

Abstract
When a long DNA molecule is stretched beyond its B-form contour length, a transition occurs in which its length increases by a factor of 1.7, with very little force increase. A quantitative model was proposed to describe this transition as force-induced melting, where double-stranded DNA is converted into single-stranded DNA. The force-induced melting model accurately describes the thermodynamics of DNA overstretching as a function of solution conditions and in the presence of DNA binding ligands. An alternative explanation suggests a transformation into S-DNA, a double-stranded form which preserves the interstrand base pairing. To determine the extent to which DNA base pairs are exposed to solution during the transition, we held DNA overstretched to different lengths within the transition in the presence of glyoxal. If overstretching involved strand separation, then force-melted basepairs would be glyoxal-modified, thus essentially permanently single-stranded. Subsequent stretches confirm that a significant fraction of the DNA melted by force is permanently melted. This result demonstrates that DNA overstretching is accompanied by a disruption of the DNA helical structure, including a loss of hydrogen bonding.
INTRODUCTION
Optical tweezers have been used extensively for studying the mechanical properties of single biomolecules, such as DNA, by allowing the molecule to be mechanically distorted under various conditions (1–6). As double-stranded DNA (dsDNA) that is torsionally unconstrained is stretched, its response appears elastic because of entropy. By further stretching dsDNA, a sudden transition occurs, as the molecule is elongated to almost twice its contour length without a strong increase in force. The resulting plateau in the force-extension curve shows the cooperative overstretching transition. Single-stranded DNA (ssDNA), when stretched at this force, is ∼1.7 times longer than the same strand wound in the B-DNA helix, and therefore ssDNA is favored by force near the transition. In addition, the mechanical work performed on the molecule within the cycle when it is stretched as dsDNA and relaxed as ssDNA is consistent with the free energy of DNA melting (7,8). Several other characteristics of the transition are consistent with the equilibrium melting nature of this transition, including its high reproducibility, its apparent independence of the pulling rate, and the small hysteresis observed upon DNA relaxation in high salt, which increases in lower salt. In contrast, stretching beyond the plateau leads to an abrupt increase in force, until complete strand separation occurs above 120 pN, at a force which is dependent upon the pulling rate (9–11).
An alternative explanation for DNA overstretching hypothesizes a secondary structural transition from B-form dsDNA into a stretched form of dsDNA termed S-DNA (10–13), in which the dsDNA unwinds and forms a ladder-like structure, with the base-stacking largely disrupted, but most of the interstrand hydrogen bonding preserved, as suggested by early computer simulation studies (14–16). In this model, the final strand separation does not occur until after the overstretching plateau, where stretching becomes nonequilibrium (14–17). The S-DNA model is difficult to test because experimentally no information is known about it, except that the energetics of the B-DNA to S-DNA transition is expected to be very much the same as for B-DNA melting (18). Attempts were made to deduce the elasticity and structural parameters for S-DNA from the small part of the DNA extension curve at forces above the plateau (19,20), or from the residual apparent DNA strand winding remaining after the plateau (2). However, because this portion of the DNA stretching curve is typically nonequilibrium (11), these parameters cannot be considered as equilibrium properties of a new dsDNA structure.
Several modeling studies were performed to deduce the structure of S-DNA. The early modeling studies (16,21) all suggested quite different S-DNA structures that were also dependent on the strand attachment (i.e., via one DNA strand at each end, e.g., via 5′5′, 3′3′, or 5′3′ ends). In addition, the calculated free energy required for reaching these structures was always approximately an order of magnitude higher than the experimentally measured mechanical work of overstretching. An additional problem with the modeled S-DNA structures is that no explanation for the experimentally observed high cooperativity of the overstretching transition was found within the B-to-S modeled transformation (16,21). More recent modeling studies (22–24) that more adequately sampled the large entropy of the melted DNA state showed that force-induced melting is an energetically much more favorable process than unstacking while retaining hydrogen bonding.
The major open question of the force-induced melting (FIM) model of DNA overstretching concerns how forces much higher than the transition plateau force, Fm, can be supported by just a few basepairing interactions remaining by the end of the transition. In future work, we will show that this is a kinetic phenomenon, fully consistent with the removal of most of the basepairing interactions during the transition (M. J. McCauley, L. Shokri, I. Rouzina, and M. C. Williams, in preparation). Another open question also concerns the kinetics of FIM. Indeed, a recent modeling study of this process (25) suggests that the melting plateau is expected to depend significantly on the pulling rate, whereas experimentally it does not. As will be shown elsewhere, FIM is indeed expected to appear quasi-equilibrium, i.e., essentially pulling rate-independent, given that large DNA fragments melt cooperatively within the continuous DNA duplex, rather than one basepair after another at the duplex/single-strand boundary.
The FIM model quantitatively predicted the dependence of the overstretching transition on solution conditions such as pH, temperature, ionic strength, and the presence of DNA binding ligands (7,8). These predictions were tested experimentally, and the results agree with the expected FIM behavior. Experiments monitoring the pH and temperature dependence of the transition reveal that the basepairs are broken as the DNA is stretched (26,27), whereas salt-dependent measurements are in agreement with thermal melting studies, and show that the two strands remain close and stretch together (5). This implies that melting occurs primarily within the internal domains rather than from the free ends. Moreover, experiments in the presence of ssDNA binding proteins or DNA binding drugs resemble those obtained in thermal melting experiments, and are quantitatively consistent with the FIM model (28–36). Although there is substantial thermodynamic evidence in favor of the FIM model, and no predictive and quantitative model for S-DNA has appeared, it has been suggested that S-DNA remains a possibility or that the model is under debate, although it may exist only as a metastable state or under specific solution conditions (2,19,20,25,38).
