

Abstract
The mechanisms regulating neutrophil transmigration of vascular endothelium are not fully elucidated, but involve neutrophil firm attachment and passage through endothelial cell–cell junctions. The goal of this study was to characterize the tangential forces exerted by neutrophils during transendothelial migration at cell–cell junctions using an in vitro laminar shear flow model in which confluent activated endothelium is grown on a microfabricated pillar substrate. The tangential forces are deduced from the measurement of pillar deflection beneath the endothelial cell–cell junction as neutrophils transmigrate. The force diagram displays an initial force increase, which coincides with neutrophil penetration into the intercellular space and formation of a gap in VE-cadherin staining. This is followed by a rapid and large increase of traction forces exerted by endothelial cells on the substrate in response to the transmigration process and the disruption of cell–cell contacts. The average maximum force exerted by an actively transmigrating neutrophil is three times higher than the force generated by an adherent neutrophil that does not transmigrate. Furthermore, we show that substrate rigidity can modify the mechanical forces induced by the transmigration of a neutrophil through the endothelium. Our data suggest that the force induced by neutrophil transmigration plays a key role in the disruption of endothelial adherens junctions.
INTRODUCTION
Neutrophil transmigration through the endothelial cell monolayer is a key process in the inflammatory response. It is a multistep cascade consisting of three overlapping steps: neutrophil initial attachment and rolling on the vascular endothelium surface, arrest and crawling (migrating) of the neutrophils to the cell–cell junctions, and transmigration itself (diapedesis) through the junction composed of the VE-cadherin complex and other proteins localized to the junctions, including PECAM-1, CD99, and JAMs (1). In addition, VE-cadherin, which localizes at cell–cell adherens junctions and forms a complex with cytosolic α-, β-, and γ-catenins, is also thought to regulate transmigration (1–4). By live-cell fluorescence imaging of endothelial cell junctions during neutrophil transmigration, previous studies have shown that leukocytes migrate within minutes through the intercellular junctions by transiently disrupting the VE-cadherin complex (2,5). The mechanism underlying rapid diapedesis and gap formation in VE-cadherin remains unclear. A number of hypotheses have been proposed. First, leukocytes induce transient disruption and disassociation of junctional complexes, such as VE-cadherin, to gain passage (1,3,6), or leukocytes actively seek areas of small preexisting gaps in VE-cadherin that expand during diapedesis (3,6,7). Alternatively, the neutrophil itself could exert sufficient force on the cell–cell junction to rupture the VE-cadherin complex and gain passage (1).
There is a paucity of data on the forces that are generated during leukocyte diapedesis (8), due perhaps to the complexity of the process as well as the lack of appropriate tools (9). In particular, previous studies have evaluated forces during neutrophil attachment to immobilized endothelial adhesion molecules (10,11). The force that drives neutrophil motility comes primarily from two sources: 1), actin polymerization at the leading edge (12); and 2), actin-myosin complexes and RhoA activity during the uropod retraction (13,14). Previous studies analyzed the forces generated by leukocytes crawling on glass, and reported forces reaching 10.7 nN (15). A more recent study that focused on the migration of neutrophils found significantly larger forces, up to 67 nN, generated during chemotaxis on the surface of polyacrylamide gel (16). Both experiments were conducted on artificial surfaces and not on the endothelial cell monolayer. These forces are generated during crawling and not transendothelial migration (TEM). Mechanical forces may also play a role in the disruption of intercellular junctions as a neutrophil penetrates the intercellular junctions, which primarily consists of VE-cadherin dimers, and squeezes between the endothelial cells. However, the force exerted by a neutrophil during diapedesis at cell–cell junctions has not yet been determined. Furthermore, the mechanical properties of leukocytes, and neutrophils in particular, have been studied with various techniques (17,18) and play a key role in determining their function in response to a specific environment (19). For instance, leukocytes have been observed to be activated and modify their mechanical properties when confined or forced to enter into microchannels (20,21). Additionally, the stiffness of leukocytes could also regulate their adhesion to the endothelium in larger blood vessels by modulating the contact area and thus the mechanical forces between leukocytes and the endothelium (22,23).
Here, we have analyzed the mechanical forces induced by the transmigration of human neutrophils through a human endothelial cell monolayer. Several techniques have been developed to measure the traction forces generated by cells and the biochemical activities associated with them (24–27). To study the relative forces generated in endothelium during neutrophil transmigration, and to characterize their potential role in this process, we used an in vitro model consisting of a surface composed of an array of flexible micropillars (Fig. 1). The micropillar system is composed of a high density of flexible poly(dimethylsiloxane) (PDMS) micropillars and has already been used in various experiments to obtain force distributions in isolated or assemblies of cells (28–30). Endothelial cells were cultured at confluence onto the micropillars, and neutrophil transendothelial cell migration was monitored by live-cell imaging under shear flow conditions that mimic blood flow in microvessels (31). By analyzing the deflection of the micropillars, we characterized the tension exerted by the endothelial cells on the substrate, and thus deduced the relative force due to the penetration of adherent neutrophils through the endothelium. In particular, the initiation of the transmigration process corresponds to a sudden and large increase of the traction forces.
FIGURE 1.

Schematic representation of the neutrophil transmigration on a micropillar substrate. HUVEC are plated on the micropillar substrate and inserted into an in vitro flow chamber that simulates blood flow. The different steps of interactions between neutrophils and endothelium are shown: (a) attachment, (b) rolling, (c) firm adhesion, and (d) migration toward endothelial junctions and transmigration. The majority of neutrophils attach to the cell surface at cell–cell junctions. Neutrophils that transmigrate induce a change of the force profile on the pillars as they pass between the intercellular junctions and then migrate beneath the monolayer.
Finally, we studied the effect of the substrate rigidity on this process. It has been shown that the compliance of the matrix strongly influences several cellular functions, including migration, adhesion, and even differentiation (32–34). Here, the rigidity of the substrate could play an important role by eventually modifying several key parameters such as cell stiffness, cell spreading, or traction forces during the interactions between the neutrophil and endothelial cells. By changing the spring constant of the micropillars (30,35), we indeed demonstrated that the substrate stiffness could play an important role in the transmigration process.
MATERIALS AND METHODS
Materials
PDMS (Sylgard 184; Dow Corning, Midland, MI) was purchased from Thermo-Fisher Scientific (Medford, MA). Monoclonal antibody (mAb) Hec-1 (murine anti-human VE-cadherin, IgG1 (36) was conjugated to Alexa 558 (Molecular Probes, Eugene, OR) following the manufacturer's directions. Alexa Fluor 488-tagged goat anti-mouse mAb (Molecular Probes) was used as a nonspecific marker to visualize the top surface of micropillars.
