

Abstract
Spontaneous magnetoencephalographic activity was recorded in awake, healthy human controls and in patients suffering from neurogenic pain, tinnitus, Parkinson's disease, or depression. Compared with controls, patients showed increased low-frequency θ rhythmicity, in conjunction with a widespread and marked increase of coherence among high- and low-frequency oscillations. These data indicate the presence of a thalamocortical dysrhythmia, which we propose is responsible for all the above mentioned conditions. This coherent θ activity, the result of a resonant interaction between thalamus and cortex, is due to the generation of low-threshold calcium spike bursts by thalamic cells. The presence of these bursts is directly related to thalamic cell hyperpolarization, brought about by either excess inhibition or disfacilitation. The emergence of positive clinical symptoms is viewed as resulting from ectopic γ-band activation, which we refer to as the “edge effect.” This effect is observable as increased coherence between low- and high-frequency oscillations, probably resulting from inhibitory asymmetry between high- and low-frequency thalamocortical modules at the cortical level.
Keywords: edge effect, θ activity, γ activity, humans
In recent years it has become evident that neuronal rhythmicity and its consequence, ensemble neuronal oscillation and resonance, are deeply related to the emergence of brain functions (1, 2). Prominent in these studies was the linking of high-frequency oscillations in the γ domain (25–50 Hz) with sensorimotor (3, 4) and cognitive functions (5–12). Although the exact mechanism by which such coherent activity ultimately results in conscious experience is still under debate, there is little doubt that slow oscillatory activity, as observed in phase four of the sleep cycle, cannot presently be correlated with cognition (9). Here we report a set of studies concerning the presence of slow thalamocortical oscillation in the θ-frequency band (4–8 Hz) in awake human patients who present a wide variety of neurological and psychiatric conditions. These results are in complete accordance with single-unit, medial thalamic recordings, made during stereotactic surgery in patients suffering from the same diseases. These recordings show the presence of low-threshold spike (LTS) bursting activity with a θ rhythmicity (13).
Methods
Subjects and Patients.
In this study, we analyzed spontaneous brain activity from nine healthy control subjects and from nine patients suffering from chronic, severe, and therapy-resistant neurological or neuropsychiatric disorders. Control subjects ranged between 24 and 45 years old; patients ranged between 28 and 73 years old. The patient population included four patients diagnosed with Parkinson's disease (three akinetic–choreatic forms and one tremulant form), one patient with tinnitus, two patients with neurogenic pain (one trigeminal and one upper-limb phantom pain), and two patients with major depression. Patients were carefully selected by using standard neurological/psychiatric diagnostic procedures.
Magnetoencephalography (MEG) Recordings and Analysis.
Magnetic recordings were obtained at our laboratory with a whole-head, 148-channel MEG system Magnes 2500 WH (Biomagnetic Technologies, San Diego). During the recording sessions, the subject was placed on a bed with the MEG recording port (Fig. 1A) surrounding the subject's head to record the magnetic fields from different angles over the head surface. Spontaneous brain activity was continuously recorded for 10 min while the subject rested with eyes closed (bandpass, 0.1–100 Hz; sample rate, 508 Hz). The EKG was simultaneously recorded digitally for off-line heart-artifact rejection.
Figure 1.

The whole-head MEG system and the cleaning procedures for MEG signals. (A) The Magnes 2500WH MEG system (Biomagnetic Technologies) shows a helmet design, including 148 signal and 11 reference channels. Recordings can be taken with subject in a seated or supine position with a noise level below 10 fT/Hz1/2. (B) Raw data were further processed to remove heart artifact and contribution from distant sources.
MEG Analysis.
Continuous MEG raw data were analyzed on a LINUX cluster computer system, by using in-house software and commercial Matlab analysis software packages.
Computational Methods.