In this study, we attempt to determine the extent to which DNA basepairs are exposed to solution during the transition, as a further test of the validity of the FIM model without relying on thermodynamic conclusions. To accomplish this, we undertook a study of λ-DNA overstretching in the presence of glyoxal, a chemical that forms a stable DNA adduct with exposed guanine residues. It introduces an additional ring to the G base, thereby sterically preventing GC basepair (bp) reannealing (39). Glyoxal was previously used to map DNA thermal melting (40,41). It is the use of glyoxal (42–46) that first allowed researchers to prove that DNA melting proceeds via steps in which individual ∼100–500 bp segments melt out cooperatively, resulting in the peaks in differential DNA melting profiles (i.e., in the df/dT vs. T curves, where f is the fraction of DNA basepairs melted). More specifically, when DNA was incubated with glyoxal at some fixed temperature close to but lower than the average melting temperature, Tm, only certain AT-rich fragments of DNA were permanently fixed in the single-stranded state with glyoxal (40,41). Determining the location of these sites, by either single-strand digestion (40,46) or electron microscopy (42,47,48), led to the identification of these low-melting sites, with the sites predicted by direct calculations for the particular DNA sequence (48).
Here we use the same approach to quantify the fraction of DNA basepairs that are exposed as DNA is overstretched. In contrast to conventional thermal melting experiments, where the fraction of DNA basepairs melted is quantified at each temperature by ultraviolet spectroscopy, in our single-molecule study, we can fix fractional DNA melting by fixing the molecular end-to-end extension at some given position within the FIM transition range. If the overstretching transition is indeed FIM, then the fractional basepair melting, f, at any extension during this transition is defined by the relative closeness of the molecular extension to either dsDNA or ssDNA length, as illustrated schematically in Fig. 1. Therefore, we expect that glyoxal exposure of DNA stretched to the fractional extension f into the plateau should result in the permanent melting of the fraction f of DNA basepairs. The latter effect should be observed in subsequent DNA stretches, in which the molecule is expected to behave as a linear combination of ds (1− f) and ss (f) fractions of DNA. In contrast, if the S-DNA model holds, the molecular extension of the DNA should change very little after glyoxal treatment, as illustrated in Fig. 1.
FIGURE 1.

Schematic diagram of expected effect of glyoxal on DNA stretching and relaxation for the FIM model and the S-DNA model. (a) Model for DNA FIM, in which DNA base-stacking and basepairing interactions are progressively broken as DNA is overstretched. (b) Expected effect of glyoxal on DNA, in which exposed basepairs are modified by glyoxal, represented by spheres attached to bases in the diagram. Although we show all exposed bases modified for simplicity, it is likely that primarily guanine bases are modified under the conditions used here. (c) In the FIM model, at forces below the overstretching force, shown here at ∼30 pN, bases that were previously modified at the overstretching force remain modified, and upon relaxation remain single-stranded, resulting in a longer DNA molecule at that force relative to the length of a fully double-stranded molecule. (d) In the S-DNA model, DNA basepairs are expected to remain hydrogen-bonded, but to unstack, as shown here. (e) Because S-DNA remains basepaired, glyoxal would not be expected to modify overstretched DNA in this model, except perhaps for some frayed DNA at the end of the molecule. Upon relaxation to 30 pN, basepairs previously converted to S-DNA revert to the normal B-form double helix. The length of DNA at 30 pN should be essentially the same as that observed without glyoxal treatment, if the S-DNA model is correct.
We show that a majority of elongated DNA basepairs become permanently single-stranded under the action of glyoxal. Because the glyoxal modification is very selective for melted DNA bases (40–47), this result demonstrates the high accessibility of the bases in their overstretched state. The glyoxal modification therefore argues in favor of the ssDNA nature of overstretched DNA, and against a basepaired S-DNA state (13–16). To find optimum conditions for the selective modification of force-melted bases, we studied this reaction as a function of glyoxal concentration, i.e., time of exposure, over a range of solution pH values of 7.5–9.7, and at two different salt conditions of 100 mM and 5 mM Na+. We found that ssDNA fixation at 100 mM Na+ requires high glyoxal concentrations of ∼0.5 M and long glyoxal exposures of ∼30 min. The reaction with glyoxal appeared to be much more efficient in low salt, whereas the pH increase had only a minor facilitating effect on this reaction. We discuss the possible reasons for these salt-dependent differences in apparent fractional DNA melting.
MATERIALS AND METHODS
The dual-beam optical-tweezers instrument used in this study consists of two counter-propagating diode lasers, each with 200 mW of 830-nm light (JDS Uniphase, San Jose, CA) that are convergently directed and focused to a small spot inside a liquid flow cell, using 60×, 1.0 numerical aperture water immersion microscope objectives (Nikon, Tokyo, Japan) that form the optical trap (49). The light leaving the trap is directed onto a lateral-effect photodiode detector (UDT Sensors, Hawthorne, CA) that determines the deflection of each beam, and outputs a voltage proportional to the force exerted on the bead in the optical trap.
To capture single DNA molecules, two 5-μm streptavidin-coated polystyrene beads (Bangs Laboratories, Fishers, IN) were held in the optical tweezers and on the end of a glass micropipette (World Precision Instrument, Sarasota, FL). A dilute solution of bacteriophage λ-DNA (∼48,500 basepairs, biotin-labeled on each 3′ terminus), typically in 10 mM HEPES, pH 7.5, and varying NaCl concentrations, was run through the cell until a single DNA molecule was captured between the two beads. The flow cell and the glass micropipette were moved using a feedback-compensated piezoelectric stage (Melles Griot, Carlsbad, CA), stretching the single DNA molecule between the beads, and resulting in a force-extension measurement, as described previously (27,50). The position measurements were converted to a measurement of the molecular extension by correcting for the trap stiffness. Each captured molecule was stretched to verify that the usual force-extension curve was obtained, and that only a single molecule was tethered.