Fabrication of the PDMS micropillar substrate
PDMS micropillar substrates were prepared according to du Roure et al. (29). Briefly, using conventional photolithography followed by a deep etching process (the “Bosch process”), silicon wafers were patterned with an array of cylindrical pits and the desired pattern was replicated in positive photoresist by photolithography. Bare parts of the wafers were then etched by the deep Si etching process down to the desired depth to obtain the negative pattern of the array. A drop of liquid PDMS was placed on the silicon template between two coverslips (thickness No. 0, 0.08–0.13 mm) and covered by a third coverslip (thickness No. 2, 0.22 mm) to obtain a thin microchip. This material was cured at 65°C for 12 h and then removed in 70% ethanol to prevent collapse of the micropillars. The ethanol was then gradually replaced by phosphate-buffered saline (PBS). In this study, 2-μm-diameter pillars of the same area-to-pillar density (22%; ratio of the post surface to the total surface) were used for all experiments, and the pillars were set at a center-to-center distance of 4 μm. Rigidity was changed by increasing the pillar height from 3.3 μm to 4.7 μm.
Fibronectin coating of the PDMS surfaces
The silicone micropillar substrate was coated with fibronectin and an anti-mouse Alexa Fluor 488 fluorescent antibody that binds nonspecifically and allowed visualization of the pillar tops: 40 μL of 1 mg/mL fibronectin in Dulbecco's phosphate buffered saline and 1 μL of Alexa488-labeled murine IgG (1 mg/mL) were deposited on the thin PDMS film, gently pressed against the entire surface with a coverslip, and incubated for 1 h at room temperature. The substrate was then rinsed with PBS and pressed against the micropillar surface in the liquid phase and incubated for 20 min. To avoid nonspecific protein adsorption on the sides of the micropillars and thus prevent cell adhesion, micropillar surfaces were saturated by immersion in PBS buffer containing 3% BSA, 0.1% Pluronics F127 (Sigma) for 1 h and then rinsed with PBS as previously described (28,37).
Endothelial and neutrophils cell culture
Human umbilical vein endothelial cells (HUVEC) were isolated and cultured as previously described (3). The HUVEC (subculture 1) were detached from the culture dish with 3 mM EGTA trypsin-free solution, washed, and placed on the micropillar substrate in three steps at 5-min intervals. This protocol resulted in a confluent monolayer as judged by robust staining of VE-cadherin at cell–cell junctions and differential interference contrast (DIC) microscopy. Confluent HUVEC on micropillar substrates were activated with 25 ng/mL tumor necrosis factor-alpha (TNF-α) for 3 h to stimulate the endothelium for more effective neutrophil transmigration as previously reported (38). In some experiments HUVEC were treated with conjugated VE-cadherin antibody 30 min before the transmigration experiment to visualize the cell–cell junctions during TEM by live-cell imaging as previously described (39). Human neutrophils (95% pure) were isolated from whole blood obtained from healthy volunteers donors by venous puncture as previously described (6). The human use committee of the Brigham and Women's Hospital approved all protocols involving the use of human subjects, and signed donor consent was obtained from all blood donors.
Endothelial cell flow transmigration assays
Coverslips containing a confluent monolayer of HUVEC on the micropillar surface were mounted into a parallel plate flow chamber as previously described (31). Freshly isolated polymorphonuclear leukocytes (PMN) were resuspended at 0.5 × 106 cells/mL and drawn across HUVEC monolayers in a parallel plate flow chamber at 0.7 dynes/cm2 as previously described (3). Neutrophils were allowed to interact with the monolayer, and transmigrated neutrophils were distinguished from those interacting with the apical surface (nontransmigrated) by live-cell imaging, as described below.
Live-cell imaging
Time-lapse, live-cell epifluorescence microscopy experiments were performed on an inverted Nikon TE-2000 microscope (equipped with 40X objective and a heating stage maintaining the temperature of 37°C) coupled to a Hamamatsu digital camera. We used DIC optics to visualize neutrophil transmigration; however, we had to observe cells through a layer of PDMS, which decreased the quality of the images. Images were recorded with MetaMorph software every 5 s for 10 min for every experiment.
Image analysis and calculation of traction forces
To quantify the area of neutrophils after transmigration, we used Image J tools to outline the borders of the neutrophils and calculated the area inside. We measured the local deformation of the pillars by using in-house-made multiparticle tracking software (29). Briefly, by fluorescently labeling only the top of the pillars, we obtained a high contrast between the top of the pillars and the background. We then used the fluorescent signals coming from each pillar to track their center of mass over time. For all experiments the drift (mechanically or thermally induced) of the microscope stage was determined and accounted for automatically. The time resolution, corresponding to the calculation of the overall stress pattern for one image, is <1 s. The resolution of the pillar displacement is on the order of 25 nm. To calculate the local tangential force, the deflection of the posts is multiplied by the spring constant according to the formula: 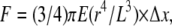 where E is the Young's modulus, r is the pillar radius, L is its length, Δx is the displacement from equilibrium, and F is the force. Depending on the spring constant, the force resolution varies. To measure the spring constant, we used dimensionally calibrated macroscopic cylinders of PDMS and measured their compression under a fixed normal strain to evaluate the Young's modulus, E. Because E depends on the PDMS cure time, we used a consistent cure time of 12 h ± 2 h at 65°C corresponding to a Young's modulus of 2 ± 0.2 MPa. By performing SEM observations, we measured the dimensions of the pillars (r, L) and calculated their spring constant according to the force-deformation equation.
where E is the Young's modulus, r is the pillar radius, L is its length, Δx is the displacement from equilibrium, and F is the force. Depending on the spring constant, the force resolution varies. To measure the spring constant, we used dimensionally calibrated macroscopic cylinders of PDMS and measured their compression under a fixed normal strain to evaluate the Young's modulus, E. Because E depends on the PDMS cure time, we used a consistent cure time of 12 h ± 2 h at 65°C corresponding to a Young's modulus of 2 ± 0.2 MPa. By performing SEM observations, we measured the dimensions of the pillars (r, L) and calculated their spring constant according to the force-deformation equation.
Confocal microscopy and image analysis
Confocal microscopy was performed on samples fixed by 4% paraformaldehyde for 10 min and then mounted with mounting medium. We used a Zeiss LSM 510 confocal microscope with a 63X oil-immersion lens. Images were acquired in three colors with a 0.2-μm step. After acquisition by Zeiss software (Thornwood, NY), the images were analyzed using Image J.
