

Abstract
This article demonstrates that the rate of mixing can regulate the rate and outcome of both biological and nonbiological autocatalytic reaction systems that display a threshold response to the concentration of an activator. Plug-based microfluidics was used to control the timing of reactions, the rate of mixing, and surface chemistry in blood clotting and its chemical model. Initiation of clotting of human blood plasma required addition of a critical concentration of thrombin. Clotting could be prevented by rapid mixing when thrombin was added near the critical concentration, and mixing also affected the rate of clotting when thrombin was added at concentrations far above the critical concentration in two clinical clotting assays for human plasma. This phenomenon was modeled by a simple mechanism—local and global competition between the clotting reaction, which autocatalytically produces an activator, and mixing, which removes the activator. Numerical simulations showed that the Damköhler number, which describes this competition, predicts the effects of mixing. Many biological systems are controlled by thresholds, and these results shed light on the dynamics of these systems in the presence of spatial heterogeneities and provide simple guidelines for designing and interpreting experiments with such systems.
INTRODUCTION
In this article, we demonstrate that mixing can regulate the outcome and kinetics of both biological and nonbiological autocatalytic reaction systems that display a threshold response to the concentration of an activator. Mixing has been shown to affect the kinetics of several nonlinear reaction systems, including inorganic reactions (1–6), competing reactions (7), and nucleation of proteins or inorganic crystals (6,8,9). This phenomenon is believed to be the result of spatial heterogeneities in the concentration of reagents (1,9,10) due to an initially nonuniform reagent distribution, or, in a stochastic system, a fluctuation in local concentrations (11,12). Systems with linear kinetics are not influenced by mixing, because the total rate of reaction is not sensitive to spatial heterogeneity in reagent distribution (Fig. 1, a and b). However, mixing influences the rate of reaction in systems with nonlinear kinetics, because the total rate of reaction depends on how the reagents are distributed (Fig. 1, c and d). For example, given two solutions with the same average concentration, a nonlinear autocatalytic reaction proceeds more rapidly in a solution with regions of low and high concentrations of the autocatalytic species than in a solution with uniformly distributed reagents (13) (Fig. 1 d). Small regions of high concentration undergo rapid autocatalysis, which builds up the local concentration of the activating species and spreads the reaction throughout the solution. In these systems, slow mixing preserves regions of high concentration and allows rapid reaction. Sufficiently fast mixing averages the concentrations before the reaction takes place, so the reaction proceeds at the lower rate that corresponds to the homogeneous solution (14). Mixing should have the greatest effect on the kinetics of reactions when the timescales for mixing and reaction are similar (7,15–17).
FIGURE 1.

Mixing can affect the rate or outcome of certain nonlinear reactions by removing spatial heterogeneities in the concentration of an activator (1,10). (a, c, and e) Hypothetical rate plots show autocatalytic production (solid lines) and degradation (dashed lines) of an activating species, C. Gray and black dots on the [C] axis indicate hypothetical low and high initial concentrations of C, respectively, and correspond to the gray and black regions shown in the test tubes below. (b, d, and f) An initially unmixed solution has a concentration of C, [C], equal to 10 in the lower subvolume of the test tube and no C in the remaining four subvolumes, so [C]i = 10 and the average initial concentration is [C]0,avg = 2. In a well mixed system, C is evenly distributed throughout the solution. The mean rate of reaction and the calculation thereof is given below each tube in b and d. (a and b) In a linear system, the rate of reaction, R, is linearly proportional to [C]. The reaction rate does not depend on the spatial distribution of activator. (c and d) In a nonlinear system, R is proportional to [C]2 or, more generally, [C]n, where n ≥ 2. Since the rate in each subvolume is no longer additive, the average rate of reaction depends strongly on the distribution of activator and thus on the rate of mixing. (e–g) In a system with threshold kinetics, the spatial distribution of activator, and thus the rate of mixing, controls the direction and outcome of the reaction in addition to its rate. (e) The threshold occurs at the concentration ([C]crit) at which the two rate curves cross and the production curve has the greater slope (diamond). Purple and yellow squares mark the upper and lower stable steady-state concentrations, respectively. (f) Supposing that [C]crit = 5, then the small black region in the left tube has an above-threshold concentration of 10. Though the remaining solution has a below-threshold concentration of 0, this test tube will react to create more C (Net +). The well mixed tube at the right has a below-threshold concentration of 2 throughout, so C will be degraded (Net −). (g) In an initially unmixed solution with a small pocket having [C] > [C]crit, slow mixing permits net production of C until the upper steady state (purple) is reached, whereas rapid mixing lowers the average C to less than the threshold and net degradation brings the system to the lower steady state (yellow).
Though the effect of mixing on autocatalytic systems has been studied, its effect on systems exhibiting threshold kinetics is less well defined (18–20). Many common mechanisms produce a threshold response to the concentration of an activator, [C] (nM) (21). For example, a bistable system may have a critical concentration of activator, [C]crit (nM), below which the forward reaction does not proceed (Fig. 1 e). However, bistability is not required. Linear production coupled to Michaelis-Menten degradation of activator or two coupled Michaelis-Menten rate laws can also produce threshold kinetics (21). In systems with threshold kinetics, mixing should determine the outcome, not just the observed rate, of reaction (22–24) (Fig. 1, f and g). When [C] is above [C]crit in a small pocket and below [C]crit in the bulk solution, mixing and reaction compete in the pocket to control the outcome of the reaction. Within the pocket, mixing decreases the concentration while the reaction simultaneously increases it. This competition is traditionally described by the unitless Damköhler number, Da = τmix/τrxn, where τmix (s) is the time required for mixing to eliminate the pocket, and τrxn (s) is the time required for a uniform solution to react. Slow mixing (large Da) permits [C] to exceed [C]crit long enough for the reaction to initiate and spread from the pocket to the bulk solution, and the system undergoes net production of activator until it reaches the upper stable steady state (Fig. 1, f and g, purple). Rapid mixing (small Da) lowers [C] in the pocket to below [C]crit before the reaction can initiate and spread, and the system undergoes net degradation of activator until it reaches the lower stable steady state (Fig. 1, f and g, yellow).
Many biological systems, including blood coagulation (25,26), are autocatalytic and exhibit threshold kinetics, yet there are few experimental reports on the influence of mixing on reactions in biological systems that are more complex than protein crystallization (8) or refolding (27). One exception is population dynamics, where the degree of mixing can strongly affect the outcome of competition between microorganisms (28–30). Coagulation, or clotting, of human blood and blood plasma is regulated by a threshold response to the concentration of activators such as tissue factor (26,31–35)—the coagulation cascade only initiates when [C] is above [C]crit (32,36–38). Support for the effect of flow (39–45) and mixing (46–49) on coagulation has been provided by previous research, but the effects of mixing have not been tested by direct experiments. In most clinical assays of coagulation, clotting is initiated by adding a small volume of an activator to a sample of blood or plasma (31), and thus the reaction begins in a state analogous to the unmixed test tube in Fig. 1 g. In this case, any effect of mixing on the rate of clotting would have clinical relevance (see Discussion).
The effect of mixing on autocatalytic, biological systems may have been masked by the difficulty of controlling the rate of mixing in macroscopic systems (10), including industrial reactors (50,51), flasks (52), and microplates (53). Also, these systems are rarely observed at reaction conditions near the threshold of activation. In clinical assays and experiments aimed at characterizing clotting, for instance, activators such as tissue factor are usually added at concentrations high enough to guarantee initiation of a reaction (i.e., [C] far above [C]crit) (31). Here, we used plug-based microfluidics (54–57) to demonstrate that the rate of mixing can regulate the reaction kinetics of two systems, clotting of human normal pooled plasma and an analogous, nonbiological chemical model system (2,37,58). Plug-based microfluidics enables precise control of reagent concentrations, surface chemistry, and mixing of reagents (54,59–63), as well as accurate observation of the kinetics of these reactions (46,55).
We demonstrate two effects of the rate of mixing on autocatalytic systems with threshold kinetics: 1), the reaction of both the complex biochemical reaction network of hemostasis (blood clotting) and its simple chemical analog can be turned “on” or “off” by adjusting the rate of mixing if the initial concentration of activator is near the critical concentration; and 2), the rate of these reactions can be slowed by rapid mixing even if the initial concentration of activator is far above the threshold. In addition, we propose a simple mechanism that explains this phenomenon based on the competition between the reaction rate and mixing rate, described by the Damköhler number, and we validate this mechanism using a two-dimensional numerical simulation of threshold kinetics occurring in a chaotically mixed plug.
MATERIALS AND METHODS
All solvents and salts used in buffers were purchased from commercial sources and used as received unless otherwise stated.