An aqueous solution of 40% glyoxal was obtained from Sigma-Aldrich (St. Louis, MO). To measure the effect of the glyoxal, ∼10 cell volumes of a buffer solution containing a fixed amount of glyoxal were added to the experimental cell until the buffer surrounding the captured DNA molecule was completely exchanged.
RESULTS AND DISCUSSION
Permanent fixation of overstretched DNA bases by glyoxal provides direct evidence of the melting nature of this transition
Shown in Fig. 2 a are the force versus extension curves for a typical individual λ-DNA molecule obtained at 100 mM Na+, pH 7.5, in the absence (black solid line) and presence (red solid line) of 0.5 M glyoxal. The DNA stretching and relaxation are performed in steps of 100 nm, and the force measurements at each extension are collected and averaged over 1 s. Thus, the processes of DNA stretching or relaxation are relatively fast and take ∼1–2 min. The addition of glyoxal to solution has only a minor effect on DNA stretching curves obtained at these pulling rates. This result shows that no guanine residues have reacted with glyoxal to provide permanently melted bases on this time scale.
FIGURE 2.

(a) Stretching (solid line) and relaxation (dashed line) curves in 10 mM HEPES (pH 7.5), 100 mM Na+ (95 mM NaCl and 5 mM NaOH), in the absence (black) and presence (color) of 0.5 M glyoxal. After introducing glyoxal into solution, the DNA molecule was stretched to a fixed position (solid red line). The translation stage was held at the corresponding extension (indicated by arrow), and after 30 min, the molecule was relaxed (dashed red line). Shown in blue are the subsequent stretching and relaxation cycle. (b) Relaxation curves are shown in color (symbol lines) after DNA has been overstretched for 30 min in the presence of glyoxal at the corresponding extensions (indicated by arrows). The best linear fits to Eq. 1 in a minimum mean-square sense are shown by solid colored lines. This was performed by selecting a set of available data points that correspond to bss > bds, before the overstretching plateau. Data are taken at 10 mM HEPES (pH 7.5), 100 mM Na+ (95 mM NaCl and 5 mM NaOH). DNA stretching and relaxation curves in the absence of glyoxal are shown as solid and dashed black lines, respectively. The solid lines in pink and cyan are DNA models for dsDNA and ssDNA, respectively.
To observe the much slower permanent fixation of the melted state of DNA by glyoxal because of its binding to unpaired G bases (44), we held DNA at particular extensions within the DNA structural transition (Fig. 2 a, red arrow) for different amounts of time, and then relaxed it. We expect that if complete fixation of melted regions were achieved, then upon relaxation and subsequent stretches, the DNA molecule would exhibit stretching behavior similar to that of ssDNA up to the fixation point, followed by the shortened transition plateau, corresponding to FIM of the remainder of the dsDNA molecule. Indeed, exposure for ∼20–30 min in a solution of 0.5 M glyoxal led to significant permanent melting of dsDNA. No permanent melting of any fraction of dsDNA was observed for the stretching forces below the plateau, independent of solution conditions or length of glyoxal exposure time. This result is in agreement with the selective fixation by glyoxal of open DNA regions (42–46). It provides direct evidence that the transition force does indeed separate the two DNA strands, including the loss of Watson-Crick hydrogen bonding.
However, a small fraction of the presumably force-melted basepairs does not become permanently fixed by glyoxal. This can be concluded because when the long-time glyoxal-exposed DNA is gradually relaxed from its fixed position, it does not immediately follow a hybrid ssDNA-dsDNA force-extension curve at that extension, but first traces back some part of the plateau, and then starts to decrease as for a slightly shorter ssDNA-dsDNA hybrid complex. All subsequent rapid stretch-release DNA cycles (Fig. 2 a, blue solid and dashed lines, respectively) retrace this first relaxation curve. This result simply indicates that no significant glyoxal binding occurs during subsequent rapid stretches of the same molecule.
Presented in Fig. 2 b are DNA stretching curves obtained under the same solution conditions as in Fig. 2 a in the absence (black) and presence (color) of 0.5 M glyoxal. The green, red, and blue curves correspond, respectively, to DNA stretched previously and held there for 30 min to 0.25, 0.50, and 0.75 fractional extension into the plateau region. We were unable to obtain significant data by holding stretched DNA at the end of the transition, i.e., at f > 0.75, because of breaking of the single DNA molecules. Presumably, complete strand separation does occur under those conditions during our long-time extension. This observation is consistent with the hypothesis that only a small fraction of DNA basepairs supports the molecule at these extensions.