RESULTS
Characterization of the traction forces induced by the endothelial cell monolayer
HUVEC were cultured on substrates composed of micropillars as detailed in Materials and Methods. We used a specific coating of the micropillars to restrict endothelial cell adhesion to the tips of the posts (see Materials and Methods section). Using microcontact printing, we delivered fibronectin from a stamp onto the tips of the posts, and adsorbed Pluronics onto the remaining unstamped regions of the array to block nonspecific protein adsorption and cell adhesion as previously described (37). This procedure results in confluent monolayers that exhibit robust staining by anti-VE-cadherin mAb (Fig. 2). VE-cadherin is a key component of adherens junctions, and its robust staining at cell–cell junctions indicated that the cells had established functional intercellular complexes. We analyzed forces generated by endothelial cells plated at different densities to obtain the range of forces and thus the mechanical background induced by those cells in the absence of neutrophils. All experiments and controls were conducted on 3-h TNF-α-activated endothelial cells. TNF activation is necessary for the transmigration to occur. First, endothelial cells were grown to confluence and the deflections of the underlying micropillars were measured. The force per pillar, averaged over 14,000 micropillars, generated by a confluent HUVEC monolayer is 1.5 ± 0.3 nN on micropillars with a 45 nN/μm-spring constant (Fig. 3). We calculated the force exerted by the endothelial cells on a per-pillar basis instead of an averaged per-cell force because the pillar density was unchanged. Then, the average force we calculated for individual human endothelial cells was 3.2 ± 0.6 nN (n = 15 cells with 7000 pillars analyzed), which is significantly larger than that for a monolayer. These results are in good agreement with our previous studies on epithelial cells and, in particular, the range of forces is similar (29,30). It is interesting that in the case of a monolayer, we obtained an average force in the endothelial junction area that was higher (5 ± 1 nN, n = 20 studies with 3000 pillars analyzed) than the mean force calculated for all the underlying pillars (1.5 ± 0.3 nN). Forces in the area of cell–cell junctions were calculated by averaging over pillars selected on each side of the intercellular junction. Basically, a two-pillar-wide band was selected around VE-cadherin positive cell–cell edges. The main part of the mechanical activity of an endothelial monolayer transmitted to the substrate was located at the cell–cell junctions. Fig. 3 shows that the histogram of the force distribution for a confluent monolayer exhibits an exponential decay. Forces below 6 nN correspond to 96% of events (Fig. 3). Few events in the area of high forces correspond to the cells generating an abnormal pattern of force. Because these cells could be abnormally adherent cells, we did not analyze any transmigration events on them. The force pattern within confluent monolayers has an important implication for transmigration experiments. In particular, the force generated by HUVECs in the area of cell–cell contacts (5 ± 1 nN) represents the force baseline against which the neutrophil transmigration force will be compared.
FIGURE 2.

HUVEC form a confluent monolayer with VE-cadherin present at cell–cell junctions. (A) DIC image of a confluent HUVEC monolayer cultured on a micropillar substrate. (B) Live-cell image of VE-cadherin staining of the endothelial junctions. (C) Merge of VE-cadherin (red channel) and DIC (green channel) images that identify intercellular junctions (red). The lack of clear focus in the red signal is due to the scattering of light by the PDMS body of the micropillars. Scale bar is 10 μm.
FIGURE 3.

Distribution of forces generated by the HUVEC monolayer on the micropillar substrate. We analyzed 14,000 pillars in three different experiments beneath 30 endothelial cells and plotted a distribution diagram.
Neutrophil transmigration triggers VE-cadherin disruption and micropillar displacement
First, we compared the kinetics of the transmigration process of neutrophils through endothelial cells on micropillar substrates versus flat substrates using the same fibronectin coating. We verified that cellular interactions, including neutrophil attachment, kinetics, and number of transmigration events, were not affected by the array of closely spaced pillars (Figs. 4 and 5). Indeed, neutrophils bound at or close to endothelial cell–cell junctions with 78 ± 8% initially attached within one neutrophil cell diameter from the VE-cadherin-labeled junctions, in a manner similar to that on glass (2). About 40% of the neutrophils that attached to the surface of the endothelial cell ultimately transmigrated. The length of time required for a transmigration event was 85 ± 20 s based on 35 cells examined in detail, which was similar to the time frame obtained with TNF-α HUVEC cultured on fibronectin-coated glass coverslips (Fig. 4; (2)). Finally, most of the transmigration events occurred in the area of contact among three cells (tricellular junctions), which was consistent with previous reports using endothelium plated on glass coverslips and laminar shear flow conditions (2,7,40). Thus it appears that neutrophil transmigration processes on a flat surface and on a micropillar surface are similar and go through the same sequence of steps.
FIGURE 4.

Neutrophil transmigration across TNF-activated HUVEC cultured on glass coverslips. The yellow arrow indicates a transmigrating neutrophil near a tricellular junction. The DIC panel shows that the neutrophil penetrates at cell–cell junctions and spreads underneath the endothelial cell body. The VE-Cad panel shows that VE-cadherin junctional staining is transiently disrupted during transmigration and then reseals after transmigration is complete. The merge of the DIC and VE-cad (merge panel) shows the relative position of a transmigrating neutrophil and the intercellular junctions rupture area, and clearly indicates that junction rupture occurs exactly in the place of transmigration.
FIGURE 5.

DIC and two-color fluorescent imaging of the endothelial monolayer during transmigration. (a) Micrographs of images. A-E are DIC images of the process of neutrophil transmigration: baseline, no neutrophil (A), a neutrophil arrests (B), extending lamellipodium (C), transmigration (D), and crawling beneath the monolayer (E). (b) Micrographs of images F-J depict the VE-cadherin junction labeling of an intact junction (F, G), the initial rupture by neutrophil (H), maximal gap in VE-cadherin (I), and resealing of VE-cadherin (J). (c) (K–O) micropillar surface with no displacement (K and L), low-force stage (M), radial displacement (N), and relaxation (O). (d) Merged images P–T represent overlays of the VE-cadherin junctional staining (green) and micropillars image (red). The forces exerted on the pillars are represented by red lines originating from the centers of the pillars. A nonmigrating neutrophil to the left of the circle is indicated by a red arrow in panel a. Scale: diameter of pillar is 2 μm, yellow bar is 10 nN.
We performed an analysis of neutrophil transmigration by acquiring images of the HUVEC and neutrophils with DIC microscopy and simultaneously captured sequential, two-color fluorescence of micropillars (Fig. 5, green) and VE-cadherin (Fig. 5, red) (essentially three-color imaging; see also Movie S1a, Movie S1b, and Data S1 in Supplementary Material). We first established the baseline position of micropillars (Fig. 5, a and c, control) before neutrophil attachment. DIC microscopy (Fig. 5 a) depicts the behavior of a neutrophil that adheres at a tricellular junction at time = 0 s (Fig. 5 a, panel B), transmigrates (panels C and D), and accumulates beneath the endothelial monolayer (Fig. 5 a, panel E; Movie S2 and Data S1). A blue circle outlines the neutrophil transmigration area. We also observed neutrophils that stably adhered but did not transmigrate, as shown in Fig. 5 a (red arrow). This can be deduced by the lack of disruption of the junctional staining of VE-cadherin as shown on Fig. 5 b. Only neutrophils that successfully transmigrate trigger transient disruption, followed by resealing of VE-cadherin staining (Fig. 5 b, panels F–J in the circle). Strikingly, during this same time period, we detected a sudden transient and reversible displacement of micropillars located directly beneath the endothelium at the site of transmigration (Fig. 5, c and d, panels N and S). The yellow arrows in panel S illustrate the vector force applied to corresponding pillars. The initial increase in pillar displacement correlated with loss of VE-cadherin staining and neutrophil migration into intercellular space (Fig. 5, panels H, M, and R).
