−1 band increasing in relative amplitude (this change was systematically observed in each of seven SADS resulting from target analysis). As explained above, the bands at 1715 and 1705 cm−1 are attributable to the 9-keto C=O groups of
with the 1715 cm−1 band increasing in relative amplitude (this change was systematically observed in each of seven SADS resulting from target analysis). As explained above, the bands at 1715 and 1705 cm−1 are attributable to the 9-keto C=O groups of 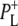 and
and 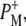 respectively, and their relative amplitudes reflect the distribution of spin density between PL and PM (70,71). The fact that the relative amplitude of the band at 1715 cm−1 increased in the
respectively, and their relative amplitudes reflect the distribution of spin density between PL and PM (70,71). The fact that the relative amplitude of the band at 1715 cm−1 increased in the 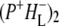 state, suggests that part of the relaxation of this state involved the positive charge becoming more localized on PL.
state, suggests that part of the relaxation of this state involved the positive charge becoming more localized on PL.
CONCLUSIONS
The processes of energy and electron transfer in the bacterial reaction center have been studied in the mid-IR after excitation in the vis/NIR. The experimental results were analyzed using a kinetic model similar to those proposed earlier for the analysis of visible region pump/probe data (7,10,11). We observe a good agreement of time constants obtained from mid-IR probe data with previous visible region probe data. On the basis of this model the mid-IR absorption spectrum of 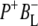 was described for the first time. A comparison of fs data with steady-state FTIR data for the states
was described for the first time. A comparison of fs data with steady-state FTIR data for the states 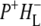 and
and 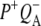 showed a good spectral consistency. Fast protein responses to the oxidation of P and the reduction of HL and QA were identified. The relaxation of the radical pair
showed a good spectral consistency. Fast protein responses to the oxidation of P and the reduction of HL and QA were identified. The relaxation of the radical pair 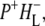 corresponding to a drop in free energy of ∼60 meV on a timescale of ∼20 ps, is shown to be correlated with an increase in localization of the electron hole on the PM half of the dimer, as well as with a relaxation of the perturbation of one or more amide C=O oscillators.
corresponding to a drop in free energy of ∼60 meV on a timescale of ∼20 ps, is shown to be correlated with an increase in localization of the electron hole on the PM half of the dimer, as well as with a relaxation of the perturbation of one or more amide C=O oscillators.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
This research was supported by the Netherlands Organization of Scientific Research (NWO) via the Foundation of Earth and Life Sciences (ALW), by the European Union (Grant MRTN-CT-2003-505069, Intro2), and by HFSP Research Grant RGP 38/2006. M.R.J. acknowledges financial support from the Biotechnology and Biological Sciences Research Council of the United Kingdom.
Editor: Feng Gai.
References
- 1.Deisenhofer, J., O. Epp, I. Sinning, and H. Michel. 1995. Crystallographic refinement at 2.3-angstrom resolution and refined model of the photosynthetic reaction center from Rhodopseudomonas viridis. J. Mol. Biol. 246:429–457. [DOI] [PubMed] [Google Scholar]
- 2.Chirino, A. J., E. J. Lous, M. Huber, J. P. Allen, C. C. Schenck, M. L. Paddock, G. Feher, and D. C. Rees. 1994. Crystallographic analyses of site-directed mutants of the photosynthetic reaction center from Rhodobacter sphaeroides. Biochemistry. 33:4584–4593. [DOI] [PubMed] [Google Scholar]
- 3.Lancaster, C. R. D., U. Ermler, and H. Michel. 1995. Anoxygenic Photosynthetic Bacteria. Kluwer Academic, Dordrecht, The Netherlands. 503–526.
- 4.Woodbury, N. W., and J. P. Allen. 1995. The pathway, kinetics and thermodynamics of electron transfer in wild type and mutant bacterial reaction centers of purple nonsulfur bacteria. In Anoxygenic Photosynthetic Bacteria. M. T. M. R. E. Blankenship, and C. E. Bauer, editors. Kluwer Academic, Dordrecht, The Netherlands. 527–557.