We see that, as expected, the apparent fraction of permanently glyoxal-modified DNA, fa, does increase with f, the expected fractional DNA melting by force. To quantify fa for each stretching condition, we fitted the preplateau portion of the curve to the linear combination of pure ss and ds curves with respect to fa:
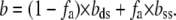 |
(1) |
Here, b(F), bds(F), and bss(F) are extensions per basepair for DNA after permanent partial melting, as well as for the pure ssDNA and dsDNA as a function of force, F. The elastic behavior of pure dsDNA was extensively characterized, and was shown to be well-described by the worm-like chain model (51):
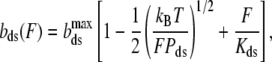 |
(2) |
where the extension per basepair, 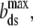 is the contour length of dsDNA, whereas Kds is the elastic modulus, and Pds is the persistence length of dsDNA. The ssDNA elasticity was also well-characterized, and was shown to be well-fitted by the extensible freely jointed chain (FJC) model (12) as:
is the contour length of dsDNA, whereas Kds is the elastic modulus, and Pds is the persistence length of dsDNA. The ssDNA elasticity was also well-characterized, and was shown to be well-fitted by the extensible freely jointed chain (FJC) model (12) as:
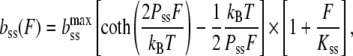 |
(3) |
where 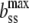 is the contour length per basepair in ssDNA, and Kss and Pss are the elastic modulus and the persistence length of ssDNA, respectively. The value of fa was estimated by finding the best linear fit to Eq. 1 in the minimum mean-square sense. To perform this fit, we used previously measured values for bds, Pds, and Kds such as 0.34 ± 0.001 nm, 48 ± 2 nm, and 1200 ± 200 nm, respectively (5). Although there are known dependencies of these parameters upon ionic strength and pH (5,52,53), changes in these values do not affect the fitting to Eq. 1 significantly. On the other hand, the FJC parameters of ssDNA, bss, Pss, and Kss, may become quite different upon glyoxal binding. Therefore, we first fitted DNA stretching curves for which the length of permanently glyoxal-melted DNA was known to Eq. 1 with respect to FJC parameters of Eq. 3. Values for ssDNA were found previously (12) (bss = 0.56 nm, Pss = 0.75 nm, and Kss = 800 pN). These values for bss and Kss are held constant, though the value for Pss was altered to 0.9 ± 0.1 nm, which is still characteristic of unmodified ssDNA, and likely reflects less hairpin formation at small extensions than according to Smith et al. (12). This result is in agreement with the observations of Johnson (46), who found that the glyoxal-bound ssDNA in electron microphotographs was indistinguishable within error from pure, thermally melted ssDNA. We then used these elastic ssDNA parameters obtained at 100 mM Na+ and pH 7.5 to fit the apparent fractional DNA melting for all other solution conditions. As discussed elsewhere (5,27), we do not expect changes in ssDNA parameters under the range of solution conditions studied to affect our conclusions significantly.
is the contour length per basepair in ssDNA, and Kss and Pss are the elastic modulus and the persistence length of ssDNA, respectively. The value of fa was estimated by finding the best linear fit to Eq. 1 in the minimum mean-square sense. To perform this fit, we used previously measured values for bds, Pds, and Kds such as 0.34 ± 0.001 nm, 48 ± 2 nm, and 1200 ± 200 nm, respectively (5). Although there are known dependencies of these parameters upon ionic strength and pH (5,52,53), changes in these values do not affect the fitting to Eq. 1 significantly. On the other hand, the FJC parameters of ssDNA, bss, Pss, and Kss, may become quite different upon glyoxal binding. Therefore, we first fitted DNA stretching curves for which the length of permanently glyoxal-melted DNA was known to Eq. 1 with respect to FJC parameters of Eq. 3. Values for ssDNA were found previously (12) (bss = 0.56 nm, Pss = 0.75 nm, and Kss = 800 pN). These values for bss and Kss are held constant, though the value for Pss was altered to 0.9 ± 0.1 nm, which is still characteristic of unmodified ssDNA, and likely reflects less hairpin formation at small extensions than according to Smith et al. (12). This result is in agreement with the observations of Johnson (46), who found that the glyoxal-bound ssDNA in electron microphotographs was indistinguishable within error from pure, thermally melted ssDNA. We then used these elastic ssDNA parameters obtained at 100 mM Na+ and pH 7.5 to fit the apparent fractional DNA melting for all other solution conditions. As discussed elsewhere (5,27), we do not expect changes in ssDNA parameters under the range of solution conditions studied to affect our conclusions significantly.
The resultant fractions of permanently melted DNA, fa, fitted for every explored f are summarized in Table 1. The DNA force-extension curves used for these fits, obtained by averaging multiple stretching curves for at least three individual DNAs for each fixation length, are presented as dashed lines in Fig. 3. The four panels of Fig. 3 represent results obtained at four different solution conditions. We see that at low f = 0.25, practically all elongated basepairs became permanently single-stranded upon glyoxal exposure under all solution conditions tested, i.e., fa ≈ f. As expected, fa grows as f increases to 0.5 and 0.75. However, in the latter cases, fa was only 0.33 ± 0.01 and 0.44 ± 0.05, respectively (see Table 1). In other words, ∼70% and ∼60% of elongated basepairs became permanently single-stranded in each case.
TABLE 1.
Permanently glyoxal-modified ssDNA fraction, fa
| Permanently glyoxal-modified ssDNA fraction, fa
|
||||
|---|---|---|---|---|
| Fractional DNA melting by force, f | pH 7.5, 100 mM Na+, and 0.5 M glyoxal | pH 8.5, 100 mM Na+, and 0.5 M glyoxal | pH 9.7, 100 mM Na+, and 0.5 M glyoxal | pH 7.5, 5 mM Na+, and 0.2 M glyoxal |
| 0.25 ± 0.01 | 0.22 ± 0.03 | 0.25 ± 0.01 | 0.25 ± 0.01 | 0.25 ± 0.01 |
| 0.50 ± 0.01 | 0.33 ± 0.01 | 0.34 ± 0.01 | 0.40 ± 0.10 | 0.52 ± 0.04 |
| 0.75 ± 0.01 | 0.44 ± 0.05 | 0.43 ± 0.01 | 0.49 ± 0.02 | 0.63 ± 0.06 |
Data are reported as mean ± SE for three or more molecules.
FIGURE 3.