The process of neutrophil penetration in between the junction was previously detailed in electron microscopy observations by Burns et al. (7) and our own DIC microscopy results (2) (Fig. 5). The maximum increase in pillar displacement (Fig. 5, panels I, N, and S) corresponded to a large increase of the disruption of cell–cell contacts. Based on the DIC microscopy live-cell imaging (Fig. 5), we also observed that the position of the neutrophil, relative to the monolayer and the top of the pillars, varied with time. The modification of the force pattern induced by endothelial cells on the substrates thus correlates with the bulk of the neutrophil passing through the cell–cell junctions. After completing transmigration, the neutrophil, located in the top left area of the blue circle in Fig. 5, causes significantly less change in micropillar displacement at the original area of transmigration. Finally, transmigrated neutrophils moved randomly beneath the endothelial monolayer. After transmigration, neutrophils continued to migrate away and induced forces on the substrate (Movie S3 and Data S1); however, the force measurements were difficult to interpret because they were due to the superposition of forces exerted by endothelial cells and neutrophils located beneath the monolayer.
Using standard optical microscopy, we could not verify the precise location of neutrophils after the transmigration. To verify the precise location of the neutrophils after transmigration, we used confocal microscopy. We analyzed 30 independent transmigration events and observed neutrophils that penetrated the endothelial monolayer and reached the bottom of the micropillar substrate (Fig. 6 A). In Fig. 6 B, the labeling of intercellular connections with a conjugated anti-VE-cadherin antibody shows that cell–cell junctions are visible above the micropillar tops. The fluorescent signal observed on some micropillars was due to nonspecific binding of the antibody on PDMS substrate.
FIGURE 6.

Confocal images of neutrophil transmigration. (A) Z-projection of a neutrophil transmigrating endothelial cells (neutrophil is shown in red, pillars in green). (B) VE-cadherin staining with a conjugated anti-VE-cadherin antibody averaged over 10 slices, shown in red. The length of the pillar was ∼5 μm for this experiment.
Neutrophil transmigration force profile
We analyzed 30 complete transmigration events using the imaging protocol described above and found that in most cases, six pillars consistently showed a distinct radial displacement pattern on both sides of the endothelial junction and had the largest absolute change in displacement. This number corresponded to an area of 10 × 6 μm (Fig. 7), which was in between the area occupied by a nonspread neutrophil ∼40 μm2 and that of a spread neutrophil ∼120 μm2 on glass (41). We also estimated the average area occupied by neutrophils after transmigration on the pillars and obtained 96 ± 12 μm2 for 30 neutrophils analyzed (see Materials and Methods). At the same time, the actual size of the gap observed in VE-cadherin staining was on average ∼4 μm. Thus, it appeared that six pillars were significantly displaced during neutrophil transmigration through a narrow gap in intercellular junctions and then when a neutrophil spread underneath the monolayer on a similar area as on flat surfaces. Therefore, in all of the experiments, only the six pillars directly beneath the transmigration site were monitored and analyzed for consistency of calculation of an average force.
FIGURE 7.

Force profile and leukocyte transmigration. (A) Cells before neutrophil transmigration. (B) Initiation of transmigration. (C) Peak force during transmigration. The length of the yellow line represents the force in the corresponding direction, with white bar = 10 nN. (D) The force profile during neutrophil transmigration is shown with (▴), and the force profile induced by a neutrophil that does not transmigrate on Fig. 5 is shown (•).
Fig. 7, A–C show the change in the displacement of the six pillars directly underneath a transmigrating neutrophil for one representative transmigration event, and Fig. 7 D depicts the plot of the elapsed time versus the average force generated on these six pillars (the triangles identify the transmigrating neutrophil). Notably, this timescale of the complete process was similar to the one obtained for neutrophil transmigration of endothelium cultured on fibronectin-coated glass coverslips (2). The time point labeled A identifies the average force per pillar (4 nN) in a resting monolayer before the neutrophil arrests (i.e., baseline force level). This force at the cell–cell contact area is randomly oriented and is comparable to the average force generated by the monolayer as noted above, reaching 5 ± 1 nN. Point B represents the time point when the dissociation of the junctions begins as a result of the neutrophil penetrating the junction, as detected by VE-cadherin staining and DIC observation. The increase of the force (Fig. 7, panel B) is correlated with an increase in the radial displacement of the pillars. We suggest that this event is caused by an initial increase of the forces exerted by endothelial cells due to their interaction with the neutrophil. Point C indicates the time of maximal strain and coincides with the neutrophil actively passing between adjacent endothelial cells. For this experiment done on a micropillar substrate with a 45 nN/μm spring constant, we observed an increase in the average force from 4 to 13 nN per pillar, and from 24 to 78 nN per neutrophil. Based on 30 experiments, the average maximum force increased from 4.8 ± 1 to 14 ± 4 nN per pillar.
Careful analysis of the DIC and fluorescent images in combination with a force diagram enabled us to understand the mechanics of neutrophil penetration of the endothelial cell–cell junctions during transmigration. The initial intercellular junction disruption, as detected by the gap in VE-cadherin staining, was a relatively low-force process (Fig. 7 D, phase B). However, frame-by-frame analysis of the DIC and two-color fluorescence images revealed that the force increased at the same time as the neutrophil started to penetrate into the intercellular space and further VE-cadherin junction dissolution was observed (Fig. 5 b–d, panels H and I, M and N, R and S). Only when the bulk of the neutrophil penetrated the cell junction of the HUVEC were large traction forces detected (Fig. 7 D, phase C). Subsequently, it appeared that the neutrophil literally pushed the endothelial cells apart for further penetration. This phenomenon was confirmed by the radial distribution of forces pointing away from the center of penetration (Fig. 5 d). Indeed, the direction of the forces confirms that the transmigration of the neutrophil induces an increase of the forces exerted by endothelial cells on the substrate in correlation with the disruption of cell–cell junctions.