- 5.van Brederode, M. E., and M. R. Jones. 2000. Reaction centres of purple bacteria. In Enzyme-Catalyzed Electron and Radical Transfer. N. S. S. A. Holzenburg, editor. Kluwer Academic/Plenum, New York. 621–676. [DOI] [PubMed]
- 6.Zinth, W., and J. Wachtveitl. 2005. The first picoseconds in bacterial photosynthesis—ultrafast electron transfer for the efficient conversion of light energy. Chemphyschem. 6:871–880. [DOI] [PubMed] [Google Scholar]
- 7.Ziolek, M., N. Pawlowicz, R. Naskrecki, and A. Dobek. 2005. Electron transfer in the reaction center of the Rb. sphaeroides R-26 studied by transient absorption. J. Phys. Chem. B. 109:18171–18176. [DOI] [PubMed] [Google Scholar]
- 8.Parson, W. W. 1996. Photosynthetic Bacterial Reaction Centers. In Protein Electron Transfer. D. S. Bendall, editor. BIOS Scientific, Oxford. 125–160.
- 9.Woodbury, N. W. T., and W. W. Parson. 1984. Nanosecond fluorescence from isolated photosynthetic reaction centers of Rhodopseudomonas sphaeroides. Biochim. Biophys. Acta. 767:345–361. [DOI] [PubMed] [Google Scholar]
- 10.Holzapfel, W., U. Finkele, W. Kaiser, D. Oesterhelt, H. Scheer, H. U. Stilz, and W. Zinth. 1989. Observation of a bacteriochlorophyll anion radical during the primary charge separation in a reaction center. Chem. Phys. Lett. 160:1–7. [Google Scholar]
- 11.Holzapfel, W., U. Finkele, W. Kaiser, D. Oesterhelt, H. Scheer, H. U. Stilz, and W. Zinth. 1990. Initial electron-transfer in the reaction center from Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. USA. 87:5168–5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt, S., T. Arlt, P. Hamm, H. Huber, T. Nagele, J. Wachtveitl, M. Meyer, H. Scheer, and W. Zinth. 1994. Energetics of the primary electron-transfer reaction revealed by ultrafast spectroscopy on modified bacterial reaction centers. Chem. Phys. Lett. 223:116–120. [Google Scholar]
- 13.van Brederode, M. E., M. R. Jones, and R. van Grondelle. 1997. Fluorescence excitation spectra of membrane-bound photosynthetic reaction centers of Rhodobacter sphaeroides in which the tyrosine M210 residue is replaced by tryptophan: evidence for a new pathway of charge separation. Chem. Phys. Lett. 268:143–149. [Google Scholar]
- 14.van Brederode, M. E., M. R. Jones, F. van Mourik, I. H. M. van Stokkum, and R. van Grondelle. 1997. A new pathway for transmembrane electron transfer in photosynthetic reaction centers of Rhodobacter sphaeroides not involving the excited special pair. Biochemistry. 36:6855–6861. [DOI] [PubMed] [Google Scholar]
- 15.van Brederode, M. E., F. van Mourik, I. H. M. van Stokkum, M. R. Jones, and R. van Grondelle. 1999. Multiple pathways for ultrafast transduction of light energy in the photosynthetic reaction center of Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. USA. 96:2054–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlodder, E., and K. Brettel. 1988. Primary charge separation in closed photosystem-II with a lifetime of 11-ns-flash-absorption spectroscopy with O-2-evolving photosystem-II complexes from Synechococcus. Biochim. Biophys. Acta. 933:22–34. [Google Scholar]