Mapping DNA FIM in the presence of glyoxal. Stretching (solid line) and relaxation (dashed line) curves are shown in the absence (black) and presence (color) of glyoxal. Shown in color are the stretch and relaxation curves after DNA has been overstretched for some time (30 min for 100 mM Na+, and 10 min for 5 mM Na+) in the presence of glyoxal at the corresponding extensions (indicated by arrows). For each relaxed curve, we averaged the relaxation curves of three or more molecules, and error bars are determined from the standard error. We used only stretches of DNA molecules with the least number of nicks by excluding all stretches from DNA molecules that broke at some point during the overstretching transition. Data are taken at (a) 10 mM HEPES (pH 7.5), 100 mM Na+ (95 mM NaCl and 5 mM NaOH), and 0.5 M glyoxal; (b) 10 mM HEPES (pH 8.5), 100 mM Na+ (95 mM NaCl and 5 mM NaOH), and 0.5 M glyoxal; (c) 10 mM HEPES (pH 9.7), 100 mM Na+ (95 mM NaCl and 5 mM NaOH), and 0.5 M glyoxal; and (d) 10 mM HEPES (pH 7.5), 5 mM Na+ (5 mM NaOH), and 0.2 M glyoxal.
In our experiments, the apparent fractional DNA melting fa was never observed to exceed its fractional elongation f. This result is consistent with the high selectivity of glyoxal fixation of the bases. In the case of thermal DNA melting, this selectivity was attributable to the heterogeneity of DNA basepair stability. Thus, at some temperatures below the average Tm, certain AT-rich segments are predominantly opened and fixed by glyoxal, whereas the rest remain closed (42,47,48). It is the extremely slow rate of the glyoxal/guanine covalent bonding that makes this reaction practically unobservable for hydrogen-bonded basepairs (44). Indeed, formaldehyde, which is analogous to glyoxal but is a much faster ssDNA fixation agent, was shown to unwind any DNA duplexes, even at room temperature (54–57). Increasing the temperature exponentially facilitates the rate of glyoxal-promoted unwinding of the DNA duplex (46). Therefore, selective fixation of the low temperature-melting segments requires very specific glyoxal exposure conditions (41,47,48). In the case of FIM, the force becomes an equivalent of the temperature, whereas the fractional extension into the plateau region, f, is an equivalent of fractional DNA melting (7). Our glyoxal exposure experiments are conducted under the condition of fixed fractional DNA melting, rather than fixed force. Thus, even for homogeneous DNA, only the force-melted basepairs are expected to be fixed. Indeed, fixation in the single-stranded state of additional bases would lead to an abrupt drop in force, which in turn should lead to an exponential slowing of further guanine fixation by glyoxal.
Gradual thermal opening of the subsequently more stable regions in λ-DNA upon temperature increase was previously monitored by glyoxal fixation and mapped on the molecule by ethidium fluorescence (46). At both 0.17 and 0.3 fractional DNA melting, most of the glyoxal-fixed regions were several hundred basepairs long, and appeared to be rather close to the molecule's center. A smaller melted fragment was also found at one of the ends of the molecule. The FIM of λ-DNA in our experiments most likely happens within the same regions, as DNA is progressively stretched into the transition, in contrast to the conclusions of Whitelam et al. (25), who proposed that DNA can be melted by force exclusively from its free ends.
Effect of solution conditions on the apparent force-melted fraction of DNA basepairs
In an attempt to understand why the apparent DNA melting remains smaller than the fractional DNA extension, we used longer glyoxal exposure times. However, the time of our experiments was limited by the drift of the optical-tweezers instrument to a few hours. During this time, no further increase in apparent binding was determined. We further tested the effect of the glyoxal reaction rate on fa by varying the solution pH from 7.5–9.7. It is known that in this pH range, the rate of glyoxal/G binding increases by ∼10-fold, whereas the binding remains essentially irreversible (44). Quantification of the permanently melted DNA fraction at 100 mM Na+ and three different pH conditions is summarized in Table 1. The data presented in Table 1 and Fig. 3, a–c, suggest that the fa for a given f grows with pH very insignificantly over this pH range.
In contrast, fa is always significantly higher and very close to f in 5 mM monovalent salt solution. In addition, higher fa in low salt can be achieved at lower glyoxal concentrations of 0.2 M, and with shorter exposure times of ∼10 min, instead of 30 min (compare the average stretching curves in Fig. 3, a and d). This more extensive reaction with glyoxal in lower salt can hardly be explained by faster glyoxal binding. Indeed, glyoxal binding to G-bases is known to be salt-independent in terms of both its kinetics and equilibrium binding constant (44,45), as expected for a neutral and nonpolar molecule. Therefore, the reason for the apparent more extensive glyoxal interaction with DNA observed in lower salt should be sought in the difference in DNA behavior, and not in its glyoxal binding.
Based on these data, it is likely that the low versus high salt situations are different not in terms of the number of guanine residues modified with glyoxal, but rather because, in high salt, some modified force-melted ssDNA regions can still partially reanneal upon DNA relaxation. Indeed, the rate of the G/glyoxal reaction is most likely still much slower (44) than our exposure time, such that only a fraction of all exposed G-residues become modified under all of the solution conditions explored in this work. As shown previously (39), reannealing of partially glyoxal-fixed DNA sequences is similar to reannealing of slightly mismatched ssDNA strands. Such reannealing occurs at temperatures lower than the Tm of the perfectly complementary strands, which depends on the fraction of modified or mismatched bases. In low salt, however, these imperfect duplexes become unstable, with the apparent Tm below room temperature. Indeed, low salt leads to a higher energetic penalty for the ds/ss boundaries (58), as well as to the lower stability of every basepair (59). Consistent with this interpretation, at 100 mM Na+, the residual melting-force plateau after glyoxal exposure becomes slightly lower than the original Fm in the absence of glyoxal. At the same time, no residual plateau is observed in the DNA relaxation curve after glyoxal exposure in 5 mM Na+, consistent with all force-exposed basepairs remaining permanently single-stranded (Table 1).