After the neutrophil completely penetrated the junction, we observed its spreading underneath the endothelial cells and a continuous local change of the force profile as it migrated under the monolayer (see Movie S3). Moreover, as the neutrophil left the area of penetration at the end of the transmigration process, we observed a local resealing of the junction and the force profile recovered its original baseline value (Figs. 5 c and 7 D).
Fate of nonmigrating neutrophils proves that neutrophils have an active role in force generation
We analyzed the force generated by neutrophils that attached to the endothelial cell surface, migrated on the apical surface to the junctions, and attempted to penetrate intercellular junctions but never did. The fate of one such neutrophil is shown in Fig. 5 A–E and identified by the red arrow. Although this neutrophil remained attached under flow conditions, there was no visible disruption of the VE-cadherin junction staining or pillar displacement in the corresponding area (Fig. 5 b, panels F–J, and Fig. 5 c for VE-cadherin staining, and panels K–O for pillar displacement). The force diagram analysis of the interactions of such neutrophils with the endothelial monolayer revealed a fluctuating pattern (Fig. 7 D, circles) in contrast to a clear-cut elevation in the force diagram for transmigration events (Fig. 7 D, triangles). Fluctuations of the force diagram may be caused by the neutrophil's attempts to project lamellipodia in the intercellular space, but we could not confirm this phenomenon because of the limitation of our optical resolution. Thus, we observed a net increase of the traction forces only when a neutrophil migrated between the cells and disrupted VE-cadherin staining.
A separate subgroup of events was represented by neutrophils that did rupture the VE-cadherin junctions but failed to transmigrate (Fig. 8). In these cases, significant forces were observed, reaching values of 6 ± 1 nN per pillar on average. This was slightly above the noise level, but could be clearly detected by the change in the distribution of force vectors from random to radial. The force exerted was still two times lower than the forces induced during a full transmigration event. It is interesting that these force values are comparable to that observed during the initial phase of a complete transmigration event (Fig. 7 D, point A). However, such neutrophils failed to complete the transmigration.
FIGURE 8.

Nonmigrating neutrophil force diagram. Some of the neutrophils project lamellipodium that rupture the intercellular junctions but then fail to transmigrate. A shows a neutrophil before an attempt to transmigrate, and B and C are force distributions during the transmigration attempt. D–F show corresponding VE-cadherin junctional staining with clear rupture on E and F. G represents the average force per pillar corresponding to A–C. Maximal average force reaches 6.3 nN per pillar and is significantly less than that observed in the events of full transmigration.
Neutrophil transmigration on substrates with different stiffness values
Finally, we addressed the possibility that the rigidity of the substrate can affect the transmigration process. By changing the length of the micropillars, one can vary their spring constant. Therefore, we studied the interaction between neutrophils and endothelial cells on substrates with the same density of micropillars but with two different rigidities: 45 nN/μm pillars and 131 nN/μm pillars (Fig. 9). In each experiment we analyzed the six pillars directly underneath the location where a neutrophil transmigrated (i.e., the penetration site). We analyzed 10 independent events for each substrate. We observed that the transmigration process exhibits a rigidity-dependent force generation mechanism because the force induced by the transmigration is higher on the more-rigid substrate (39 ± 6 nN) than on the soft one (14 ± 4 nN). The force exerted on the pillars at the peak phase of the transmigration (i.e., maximum force) is threefold greater than the baseline for each substrate rigidity and reaches 250 ± 42 nN per neutrophil for the rigid pillars compared to 84 ± 24 nN for endothelial cells alone.
FIGURE 9.

Force profile of transmigration on micropillars substrates with different rigidities. Average force per pillar recorded underneath the neutrophil transmigration site. (Light gray) 45 nN/μm pillars; (dark gray) 131 nN/μm pillars. Six pillars were analyzed in each experiment, and 10 experiments were conducted for each rigidity. p < 0.05 for before transmigration versus peak force for both stiffnesses.
DISCUSSION
In this study we investigated the role of the mechanical interactions that occur between neutrophils and endothelial cells during the transmigration process. By combining the use of microfabricated substrates and an in vitro human cell culture assay, we analyzed the different steps of the TEM in terms of mechanical traction forces. We performed experiments in a well-characterized in vitro flow adhesion model that mimics physiological laminar shear flow conditions (2,3,38), and cultured cells on a substrate made of a closely spaced array of flexible micropillars. To our knowledge, this is the first study to provide quantitative results regarding the forces transmitted by neutrophils to endothelial cells during transmigration, and to address their implication in the disruption of cell–cell junctions.
First, the analysis of the traction forces generated by endothelial cells in the absence of neutrophils was necessary to measure the force deviation induced by the transmigration. It is interesting that the results revealed that the mechanical activity within a confluent monolayer was localized mainly at cell–cell junctions. The average force exerted on the substrate and located under cell–cell junctions was 5 nN, which was more than three times larger than the force averaged over the entire monolayer (∼1.5 nN). These data were consistent with our previous observations on epithelial cells (29). This indicates that cell–cell junctions should play an important role in the mechanical integrity of an endothelial monolayer, in particular by regulating the cell–substrate interactions. Hence, the low basal value of the forces that were generated by the endothelial cells allowed us to detect changes in the force profile at the intercellular junctions during transmigration.
Neutrophils transmigrated normally through our hybrid HUVEC-micropillar flow system model and followed the well-described multistep adhesion cascade paradigm (1,2). The analysis of the neutrophil diapedesis force profile revealed a precise sequence of events. The first events corresponded to the adhesion of neutrophils onto the endothelial cells, followed by neutrophil penetration at the intercellular space. It is interesting that a low force was measured during this event despite the fact that this step correlated with the disruption or the formation of opening gaps at intercellular junctions as detected by VE-cadherin staining. Further force increase correlated with the size of the junction disruption (Fig. 5 b) as the neutrophil migrated between the endothelial cells. We observed a clear radial force distribution induced by the transmigration on the underlying pillars that correlated with the location of the neutrophil body. The maximal force, which corresponded to the displacements within the estimated area of interaction between the neutrophil and the monolayer, could reach up to 250 nN per neutrophil. The overall time frame for this process was ∼60 s as measured from the force increase initiation to the peak force. After that, the neutrophil spread underneath the endothelial cell surface and the force profile became randomly distributed at the original area of diapedesis. The last step correlated with the resealing of VE-cadherin within the junction (see Fig. 5 and Movie S1a, Movie S1b, and Data S1 for the detailed process).
Thus, the force profile in the area of transmigration appears to be a good indicator of the state of the diapedesis process. Forces were initially distributed randomly in the zone of neutrophil penetration as the endothelial cells in the monolayer acted in synergy and did not exert large differential forces in the cell junction area. When the bulk of the migrating neutrophil reached the intercellular space, the force profile changed dramatically: the magnitude of forces increased and their direction exhibited a radial pattern (Fig. 7). This radial orientation clearly indicated that the forces were due to endothelial cells and the rupture of cell–cell junctions in response to the neutrophil transmigration.