MEG data have a low signal-to-noise ratio and a large dynamic range, which make it a challenging data analysis problem. Although spectral analysis is an old subject, with a number of different techniques currently in use in EEG and MEG literature, we have benefited substantially from the usage of a modern framework for performing spectral analysis, the multitaper technique (14). This technique elegantly resolves the problems of bias and variance in spectral estimation, by averaging over a set of orthogonal basis functions, the so-called Slepian sequences (15). These sequences wk(t) form a sequence of orthogonal functions, defined on the time interval t = 1, 2, … , T. They are parameterized by a bandwidth parameter W, such that there are K = [2WT] basis functions with spectra that are confined to a frequency band [f − W, f + W] around the frequency of interest, f. These methods have recently been applied to the analysis of neurobiological time series (16).
For a given data sequence x(t), the basic quantity involved in the spectral estimation is the tapered Fourier transform:
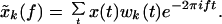 |
1 |
In terms of these quantities, a direct estimate of the spectrum is given by:
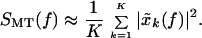 |
2 |
Note that the average across tapers reduces the variance by a factor of 1/K. This is the basic estimate for spectra that we have employed in our work. Spectral estimates were performed on data windows 5 s long, with a bandwidth parameter 2W = 0.8 Hz, leading to a time bandwidth product 2WT = 4 and K = 3. The estimates were obtained with a moving window, and the logarithms of the spectra were averaged across windows to improve the stability of the estimate.
Another quantity that is presented in the paper is the cross-correlation between spectral amplitudes at different frequencies. These were obtained by computing the multitaper spectrogram SMT(f, t), and using a moving analysis window, and then computing the correlation coefficient of the two time series, log(SMT(f1, t)) and log(SMT(f2, t)), with their means removed. By performing this computation for a two-dimensional grid of points in f1, f2 space, a two-dimensional image of spectral correlations was generated.
Removal of Cardiac and Distant Source Artifacts.
Because the experiments were performed with a magnetometer rather than a gradiometer, the raw data displayed noise arising from magnetic fields and originating from the heart and other distant sources (Fig. 1). The cardiac artifacts were removed by a careful time-domain modeling of the cardiac spikes on a channel-by-channel basis. The locations of the spikes were accurately determined by using an auxiliary EKG measurement, on which spike times were extracted by using a matched filter to the spike shape, which was interpolated to a tenth of the sample spacing. These spike times were used to extract interpolated spike shapes from the various channels, which were used to create smooth templates that were then subtracted from the data. The validity of the subtraction was estimated by computing the multitaper cross-coherence with the EKG time series; typically no significant coherence with the EKG time series was found in the residual time series. The reference channels were subtracted in the frequency domain by using a linear model for the contribution of the reference channels on the MEG data. The coefficients of the linear model were estimated by using a multitaper transfer function estimate. Thus, if y(t) was a data channel and x(t) was a reference channel, the subtracted spectral coefficients for use in the spectral estimate were defined as:
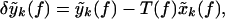 |
3 |
where
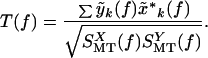 |
4 |
To improve the stability of the estimate T(f), the cross-spectra in the numerator as well as the spectra in the denominator were further averaged across the moving analysis window.
Results
To determine whether a clear grouping could be obtained from the MEG measurements in control subjects and in patients, a set of recordings of spontaneous activity were obtained, and the 18 sets of results were analyzed, such that an unbiased plot could be drawn. The recordings obtained from a control and from a patient (Fig. 2 A and B) illustrate the differences in overall frequency content for the rostral and caudal halves of the brain. Note that the peak frequencies are clearly different in these two subjects, with the difference in low-frequency activity being most clearly recognizable in the caudal pole. This result is to be expected, because both sets of recordings were obtained when the subjects' eyes were closed. Under these conditions, the α rhythm is very prominent in the caudal pole (17). We interpret the results obtained in the patient as a shift from a normal α rhythm to a robust low-frequency θ rhythmicity. The differences in the rostral pole are less prominent, in agreement with the fact that under certain circumstances, θ rhythmicity is observed in normal individuals (18, 19). However, the issue of coherence, as will be detailed, helps to separate “normal” from “abnormal” θ-band frequencies.