Microfluidic experiments
Fluorinert FC-70, FC-3283, and FC-77 were obtained from 3M (St. Paul, MN). Synthesis of the surfactant RfOEG (CF3(CF2)7CH2O(CH2CH2O)3H) will be described in detail in a future publication. 1H,1H,2H,2H-perfluoro-1-octanol was purchased from Alfa Aesar (Heysham, Lancashire, England). Teflon AF (Poly(4,5-difluoro-2,2-bis(trifluoromethyl)-1,3-dioxole-co-tetrafluoroethylene)) was purchased from Aldrich (St. Louis, MO). Silane ((tridecafluoro-1,1,2,2,-tetrahydrooctyl)-1 trichlorosilane) was obtained from United Chemical Technologies (Bristol, PA). Poly(dimethylsiloxane) (PDMS) (Sylgard 184 Silicone Elastomer kit) was obtained from Dow Corning (Midland, MI). Teflon (PTFE) tubing (200 μm i.d., 250 μm o.d.; 400 μm i.d., 550 μm o.d.) was received from Zeus (Orangeburg, SC). Thirty-gauge Teflon tubing was obtained from Weico Wire & Cable (Edgewood, NY). Gastight syringes were obtained from Hamilton (Reno, NV). Polyimide-coated glass capillary tubing (50 μm i.d., 150 μm o.d., 10 μm external coating) was obtained from Polymicro Technologies (Phoenix, AZ).
Plasma experiments
Human normal pooled plasma (NPP) (platelet-poor, 1 mL aliquots) was obtained from George King Biomedical (Overland Park, KS) and stored at −80°C until needed. α-thrombin from human plasma (citrate-free, formulated at 3.13 mg/mL in 20 mM Tris-HCl, 100 mM NaCl, pH 7.4) was obtained from CalBioChem (La Jolla, CA). Bovine serum albumin (BSA) (fraction V, 99%) was obtained from Sigma (St. Louis, MO). CaCl2·6H20 (certified) was obtained from Fisher Scientific (Rochester, NY). The fluorogenic substrate tert-butyloxycarbonyl-β-benzyl-Asp-Pro-Arg-4-methyl-coumarin-7-amide (MCA-sub) was obtained from Peptides International (Louisville, KY), and bis-(p-tosyl-Gly-Pro-Arg amide)-Rhodamine 110 (Rho-sub) and Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (DHPE) were obtained from Molecular Probes/Invitrogen (Eugene, OR). 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC) and L-α-phosphatidylserine (PS) from porcine brain were bought from Avanti Polar Lipids (Alabaster, AL). ThromboMax HS with calcium and Alexin were obtained from Trinity Biotech (Bray, Ireland).
Chemical model experiments
All reagents required for the chemical model solutions were obtained as described in Kastrup et al. (37). Acetic acid (glacial, certified ACS plus) was obtained from Fisher Scientific.
Fabrication of microfluidic devices
PDMS devices were fabricated as described (46), with the following modifications. Devices were rendered hydrophobic and fluorophilic by using the protocol described by Roach et al. (62). After sealing, the device was baked at 110°C for 1 h. Then, silane vapor was flowed into the device for 1–2 h by using pressure from dry N2. Next, devices were baked at 110°C for 1 h, after which the channels were rinsed thoroughly with a mixture of 10:1 (v/v) FC-3283/1H,1H,2H,2H-perfluoro-1-octanol and then baked overnight at 60°C. Silane solution was stored wrapped in parafilm to prevent humid air from quenching the silane. To coat the silanized channels with amorphous Teflon, channels were filled with a 2.5% (m/v) solution of Teflon AF in FC-77, baked at 60°C for 4 h until the channels were almost dry, and rinsed with FC-3283 to remove excess Teflon from all inlet areas. Finally, the devices were baked at 60°C overnight.
A hydrophilic glass capillary (50 μm i.d., 150 μm o.d.) merging junction was added to the PDMS device for inflow of a reagent stream, as described previously (46). The polyimide coating was left in place on the tubing and did not affect fluid merging.
For observation of rapidly flowing plugs for an extended period of time (for experiments with NPP only), a second connection was made at the end of the PDMS channel to a length of Teflon tubing. The PDMS was cut perpendicular to the channel so that the rectangular channel cross-section was visible. A 1- to 2-cm length of small PTFE tubing (200 μm i.d., 250 μm o.d.) was attached to the end of a 90-cm length of large PTFE tubing (400 μm i.d., 550 μm o.d.) by using partially cured PDMS. The connection was immediately dried by baking at 110°C for ∼5 min. The small tubing was cut carefully to obtain smooth edges and thus prevent clotted plasma from adhering to the entrance of the tubing during experiments. The open end of the small tubing was then inserted a short distance (3–6 mm) into the end of the PDMS channel and sealed into place with partially cured PDMS. The connection was immediately dried by baking at 110°C for ∼5 min. Over-drying the devices at this stage decreased the effectiveness of the Teflon AF coating. Devices were stored at 60°C until use and were cooled to room temperature for experiments.
Initiating clotting of human plasma with thrombin in a microfluidic device
The general procedure for plug-based microfluidics experiments has been reviewed previously (54). The carrier fluid in all microfluidic experiments consisted of Fluorinert FC-70 and 1 mg/mL RfOEG. Plasma was thawed at room temperature before experiments and used within 6 h. All aqueous solutions for plasma experiments were prepared in a Tris buffer (20 mM Tris-HCl, 100 mM NaCl, pH 7.4). Rho-sub, a green fluorogenic substrate for thrombin, was prepared as a 10-mM stock solution in dimethylsulfoxide according to package instructions. This molecule aggregates easily in aqueous solution but was found to be soluble when prepared as a 75-μM solution in the presence of 22.2 mM CaCl2. Negatively charged phosphatidylcholine (PC)/PS lipid vesicles (5 mg/mL; 4:1 mol ratio DLPC/porcine L-α-PS) containing Texas Red DHPE were prepared as described previously (37). To minimize surface adsorption of thrombin, 2 mg/mL BSA was included in the thrombin solution from its first dilution.
Plugs of recalcified and relipidated NPP were formed on-chip by combining a stream of NPP with two identical aqueous streams of solution of CaCl2/Rho-sub/lipids (22.2 mM CaCl2, 75 μM Rho-sub, and 0.5 mg/mL PC/PS lipid vesicles prepared in Tris buffer) in a 2:1:1 volume ratio. These plugs were completely mixed before they reached the merging junction, where they were merged with a droplet containing human thrombin (0–6 nM) in Tris buffer. The final [Rho-sub] in the mixed plugs of plasma was 28 μM.
The experiment to determine the experimental critical concentration, [C]i,crit, of thrombin (Fig. 2 b) was conducted in a device with straight smooth channels (280 × 300-μm2 cross section) after the merging junction. The flow rates for the carrier fluid, left inflow of CaCl2/Rho-sub/lipids, center inflow of CaCl2/Rho-sub/lipids, NPP, and thrombin solutions were 3.0, 0.075, 0.075, 0.15, and 0.1 μL/min, respectively.
FIGURE 2.

Rapid mixing prevents clotting in human NPP near the reaction threshold. (a) Schematic of the device used to merge droplets of an activator, C (thrombin), with plugs of recalcified, relipidated NPP and a fluorogenic substrate for thrombin. (b) In a straight, smooth channel, the fraction of plugs (n ≥ 30) that clot increased from 0 to 1 as the concentration of thrombin, [C]i, injected into the NPP plug increased from 0.0 to 0.6 nM (2 parts 0.0–0.6 nM thrombin/6 parts recalcified, relipidated NPP with substrate, by volume). Black line shows a sigmoidal curve drawn to guide the eye. (c and d) Wide-field fluorescence and brightfield microphotographs of plugs with clotted (clumps of particles, bright fluorescence) and unclotted (no clumps, no fluorescence) NPP flowing through a microfluidic device after merging with 0.6 nM (+, *, **) or 0.1 nM (−) [C]i. Blue symbols correspond to those in e. Black arrows indicate direction of fluid flow. Scale bars, 300 μm. (c) Slowly flowing (0.37 mm/s) plugs are visible in the channel 20 min after merging. Those receiving 0.6 nM (+) and 0.1 nM (−) [C]i all clotted or remained unclotted, respectively. (d) Faster flowing plugs were monitored in Teflon tubing 20 min after merging. Those receiving [C]i ≈ [C]i,crit either clotted (*) or did not (**), depending on the mixing rate. (e) In a winding, bumpy channel, the fraction of plugs that clotted within 20 min decreased with increasing mixing rate when [C]i ≈ [C]i,crit (0.6 nM thrombin).
The experiment to show the effect of mixing (Fig. 2, c–e) was conducted in a device with winding, bumpy channels (280 × 270-μm2 cross section) after the merging junction. The flow rates for the carrier fluid, left inflow of CaCl2/Rho-sub/lipids, center inflow of CaCl2/Rho-sub/lipids, NPP, and thrombin solutions were 1.5, 0.075, 0.0375, 0.0375, and 0.05 μL/min, respectively, at the slowest total flow rate (0.37 mm/s). These flow rates were scaled up as necessary to increase the total flow rate while maintaining the individual flow rates in a constant ratio. Plugs of plasma eventually were flowed from the channel into Teflon tubing, because they began to adhere to the walls of the PDMS channel after 50–80 cm of travel. We believe that this adhesion was due to depletion of the RfOEG surfactant from the carrier fluid, which will be characterized further in a future article. To image plugs clearly in Teflon tubing, the tubing was submerged in FC-70.
Initiating “clotting” in the chemical model with acid in a microfluidic device
The solutions for the chlorite-thiosulfate reaction (58) were prepared fresh daily as described previously (37). Acetic acid solutions were prepared by diluting a 1.0 M stock solution of acid with Millipore water to the desired concentration, and pH values were remeasured daily for each solution.