A secondary effect could also contribute to the low and high salt differences in fa. The DNA melting partially occurs from its free ends by peeling off of the strand that is not under tension. In high salt (100 mM Na+), this strand will most likely form its own intrastrand secondary structure in the form of hairpins, in which G-residues will be protected. Even though the G-residues in its complementary DNA strand will be permanently modified upon glyoxal exposure, such protection will quite likely lead to the ability of more DNA regions to reanneal upon DNA relaxation, thereby leading to a smaller apparent fraction of permanently glyoxal-modified DNA. In contrast, in low salt, most of the ssDNA hairpins will become unstable and unfold (for the reasons mentioned above), leading to modification of the G-residues in these regions. This phenomenon may also explain why a smaller fraction of all basepairs melted by stretching become permanently modified by glyoxal as the DNA is stretched further into the plateau (see Table 1). Although the least stable AT-rich regions melt first upon DNA stretching in the middle of λ-DNA, further DNA stretching leads to more melting from the ends, and therefore to more hairpin-protected basepairs and less permanent melting. These features are specific for the λ-DNA sequence, and therefore will likely vary with the DNA sequence studied.
CONCLUSIONS
In this study, we show that a majority of DNA basepairs elongated by force during the overstretching transition become permanently single-stranded when exposed for a sufficient time to glyoxal. Because glyoxal is known to covalently bind only solution-exposed guanine bases (42–45), this result provides direct evidence that the elongated basepairs have lost their Watson-Crick hydrogen bonding, i.e., they are melted. According to all modeling studies, most of the interstrand basepairing is expected to be preserved in S-DNA (14–17). Indeed, the length of the overstretched DNA is close to the length of the stretched single DNA strand. This implies that independent of the details of the proposed S-DNA structure, most of its stacking interactions should be destroyed, whereas the double-stranded structure relies on interstrand hydrogen bonding. Thus our results strongly argue in favor of the melting nature of the overstretching transition in DNA, and against a double-stranded form of overstretched DNA, i.e., S-DNA.
In addition, we show that, as with thermal melting, in FIM, glyoxal is an appropriate agent to bind and to fix permanently and exclusively the single-stranded regions, while not unwinding the double-stranded regions of DNA. This result is reinforced by the specific setup of our experiment, in which fractional DNA melting by force is controlled (as opposed to the control of force or temperature). Such a setup ensures that permanent fixation is achieved for only the equilibrium force-melted basepairs, because any additional glyoxal binding leads to an abrupt drop in force, associated with an exponential drop in the already extremely slow G/glyoxal reaction rate. This makes our approach for the study of force-melting of DNA with glyoxal reliable and quantitative. We also show that this selective glyoxal fixation of force-melted DNA remains valid in the broad range of solution pH of 7.5–9.7, and for the half to a few hours of glyoxal exposure time at room temperature and ∼0.5 M glyoxal concentrations. According to this study, the apparent glyoxal-fixed fraction of DNA is a better measure of fractional force-melted DNA in low (5 mM Na+) rather than in high (100 mM Na+) salt, because of the partial reannealing of the modified DNA strands in the latter case.
Acknowledgments
This work was supported in part by National Institutes of Health grant GM 072462 and National Science Foundation grant MCB-0744456.
Editor: Taekjip Ha.
References
- 1.Bustamante, C., S. B. Smith, J. Liphardt, and D. Smith. 2000. Single-molecule studies of DNA mechanics. Curr. Opin. Struct. Biol. 10:279–285. [DOI] [PubMed] [Google Scholar]
- 2.Bustamante, C., Z. Bryant, and S. B. Smith. 2003. Ten years of tension: single-molecule DNA mechanics. Nature. 421:423–427. [DOI] [PubMed] [Google Scholar]
- 3.Strick, T., J. Allemand, V. Croquette, and D. Bensimon. 2000. Twisting and stretching single DNA molecules. Prog. Biophys. Mol. Biol. 74:115–140. [DOI] [PubMed] [Google Scholar]
- 4.Allemand, J.-F., D. Bensimon, and V. Croquette. 2003. Stretching DNA and RNA to probe their interactions with proteins. Curr. Opin. Struct. Biol. 13:266–274. [DOI] [PubMed] [Google Scholar]
- 5.Wenner, J. R., M. C. Williams, I. Rouzina, and V. A. Bloomfield. 2002. Salt dependence of the elasticity and overstretching transition of single DNA molecules. Biophys. J. 82:3160–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams, M. C., and I. Rouzina. 2002. Force spectroscopy of single DNA and RNA molecules. Curr. Opin. Struct. Biol. 12:330–336. [DOI] [PubMed] [Google Scholar]
- 7.Rouzina, I., and V. A. Bloomfield. 2001. Force-induced melting of the DNA double helix 1. Thermodynamic analysis. Biophys. J. 80:882–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rouzina, I., and V. A. Bloomfield. 2001. Force-induced melting of the DNA double helix. 2. Effect of solution conditions. Biophys. J. 80:894–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hegner, M., S. B. Smith, and C. Bustamante. 1999. Polymerization and mechanical properties of single RecA-DNA filaments. Proc. Natl. Acad. Sci. USA. 96:10109–10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clausen-Schaumann, H., M. Rief, C. Tolksdorf, and H. E. Gaub. 2000. Mechanical stability of single DNA molecules. Biophys. J. 78:1997–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rief, M., M. Gautel, F. Oesterhelt, J. M. Fernandez, and H. E. Gaub. 1997. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 276:1109–1112. [DOI] [PubMed] [Google Scholar]