Figure 2.

Power spectra for control subjects and patients. Spectra are shown for one representative control subject and for one Parkinson patient (A and B); and for the average of all controls and all patients (C–E). The power was averaged over all rostral MEG channels (A and C), over all caudal channels (B and D), and averaged over all 148 channels for the entire brain (E). Note the shift toward the θ range and the increase in global power in the patient population. Also note small bumps in the 5- to 10-Hz range in the patients, indicating distinct peaks, depending on the pathology or level of severity. Dashed line indicates maximum of averaged α activity in controls.
Average power spectra of the recordings obtained from all of the patients and all of the control subjects are shown in Fig. 2C for the rostral pole, in Fig. 2D for the caudal poles, and in Fig. 2E for the aggregate of all channels in the control and patient group. Note that the two individual recordings in Fig. 2 A and B and the aggregate plot in E illustrate the same characteristics with respect to frequency and rostrocaudal location. Comparing the average power spectra obtained from all of the patients with those obtained from all of the control subjects indicates, once again, a decrease in α power and an increase in lower-frequency activity in the θ range, as well as an increase in global power. The latter would be expected if the overall coherence had increased in the patient group.
These findings were confirmed by plotting total power in the 5- to 15-Hz band against the power ratio between the 5- to 10-Hz band and between the 10- to 15-Hz band (Fig. 3). This choice was motivated by the results from the principal component analysis of all power spectra. In this case, all of the subjects were directly compared with one other, creating an unbiased grouping of the population. Note that the patients tend to be located over a wide area in low-frequency space with increased global power, whereas normal subjects are clustered in the higher-frequency space with less global power.
Figure 3.

Grouping of power spectral traces, indicating the distribution of all control subjects (blue dots) and all patients (red dots). The ratio 5–10 Hz power/10–15 Hz power is plotted against the total 5–15 Hz power averaged over MEG channels for the rostral pole (A), for the caudal pole (B), and for the entire brain (C). Patients' disorders are specified as T, tinnitus, D, major depression, P, neurogenic pain, and M, Parkinson's.
To test whether the increased power in the θ band comes with an increased coherence inside low frequencies, as well as between low and high frequencies, correlation plots were computed for individual patients and control subjects. In Fig. 4, the correlation for a patient and a control are illustrated with the correlation for the average of all control and patient groups. The increase in θ power is in complete accordance with the presence of LTS bursting activity, with θ rhythmicity in the medial thalamus of patients with the same diseases, as demonstrated by single-unit recordings during stereotactic surgery (13).
Figure 4.

Correlation plots of power spectra over a period of 10 min. (A) One representative control subject. (B) One Parkinson patient. (C) The average of all controls. (D) The average of all patients. For clarity, the thickness of the white diagonal indicating the self-correlation was reduced. Note the wide range of correlation (from θ to γ) in the patient group.
Moreover, the correlation shows harmonics at γ frequency, indicating an edge effect (11). If certain cortical structures in the brain are forced to generate γ frequencies in a continuous and stereotyped manner, the brain generates cognitive experiences and motor behavior, in the absence of context with the external world and without the intentionality that normally characterizes human function. We therefore propose that this edge effect, γ-band activity is responsible for the positive symptoms reported by these patients.
Discussion
Thalamocortical Dysrhythmia: Low-Frequency Rhythmicity and the Edge Effect.
The results we describe in this paper indicate that a common mechanism is operant, and that, depending on its localization in the thalamocortical network, it may produce dysfunctions and symptoms ascribed to various common neurological or psychiatric conditions.