Plugs of the chemical model reaction mixture were formed on-chip by combining three solutions: 1 part 0.6 M NaClO2 (pH 11.6); 1 part NaOH (pH 10.5 in Millipore water); and 2 parts 0.1 M Na2S2O3, 0.03 M sodium alginate, and bromophenol blue. The plugs were completely mixed before they reached the merging junction, where they merged with a droplet containing dilute acetic acid (pH 3.37–3.04).
The experiment to determine [C]i,crit of acid (Fig. 3 b) was conducted in a device with straight, smooth channels (280 × 300-μm2 cross section) after the merging junction. The flow rates for the carrier fluid, Na2S2O3/alginate/bromophenol blue, NaOH, NaClO2, and acetic acid solutions were 6.0, 0.3, 0.15, 0.15, and 0.25 μL/min, respectively.
FIGURE 3.

Rapid mixing prevents “clotting” in an autocatalytic chemical system with threshold kinetics. (a) Schematic of the device design used to merge droplets containing the activator, C, (CH3COOH) with plugs containing the reagents of the chlorite-thiosulfate reaction system. (b) In a straight channel, the fraction of plugs that are “clotted” after 1 min of reaction (n = 40) increased from 0 to 1 as [C]i (H+ from acetic acid) increased from 600 to 900 μM. Trial 1 data were taken the same day as the data in d. Black line shows a sigmoidal curve drawn to guide the eye. (c) Microphotographs show yellow “clotted” plugs and purple “unclotted” plugs flowing through a microfluidic device after merging with 660 μM H+, the experimental [C]i,crit. Scale bars, 300 μm. (d) In a winding, bumpy channel, the fraction of plugs that clot decreased with increasing mixing rate when [C]i ≈ [C]i,crit = 660 μM.
The experiment to show the effect of mixing (Fig. 3, c and d) was conducted in a device with winding, bumpy channels (277 × 392-μm2 cross section) after the merging junction. The flow rates for the carrier fluid, Na2S2O3/alginate/bromophenol blue, NaOH, NaClO2, and acetic acid solutions were 3.0, 0.15, 0.075, 0.075, and 0.1 μL/min, respectively, at the slowest total flow rate (0.52 mm/s). These flow rates were scaled up as necessary to increase the total flow rate while maintaining the individual flow rates in a constant ratio.
Conducting microfluidic APTT and PT assays
To vary mixing time for the activated partial thromboplastin time (APTT) and prothrombin time (PT) assays, we used four devices with different channel geometries after the merging junction. In the APTT assay, devices with straight, smooth channels with a cross section of 260 × 265 μm2 were used to achieve slow mixing (see Fig. 6 b, black circles). Devices with straight, bumpy channels with a cross section of 260 × 290 μm2 were used to achieve moderate mixing (see Fig. 6 b, blue triangles). Devices with winding, bumpy channels with a cross section of 270 × 270 μm2 were used to achieve rapid mixing (see Fig. 6 b, red squares). The microfluidic APTT assay was conducted at room temperature, as previously described (46), using Alexin as the activator.
FIGURE 6.

Rapid mixing slows clotting of human NPP with average [C] ≫ [C]crit. (a and c) Schematic drawings of device designs used for microfluidic APTT (a) and PT (c) assays. (b and d) Increased mixing rate led to decreased reaction time in both APTT (b) and PT (d) assays. See Materials and Methods for a detailed description of device designs and experimental setup.
The PT assay was performed at room temperature using ThromboMax HS with calcium as the activator (a lyophilized extract of rabbit brain with buffer, stabilizers, and CaCl2), which was reconstituted per package instructions in Millipore water and prepared fresh every 4 weeks. Procedure and device designs were similar to those used in the APTT assay (described above). Plugs were formed from the activator alone and were merged with a droplet of NPP solution (150 μM MCA-substrate in NPP). In the PT assay, devices with straight, smooth channels with a cross section of 260 × 265 μm2 were used to achieve slow mixing (see Fig. 6 d, black circles). Devices with winding, bumpy channels with a cross section of 270 × 270 μm2 were used to achieve moderate mixing (see Fig. 6 d, red squares). Devices with winding, bumpy channels with a cross section of 170 × 145 μm2 were used to achieve rapid mixing (see Fig. 6 d, orange inverted triangles). In the PT assay, flow rates in experiments with slow and moderate mixing were 6.0, 0.6, and 0.3 μL/min for the carrier fluid, activator, and NPP solutions, respectively. Flow rates for experiments with rapid mixing were 4.0, 0.6, and 0.3 μL/min, respectively. The linear flow rate was faster in the experiments with rapid mixing than in the experiments with slow and moderate mixing because the cross-sectional area of the channel used for rapid mixing was smaller. MCA-sub, a blue fluorogenic substrate for thrombin, was used to detect clotting in the PT assay, and clotting in the APTT assay was detected by the appearance of fibrin in brightfield images.
RESULTS
To control mixing, we used plug-based microfluidics (54). In this approach, reagents are compartmentalized in microscopic aqueous droplets that are surrounded by fluorinated carrier fluid. This compartmentalization is essential for controlling timing of reactions, rate of mixing, and surface chemistry. Compartmentalization also eliminates carry-over from one reaction to the next, a critical feature when studying autocatalytic reactions (6). In addition, compartmentalization prevents interaction of reagents with the walls of the channels, important for dealing with blood coagulation. We designed a microfluidic device to control mixing rate by changing channel geometry and total flow rate (60,61,63). In plugs, mixing is not chaotic during flow through straight channels, whereas flow through winding channels induces mixing by chaotic advection (61,64–66). Thus, everything else being equal, mixing is faster in winding channels than in straight ones (61). The timescale of chaotic mixing, tmix (s), scales with flow rate (17,61,67) as follows:
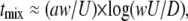 |
(1) |
where U (mm/s) is the linear flow rate, a is the dimensionless plug length (plug length/plug width), w (mm) is the diffusion distance (roughly the channel width), and D (m2/s) is the diffusion coefficient of the species to be mixed. Bumps were incorporated on the walls of the winding channel to accelerate recirculation of viscous solutions and thus enhance mixing (63).
The microfluidic PDMS devices used to study clotting have been described previously (46) (Fig. 2 a). Briefly, plugs containing all necessary reagents except the activator were formed in a stream of fluorocarbon carrier fluid and merged with a droplet of activator with concentration [C]i emerging from a 50-μm-i.d. glass capillary. The fluid in the merged plugs was mixed, and the rate and outcome of the reaction were observed. The dimensions of the cross section of the channel were ∼280 × 280 μm2. To minimize sticking of clotted plasma to PDMS surfaces, channel walls were coated with Teflon AF (see Materials and Methods), and all tests were performed at room temperature (21–25°C).
For experiments using normal pooled plasma, brightfield microscopy was used to monitor the formation of fibrin, and fluorescence microscopy was used to visualize the thrombin-induced cleavage of a fluorescent substrate (Rho-sub). The experimental set-up was sensitive to as little as 0.1 nM thrombin (Fig. S1 in Supplementary Material, Data S1). Plugs were considered clotted when clumps of lipids and fibrin became visible in brightfield, and they were monitored for at least 2 min after the recorded clot time to confirm the observation. Plugs that did not clot were flowed from the microfluidic PDMS channel into 400-i.d. PTFE tubing and observed for at least 20 min to confirm the observation using the fluorescence intensity. Movement into the tubing did not affect clotting dynamics, because mixing and reaction initiation were complete before plugs entered the tubing. Total flow rates were kept low in all experiments to avoid activation of platelets by shear (in all, shear rates at the wall ranged from 5 to 50 s−1). In these experiments, the spontaneous clot time of NPP was >20 min.
Initiation of clotting of normal pooled plasma by thrombin displays threshold kinetics
In experiments using NPP, the activator of clotting, C, was thrombin, a plasma protein central to the coagulation cascade (26,31). We wished to establish whether initiation of clotting displays a threshold response to the concentration of thrombin (68) and to determine the initial concentration of added activator that was required to initiate clotting, [C]i,crit. In these experiments, plugs of recalcified, relipidated NPP were merged with thrombin at varying concentrations. The volume ratio of all solutions was maintained, and only the initial concentration of thrombin was varied. The final plugs contained 8.3 mM CaCl2 and 0.2 mg/mL DLPC-PS lipid vesicles, NPP diluted 3:8 (NPP/total volume), and thrombin. To induce slow mixing, the merged plugs were flowed through a straight channel at a constant total flow rate of 0.67 mm/s. Clotting occurred only when plugs were merged with a droplet of activator with a concentration, [C]i, of at least 0.3 nM thrombin (Fig. 2 b). Therefore, [C]i,crit = 0.3 nM. Thrombin-activated clotting occurred within 3–10 min. This experimental [C]i,crit is consistent with previous estimates (69) (see Discussion). According to the proposed mechanism, [C]i of the droplet should be considered rather than the average concentration of the newly formed plug at time t = 0, [C]0,avg, because the mixing rate and the initial reaction rate, which is set by [C]i, control the kinetics and outcome of the reaction in the entire plug.