- 12.Smith, S. B., Y. Cui, and C. Bustamante. 1996. Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded DNA molecules. Science. 271:795–799. [DOI] [PubMed] [Google Scholar]
- 13.Cluzel, P., A. Lebrun, C. Heller, R. Lavery, J.-L. Viovy, D. Chatenay, and F. Caron. 1996. DNA: an extensible molecule. Science. 271:792–794. [DOI] [PubMed] [Google Scholar]
- 14.Konrad, M. W., and J. I. Bolonick. 1996. Molecular dynamics simulation of DNA stretching is consistent with the tension observed for extension and strand separation and predicts a novel ladder structure. J. Am. Chem. Soc. 118:10989–10994. [Google Scholar]
- 15.Kosikov, K. M., A. A. Gorin, V. B. Zhurkin, and W. K. Olson. 1999. DNA stretching and compression: large-scale simulations of double helical structures. J. Mol. Biol. 289:1301–1326. [DOI] [PubMed] [Google Scholar]
- 16.Lebrun, A., and R. Lavery. 1996. Modelling extreme stretching of DNA. Nucleic Acids Res. 24:2260–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cizeau, P., and J. L. Viovy. 1997. Modeling extreme extension of DNA. Biopolymers. 42:383–385. [Google Scholar]
- 18.Marko, J. F. 1998. DNA under high tension: overstretching, undertwisting, and relaxation dynamics. Phys. Rev. E. 57:2134–2149. [Google Scholar]
- 19.Cocco, S., J. Yan, J. F. Leger, D. Chatenay, and J. F. Marko. 2004. Overstretching and force-driven strand separation of double-helix DNA. Phys. Rev. E. 70:011910. [DOI] [PubMed] [Google Scholar]
- 20.Storm, C., and P. C. Nelson. 2003. Theory of high-force DNA stretching and overstretching. Phys. Rev. E. 67:051906. [DOI] [PubMed] [Google Scholar]
- 21.Gorin, A. A., V. B. Zhurkin, and W. K. Olson. 1995. B-DNA twisting correlates with base-pair morphology. J. Mol. Biol. 247:34–48. [DOI] [PubMed] [Google Scholar]
- 22.Harris, S. A., Z. A. Sands, and C. A. Laughton. 2005. Molecular dynamics simulations of duplex stretching reveal the importance of entropy in determining the biomechanical properties of DNA. Biophys. J. 88:1684–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piana, S. 2005. Structure and energy of a DNA dodecamer under tensile load. Nucleic Acids Res. 33:7029–7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heng, J. B., A. Aksimentiev, C. Ho, P. Marks, Y. V. Grinkova, S. Sligar, K. Schulten, and G. Timp. 2006. The electromechanics of DNA in a synthetic nanopore. Biophys. J. 90:1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitelam, S., S. Pronk, and P. L. Geissler. 2008. There and (slowly) back again: entropy-driven hysteresis in a model of DNA overstretching. Biophys. J. 94:2452–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams, M. C., J. R. Wenner, I. Rouzina, and V. A. Bloomfield. 2001. Entropy and heat capacity of DNA melting from temperature dependence of single molecule stretching. Biophys. J. 80:1932–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams, M. C., J. R. Wenner, I. Rouzina, and V. A. Bloomfield. 2001. Effect of pH on the overstretching transition of double-stranded DNA: evidence of force-induced DNA melting. Biophys. J. 80:874–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pant, K., R. L. Karpel, I. Rouzina, and M. C. Williams. 2005. Salt dependent binding of T4 gene 32 protein to single and double-stranded DNA: single molecule force spectroscopy measurements. J. Mol. Biol. 349:317–330. [DOI] [PubMed] [Google Scholar]
- 29.Pant, K., R. L. Karpel, I. Rouzina, and M. C. Williams. 2004. Mechanical measurement of single-molecule binding rates: kinetics of DNA helix-destabilization by T4 gene 32 protein. J. Mol. Biol. 336:851–870. [DOI] [PubMed] [Google Scholar]
- 30.Pant, K., R. L. Karpel, and M. C. Williams. 2003. Kinetic regulation of single DNA molecule denaturation by T4 gene 32 protein structural domains. J. Mol. Biol. 327:571–578. [DOI] [PubMed] [Google Scholar]
- 31.Rouzina, I., K. Pant, R. L. Karpel, and M. C. Williams. 2005. Theory of electrostatically regulated binding of T4 gene 32 protein to single- and double-stranded DNA. Biophys. J. 89:1941–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shokri, L., B. Marintcheva, C. C. Richardson, I. Rouzina, and M. C. Williams. 2006. Single molecule force spectroscopy of salt-dependent bacteriophage T7 gene 2.5 protein binding to single-stranded DNA. J. Biol. Chem. 281:38689–38696. [DOI] [PubMed] [Google Scholar]
- 33.Vladescu, I. D., M. J. McCauley, I. Rouzina, and M. C. Williams. 2005. Mapping the phase diagram of single DNA molecule force-induced melting in the presence of ethidium. Phys. Rev. Lett. 95:158102. [DOI] [PubMed] [Google Scholar]
- 34.Krautbauer, R., S. Fischerländer, S. Allen, and H. E. Gaub. 2002. Mechanical fingerprints of DNA drug complexes. Single Molecules. 3:97–103. [Google Scholar]
- 35.Mihailovic, A., I. Vladescu, M. McCauley, E. Ly, M. C. Williams, E. M. Spain, and M. E. Nunez. 2006. Exploring the interaction of ruthenium(II) polypyridyl complexes with DNA using single-molecule techniques. Langmuir. 22:4699–4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vladescu, I. D., M. J. McCauley, M. E. Nunez, I. Rouzina, and M. C. Williams. 2007. Quantifying force-dependent and zero-force DNA intercalation by single-molecule stretching. Nat. Methods. 4:517–522. [DOI] [PubMed] [Google Scholar]
- 37.Reference deleted in proof.