From a functional point of view, the common link among these different medical conditions relates to electrophysiological properties of the thalamocortical loops involved (20). Thus, low-frequency, coherent electrical activity with wide hemispheric representation is common in all of the patients studied. This low-frequency, thalamocortical activity has two characteristics that distinguish it from the θ rhythmicity present under normal waking conditions. The first, and most important, is the presence of a persistent low-frequency, thalamocortical resonance during the awake state. The second characteristic is its wide coherence over the recorded channels. We must emphasize here that low frequencies are not themselves pathological; they occur as thalamocortical synchronization (21, 22) continuously during δ sleep (20, 23), and transiently during wakefulness, under specific conditions of mental and emotional activity (18, 19, 24–26). Rather, we are dealing here with an ongoing low-frequency activity that is present during the entire day and that continuously modifies and limits the dynamic organization of the brain; it does so all the more efficiently because it produces large-scale coherence.
Thus, we postulate that an edge effect generates the high-frequency, γ-band activity that is the origin for the appearance of the clinical symptoms and signs (Fig. 5). The basic hypothesis, as illustrated in Fig. 5, proposes that protracted hyperpolarization of a specific nucleus will result in low-frequency oscillation at the θ-frequency band. Such oscillation, by activating return corticothalamic pathways, will entrain, through the reticular nucleus and through direct thalamic activation, the nonspecific system. The result is the promotion of large-scale, low-frequency oscillatory coherence. At the cortical level, the reduction of lateral inhibition promotes coherent γ-frequency oscillation and thus positive symptoms. Note that both deep brain stimulation and thalamic lesions aim at reducing the nonspecific component of this pathway. Our proposal regarding the γ-band oscillation is based on the many studies indicating that perceptual, cognitive, and motor experiences are associated with such activity (3–12, 27–30). The term “edge effect” was coined in the consideration of the aura that accompanies migraine attacks and is seen most notably in the visual cortex. During this condition, a wave of depolarization generating the scotoma is surrounded by an edge of excitation that produces the bright visual illumination known as a “halo” (31). This halo is the manifestation of the interface between the area of depolarization and the unaffected area that surrounds the malfunction site.
Figure 5.

Diagram of the thalamocortical circuits that support the positive symptoms hypothesis. Two thalamocortical systems are shown, the specific pathway (yellow) to layer IV of the cortex that activates layer VI cortical neurons; and feed-forward inhibition through inhibitory cortical interneurons (red). Collaterals of these projections produce thalamic feedback inhibition through the reticular nucleus (red at thalamic level). The return pathway (circular arrow on the right) re-enters this oscillation to specific- and reticularis-thalamic nuclei through layer VI pyramidal cells (blue). The second loop shows nonspecific nuclei (green), projecting to the most superficial layer of the cortex and giving collaterals to the reticular nucleus. The conjunction of the specific and nonspecific loops is proposed to generate temporal coherence (Center). Protracted thalamic cell hyperpolarization by altered synaptic input triggers low-frequency neuronal oscillation (Center). Either disfacilitation, as occurs after deafferentation (as in neurogenic pain or tinnitus), or excess inhibition caused by pallidal over-activity (as in Parkinson's disease), hyperpolarize the cells sufficiently to deinactivate T-type calcium channels, resulting in thalamic oscillation at θ range. Such oscillation can entrain corticothalamic loops (Left), generating increased coherence, as observed in this study. At the cortical level, low-frequency activation of cortico–cortical inhibitory interneurons, by reducing lateral inhibitory drive, can result in high-frequency, coherent activation of neighboring cortical modules, the “edge effect” (Right).