Rapid mixing prevents clotting of normal pooled plasma when the initial concentration of activator is near the critical concentration
To characterize the effect of the mixing rate on reaction rate (clot time) in NPP, plugs of NPP were merged with thrombin at [C]i ≈ [C]i,crit, and the resulting plugs were flowed through a winding, bumpy channel at varying flow rates. In this device, [C]i = 0.3 nM resulted in very slow clotting, and observation of plugs for the necessary time period was difficult. Therefore, the experimental [C]i,crit used here was 0.6 nM thrombin. When [C]i ≈ [C]i,crit, clotting occurred in at least 90% of plugs under slow mixing conditions (flow rate ≤1 mm/s) (Fig. 2, c and d, left), but was slowed or prevented under fast mixing conditions. At rapid flow rates of 1.5 mm/s and 1.8 mm/s, the fraction of plugs that clotted decreased to 12–60% and to 10% (Fig. 2 d, right), respectively. At lower [C]i (0.1 nM, Fig. 2 c, right), <10% of plugs clotted, and at higher [C]i (1.2 nM, image not shown), >88% of plugs clotted, independent of the mixing rate.
Rapid mixing also prevents “clotting” of the chemical model system when the initial concentration of activator is near the critical concentration
We wished to test whether the results observed for clotting of human plasma could be extended to other systems with threshold kinetics and to eliminate the possibility that these results were due to some unknown feature of the coagulation cascade. We used a simple chemical model system of coagulation to achieve these goals. For experiments using the chemical model system, the activator of “clotting”, C, was the hydronium ion, abbreviated here as H+, from acetic acid. The set-up and solutions used in the chemical model system have been described previously (37,58). Briefly, the ∼80 reactions of the coagulation cascade were simplified into three functional modules—autocatalytic production of C, linear consumption of C, and precipitation at high [C]. In the chemical model system, these three modules were represented by three chemical reactions (37,58). The rate of chlorite-thiosulfate reaction we used is known to decrease in response to increased stirring rate (2). The initial solution is basic, and becomes acidic once the reaction occurs. Sodium alginate was used to represent the precipitation module; at high [H+], the solution becomes a gel, which mimics the clotting of human blood. The reaction was initiated by merging droplets of acetic acid with plugs of reaction solution (Fig. 3 a). “Clotting” was visualized by the transition of a pH indicator, bromophenol blue, from purple to yellow. “Clot” time was defined as the time from merging until the entire plug turned yellow. The same microfluidic device design used in experiments with NPP was used for experiments with the chemical model system.
To determine the experimental [C]i,crit in the chemical model system, plugs of the chemical model solution were merged with acetic acid at varying concentrations. The resulting plugs were flowed through a straight channel at a constant total flow rate of 1.3 mm/s to ensure slow mixing. “Clotting” occurred only when [C]i was >∼650–750 μM H+ (pH 3.12–3.19) (Fig. 3 b), and the experimental [C]i,crit used here was 660 μM H+. Acid-activated “clotting” was complete within 30 s after merging. To characterize the effect of mixing on the reaction rate, plugs of the chemical model solution were merged with acetic acid at [C]i ≈ [C]i,crit, and resulting plugs were flowed through a winding, bumpy channel at varying flow rates. When the [C]i ≈ [C]i,crit, “clotting” occurred under slow mixing conditions (flow rate <1 mm/s) but did not occur under fast mixing conditions (flow rate >1.5 mm/s) (Fig. 3, c and d). Plugs for which [C]i > [C]i,crit “clotted” under all flow rates, whereas those for which [C]i < [C]i,crit did not “clot” at any flow rate.
Based on these results and those obtained with human blood plasma, we propose that the outcome of the clotting reaction in each case is regulated by a simple mechanism—the competition between mixing rate and reaction rate. When the small droplet of activator from the capillary merges with the larger plug in the channel, the droplet becomes a pocket of high [C] in the plug. As suggested in Fig. 1 g, when mixing is slow compared to reaction (high Da), even a small pocket of [C] at or above [C]i,crit can initiate clotting that spreads throughout the plug (Figs. 2, c and d, and 3 c, left panels). However, when mixing is fast compared to reaction (low Da), the activator will be redistributed evenly throughout the total volume (Fig. 1, f and g) so that the average [C] of the whole plug will be below [C]i,crit, and clotting does not occur (Figs. 2 d and 3 c, right).
Design and characterization of the numerical simulation
To validate the proposed mechanism, we created a numerical simulation of mixing and reaction of an activating species with autocatalysis and threshold kinetics inside a plug by using Comsol Multiphysics 3.3 (Stockholm, Sweden). This simulation incorporates two interacting time-dependent physics modules—incompressible Navier-Stokes (fluid flow) and convection-diffusion (activator reaction and mixing). To model two-dimensional fluid recirculation in a plug moving through a straight or winding channel with average plug velocity U, the velocity of the top, Vtop (mm/s), and bottom, Vbot (mm/s), edges of the plug were either set to a constant velocity, −Vel = −U, for simple recirculation in a straight channel, or set to a sine-squared function with maximum amplitude of −Vel = −2U, which is known to lead to chaotic mixing (Fig. 4, a–c) (66). The fluid recirculation produced by the straight-channel model (Fig. 4 c, second and fourth images from left, and Movie S1) was in agreement with previous straight-channel simulations of plug flow that used this (70) and other methods (71,72). Mixing was optimized in the simulated winding channel (Movie S2) by varying the distance the plug traveled per period, P (66). The distribution of massless tracers (Fig. S2 in Data S1) became most uniform at long times for P = 3.5 mm (seven times the length of the plug), as did that of particles with a defined diffusion coefficient (Fig. S3 in Data S1). This model does not include physical bending of the plug as it passes a turn in a channel nor the consequent crossing of fluid streamlines. Thus, mixing in real channels is expected to be more effective than in this simulation.
FIGURE 4.

A two-dimensional numerical simulation of chaotic mixing and molecular diffusion in a plug. (a) Fluid flow inside a rightward-moving plug was simulated by moving the top and bottom edges (dark blue lines) of the plug-shaped region leftward with a defined velocity profile. To simulate chaotic mixing as in a winding channel, the velocity profile along the bottom edge, Vbot, is a sine-squared wave, phase-shifted from the profile along the top edge, Vtop (boxed equations) (66). (b) Drawing shows plugs in a winding channel with period τ and distance per period P. This geometry was not drawn into the simulation. (c) Arrow plots produced by the simulation show the flow profile in plugs at time points t = 0, τ/4, τ/2, and 3τ/4, corresponding to the plugs illustrated in b. (d) Chaotic mixing was completed more quickly in plugs flowing at a higher velocity. The left column shows the initial condition (t = 0 s), with 8 nM [C]i (red) at the back of the plug. The concentration scale for all four images is located beneath. The right column shows plugs after 10 s of flow at Vel = 0.35, 1.6, or 4.0 mm/s. Note that the region of highest concentration moves around inside the plug due to chaotic fluid flow and fluid recirculation during mixing. In this experiment, the diffusion constant for C was D = 5 × 10−11 m2/s, P = 3.5 mm, and the clotting reaction was turned off. (e) Measured mixing time decreased with plug velocity (0.5 × Vel) in the simulation. The solid line shows the curve predicted by the chaotic mixing scaling law (61) and was calculated relative to the observed mixing time of 7 s at a flow rate (Vel/2) of 2 mm/s.
Mixing time in the winding-channel simulation decreased with increasing flow rate, as predicted by Eq. 1 (Figs. 4, d and e, and S4) (61), a trend that was not explicitly built into the simulation. Mixing time was defined as the time required for a region of high [C] at the rear of the plug (Fig. 4 d, t = 0 s) to become evenly distributed, specifically for the range (max–min) (Fig. 4 e, triangles) or standard deviation (Fig. 4 e, asterisks) of [C] across the area of the plug to be reduced to 10% of its initial value. The curve predicted by the scaling law (Fig. 4 e, solid line) was normalized to the observed mixing time (7 s) for 4 mm/s flow velocity (see text and Eq. 9S in Data S1). The close agreement of the predicted and observed values validates the chaotic nature of the mixing in the simulation, which was determined by the equations used for Vtop and Vbot (66). Further details about the simulation, including model set-up and characterization of the mixing in the simulated plug, can be found in Data S1.
To simulate the effect of mixing on the reaction rate and kinetics of blood clotting, reaction kinetics inside the plug were defined by a modified version of a modular model of clotting described previously (37,73):
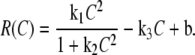 |
(2) |
Second-order autocatalytic production and linear degradation of C create a threshold concentration for the reaction (Fig. S5 in Data S1). To provide an upper stable steady state appropriate for kinetics of blood clotting, the production term was modified in this work from a simple quadratic term to a fraction with limit equal to k1/k2 as [C] approaches infinity. See Data S1 for values of the kinetic rate constants. The three steady states in this model were 5.0 pM (lower stable state, “unclotted”), 0.9959 nM (unstable, [C]crit), and 1.1 μM (upper stable state; “clotted”) (31,69,73).
The initial condition was designed to approximate an experimental plug immediately after merging with a droplet of activator. Thirty percent of the area at the back of the plug initially contained [C]i = 0–10 nM, whereas the remainder of the plug initially contained a very small [C] corresponding to the “unclotted” steady state (Fig. 5 a). In the following discussion, “clotting” indicates convergence of [C] on the upper steady state (1.1 μM). In this model, it could be known with certainty whether a plug “clotted” or not by observing whether the average [C] converges on the “clotted” steady-state value (1.1 μM) or on the “unclotted” value (5 pM). The “clot time” was defined as the time at which a tangent to the initial slope of median [C] versus time intersected with a tangent to the maximum slope of generation of C (69), i.e., the time at which the rapid increase in median [C] began (Fig. S6 in Data S1).