- 17.Groot, M. L., E. J. G. Peterman, P. J. M. van Kan, I. H. M. van Stokkum, J. P. Dekker, and R. van Grondelle. 1994. Temperature-dependent triplet and fluorescence quantum yields of the photosystem-II reaction-center described in a thermodynamic model. Biophys. J. 67:318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Stokkum, I. H. M., L. M. P. Beekman, M. R. Jones, M. E. van Brederode, and R. van Grondelle. 1997. Primary electron transfer kinetics in membrane-bound Rhodobacter sphaeroides reaction centers: a global and target analysis. Biochemistry. 36:11360–11368. [DOI] [PubMed] [Google Scholar]
- 19.Hamm, P., M. Zurek, W. Mantele, M. Meyer, H. Scheer, and W. Zinth. 1995. Femtosecond infrared-spectroscopy of reaction centers from Rhodobacter sphaeroides between 1000 and 1800 cm(-1). Proc. Natl. Acad. Sci. USA. 92:1826–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamm, P., and W. Zinth. 1995. Ultrafast initial reaction in bacterial photosynthesis revealed by femtosecond infrared-spectroscopy. J. Phys. Chem. 99:13537–13544. [Google Scholar]
- 21.Maiti, S., B. R. Cowen, R. Diller, M. Iannone, C. C. Moser, P. L. Dutton, and R. M. Hochstrasser. 1993. Picosecond infrared studies of the dynamics of the photosynthetic reaction center. Proc. Natl. Acad. Sci. USA. 90:5247–5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maiti, S., G. C. Walker, B. R. Cowen, R. Pippenger, C. C. Moser, P. L. Dutton, and R. M. Hochstrasser. 1994. Femtosecond coherent transient infrared-spectroscopy of reaction centers from Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. USA. 91:10360–10364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker, G. C., S. Maiti, B. R. Cowen, C. C. Moser, P. L. Dutton, and R. M. Hochstrasser. 1994. Time resolution of electronic-transitions of photosynthetic reaction centers in the infrared. J. Phys. Chem. 98:5778–5783. [Google Scholar]
- 24.Wynne, K., G. Haran, G. D. Reid, C. C. Moser, P. L. Dutton, and R. M. Hochstrasser. 1996. Femtosecond infrared spectroscopy of low-lying excited states in reaction centers of Rhodobacter sphaeroides. J. Phys. Chem. 100:5140–5148. [Google Scholar]
- 25.Haran, G., K. Wynne, C. C. Moser, P. L. Dutton, and R. M. Hochstrasser. 1996. Level mixing and energy redistribution in bacterial photosynthetic reaction centers. J. Phys. Chem. 100:5562–5569. [Google Scholar]
- 26.Wang, H. Y., S. Lin, J. P. Allen, J. C. Williams, S. Blankert, C. Laser, and N. W. Woodbury. 2007. Protein dynamics control the kinetics of initial electron transfer in photosynthesis. Science. 316:747–750. [DOI] [PubMed] [Google Scholar]
- 27.Groot, M. L., L. J. G. W. van Wilderen, D. S. Larsen, M. A. van der Horst, I. H. M. van Stokkum, K. J. Hellingwerf, and R. van Grondelle. 2003. Initial steps of signal generation in photoactive yellow protein revealed with femtosecond mid-infrared spectroscopy. Biochemistry. 42:10054–10059. [DOI] [PubMed] [Google Scholar]
- 28.Groot, M. L., J. Breton, L. J. G. W. van Wilderen, J. P. Dekker, and R. van Grondelle. 2004. Femtosecond visible/visible and visible/mid-IR pump-probe study of the photosystem II core antenna complex CP47. J. Phys. Chem. B. 108:8001–8006. [Google Scholar]
- 29.Feher, G., and M. Y. Okamura. 1978. Chemical composition and properties of reaction centers. In The Photosynthetic Bacteria. R. K. Clayton and W. R. Sistrom, editors. Plenum Press, New York. 349–386.