In the proposed scenario, the neurological or psychiatric manifestations of the patients are conditioned by the localization of the primary lesion. Thus, in the case of Parkinsonism, in which low-frequency activity was reported (32–34), excess inhibition, produced by hyperactive pallidal input onto the motor thalamus, produces hyperpolarization of thalamic relay cells, with the consecutive de-inactivation of T-channels (35) and the appearance of low-threshold calcium spiking and low-frequency oscillation (36–38). This oscillation produces then the edge effect, which generates the clinical Parkinsonian manifestations (32). We know this to be the case, because the reduction of thalamic overinhibition, by surgical decrease of the pallidal output to the thalamus, diminishes or suppresses Parkinsonian manifestations (13). This therapeutic goal can be reached by a radio-frequency lesion and a chronic stimulation device that causes a continuous depolarization block (39, 40), or by pharmacological treatment to reduce excess inhibition (41, 42). In other words, reduction of the thalamic overinhibition suppresses the thalamocortical dysrhythmia that is responsible for the clinical symptomatology. This view is directly supported by single-unit recordings taken during stereotactic operations that show the presence of LTS bursting activity in the pallidal-recipient, motor-thalamic nuclei of Parkinsonian patients (43).
A similar case may be made for the other patients in this study. Indeed, in the case of neurogenic pain, depression, and tinnitus, a persistent and coherent θ-thalamocortical oscillatory activity was also observed in this study. In these three clinical situations, but also in epilepsy, obsessive–compulsive disease, dystonia, and spasticity, medial thalamic LTS θ-rhythmic activity was shown, as well as the possibility of reducing the symptoms by a stereotactic intervention at the medial thalamic level (13). In the field of psychiatry (44), in addition to obsessive–compulsive disease and depression, one may also remember that low-frequency activity has been known to be recorded in schizophrenic patients (45), although a thalamocortical dysrhythmia was never considered as a mechanism.
The Issue of Localization and Dynamics.
As described above, the basic idea infers that the symptomatology presented by the patients ultimately relates to the overall localization of the low-frequency activity. We do not present a purely phrenological description; rather, what we observed is an abnormal distribution and coherence of low-frequency activity over wide areas of the brain, anterior as well as posterior, and the persistence of this phenomenon throughout the recording session. Thus, in the case of a depressed patient, stimuli that may produce short-lived sadness in normal individuals may have a dynamic time course that prolongs the normal emotional experience into long-lasting depressive periods. Such experience may occur even after exposure to stimuli that are not normally depressant. Similar conclusions may be reached concerning all of the other thalamocortical dysrhythmias described here, such as the exacerbation of neurogenic pain by nonpainful, tactile, or proprioceptive stimuli.
An amplification of the symptomatology caused by fear and stress is also recognized by patients suffering from the various positive symptoms described in this paper. In view of the widespread distribution of the coherent thalamocortical θ-activity described here, we may also propose that the large associational–mesocortical system is more relevant than lateral unimodal cortical areas. The areas where we expect to see maximal low-frequency activity are the cingulate, medial prefrontal, and orbitofrontal cortices for neuropsychiatric symptoms; the supplementary motor and cingulate areas for Parkinson's disease; the insular, parietal opercular, and cingular cortices for neurogenic pain; and the medial temporal areas for tinnitus (13).
In terms of thalamocortical dynamics, the medial thalamic nuclei might be seen as the best candidates in view of their better coherence abilities (46). We have, however, proposed that thalamic dynamics relates essentially to the temporal interactions between the “content, specific or lateral” and the “context, nonspecific or medial” thalamocortical systems (9, 20). In this sense, the relevance of the generation of the thalamocortical dysrhythmia must be ascribed to both systems.
Origin of the Thalamocortical Dysrhythmia: “Bottom-Up Versus Top-Down” Mechanism.
The basic assumption in this discussion is that the thalamocortical dysrhythmia is central and that the abnormal condition is brought about by changes in intrinsic, voltage-gated ionic conductances at the level of thalamic relay cells (36), namely, the deinactivation of T channels by cell membrane hyperpolarization (35). LTS bursts are produced and lock the related thalamocortical circuits in low-frequency resonance. Low-frequency loops interact at the cortical level with high-frequency ones, giving rise to the edge effect and the generation of a positive symptom. In tinnitus, peripheral neurogenic pain, Parkinson's disease, and some neuropsychiatric disorders with striatal origin, the dysrhythmic mechanism is triggered bottom-up, i.e., from the thalamus toward the cortex. In other situations, such as epilepsy, neuropsychiatric conditions of cortical origin, and central cortical neurogenic pain, the mechanism may be top-down, triggered by a reduction of the corticothalamic input. Both bottom-up and top-down situations result in excess inhibition or disfacilitation, generating thalamic cell membrane hyperpolarization and low-frequency oscillation.