FIGURE 5.

Rapid mixing prevents “clotting” in a numerical simulation of blood clotting, using an autocatalytic reaction with threshold kinetics. (a) At t = 0 s, the shaded region at the rear of the simulated plug has [C] = [C]i, whereas the remainder of the plug has [C] = 5 pM. (b) Images of plugs in a simulated winding channel with [C]i = 3 nM. The left plug shows the initial condition (t = 0 s). The slowly flowing plug (Vel = 0.35 mm/s) begins to “clot”; the quickly flowing plug (Vel = 3.5 mm/s) does not. The moving band upon which clotting is occurring is visible in the middle plug, though at the early time point that was chosen for clarity (t = 45 s), only a small amount of C has been made (concentration scale is above the plugs). (c) For [C]i = 3 nM, median [C] rises to the upper steady state (1.1 μM, “clotted”) or falls to the lower steady state (5 pM; “unclotted”) depending on the velocity of the plug. (d) A plot of reaction outcome (1 = “clotted”; 0 = “unclotted”) versus plug velocity shows that increasing the mixing rate shifts the system from “clotting” to not “clotting.” (e) “Clot” times for [C]i = 3.0 nM (circles) grow with faster flow until 2.5 mm/s and then jump to infinity; “clot” times for [C]i = 8.0 nM (triangles) increase steadily. (f) Locally, on a band of activator, “clotting” occurs if and only if τdiff > τrxn (Dalocal > 1). τdiff decreases exponentially with time due to chaotic mixing, as shown for P = 3.5 mm, d = 5 × 10−11 m2/s, d0 = 200 μm, and U = 1 mm/s. τrxn is constant and is determined by [C]i, here 3.0 nM. “Clotting” occurs globally in the plug if and only if tlocal > tcommit (Daglobal).
Rapid mixing prevents simulated clotting when the initial concentration of activator is near the critical concentration
In a simulated straight channel with Vel = 1 mm/s, the observed [C]i,crit was 3.0 nM (Fig. S7 in Data S1). “Clotting” occurred within 300 s. The observed [C]i,crit exceeds the mathematically defined [C]crit of 0.9959 nM due to the presence of recirculation and diffusional mixing in the flowing plug. To clarify, [C]crit is defined by the rate equations for the reaction and is the theoretical critical concentration under perfectly-mixed conditions. [C]i,crit is the experimental critical concentration, and since instantaneous perfect mixing is not possible experimentally, its value is affected by the rate of mixing and the initial distribution of reagents. Closer agreement between [C]i,crit and [C]crit is achieved when [C]i,crit is measured under conditions of minimal dispersal of C from the pocket and when the fraction of the total volume having [C]i at t = 0 is large enough to persist at least briefly in the presence of diffusion. Although fluid recirculation and diffusion are slow, these forces are nonetheless capable of eliminating some pockets of high [C] before they react. This difference was demonstrated in the simulation when “clotting” occurred in the simulated plug without flow (no recirculation) with [C]i ≥ 1.0 nM (data not shown).
In the simulation, “clotting” was completely prevented by rapid mixing when [C]i ≈ [C]i,crit (Fig. 5, c–e). In a simulated smooth, winding channel, plugs with [C]i = 3.0 nM clotted only under slow mixing (Vel ≤ 2.5 mm/s), not under fast mixing (Vel ≥ 3.0 mm/s) (Fig. 5, c and d, Movie S3, Movie S3 b, and Movie S4, and Movie S4 b). The mixing rate increased by only 15% (mixing time from 11.0 s to 9.5 s) between 2.5 mm/s and 3.0 mm/s (Fig. 4 f). “Clot times” at [C]i = 3.0 nM extended gradually under faster mixing, then jumped suddenly to infinity (Fig. 5 e, circles). As expected, “clotting” always occurred at high [C]i (8 nM) and never occurred at low [C]i (1 nM), independent of the mixing rate (Fig. S8 in Data S1).
Global Da predicts outcome of the reaction when the initial concentration of activator is near the critical concentration
We observed that the mixing rate necessary to prevent the reaction increased as the inequality [C]i > [C]i,crit in the pocket increased. This relation can be predicted by the Damköhler number, Da, when applied appropriately to the competition between reaction and mixing as the pocket of activator is stretched into the ever-thinning bands in the chaotically mixed plug (Fig. S9 in Data S1) (59,66). It is useful to think of this competition as occurring simultaneously at two lengthscales (7)—locally on the band, and globally in the entire solution. On a band with concentration [C]b (nM) and width d (mm), Dalocal predicts the outcome of the reaction on the band by comparing the timescales of diffusion and reaction as follows:
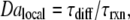 |
(3) |
where τdiff = d2/2D (s) is the time required for molecular diffusion of C with diffusion constant D (m2/s), and τrxn (s) is the time required for a uniform solution of [C]b to react, as predicted by Eq. 2 (Fig. S10 in Data S1). Because mixing is chaotic, d decreases exponentially with the number of chaotic mixing cycles, n, and thus with time, t (60,61):
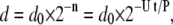 |
(4) |
where d0 is the width of the band at t = 0, U is the linear flow velocity, and P is the distance per period (mm). On the band, reaction will proceed to the upper steady state if Dalocal > 1, that is, if the rate of reaction that is producing C on the band is greater than the rate of diffusion that is removing C from the band (τrxn < τdiff). At the critical Dalocal = 1, we can solve Eq. 3 for the time of transition from Dalocal > 1 to Dalocal < 1, tlocal, as follows:
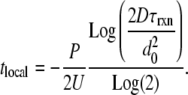 |
(5) |
The variable tlocal (s) indicates the time at which chaotic mixing has thinned the bands enough that diffusion becomes faster than reaction (Fig. 5 f) (7). See Data S1 for derivation and calculated values of Eq. 5. In a system with threshold kinetics at t > tlocal, diffusion prevents reaction on the band by lowering [C]b to <[C]crit.
Next, we consider the global scale of reaction and mixing in the entire solution, or plug. Globally, the solution will proceed to the upper steady state only if the average [C] throughout the plug becomes >[C]crit. This transition occurs at a time tcommit (s). This time is calculated by taking a ratio of the [C] that needs to be produced in the plug to reach [C]crit, divided by the total rate at which C is produced in the plug:
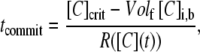 |
(6) |
where Volf = Volband/Voltot is the fraction of the volume occupied by the band, Volband (mm3), over the total volume, Voltot (mm3), and [C]i,b is the initial concentration on the band. We assume that Volf remains constant over time, neglecting the concentration gradient produced by diffusion at the edge of the band (15). This equation requires that the average [C] in the plug is <[C]crit initially. R([C](t)) (nM/s) is the total rate at which C is produced in the plug as the average [C] increases from [C]i,b × Volf to [C]crit. See Data S1 for a brief derivation of Eq. 6 including methods of approximating R([C](t)) (Eq. 10S), and for calculated values of tcommit (Fig. S11 in Data S1). The higher the initial concentration [C]i,b and the higher the volume fraction of the band, the less C needs to be produced to reach [C]crit and the smaller the value of tcommit. Similarly, the faster the rate of production R([C](t)), the smaller the value of tcommit.
To predict the outcome of the clotting reaction, the ratio Daglobal can be used to determine whether the length of time required for the average [C] to reach [C]crit, tcommit, falls within the length of time during which reaction is possible, t ≤ tlocal, as follows:
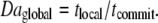 |
(7) |
Daglobal > 1 indicates that the global reaction will proceed to the upper steady state, because the time required for the average [C] to reach [C]crit is less than the time after which diffusion prevents local reaction. tcommit is set by the reaction kinetics and the initial concentration and distribution of C, whereas tlocal is set by these parameters, as well as by the rates of chaotic mixing and diffusion. In the simulation, we find that the global reaction will proceed to the upper steady state, i.e., clot, if and only if Daglobal > ∼2.5 (Fig. S12 in Data S1).
We can solve Eq. 7 for any of the parameters that contribute to Da and predict the outcome of the reaction as that parameter varies (24). For example, at Daglobal = 1 for all else fixed, the minimum flow rate that will prevent clotting in the overall solution can be predicted as follows:
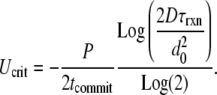 |
(8) |
The critical flow rate, Ucrit (mm/s), predicted by Eq. 8 was within a factor of 2 of the Ucrit observed in the numerical simulation at all values of [C]i except 2.2 nM, where it was within a factor of 3. See Data S1 for a brief derivation of Eq. 8 and calculated values of Ucrit.
Rapid mixing slows simulated and experimental clotting when the initial concentration of activator is far above the threshold
The behavior of the system upon exposure to a high initial concentration of activator is of interest, because many experiments are performed this way (31). In an autocatalytic system with threshold kinetics (Fig. 1 e), the mixing rate controls only the rate, not the outcome, of the reaction, if the initial concentration of activator is far above the critical concentration (22). In this case, the system behaves as a system without threshold kinetics (Fig. 1, c and d). In agreement with the qualitative predictions in Fig. 1 d, in the simulation, rapid mixing slowed “clotting” when [C]i was far above [C]crit. At [C]i = 8.0 nM in the simulation, faster mixing produced a linear increase in “clot” time (Fig. 5 e, triangles), without preventing the reaction. This system would not be prevented from “clotting” even at an infinitely high mixing rate, because [C] in the well-mixed, unreacted bulk solution, [C]0,avg, is >[C]crit (30% area contains 8.0 nM C initially, so [C]0,avg = 30% × 8.0 nM = 2.4 nM > 0.9959 nM). Mixing, and more specifically, Da, controls both the outcome and rate of the reaction only in cases where [C] > [C]crit in one or many pockets and the average [C] < [C]crit, as is the case in the simulation for [C]i = 3 nM.