- 30.Ridge, J. P., M. E. van Brederode, M. G. Goodwin, R. van Grondelle, and M. R. Jones. 1999. Mutations that modify or exclude binding of the QA ubiquinone and carotenoid in the reaction center from Rhodobacter sphaeroides. Photosynth. Res. 59:9–26. [Google Scholar]
- 31.McAuley, K. E., P. K. Fyfe, J. P. Ridge, R. J. Cogdell, N. W. Isaacs, and M. R. Jones. 2000. Ubiquinone binding, ubiquinone exclusion, and detailed cofactor conformation in a mutant bacterial reaction center. Biochemistry. 39:15032–15043. [DOI] [PubMed] [Google Scholar]
- 32.Breton, J., M. C. Wakeham, P. K. Fyfe, M. R. Jones, and E. Nabedryk. 2004. Characterization of the bonding interactions of Q(B) upon photoreduction via A-branch or B-branch electron transfer in mutant reaction centers from Rhodobacter sphaeroides. Biochim. Biophys. Acta. 1656:127–138. [DOI] [PubMed] [Google Scholar]
- 33.Okamura, M. Y., R. A. Isaacson, and G. Feher. 1975. Primary acceptor in bacterial photosynthesis—obligatory role of ubiquinone in photoactive reaction centers of Rhodopseudomonas spheroides. Proc. Natl. Acad. Sci. USA. 72:3491–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woodbury, N. W., W. W. Parson, M. R. Gunner, R. C. Prince, and P. L. Dutton. 1986. Radical pair energetics and decay mechanisms in reaction centers containing anthraquinones, naphthoquinones or benzoquinones in place of ubiquinone. Biochim. Biophys. Acta. 851:6–22. [DOI] [PubMed] [Google Scholar]
- 35.van Stokkum, I. H. M., D. S. Larsen, and R. van Grondelle. 2004. Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta. 1657:82–104. [DOI] [PubMed] [Google Scholar]
- 36.Lozovoy, V. V., I. Pastirk, M. G. Comstock, and M. Dantus. 2001. Cascaded free-induction decay four-wave mixing. Chem. Phys. 266:205–212. [Google Scholar]
- 37.Mitsumori, Y., H. Fukushima, M. Ogura, T. Kuroda, S. Koshihara, F. Minami. 1996. Excitonic free induction decay studied with femtosecond pulse pairs. Luminescence. 66-67:81–83. [Google Scholar]
- 38.Holzwarth, A. R., and M. G. Muller. 1996. Energetics and kinetics of radical pairs in reaction centers from Rhodobacter sphaeroides. A femtosecond transient absorption study. Biochemistry. 35:11820–11831. [DOI] [PubMed] [Google Scholar]
- 39.Nabedryk, E. 1996. Light-induced Fourier transform infrared difference spectroscopy of the primary electron donor in photosynthetic reaction centers. In Infrared Spectroscopy of Biomolecules. H. H. Mantsch and D. Chapman, editors. Wiley-Liss, New York. 39–81.
- 40.Bagley, K. A., E. Abresch, M. Y. Okamura, G. Feher, M. Bauscher, W. Mantele, E. Nabedryk, and J. Breton. 1990. FTIR studies of the D+QA− and D+QB− states in reaction centers from Rb. Sphaeroides. In Current Research in Photosynthesis, Vol. I. M. Baltscheffsky, editor. Kluwer Academic Publishers, Dordrecht, The Netherlands. I.1.77–I.1.80.
- 41.Breton, J., D. L. Thibodeau, C. Berthomieu, W. Mantele, A. Vermeglio, and E. Nabedryk. 1991. Probing the primary quinone environment in photosynthetic bacterial reaction centers by light-induced FTIR difference spectroscopy. FEBS Lett. 278:257–260. [DOI] [PubMed] [Google Scholar]
- 42.Groot, M. L., N. P. Pawlowicz, L. J. G. W. van Wilderen, J. Breton, I. H. M. van Stokkum, and R. van Grondelle. 2005. Initial electron donor and acceptor in isolated Photosystem II reaction centers identified with femtosecond mid-IR spectroscopy. Proc. Natl. Acad. Sci. USA. 102:13087–13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nabedryk, E., S. Andrianambinintsoa, D. Dejonghe, and J. Breton. 1995. FTIR spectroscopy of the photoreduction of the bacteriopheophytin electron-acceptor in reaction centers of Rhodobacter sphaeroides and Rhodopseudomonas viridis. Chem. Phys. 194:371–378. [Google Scholar]
- 44.Mantele, W. G., A. M. Wollenweber, E. Nabedryk, and J. Breton. 1988. Infrared spectroelectrochemistry of bacteriochlorophylls and bacteriopheophytins—implications for the binding of the pigments in the reaction center from photosynthetic bacteria. Proc. Natl. Acad. Sci. USA. 85:8468–8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nabedryk, E., K. A. Bagley, D. L. Thibodeau, M. Bauscher, W. Mantele, and J. Breton. 1990. A protein conformational change associated with the photoreduction of the primary and secondary quinones in the bacterial reaction center. FEBS Lett. 266:59–62. [DOI] [PubMed] [Google Scholar]
- 46.Thibodeau, D. L., J. Breton, C. Barthomieu, W. Mantele, and E. Nabedryk. 1990. Reaction Centres of Photosynthetic Bacteria. Springer, Berlin. 87–98.