Relation to Other Imaging Technologies.
A proper analysis of the thalamocortical dysrhythmia must ultimately be implemented with a technique that is fast enough to distinguish among the different thalamocortical frequencies (4–50 Hz), and it must have sufficient spatial resolution to localize accurately all sites involved. These criteria seem to be ideally fulfilled by MEG (29). However, considering the enormous diagnostic relevance of both PET and functional MRI, the best approach may be to combine these three noninvasive tools, such that their relative advantages may coordinate to optimize understanding and diagnosis of these abnormal conditions. When comparing our results with those obtained from PET recordings (47, 48), it seems evident that low-frequency activity in MEG correlates well with hypometabolism in PET, which is not altogether surprising, in view of the decreased electrical activity accompanying calcium-dependent potassium conductances (49). PET may demonstrate high-frequency, edge-effect activation areas as hypermetabolic, and low-frequency areas as hypometabolic—a proposition already supported by reports of both hyper- and hypometabolic areas in the thalamus and cortex of patients suffering from neurogenic pain (50). On the other hand, functional MRI may prove to be a powerful tool for localizing areas that harbor elevated γ activity, if such elevation leads to elevated metabolic activity.
Acknowledgments
We thank J. Schulman for the careful handling of subject/patient recordings. The excellent assistance of M. Villegas, R. Jagow, and A. Polyakov is appreciated. For the careful patient selection, we are indebted to Drs. R. Cancro, A. Rezai, J. T. Roland, and Siegemund. The contributions of Drs. A. Mogilner, M. Magnin, and A. Morel are also appreciated. We acknowledge the support of Biomagnetic Technologies, Inc. for this project. This study was supported by Lucent Technologies, The Charles A. Dana Foundation, the National Institutes of Health (NINDS13742), and the New York University Medical Center General Clinical Research Center (NIH–NCRR M01 RR00096).
Abbreviations
- LTS
low-threshold spike
- MEG
magnetoencephalography
- PET
positron emission tomography
References
- 1.Llinás R. Science. 1988;242:1654–1664. doi: 10.1126/science.3059497. [DOI] [PubMed] [Google Scholar]
- 2.Llinás R, Ribary U, Joliot M, Wang X J. In: Temporal Coding in the Brain. Buzsaki G, Llinás R, Singer W, Berthoz A, Christen Y, editors. Berlin: Springer; 1994. pp. 251–272. [Google Scholar]
- 3.Ahissar E, Vaaida E. Proc Natl Acad Sci USA. 1990;87:8935–8939. doi: 10.1073/pnas.87.22.8935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murthy V N, Fetz E E. Proc Natl Acad Sci USA. 1992;89:5670–5674. doi: 10.1073/pnas.89.12.5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.von der Marlsburg C. Internal Report. Göttingen, Germany: Max Planck Institute for Biophysical Chemistry; 1981. [Google Scholar]
- 6.Eckhorn R, Bauer R, Jordan W, Brosch M, Kruse W, Munk M, Reitboeck H J. Biol Cybern. 1988;60:121–130. doi: 10.1007/BF00202899. [DOI] [PubMed] [Google Scholar]