Mixing also altered the rate of clotting of human NPP when the average [C] was above [C]i,crit (Figs. 6 and S13). We used microfluidic analogs of common clinical clotting assays, the APTT (Fig. 6 b) and the PT (Fig. 6 d), to examine clotting of NPP at high average [C]. To vary the mixing time for the PT and APTT assay experiments, the channel geometry after the merging junction was varied (see Materials and Methods). Faster mixing led to slower reaction in both of these assays. Though this result was shown at room temperature, mixing will likely have a similar effect on clotting at 37°C, the temperature at which clotting assays are usually performed. This result demonstrates the importance of mixing rate to the accuracy and reproducibility of clinical clotting assays, which are highly variable between labs and must be standardized by using ratios of clot times (74,75). Variability is also common in the same lab over time. One explanation could be that mixing rates vary between clinical instruments and change further as the instrument ages. These results indicate that even a slight decrease in mixing rate could shorten the reaction time significantly, which implies that variable mixing rates in clinical instruments could impact their accuracy.
DISCUSSION
The results of the experiments and simulations presented here demonstrate the significant influence of mixing on the kinetics of autocatalytic systems with a threshold response to the concentration of activator, including biological systems such as blood coagulation, and provide insight into the slowing of autocatalytic chemical reactions by rapid mixing (1,6,9,17,22,23). The global Damköhler number derived here includes the effects of both chaotic mixing and molecular diffusion and can be used to predict the outcome of autocatalytic and threshold-limited reactions at various initial concentrations of activator and flow rates. The Damköhler number has been used previously to describe the effect of mixing on theoretical and chemical systems with autocatalysis or bistability (22,76). For these nonlinear systems, the classical parameters T, [C], and k are not sufficient to describe the reaction rates in detail (1). These results indicate that mixing should be considered (55) when describing the kinetics of autocatalytic biological systems.
These results are the first experimental evidence that a threshold exists for activation of plasma by thrombin. This is a true threshold, because there is no response (no fibrin formation; no thrombin formation detected by fluorescent substrate) below a finite, nonzero concentration of activator (33). Though shown at room temperature, the observed critical concentration of thrombin necessary to initiate clotting in NPP (0.3–0.6 nM [C]i) is consistent with predictions obtained at 37°C (69) (see below). Thrombin is the primary activator of platelets (25) and is part of a positive-feedback loop with platelets that is required for clot formation (33). In whole blood, ∼0.5 nM thrombin is present at the time of platelet activation (77). The threshold observed in these experiments may correspond to the threshold for platelet activation, though this is speculative and has not been tested in these experiments, which used platelet-poor plasma (∼400 platelets present in each 40-nL plug). The observed critical concentration of thrombin is also close to the 0.8 nM concentration (77) that is present at the inception of factor Va generation, an important feedback step, and factor XIII activation, though the latter is less relevant here.
The effect of mixing on the rate of clotting may explain some of the variability seen in routine clinical clotting tests, which are often performed in a simple U-shaped chamber and mixed by parabolic motion of a magnetic bead. Normal test results have a fairly broad range and vary between instruments (74,75). Stochastic initiation of blood clotting has also been observed previously in reactions carried out in microtiter plates (48,49). Though much of the clinical variation may be due to preanalytical errors or variables (78), our results show that some variability in the clinical and well-plate experiments could be attributed to variability in mixing in each experiment. Fortunately, control of mixing can likely be improved with relatively small modifications to existing instrumentation, and may improve the ability of physicians to distinguish healthy patients from those with small but critical shifts in their clotting dynamics.
These results indicate that blood clotting probably occurs in response to the initial concentration of added activator, [C]i, not the well-mixed concentration, [C]0,avg (Fig. 1). Since blood clotting is often initiated experimentally by the addition of a small volume of concentrated activator (i.e., CaCl2 in APTT assays, TF solution in PT assays, or plasma proteins such as thrombin, factor Xa, XIIa, etc.), the rate of mixing will have a large influence on the rate or outcome of the coagulation reactions. In many experiments, a 4-mL blood sample is drawn directly into a tube that contains <35 μL of concentrated activator solution at the bottom, and mixing is performed by inversion or vortexing. In such an experiment, the concentrated activator is probably mixed into the bulk relatively slowly. Thus, though 5.3 pM ([C]0,avg) amounts of thrombin added directly to NPP have been shown to cause clotting (69), the clot-inducing concentration may have actually been the 0.6 nM initial value ([C]i, assuming 35 μL initial and 4 mL final volume), which is near the critical concentration shown here. The importance of the initial concentration of activator and the mixing rate should also be pertinent to any autocatalytic or threshold-bearing system that is typically activated by using small volumes, including classical inorganic, nonlinear reaction systems (1,2) and larger lengthscale systems, such as ozone depletion in the atmosphere and plankton production in the ocean (76).
It is interesting that when systems with threshold dynamics were held near their threshold, the outcome in each case appeared to be dictated by subtle factors that vary from trial to trial. In experiments with blood plasma and in the chemical model, plugs that merged with activator near the critical concentration, and if applicable, near the critical flow rate, clotted somewhat stochastically. Only 40–60% of plugs clotted, and the remaining plugs remained completely unclotted in response to the activator. We observed this percentage to be roughly constant for >30 min (300–1000 plugs; data not shown). Which plugs would clot under these conditions was not predictable. Because mixing in every plug is highly reproducible and controlled by deterministic chaotic advection (61), we attribute this stochastic behavior to either slight variation in the concentration of reagents in plugs, or to truly stochastic higher-order processes, well established in nucleation (12,79) but not yet documented in clotting. A possible candidate for such a process is aggregation of platelets. Because many mechanisms can lead to threshold behavior, these results are likely applicable to many chemical and biological systems. Other apparently stochastic biological behaviors may be operating under a similar mechanism; they may be functioning very near some threshold and thus may be susceptible to otherwise insignificant variations in their environment.
Here, we also present an easy-to-use numerical simulation of simultaneous chaotic mixing and chemical reaction occurring inside a plug. Defining the flow velocities along the top and bottom edges of the plug provides a simple and direct method to simulate the fluid flow inside the nonwetting plug in a time-continuous manner (66). Because the area of interest for the reaction is confined to the plug itself, it is not necessary to include the fluid dynamics of the surrounding carrier fluid or channel walls in the simulation, nor to define flow profiles at discontinuous points in a winding channel (80). This approach reduces computational cost, though it does neglect the additional mixing that results from changes in the shape of an experimental plug in response to channel geometry (81) and could not incorporate chemical species that travel between the two liquid phases (82,83). The current version includes a single species of reactant, but the simulation could easily be scaled up to include multiple chemical species or to include heat evolution and transfer. This type of numerical simulation should prove useful for understanding the mechanics of chemical or biological reactions inside plugs.
The findings from this study may be applicable to a range of chemical and biological systems. A number of different mechanisms can lead to threshold or nonlinear kinetics (21) in addition to the autocatalytic mechanism used here, making the results general beyond systems in which thresholds arise due to autocatalysis. These results indicate that the dynamics of any system controlled by nonlinear kinetics or a threshold concentration should be more sensitive to the initial, local concentration of activator than to the average concentration of activator throughout the system, be it a cellular compartment, tissue section, or fluid sample. In addition, the behavior of such a system should be dependent on the rate at which the activator is mixed into the sample, either by diffusion, convection, or active transport. The effect of the initial local concentration and the rate of mixing will be especially important in a system where an activating species is initially produced in a small or confined pocket, such as inside a cellular organelle, atop a membrane, or within a neurological or immunological synapse. The crowded environment found in biological milieu should amplify this effect by decreasing the rate of diffusion and material transport in the space near the concentrated pocket of activator. This study suggests an exciting opportunity—which we believe is already widely used in biological systems—to control both the rate and outcome of chemical and biological processes simply by controlling the rate of transport phenomena, such as mixing and diffusion.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
This work was supported in part by ONR under Grant No. N00014-03-10482, the NIBIB under Grant No. ROI EB001903-04, and by the Camille Dreyfus Teacher-Scholar Awards Program. R.F.I. is a Cottrell Scholar of Research Corporation and an A. P. Sloan Research Fellow. Some of this work was performed at the MRSEC microfluidic facility (funded by the NSF). We thank Jessica M. Price for contributions in writing and editing this manuscript.
Editor: Petra Schwille.