To exclude the possibility that the force we observed was due to the induction of endothelial contraction in response to neutrophil adhesion, and not to the neutrophil diapedesis itself, we studied the fate of neutrophils that formed strong adhesions to the intercellular space area but did not penetrate the junctions. These neutrophils did not rupture the junctions, as deduced by the lack of change in VE-cadherin staining, and did not induce a force increase. These data support our conclusion that the increase in tensile forces measured under the endothelial cells is most likely caused by an extension of a neutrophil lamellipodia into the junction, as shown in confocal analysis (Fig. 6) and by previous reports (2). In addition, the analysis of the force diagram after the transmigration revealed a continuous deformation of the substrate as neutrophils migrated beneath the monolayer and interacted with both the basolateral surface of endothelium and the pillars. Another important piece of evidence in support of an active role of neutrophils in force generation was shown by the force diagram obtained from the adhesion of neutrophils on the endothelial monolayer that ruptured VE-cadherin junctions between the cells but did not transmigrate. A small dissociation of VE-cadherin junctions was observed, and was comparable to that observed in the first steps of a complete transmigration event. Consequently, the resulting force is just 1–2 nN per pillar above the force generated by endothelial cells without neutrophils. Here, we established that the forces generated during diapedesis proceeded in at least two distinct mechanical phases: first, a low-force dependent mechanism of around 6 ± 1 nN that involves an initial increase in the opening of the junction, and second, a high-force phase (∼13 nN) in which the bulk of the neutrophil's body penetrates, causing a large dissociation of cell–cell junctions concomitant with a transient loss of VE-cadherin staining. These forces respectively correspond to ∼1-2 and ∼7 nN per pillar above the force baseline defined by the forces exerted by endothelial cells in the absence of neutrophils. This sequential process involves complex events that regulate interactions between the mechanical and adhesive properties of both neutrophils and endothelial cells. In particular, previous studies have shown that transendothelial neutrophil migration is regulated by endothelial cell-dependent cytoskeletal mechanisms, and a variety of intracellular signaling steps contribute to this process (39,43,44). Our findings illustrate that transmigration may be governed by a combination of different force-generation mechanisms, and thus the increase of the forces probably requires a remodeling of the actin cytoskeleton and actin-myosin contractile machinery in both neutrophils and endothelial cells. In particular, one would expect that transmigration induces a reinforcement of the adhesion of endothelial cells onto the substrate and the actin cytoskeleton to help the neutrophils migrate across the monolayer. Such a cytoskeleton reorganization of endothelial cells could also play a role at the end of the process to allow the penetration of the neutrophil body through the junctions. The increase of the traction forces exerted by endothelial cells during transmigration clearly demonstrates that an active process is involved in locally remodeling the adhesive contacts on fibronectin-coated pillars, as previously shown in other cellular systems (45). Furthermore, transient disruption of VE-cadherin–mediated cell–cell adhesion is also an important event in the process of neutrophil transmigration (2,3,46). In agreement with previous studies (47), our results suggest that a “tug of war” may occur between cell–cell and cell–substrate adhesions, and the structure that is able to develop the most tension on its ligand in response to neutrophil adhesion “wins.” However, further experiments need to be done to directly correlate the remodeling of the different adhesions and the corresponding mechanical forces.
Furthermore, our interpretation is supported by the fact that neutrophils can generate higher forces during transmigration of the endothelium cultured on stiffer micropillar substrates. The average maximal force, calculated as an average of the sum of six moduli of force vectors induced on pillars under the transmigration site at peak force, increases from 78 to ∼250 nN as the substrate rigidity varies from 45 up to 131 nN/μm. This change corresponds to a change from 0.026 pJ to 0.08 pJ in terms of energy spent, calculated as a sum of energies stored in deflected pillars, with a total increase of 0.054 pJ per neutrophil on rigid substrate compared to soft substrate. The increase of the energy barrier with the stiffness of the substrate could reflect that the neutrophil transmigration affects the mechanical interactions of endothelial cells. Indeed, many cell types respond to an increase of substrate rigidity by forming well-defined and stable focal adhesions and thus remodeling their cytoskeleton (48). Again, our findings point out that the observed neutrophil migration could be associated with endothelial actin polymerization and myosin II reorganization, as suggested by previous studies (43,49). The force increase with the substrate stiffness could be attributed to a higher resistance of endothelial cells to the cell transmigration on rigid substrates due to an increase of cytoskeleton contractility and stronger adhesion complexes between endothelial cell and pillars. Along the same line, it is of interest to compare our results with the calculated force necessary to break the bonds of VE-cadherin at junctions. From our observations, the average size of the VE-cadherin gap is ∼4 μm at maximal force (2), and we estimate from the literature that the VE-cadherin concentration in the intercellular space is ∼5 × 103 dimers/μm2 (50), assuming that the height of the intercellular connection is 1 μm and a 50 pN for the unbinding force (51). This leads to an estimated force of around ∼1 μN for simultaneous unbinding of the cadherin-cadherin bonds. Our measurements on substrates with different rigidities yielded lower values but in the same range of amplitudes, from 0.08 up to 0.25 μN. These differences could be explained by the fact that the adhesions at the junctions are not broken at the same time, but certainly in a sequential process. The source of the process that induces a dissociation of cell–cell junctions remains unclear. It could be due to actin polymerization at the front edge of the neutrophil in conjunction with biochemical interactions between the neutrophil and endothelial cells that trigger phosphorylation of the cytoplasmic tail of VE-cadherin (46). This process could affect signaling pathways that govern cell–cell adhesive interactions.
In summary, we measured the tangential force exerted by the interaction of neutrophils with a confluent TNF-α-activated endothelial cell monolayer. We demonstrated that neutrophil transmigration through the intercellular junctions is a multistep process with a clear mechanical signature. Our assay, which combines an in vitro flow adhesion model that mimics physiological shear flow conditions and a micro-force sensor array, enabled us to characterize and quantify the mechanical interactions between neutrophils and endothelial cells that occur during diapedesis. Finally, by changing the substrate rigidity, we obtained indirect evidence that the modulation of cytoskeleton contractility changes the transmigration force profile. This suggests that competition between cell–substrate adhesions and cell–cell junctions plays a key role in determining whether neutrophils successfully transmigrate through an endothelial cell monolayer.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
The authors thank A. Buguin, A. Saez, and P. Silberzan for their help in microfabrication and force measurements; Prof. C. Forbes Dewey and J.P. Urbanski from Hatsopoulos Microfluids Laboratory at MIT for providing equipment and laboratory space to prepare the PDMS microchips; Dr. Gail Newton, Brigham and Women's Hospital, for providing technical assistance for the experiments; and Kay Case, Vanessa Davis, and Deanna Lamont for providing well-characterized HUVEC.