- 47.Nabedryk, E., B. A. Tavitian, W. Mantele, W. Kreutz, and J. Breton. 1987. Progress in Photosynthesis Research. Kluwer. Dordrecht. 177–180.
- 48.Nabedryk, E., B. A. Tavitian, W. Mantele, and J. Breton. 1985. Spectroscopy of Biological Molecules. Wiley-Liss, New York. 367–369.
- 49.Mantele, W., E. Nabedryk, B. A. Tavitian, W. Kreutz, and J. Breton. 1985. Light-induced Fourier-transform infrared (FTIR) spectroscopic investigations of the primary donor oxidation in bacterial photosynthesis. FEBS Lett. 187:227–232. [Google Scholar]
- 50.Wakeham, M. C., M. G. Goodwin, C. McKibbin, and M. R. Jones. 2003. Photo-accumulation of the P(+)Q(B)(−) radical pair state in purple bacterial reaction centres that lack the Q(A) ubiquinone. FEBS Lett. 540:234–240. [DOI] [PubMed] [Google Scholar]
- 51.Breton, J., E. Nabedryk, and W. W. Parson. 1992. A new infrared electronic-transition of the oxidized primary electron-donor in bacterial reaction centers— a way to assess resonance interactions between the bacteriochlorophylls. Biochemistry. 31:7503–7510. [DOI] [PubMed] [Google Scholar]
- 52.Breton, J., E. Nabedryk, and A. Clerici. 1999. Light-induced FTIR difference spectroscopy of photosynthetic charge separation between 9000 and 250 cm(-1). Vib. Spectrosc. 19:71–75. [Google Scholar]
- 53.Reimers, J. R., and N. S. Hush. 1995. Nature of the ground and first excited-states of the radical cations of photosynthetic bacterial reaction centers. Chem. Phys. 197:323–332. [Google Scholar]
- 54.Breton, J., and E. Nabedryk. 1998. Proton uptake upon quinone reduction in bacterial reaction centers: IR signature and possible participation of a highly polarizable hydrogen bond network. Photosynth. Res. 55:301–307. [Google Scholar]
- 55.Lockhart, D. J., and S. G. Boxer. 1987. Magnitude and direction of the change in dipole-moment associated with excitation of the primary electron-donor in Rhodopseudomonas sphaeroides reaction centers. Biochemistry. 26:664–668. [Google Scholar]
- 56.Lockhart, D. J., and S. G. Boxer. 1988. Stark-effect spectroscopy of Rhodobacter sphaeroides and Rhodopseudomonas viridis reaction centers. Proc. Natl. Acad. Sci. USA. 85:107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Breton, J., M. Bauscher, C. Berthomieu, D. L. Thibodeau, S. Andiranambinintsoa, D. Dejonghe, W. Mantele, and E. Nabedryk. 1991. Spectroscopy of Biological Molecules. The Royal Society of Chemistry, Cambridge. 43–46.