References
- 1.Epstein, I. R. 1995. The consequences of imperfect mixing in autocatalytic chemical and biological systems. Nature. 374:321–327. [DOI] [PubMed] [Google Scholar]
- 2.Nagypal, I., and I. R. Epstein. 1986. Systematic design of chemical oscillators. 37. Fluctuations and stirring rate effects in the chlorite thiosulfate reaction. J. Phys. Chem. 90:6285–6292. [Google Scholar]
- 3.Gyorgyi, L., and R. J. Field. 1989. Aperiodicity resulting from 2-cycle coupling in the Belousov-Zhabotinskii reaction. 3. Analysis of a model of the effect of spatial inhomogeneities at the input ports of a continuous-flow stirred tank reactor. J. Chem. Phys. 91:6131–6141. [Google Scholar]
- 4.Roux, J. C., P. Dekepper, and J. Boissonade. 1983. Experimentalevidence of nucleation induced transition in a bistable chemical system. Phys. Lett. A. 97:168–170. [Google Scholar]
- 5.Hlavacova, J., and P. Sevcik. 1994. Numerical study of concentration fluctuations and stirring effects in a batch reactor. Comput. Chem. 18:21–25. [Google Scholar]
- 6.Gerdts, C. J., D. E. Sharoyan, and R. F. Ismagilov. 2004. A synthetic reaction network: chemical amplification using nonequilibrium autocatalytic reactions coupled in time. J. Am. Chem. Soc. 126:6327–6331. [DOI] [PubMed] [Google Scholar]
- 7.Bourne, J. R. 2003. Mixing and the selectivity of chemical reactions. Org. Process Res. Dev. 7:471–508. [Google Scholar]
- 8.Chen, D. L., C. J. Gerdts, and R. F. Ismagilov. 2005. Using microfluidics to observe the effect of mixing on nucleation of protein crystals. J. Am. Chem. Soc. 127:9672–9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kondepudi, D. K., and K. Asakura. 2001. Chiral autocatalysis, spontaneous symmetry breaking, and stochastic behavior. Acc. Chem. Res. 34:946–954. [DOI] [PubMed] [Google Scholar]
- 10.Epstein, I. R. 1990. Physical chemistry: shaken, stirred—but not mixed. Nature. 346:16–17. [Google Scholar]
- 11.Dykman, M. I., E. Mori, J. Ross, and P. M. Hunt. 1994. Large fluctuations and optimal paths in chemical kinetics. J. Chem. Phys. 100:5735–5750. [Google Scholar]
- 12.Ferrone, F. A., J. Hofrichter, and W. A. Eaton. 1985. Kinetics of sickle hemoglobin polymerization. 2. A double nucleation mechanism. J. Mol. Biol. 183:611–631. [DOI] [PubMed] [Google Scholar]
- 13.Tyson, J. J., and J. P. Keener. 1988. Singular perturbation theory of traveling waves in excitable media. Physica D. 32:327–361. [Google Scholar]
- 14.Hlavacova, J., and P. Sevcik. 1994. Coherence between the stirring effect in bimolecular reactions and the Belousov-Zhabotinskii reaction in the closed batch reactor. J. Phys. Chem. 98:6304–6307. [Google Scholar]
- 15.Ott, R. J., and P. Rys. 1975. Chemical selectivities disguised by mass diffusion. 1. Simple model of mixing disguised reactions in solution: 1st communication on selectivity of chemical processes. Helv. Chim. Acta. 58:2074–2093. [Google Scholar]
- 16.Villermaux, J. 1991. Mixing effects on complex chemical reactions in a stirred reactor. Rev. Chem. Eng. 7:51–108. [Google Scholar]
- 17.Ottino, J. M. 1994. Mixing and chemical reactions: a tutorial. Chem. Eng. Sci. 49:4005–4027. [Google Scholar]
- 18.Ruoff, P. 1992. Chaos in batch Belousov-Zhabotinsky systems. J. Phys. Chem. 96:9104–9106. [Google Scholar]
- 19.Vanag, V. K., and D. P. Melikhov. 1995. Asymmetrical concentration fluctuations in the autocatalytic bromate-bromide-catalyst reaction and in the oscillatory Belousov-Zhabotinsky reaction in closed reactor: stirring effects. J. Phys. Chem. 99:17372–17379. [Google Scholar]
- 20.Hlavacova, J., and P. Sevcik. 1993. Concentration fluctuations and the simulation of stirring effects in the Belousov-Zhabotinskii reaction. Chem. Phys. Lett. 201:242–246. [Google Scholar]
- 21.Tyson, J. J., K. C. Chen, and B. Novak. 2003. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 15:221–231. [DOI] [PubMed] [Google Scholar]
- 22.Neufeld, Z., P. H. Haynes, and T. Tel. 2002. Chaotic mixing induced transitions in reaction-diffusion systems. Chaos. 12:426–438. [DOI] [PubMed] [Google Scholar]
- 23.Zorzano, M. P., D. Hochberg, and F. Moran. 2006. Consequences of imperfect mixing: the Gray-Scott model. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 74:057102. [DOI] [PubMed] [Google Scholar]
- 24.Marion, G., X. R. Mao, E. Renshaw, and J. L. Liu. 2002. Spatial heterogeneity and the stability of reaction states in autocatalysis. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 66:051915. [DOI] [PubMed] [Google Scholar]
- 25.Mann, K. G., K. Brummel-Ziedins, T. Orfeo, and S. Butenas. 2006. Models of blood coagulation. Blood Cells Mol. Dis. 36:108–117. [DOI] [PubMed] [Google Scholar]
- 26.Mann, K. G. 1999. Biochemistry and physiology of blood coagulation. Thromb. Haemost. 82:165–174. [PubMed] [Google Scholar]
- 27.Mannall, G. J., N. J. Titchener-Hooker, H. A. Chase, and P. A. Dalby. 2006. A critical assessment of the impact of mixing on dilution refolding. Biotechnol. Bioeng. 93:955–963. [DOI] [PubMed] [Google Scholar]
- 28.Karolyi, G., A. Pentek, I. Scheuring, T. Tel, and Z. Toroczkai. 2000. Chaotic flow: the physics of species coexistence. Proc. Natl. Acad. Sci. USA. 97:13661–13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin, A. P. 2003. Phytoplankton patchiness: the role of lateral stirring and mixing. Prog. Oceanogr. 57:125–174. [Google Scholar]
- 30.Kerr, B., M. A. Riley, M. W. Feldman, and B. J. M. Bohannan. 2002. Local dispersal promotes biodiversity in a real-life game of rock-paper-scissors. Nature. 418:171–174. [DOI] [PubMed] [Google Scholar]
- 31.Mann, K. G., K. Brummel, and S. Butenas. 2003. What is all that thrombin for? J. Thromb. Haemost. 1:1504–1514. [DOI] [PubMed] [Google Scholar]
- 32.van 't Veer, C., and K. G. Mann. 1997. Regulation of tissue factor initiated thrombin generation by the stoichiometric inhibitors tissue factor pathway inhibitor, antithrombin-III, and heparin cofactor-II. J. Biol. Chem. 272:4367–4377. [DOI] [PubMed] [Google Scholar]
- 33.Jesty, J., and E. Beltrami. 2005. Positive feedbacks of coagulation: their role in threshold regulation. Arterioscler. Thromb. Vasc. Biol. 25:2463–2469. [DOI] [PubMed] [Google Scholar]
- 34.Beltrami, E., and J. Jesty. 1995. Mathematical analysis of activation thresholds in enzyme-catalyzed positive feedbacks: application to the feedbacks of blood coagulation. Proc. Natl. Acad. Sci. USA. 92:8744–8748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jesty, J., E. Beltrami, and G. Willems. 1993. Mathematical analysis of a proteolytic positive-feedback loop: dependence of lag time and enzyme yields on the initial conditions and kinetic parameters. Biochemistry. 32:6266–6274. [DOI] [PubMed] [Google Scholar]
- 36.Kastrup, C. J., F. Shen, M. K. Runyon, and R. F. Ismagilov. 2007. Characterization of the threshold response of initiation of blood clotting to stimulus patch size. Biophys. J. 93:2969–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kastrup, C. J., M. K. Runyon, F. Shen, and R. F. Ismagilov. 2006. Modular chemical mechanism predicts spatiotemporal dynamics of initiation in the complex network of hemostasis. Proc. Natl. Acad. Sci. USA. 103:15747–15752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beltrami, E., and J. Jesty. 2001. The role of membrane patch size and flow in regulating a proteolytic feedback threshold on a membrane: possible application in blood coagulation. Math. Biosci. 172:1–13. [DOI] [PubMed] [Google Scholar]
- 39.Turitto, V. T., and C. L. Hall. 1998. Mechanical factors affecting hemostasis and thrombosis. Thromb. Res. 92:S25–S31. [DOI] [PubMed] [Google Scholar]
- 40.Slack, S. M., Y. W. Cui, and V. T. Turitto. 1993. The effects of flow on blood-coagulation and thrombosis. Thromb. Haemost. 70:129–134. [PubMed] [Google Scholar]
- 41.Goldsmith, H. L., and V. T. Turitto. 1986. Rheological aspects of thrombosis and hemostasis: basic principles and applications. ICTH report: Subcommittee on Rheology of the International Committee on Thrombosis and Hemostasis. Thromb. Haemost. 55:415–435. [PubMed] [Google Scholar]
- 42.Turitto, V. T. 1982. Blood viscosity, mass-transport, and thrombogenesis. Prog. Hemost. Thromb. 6:139–177. [PubMed] [Google Scholar]