- 7.Gray C M, Singer W. Proc Natl Acad Sci USA. 1989;86:1698–1702. doi: 10.1073/pnas.86.5.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crick F, Koch C. Cold Spring Harbor Symp Quant Biol. 1990;55:953–962. doi: 10.1101/sqb.1990.055.01.089. [DOI] [PubMed] [Google Scholar]
- 9.Llinás R, Pare D. Neuroscience. 1991;44:521–535. doi: 10.1016/0306-4522(91)90075-y. [DOI] [PubMed] [Google Scholar]
- 10.Joliot M, Ribary U, Llinás R. Proc Natl Acad Sci USA. 1994;91:11748–11751. doi: 10.1073/pnas.91.24.11748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Llinás R, Ribary U, Contreras D, Pedroarena C. Philos Trans R Soc London B. 1998;353:1841–1849. doi: 10.1098/rstb.1998.0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ribary U, Cappell J, Mogilner A, Hund-Georgiadis M, Kronberg E, Llinás R. Adv Neurol. 1999;81:49–56. [PubMed] [Google Scholar]
- 13.Jeanmonod D, Magnin M, Morel A. Brain. 1996;119:363–375. doi: 10.1093/brain/119.2.363. [DOI] [PubMed] [Google Scholar]
- 14.Thomson D J. Proc IEEE. 1982;70:1055–1096. [Google Scholar]
- 15.Slepian D, Pollak H O. Bell Syst Techn J. 1961;40:43–63. [Google Scholar]
- 16.Mitra P P, Pesaran B. Biophys J. 1999;76:691–708. doi: 10.1016/S0006-3495(99)77236-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niedermeyer E, Lopes da Silva F. Electroencephalography; Basic Principles, Clinical Applications and Related Fields. Baltimore: Urban and Schwarzenberg; 1982. [Google Scholar]
- 18.Sasaki K, Nambu A, Tsujimoto T, Matsuzaki R, Kyuhou S, Gemba H. Cognit Brain Res. 1996;5:165–174. doi: 10.1016/s0926-6410(96)00053-5. [DOI] [PubMed] [Google Scholar]
- 19.Machleidt W, Gutjahr L, Muegge A. Grundgefuehle: Phaenomenologie, Psychodynamik und EEG Spektralanalytik. Berlin: Springer; 1989. [Google Scholar]
- 20.Llinás R, Ribary U. Proc Natl Acad Sci USA. 1993;90:2078–2081. doi: 10.1073/pnas.90.5.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Contreras D, Destexhe A, Sejnowski T J, Steriade M. Science. 1996;274:771–774. doi: 10.1126/science.274.5288.771. [DOI] [PubMed] [Google Scholar]
- 22.Contreras D, Steriade M. Neuroscience. 1997;76:11–24. doi: 10.1016/s0306-4522(96)00393-4. [DOI] [PubMed] [Google Scholar]
- 23.Steriade M, Curró Dossi R, Nuñez A. J Neurosci. 1991;11:3200–3217. doi: 10.1523/JNEUROSCI.11-10-03200.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarnthein J, Petsche H, Rappelsberger P, Shaw G L, von Stein A. Proc Natl Acad Sci USA. 1998;95:7092–7096. doi: 10.1073/pnas.95.12.7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lisman J E, Idiart M A P. Science. 1995;267:1512–1515. doi: 10.1126/science.7878473. [DOI] [PubMed] [Google Scholar]
- 26.Schack B, Grieszback G, Krause W. Int J Psychophysiol. 1999;3(1):219–240. doi: 10.1016/s0167-8760(98)00052-x. [DOI] [PubMed] [Google Scholar]
- 27.Galambos R, Makeig S, Talmadroff P J. Proc Natl Acad Sci USA. 1981;78:2643–2647. doi: 10.1073/pnas.78.4.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engel A K, Konig P, Singer W. Proc Natl Acad Sci USA. 1991;88:9136–9140. doi: 10.1073/pnas.88.20.9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ribary U, Ioannides A A, Singh K D, Hasson R, Bolton J P R, Lado R, Mogilner A, Llinás R. Proc Natl Acad Sci USA. 1991;88:11037–11041. doi: 10.1073/pnas.88.24.11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghoose G M, Freeman R D. J Neurophysiol. 1992;68:1558–1574. doi: 10.1152/jn.1992.68.5.1558. [DOI] [PubMed] [Google Scholar]