- 58.Breton, J., J. R. Burie, C. Berthomieu, G. Berger, and E. Nabedryk. 1994. The binding-sites of quinones in photosynthetic bacterial reaction centers investigated by light-induced FTIR difference spectroscopy—assignment of the Q(a) vibrations in Rhodobacter sphaeroides using O-18-labeled or C-13-labeled ubiquinone and vitamin-K-1. Biochemistry. 33:4953–4965. [DOI] [PubMed] [Google Scholar]
- 59.Breton, J., and E. Nabedryk. 1996. Protein-quinone interactions in the bacterial photosynthetic reaction center: light-induced FTIR difference spectroscopy of the quinone vibrations. Biochim. Biophys. Acta. 1275:84–90. [Google Scholar]
- 60.Breton, J., E. Nabedryk, J. P. Allen, and J. C. Williams. 1997. Electrostatic influence of Q(A) reduction on the IR vibrational mode of the 10a-ester C=O of H-A demonstrated by mutations at residues Glu L104 and Trp L100 in reaction centers from Rhodobacter sphaeroides. Biochemistry. 36:4515–4525. [DOI] [PubMed] [Google Scholar]
- 61.Hamm, P., K. A. Gray, D. Oesterhelt, R. Feick, H. Scheer, and W. Zinth. 1993. Subpicosecond emission studies of bacterial reaction centers. Biochim. Biophys. Acta. 1142:99–105. [Google Scholar]
- 62.Arlt, T., S. Schmidt, W. Kaiser, C. Lauterwasser, M. Meyer, H. Scheer, and W. Zinth. 1993. The accessory bacteriochlorophyll—a real electron carrier in primary photosynthesis. Proc. Natl. Acad. Sci. USA. 90:11757–11761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jia, Y. W., D. M. Jonas, T. H. Joo, Y. Nagasawa, M. J. Lang, and G. R. Fleming. 1995. Observation of ultrafast energy transfer from the accessory bacteriochlorophylls to the special pair in photosynthetic reaction centers. J. Phys. Chem. 99:6263–6266. [Google Scholar]
- 64.Beekman, L. M. P., R. W. Visschers, R. Monshouwer, M. Heerdawson, T. A. Mattioli, P. McGlynn, C. N. Hunter, B. Robert, I. H. M. van Stokkum, R. van Grondelle, and M. R. Jones. 1995. Time-resolved and steady-state spectroscopic analysis of membrane-bound reaction centers from Rhodobacter sphaeroides—comparisons with detergent-solubilized complexes. Biochemistry. 34:14712–14721. [DOI] [PubMed] [Google Scholar]
- 65.Robert, B., and M. Lutz. 1988. Proteic events following charge separation in the bacterial reaction center: resonance Raman spectroscopy. Biochemistry. 27:5108–5114. [Google Scholar]
- 66.Breton, J., and E. Nabedryk. 1993. S0–T1 infrared difference spectrum of the triplet-state of the primary electron-donor in Rhodobacter sphaeroides photosynthetic bacterial reaction centers. Chem. Phys. Lett. 213:571–575. [Google Scholar]
- 67.Hartwich, G., C. Geskes, H. Scheer, J. Heinze, and W. Mantele. 1995. Fourier transform infrared spectroscopy of electrogenerated anions and cations of metal-substituted bacteriochlorophyll a. J. Am. Chem. Soc. 117:7784–7790. [Google Scholar]
- 68.Breton, J., J. Burie, C. Berthomieu, D. L. Thibodeau, S. Andiranambinintsoa, D. Dejonghe, G. Berger, and E. Nabedryk. 1992. Exploring the primary electron acceptor QA. In The Photosynthetic Bacterial Reaction Center II. Structure, Spectroscopy and Dynamics. J. Breton, editor. Plenum Press, New York. 155–162.
- 69.Adolphs, J., T. Renger, F. Muh, and M. Madjet. 2007. Theory of optical spectra—how proteins control excitation energy transfer. Photosynth. Res. 91:161–162. [Google Scholar]
- 70.Brudler, R., H. J. M. de Groot, W. B. S. van Liemt, W. F. Steggerda, R. Esmeijer, P. Gast, A. J. Hoff, J. Lugtenburg, and K. Gerwert. 1994. Asymmetric binding of the 1-C=0 and 4-C=0 groups of Q(a) in Rhodobacter sphaeroides-R26 reaction centers monitored by Fourier-transform infrared-spectroscopy using site-specific isotopically labeled ubiquinone-10. EMBO J. 13:5523–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vos, M. H., M. R. Jones, C. N. Hunter, J. Breton, J. C. Lambry, and J. L. Martin. 1994. Coherent dynamics during the primary electron-transfer reaction in membrane-bound reaction centers of Rhodobacter sphaeroides. Biochemistry. 33:6750–6757. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.