- 43.Ataullakhanov, F. I., and M. A. Panteleev. 2005. Mathematical modeling and computer simulation in blood coagulation. Pathophysiol. Haemost. Thromb. 34:60–70. [DOI] [PubMed] [Google Scholar]
- 44.Ermakova, E. A., M. A. Panteleev, and E. E. Shnol. 2005. Blood coagulation and propagation of autowaves in flow. Pathophysiol. Haemost. Thromb. 34:135–142. [DOI] [PubMed] [Google Scholar]
- 45.Runyon, M. K., B. L. Johnson-Kerner, C. J. Kastrup, T. G. Van Ha, and R. F. Ismagilov. 2007. Propagation of blood clotting in the complex biochemical network of hemostasis is described by a simple mechanism. J. Am. Chem. Soc. 129:7014–7015. [DOI] [PubMed] [Google Scholar]
- 46.Song, H., H. W. Li, M. S. Munson, T. G. Van Ha, and R. F. Ismagilov. 2006. On-chip titration of an anticoagulant argatroban and determination of the clotting time within whole blood or plasma using a plug-based microfluidic system. Anal. Chem. 78:4839–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hathcock, J. J. 2006. Flow effects on coagulation and thrombosis. Arterioscler. Thromb. Vasc. Biol. 26:1729–1737. [DOI] [PubMed] [Google Scholar]
- 48.Lo, K., and S. L. Diamond. 2004. Blood coagulation kinetics: high throughput method for real-time reaction monitoring. Thromb. Haemost. 92:874–882. [DOI] [PubMed] [Google Scholar]
- 49.Lo, K., W. S. Denney, and S. L. Diamond. 2005. Stochastic modeling of blood coagulation initiation. Pathophysiol. Haemost. Thromb. 34:80–90. [DOI] [PubMed] [Google Scholar]
- 50.Kantarci, N., F. Borak, and K. O. Ulgen. 2005. Bubble column reactors. Process Biochem. 40:2263–2283. [Google Scholar]
- 51.Gerson, D. F., and M. M. Kole. 2001. Quantitative measurements of mixing intensity in shake-flasks and stirred tank reactors: use of the Mixmeter, a mixing process analyzer. Biochem. Eng. J. 7:153–156. [DOI] [PubMed] [Google Scholar]
- 52.Buchs, J. 2001. Introduction to advantages and problems of shaken cultures. Biochem. Eng. J. 7:91–98. [DOI] [PubMed] [Google Scholar]
- 53.Weiss, S., G. T. John, I. Klimant, and E. Heinzle. 2002. Modeling of mixing in 96-well microplates observed with fluorescence indicators. Biotechnol. Prog. 18:821–830. [DOI] [PubMed] [Google Scholar]
- 54.Song, H., D. L. Chen, and R. F. Ismagilov. 2006. Reactions in droplets in microflulidic channels. Angew. Chem. Int. Ed. 45:7336–7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song, H., and R. F. Ismagilov. 2003. Millisecond kinetics on a microfluidic chip using nanoliters of reagents. J. Am. Chem. Soc. 125:14613–14619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tice, J. D., A. D. Lyon, and R. F. Ismagilov. 2004. Effects of viscosity on droplet formation and mixing in microfluidic channels. Anal. Chim. Acta. 507:73–77.17186061 [Google Scholar]
- 57.Tice, J. D., H. Song, A. D. Lyon, and R. F. Ismagilov. 2003. Formation of droplets and mixing in multiphase microfluidics at low values of the Reynolds and the capillary numbers. Langmuir. 19:9127–9133. [Google Scholar]
- 58.Runyon, M. K., B. L. Johnson-Kerner, and R. F. Ismagilov. 2004. Minimal functional model of hemostasis in a biomimetic microfluidic system. Angew. Chem. Int. Ed. 43:1531–1536. [DOI] [PubMed] [Google Scholar]
- 59.Bringer, M. R., C. J. Gerdts, H. Song, J. D. Tice, and R. F. Ismagilov. 2004. Microfluidic systems for chemical kinetics that rely on chaotic mixing in droplets. Philos. Trans. R. Soc. Lond. A. 362:1087–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ottino, J. M., and S. Wiggins. 2004. Introduction: mixing in microfluidics. Philos. Trans. R. Soc. Lond. A. 362:923–935. [DOI] [PubMed] [Google Scholar]
- 61.Song, H., M. R. Bringer, J. D. Tice, C. J. Gerdts, and R. F. Ismagilov. 2003. Experimental test of scaling of mixing by chaotic advection in droplets moving through microfluidic channels. Appl. Phys. Lett. 83:4664–4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roach, L. S., H. Song, and R. F. Ismagilov. 2005. Controlling nonspecific protein adsorption in a plug-based microfluidic system by controlling interfacial chemistry using fluorous-phase surfactants. Anal. Chem. 77:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liau, A., R. Karnik, A. Majumdar, and J. H. D. Cate. 2005. Mixing crowded biological solutions in milliseconds. Anal. Chem. 77:7618–7625. [DOI] [PubMed] [Google Scholar]
- 64.Wiggins, S., and J. M. Ottino. 2004. Foundations of chaotic mixing. Philos. Trans. R. Soc. Lond. A. 362:937–970. [DOI] [PubMed] [Google Scholar]
- 65.Leong, C. W., and J. M. Ottino. 1989. Experiments on mixing due to chaotic advection in a cavity. J. Fluid Mech. 209:463–499. [Google Scholar]
- 66.Ottino, J. M., C. W. Leong, H. Rising, and P. D. Swanson. 1988. Morphological structures produced by mixing in chaotic flows. Nature. 333:419–425. [Google Scholar]
- 67.Stroock, A. D., S. K. W. Dertinger, A. Ajdari, I. Mezic, H. A. Stone, and G. M. Whitesides. 2002. Chaotic mixer for microchannels. Science. 295:647–651. [DOI] [PubMed] [Google Scholar]
- 68.Basmadjian, D., M. V. Sefton, and S. A. Baldwin. 1997. Coagulation on biomaterials in flowing blood: some theoretical considerations. Biomaterials. 18:1511–1522. [DOI] [PubMed] [Google Scholar]
- 69.Butenas, S., T. Orfeo, M. T. Gissel, K. E. Brummel, and K. G. Mann. 2004. The significance of circulating factor IXa in blood. J. Biol. Chem. 279:22875–22882. [DOI] [PubMed] [Google Scholar]
- 70.Tanthapanichakoon, W., N. Aoki, K. Matsuyama, and K. Mae. 2006. Design of mixing in microfluidic liquid slugs based on a new dimensionless number for precise reaction and mixing operations. Chem. Eng. Sci. 61:4220–4232. [Google Scholar]
- 71.Handique, K., and M. A. Burns. 2001. Mathematical modeling of drop mixing in a slit-type microchannel. J. Micromech. Microeng. 11:548–554. [Google Scholar]
- 72.Kashid, M. N., I. Gerlach, S. Goetz, J. Franzke, J. F. Acker, F. Platte, D. W. Agar, and S. Turek. 2005. Internal circulation within the liquid slugs of a liquid-liquid slug-flow capillary microreactor. Ind. Eng. Chem. Res. 44:5003–5010. [Google Scholar]
- 73.Runyon, M. K., C. J. Kastrup, B. Johnson-Kerner, T. G. V. Ha, and R. F. Ismagilov. 2008. The effects of shear rate on propagation of blood clotting determined using microfluidics and numerical simulations. J. Am. Chem. Soc. 130:3458–3464. [DOI] [PubMed] [Google Scholar]
- 74.Doherty, T. M., R. M. Shavelle, and W. J. French. 2005. Reproducibility and variability of activated clotting time measurements in the cardiac catheterization laboratory. Catheter. Cardiovasc. Interv. 65:330–337. [DOI] [PubMed] [Google Scholar]
- 75.Nutescu, E. A. 2004. Point of care monitors for oral anticoagulant therapy. Semin. Thromb. Hemost. 30:697–702. [DOI] [PubMed] [Google Scholar]
- 76.Tel, T., A. de Moura, C. Grebogi, and G. Karolyi. 2005. Chemical and biological activity in open flows: a dynamical system approach. Phys. Rep. 413:91–196. [Google Scholar]
- 77.Brummel, K. E., S. G. Paradis, S. Butenas, and K. G. Mann. 2002. Thrombin functions during tissue factor-induced blood coagulation. Blood. 100:148–152. [DOI] [PubMed] [Google Scholar]
- 78.Bonini, P., M. Plebani, F. Ceriotti, and F. Rubboli. 2002. Errors in laboratory medicine. Clin. Chem. 48:691–698. [PubMed] [Google Scholar]
- 79.Szabo, A. 1988. Fluctuations in the polymerization of sickle hemoglobin: a simple analytic model. J. Mol. Biol. 199:539–542. [DOI] [PubMed] [Google Scholar]
- 80.Stone, Z. B., and H. A. Stone. 2005. Imaging and quantifying mixing in a model droplet micromixer. Phys. Fluids. 17:063103. [Google Scholar]
- 81.Muradoglu, M., and H. A. Stone. 2005. Mixing in a drop moving through a serpentine channel: a computational study. Phys. Fluids. 17:073305. [Google Scholar]
- 82.Kashid, M. N., D. W. Agar, and S. Turek. 2007. CFD modelling of mass transfer with and without chemical reaction in the liquid-liquid slug flow microreactor. Chem. Eng. Sci. 62:5102–5109. [Google Scholar]
- 83.Burns, J. R., and C. Ramshaw. 2001. The intensification of rapid reactions in multiphase systems using slug flow in capillaries. Lab Chip. 1:10–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.