- 31.Russell M B, Olesen J. Brain. 1996;119:355–361. doi: 10.1093/brain/119.2.355. [DOI] [PubMed] [Google Scholar]
- 32.Volkmann J, Joliot M, Mogilner A, Ioannides A A, Lado F, Fazzini E, Ribary U, Llinás R. Neurology. 1996;46:1359–1370. doi: 10.1212/wnl.46.5.1359. [DOI] [PubMed] [Google Scholar]
- 33.Soikkeli R, Partanen J, Soininen H, Paakonen A, Riekkinen P S. Electroencephalogr Clin Neurophysiol. 1991;79:159–165. doi: 10.1016/0013-4694(91)90134-p. [DOI] [PubMed] [Google Scholar]
- 34.England A C, Schwab R S, Peterson E. Electroencephalogr Clin Neurophysiol. 1959;11:723–731. doi: 10.1016/0013-4694(59)90111-7. [DOI] [PubMed] [Google Scholar]
- 35.Tsien R W, Lipscombe D, Madison D V, Bley K R, Fox A P. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- 36.Jahnsen H, Llinás R. J Physiol (London) 1984;349:205–226. doi: 10.1113/jphysiol.1984.sp015153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCormick D A, Pape H. J Physiol (London) 1990;431:291–318. doi: 10.1113/jphysiol.1990.sp018331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCormick D A. J Neurophysiol. 1991;66:1176–1189. doi: 10.1152/jn.1991.66.4.1176. [DOI] [PubMed] [Google Scholar]
- 39.Rezai A R, Hutchinson W, Lozano A M. In: Neurosurgical Operative Atlas. Rengachary S S, Wilkins R H, editors. Vol. 8. Chicago: American Association of Neurological Surgeons; 1999. pp. 195–207. [Google Scholar]
- 40.Limousin P, Krack P, Pollack P, Benazzouz A, Ardouin C, Hoffmann D, Benabid A L. N Engl J Med. 1998;339:1105–1111. doi: 10.1056/NEJM199810153391603. [DOI] [PubMed] [Google Scholar]
- 41.Lang A E, Lozano A M. N Engl J Med. 1998;339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- 42.Lang A E, Lozano A M. N Engl J Med. 1998;339:1130–1143. doi: 10.1056/NEJM199810153391607. [DOI] [PubMed] [Google Scholar]
- 43.Raeva S, Vainberg N, Dubinin V. Neuroscience. 1999;88:365–376. doi: 10.1016/s0306-4522(98)00228-0. [DOI] [PubMed] [Google Scholar]
- 44.John E R, Prichep L S, Fridman J, Easton P. Science. 1988;239:162–169. doi: 10.1126/science.3336779. [DOI] [PubMed] [Google Scholar]
- 45.Canive J M, Lewine J D, Edgar J C, Davis J T, Torres F, Roberts B, Graeber D, Orrison W W, Tuason V B. Psychopharmacol Bull. 1996;32:741–750. [PubMed] [Google Scholar]
- 46.Morrison R S, Dempsey E W. Am J Physiol. 1942;135:281–292. [Google Scholar]
- 47.Buchsbaum M S, Hazlett E A. Int Rev Psychiatr. 1997;9:339–354. [Google Scholar]
- 48.Perlmutter J S, Raichle M E. Neurology. 1985;35:1127–1134. doi: 10.1212/wnl.35.8.1127. [DOI] [PubMed] [Google Scholar]
- 49.Latorre R, Oberhauser A, Labarca P, Alvarez O. Annu Rev Physiol. 1989;51:385. doi: 10.1146/annurev.ph.51.030189.002125. [DOI] [PubMed] [Google Scholar]
- 50.Hsieh J C, Belfrage M, Stone-Elander S, Hansson P, Ingvar M. Pain. 1995;63:225–236. doi: 10.1016/0304-3959(95)00048-W. [DOI] [PubMed] [Google Scholar]