This work was supported by the Journal of Cell Science Traveling Fellowship Award 2006 (A.R.), Ligue contre le Cancer (B.L.), Programme Nanosciences et Nanotechnologies/l'Agence Nationale de la Recherche (ANR) (B.L.), and National Institutes of Health grants HL36028 and HL53993 (F.W.L.). A.R. acknowledges the ANR for postdoctoral grants.
Editor: Jason M. Haugh.
References
- 1.Luscinskas, F. W., S. Ma, A. Nusrat, C. A. Parkos, and S. K. Shaw. 2002. Leukocyte transendothelial migration: a junctional affair. Semin. Immunol. 14:105–113. [DOI] [PubMed] [Google Scholar]
- 2.Shaw, S. K., P. S. Bamba, B. N. Perkins, and F. W. Luscinskas. 2001. Real-time imaging of vascular endothelial-cadherin during leukocyte transmigration across endothelium. J. Immunol. 167:2323–2330. [DOI] [PubMed] [Google Scholar]
- 3.Allport, J. R., W. A. Muller, and F. W. Luscinskas. 2000. Monocytes induce reversible focal changes in vascular endothelial cadherin complex during transendothelial migration under flow. J. Cell Biol. 148:203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lampugnani, M. G., M. Corada, P. Andriopoulou, S. Esser, W. Risau, and E. Dejana. 1997. Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J. Cell Sci. 110:2065–2077. [DOI] [PubMed] [Google Scholar]
- 5.Su, W. H., H. I. Chen, and C. J. Jen. 2002. Differential movements of VE-cadherin and PECAM-1 during transmigration of polymorphonuclear leukocytes through human umbilical vein endothelium. Blood. 100:3597–3603. [DOI] [PubMed] [Google Scholar]
- 6.Allport, J. R., H. T. Ding, A. Ager, D. A. Steeber, T. F. Tedder, and F. W. Luscinskas. 1997. L-selectin shedding does not regulate human neutrophil attachment, rolling, or transmigration across human vascular endothelium in vitro. J. Immunol. 158:4365–4372. [PubMed] [Google Scholar]
- 7.Burns, A. R., R. A. Bowden, S. D. MacDonell, D. C. Walker, T. O. Odebunmi, E. M. Donnachie, S. I. Simon, M. L. Entman, and C. W. Smith. 2000. Analysis of tight junctions during neutrophil transendothelial migration. J. Cell Sci. 113:45–57. [DOI] [PubMed] [Google Scholar]
- 8.Muller, W. A. 2003. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 24:327–334. [DOI] [PubMed] [Google Scholar]
- 9.Johnson-Leger, C., M. Aurrand-Lions, and B. A. Imhof. 2000. The parting of the endothelium: miracle, or simply a junctional affair? J. Cell Sci. 113:921–933. [DOI] [PubMed] [Google Scholar]
- 10.House, S. D., and H. H. Lipowsky. 1988. In vivo determination of the force of leukocyte-endothelium adhesion in the mesenteric microvasculature of the cat. Circ. Res. 63:658–668. [DOI] [PubMed] [Google Scholar]
- 11.Zhang, X. H., A. Chen, D. De Leon, H. Li, E. Noiri, V. T. Moy, and M. S. Goligorsky. 2004. Atomic force microscopy measurement of leukocyte-endothelial interaction. Am. J. Physiol. Heart Circ. Physiol. 286:H359–H367. [DOI] [PubMed] [Google Scholar]
- 12.Pollard, T. D., and G. G. Borisy. 2003. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 112:453–465. [DOI] [PubMed] [Google Scholar]
- 13.Xu, J. S., F. Wang, A. Van Keymeulen, P. Herzmark, A. Straight, K. Kelly, Y. Takuwa, N. Sugimoto, T. Mitchison, and H. R. Bourne. 2003. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell. 114:201–214. [DOI] [PubMed] [Google Scholar]
- 14.Eddy, R. J., L. M. Pierini, F. Matsumura, and F. R. Maxfield. 2000. Ca2+-dependent myosin II activation is required for uropod retraction during neutrophil migration. J. Cell Sci. 113:1287–1298. [DOI] [PubMed] [Google Scholar]
- 15.Guilford, W. H., R. C. Lantz, and R. W. Gore. 1995. Locomotive forces produced by single leukocytes in vivo and in vitro. Am. J. Physiol. Cell Physiol. 37:C1308–C1312. [DOI] [PubMed] [Google Scholar]
- 16.Smith, L. A., H. Aranda-Espinoza, J. B. Haun, M. Dembo, and D. A. Hammer. 2007. Neutrophil traction stresses are concentrated in the uropod during migration. Biophys. J. 92:L58–L60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hochmuth, R. M., H. P. Tingbeall, B. B. Beaty, D. Needham, and R. Transontay. 1993. Viscosity of human neutrophils undergoing small deformations. Biophys. J. 64:1596–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roca-Cusachs, P., I. Almendros, R. Sunyer, N. Gavara, R. Farre, and D. Navajas. 2006. Rheology of passive and adhesion-activated neutrophils probed by atomic force microscopy. Biophys. J. 91:3508–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Worthen, G. S., B. Schwab, E. L. Elson, and G. P. Downey. 1989. Mechanics of stimulated neutrophils—cell stiffening induces retention in capillaries. Science. 245:183–186. [DOI] [PubMed] [Google Scholar]
- 20.Yap, B., and R. D. Kamm. 2005. Cytoskeletal remodeling and cellular activation during deformation of neutrophils into narrow channels. J. Appl. Physiol. 99:2323–2330. [DOI] [PubMed] [Google Scholar]
- 21.Yap, B., and R. D. Kamm. 2005. Mechanical deformation of neutrophils into narrow channels induces pseudopod projection and changes in biomechanical properties. J. Appl. Physiol. 98:1930–1939. [DOI] [PubMed] [Google Scholar]
- 22.Wojcikiewicz, E. P., X. Zhang, A. Chen, and V. T. Moy. 2003. Contributions of molecular binding events and cellular compliance to the modulation of leukocyte adhesion. J. Cell Sci. 116:2531–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spillmann, C. M., E. Lomakina, and R. E. Waugh. 2004. Neutrophil adhesive contact dependence on impingement force. Biophys. J. 87:4237–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harris, A. K., P. Wild, and D. Stopak. 1980. Silicone rubber substrata: a new wrinkle in the study of cell locomotion. Science. 208:177–179. [DOI] [PubMed] [Google Scholar]