- 38.Strick, T. R., M. N. Dessinges, G. Charvin, N. H. Dekker, J. F. Allemand, D. Bensimon, and V. Croquette. 2003. Stretching of macromolecules and proteins. Rep. Prog. Phys. 66:1–45. [Google Scholar]
- 39.Hutton, J. R., and J. G. Wetmur. 1973. Effect of chemical modification on the rate of renaturation of deoxyribonucleic acid. Deaminated and glyoxalated deoxyribonucleic acid. Biochemistry. 12:558–563. [DOI] [PubMed] [Google Scholar]
- 40.Pavlov, V. M., Y. L. Lyubchenko, A. S. Borovik, and Y. S. Lazurkin. 1977. Specific fragmentation of T7 phage DNA at low-melting sites. Nucleic Acids Res. 4:4053–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lyamichev, V. I., I. G. Panyutin, D. I. Cherny, and L. Lyubchenko Yu. 1983. Localization of low-melting regions in phage T7 DNA. Nucleic Acids Res. 11:2165–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borovik, A. S., Y. A. Kalambet, Y. L. Lyubchenko, V. T. Shitov, and E. I. Golovanov. 1980. Equilibrium melting of plasmid ColE1 DNA: electron-microscopic visualization. Nucleic Acids Res. 8:4165–4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borovik, A. S., L. Liubchenko Iu, B. S. Naroditskii, and T. I. Tikhonenko. 1984. Electron microscope maps of SA7 DNA melting. Mol. Biol. (Mosk.). 18:1634–1638. [PubMed] [Google Scholar]
- 44.Broude, N. E., and E. I. Budowsky. 1971. The reaction of glyoxal with nucleic acid components. III. Kinetics of the reaction with monomers. Biochim. Biophys. Acta. 254:380–388. [DOI] [PubMed] [Google Scholar]
- 45.Broude, N. E., and E. I. Budowsky. 1973. The reaction of glyoxal with nucleic acid components. V. Denaturation of DNA under the action of glyoxal. Biochim. Biophys. Acta. 294:378–384. [DOI] [PubMed] [Google Scholar]
- 46.Johnson, D. 1975. A new method of DNA denaturation mapping. Nucleic Acids Res. 2:2049–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lyamichev, V. I., I. G. Panyutin, and L. Lyubchenko Yu. 1982. Gel electrophoresis of partially denatured DNA. Retardation effect: its analysis and application. Nucleic Acids Res. 10:4813–4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lyubchenko Y. L., A. Y. Kalambet, V. I. Lyamichev, and A. S. Borovik. 1982. A comparison of experimental and theoretical melting maps for replicative form of phi X174 DNA. Nucleic Acids Res. 10:1867–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCauley, M., P. R. Hardwidge, L. J. Maher 3rd, and M. C. Williams. 2005. Dual binding modes for an HMG domain from human HMGB2 on DNA. Biophys. J. 89:353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCauley, M. J., and M. C. Williams. 2007. Mechanisms of DNA binding determined in optical tweezers experiments. Biopolymers. 85:154–168. [DOI] [PubMed] [Google Scholar]
- 51.Odijk, T. 1995. Stiff chains and filaments under tension. Macromolecules. 28:7016–7018. [Google Scholar]
- 52.Marko, J. F., and E. D. Siggia. 1995. Stretching DNA. Macromolecules. 28:8759–8770. [Google Scholar]
- 53.Bustamante, C., J. F. Marko, E. D. Siggia, and S. Smith. 1994. Entropic elasticity of lambda-phage DNA. Science. 265:1599–1600. [DOI] [PubMed] [Google Scholar]
- 54.McGhee, J. D., and P. H. von Hippel. 1974. Theoretical aspects of DNA-protein interactions: co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 86:469–489. [DOI] [PubMed] [Google Scholar]
- 55.McGhee, J. D., and P. H. von Hippel. 1975. Formaldehyde as a probe of DNA structure. I. Reaction with exocyclic amino groups of DNA bases. Biochemistry. 14:1281–1296. [DOI] [PubMed] [Google Scholar]
- 56.Lukashin, A. V., A. V. Vologodskii, M. D. Frank-Kamenetskii, and Y. L. Lyubchenko. 1976. Fluctuational opening of the double helix as revealed by theoretical and experimental study of DNA interaction with formaldehyde. J. Mol. Biol. 108:665–682. [DOI] [PubMed] [Google Scholar]
- 57.McGhee, J. D. 1976. Theoretical calculations of the helix-coil transition of DNA in the presence of large, cooperatively binding ligands. Biopolymers. 15:1345–1375. [DOI] [PubMed] [Google Scholar]
- 58.Kozyavkin, S. A., S. M. Mirkin, and B. R. Amirikyan. 1987. The ionic strength dependence of the cooperativity factor for DNA melting. J. Biomol. Struct. Dyn. 5:119–126. [DOI] [PubMed] [Google Scholar]
- 59.Frank-Kamenetskii, F. 1971. Simplification of the empirical relationship between melting temperature of DNA, its GC content and concentration of sodium ions in solution. Biopolymers. 10:2623–2624. [DOI] [PubMed] [Google Scholar]