- 25.Munevar, S., Y. Wang, and M. Dembo. 2001. Traction force microscopy of migrating normal and H-ras transformed 3T3 fibroblasts. Biophys. J. 80:1744–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galbraith, C. G., and M. P. Sheetz. 1997. A micromachined device provides a new bend on fibroblast traction forces. Proc. Natl. Acad. Sci. USA. 94:9114–9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balaban, N. Q., U. S. Schwarz, D. Riveline, P. Goichberg, G. Tzur, I. Sabanay, D. Mahalu, S. Safran, A. Bershadsky, L. Addadi, and B. Geiger. 2001. Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates. Nat. Cell Biol. 3:466–472. [DOI] [PubMed] [Google Scholar]
- 28.Tan, J. L., J. Tien, D. M. Pirone, D. S. Gray, K. Bhadriraju, and C. S. Chen. 2003. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc. Natl. Acad. Sci. USA. 100:1484–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.du Roure, O., A. Saez, A. Buguin, R. H. Austin, P. Chavrier, P. Silberzan, and B. Ladoux. 2005. Force mapping in epithelial cell migration. Proc. Natl. Acad. Sci. USA. 102:2390–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saez, A., A. Buguin, P. Silberzan, and B. Ladoux. 2005. Is the mechanical activity of epithelial cells controlled by deformations or forces? Biophys. J. 89:L52–L54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luscinskas, F. W., G. S. Kansas, H. Ding, P. Pizcueta, B. E. Schleiffenbaum, T. F. Tedder, and M. A. Gimbrone. 1994. Monocyte rolling, arrest and spreading on IL-4-activated vascular endothelium under flow is mediated via action of L-selectin, β1-integrins, and β2-integrins. J. Cell Biol. 125:1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lo, C. M., H. B. Wang, M. Dembo, and Y. L. Wang. 2000. Cell movement is guided by the rigidity of the substrate. Biophys. J. 79:144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giannone, G., B. J. Dubin-Thaler, H. G. Dobereiner, N. Kieffer, A. R. Bresnick, and M. P. Sheetz. 2004. Periodic lamellipodial contractions correlate with rearward actin waves. Cell. 116:431–443. [DOI] [PubMed] [Google Scholar]
- 34.Engler, A. J., S. Sen, H. L. Sweeney, and D. E. Discher. 2006. Matrix elasticity directs stem cell lineage specification. Cell. 126:677–689. [DOI] [PubMed] [Google Scholar]
- 35.Saez, A., M. Ghibaudo, A. Buguin, P. Silberzan, and B. Ladoux. 2007. Rigidity-driven growth and migration of epithelial cells on microstructured anisotropic substrates. Proc. Natl. Acad. Sci. USA. 104:8281–8286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Corada, M., F. Liao, M. Lindgren, M. G. Lampugnani, F. Breviario, R. Frank, W. A. Muller, D. J. Hicklin, P. Bohlen, and E. Dejana. 2001. Monoclonal antibodies directed to different regions of vascular endothelial cadherin extracellular domain affect adhesion and clustering of the protein and modulate endothelial permeability. Blood. 97:1679–1684. [DOI] [PubMed] [Google Scholar]
- 37.Ganz, A., M. Lambert, A. Saez, P. Silberzan, A. Buguin, R. M. Mege, and B. Ladoux. 2006. Traction forces exerted through N-cadherin contacts. Biol. Cell. 98:721–730. [DOI] [PubMed] [Google Scholar]
- 38.Sircar, M., P. F. Bradfield, M. Aurrand-Lions, R. J. Fish, P. Alcaide, L. Yang, G. Newton, D. Lamont, S. Sehrawat, T. Mayadas, T. W. Liang, C. A. Parkos, B. A. Imhof, and F. W. Luscinskas. 2007. Neutrophil transmigration under shear flow conditions in vitro is junctional adhesion molecule-C independent. J. Immunol. 178:5879–5887. [DOI] [PubMed] [Google Scholar]
- 39.Yang, L., J. R. Kowalski, X. Zhan, S. M. Thomas, and F. W. Luscinskas. 2006. Endothelial cell cortactin phosphorylation by src contributes to polymorphonuclear leukocyte transmigration in vitro. Circ. Res. 98:394–402. [DOI] [PubMed] [Google Scholar]
- 40.Burns, A. R., D. C. Walker, E. S. Brown, L. T. Thurmon, R. A. Bowden, C. R. Keese, S. I. Simon, M. L. Entman, and C. W. Smith. 1997. Neutrophil transendothelial migration is independent of tight junctions and occurs preferentially at tricellular corners. J. Immunol. 159:2893–2903. [PubMed] [Google Scholar]
- 41.Coughlin, M. F., and G. W. Schmid-Schonbein. 2004. Pseudopod projection and cell spreading of passive leukocytes in response to fluid shear stress. Biophys. J. 87:2035–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reference deleted in proof.
- 43.Saito, H., Y. Minamiya, S. Saito, and J. Ogawa. 2002. Endothelial Rho and Rho kinase regulate neutrophil migration via endothelial myosin light chain phosphorylation. J. Leukoc. Biol. 72:829–836. [PubMed] [Google Scholar]
- 44.Garcia, J. G., A. D. Verin, M. Herenyiova, and D. English. 1998. Adherent neutrophils activate endothelial myosin light chain kinase: role in transendothelial migration. J. Appl. Physiol. 84:1817–1821. [DOI] [PubMed] [Google Scholar]
- 45.Choquet, D., D. P. Felsenfeld, and M. P. Sheetz. 1997. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. 88:39–48. [DOI] [PubMed] [Google Scholar]
- 46.Allingham, M. J., J. D. van Buul, and K. Burridge. 2007. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J. Immunol. 179:4053–4064. [DOI] [PubMed] [Google Scholar]
- 47.Guo, W. H., M. T. Frey, N. A. Burnham, and Y. L. Wang. 2006. Substrate rigidity regulates the formation and maintenance of tissues. Biophys. J. 90:2213–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Discher, D. E., P. Janmey, and Y. L. Wang. 2005. Tissue cells feel and respond to the stiffness of their substrate. Science. 310:1139–1143. [DOI] [PubMed] [Google Scholar]
- 49.Cooper, D., F. P. Lindberg, J. R. Gamble, E. J. Brown, and M. A. Vadas. 1995. Transendothelial migration of neutrophils involves integrin-associated protein (CD47). Proc. Natl. Acad. Sci. USA. 92:3978–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baumgartner, W., and D. Drenckhahn. 2002. Plasmalemmal concentration and affinity of mouse vascular endothelial cadherin, VE-cadherin. Eur. Biophys. J. 31:532–538. [DOI] [PubMed] [Google Scholar]
- 51.Baumgartner, W., P. Hinterdorfer, W. Ness, A. Raab, D. Vestweber, H. Schindler, and D. Drenckhahn. 2000. Cadherin interaction probed by atomic force microscopy. Proc. Natl. Acad. Sci. USA. 97:4005–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.