

Abstract
We quantified the amount of amyloid β-peptide (Aβ) immunoreactivity as well as amyloid deposits in a large cohort of transgenic mice overexpressing the V717F human amyloid precursor protein (APPV717F+/− TG mice) with no, one, or two mouse apolipoprotein E (Apoe) alleles at various ages. Remarkably, no amyloid deposits were found in any brain region of APPV717F+/− Apoe−/− TG mice as old as 22 mo of age, whereas age-matched APPV717F +/− Apoe+/− and Apoe+/+ TG mice display abundant amyloid deposition. The amount of Aβ immunoreactivity in the hippocampus was also markedly reduced in an Apoe gene dose-dependent manner (Apoe+/+ > Apoe+/− ≫ Apoe−/−), and no Aβ immunoreactivity was detected in the cerebral cortex of APPV717F+/− Apoe−/− TG mice at any of the time points examined. The absence of apolipoprotein E protein (apoE) dramatically reduced the amount of both Aβ1–40 and Aβ1–42 immunoreactive deposits as well as the resulting astrogliosis and microgliosis normally observed in APPV717F TG mice. ApoE immunoreactivity was detected in a subset of Aβ immunoreactive deposits and in virtually all thioflavine-S-fluorescent amyloid deposits. Because the absence of apoE alters neither the transcription or translation of the APPV717F transgene nor its processing to Aβ peptide(s), we postulate that apoE promotes both the deposition and fibrillization of Aβ, ultimately affecting clearance of protease-resistant Aβ/apoE aggregates. ApoE appears to play an essential role in amyloid deposition in brain, one of the neuropathological hallmarks of Alzheimer's disease.
Individuals who inherit one or two ɛ4 alleles of the human apolipoprotein E (APOE) gene have an increased risk of developing and an earlier age of onset of Alzheimer's disease (AD), when compared to individuals who inherit the more common ɛ3 allele (1, 2). In addition, the ɛ2 allele is associated with reduced risk of developing and/or a later age of onset of AD and thus may constitute a protective factor (3). Recently, several polymorphisms in the APOE gene promoter have also been linked to AD risk (4, 5), and, in general, these promoter variants result in an increase in transcriptional activity when studied in isolated expression systems (6). Of all the genes so far found to contribute to the risk of developing AD, APOE4 is by far the most important, because it has been estimated to increase the age-adjusted relative risk by 3- to 10-fold in heterozygotes and homozygotes, respectively (1–7). Nevertheless, individuals who are homozygous for the ɛ4 allele do not invariably develop AD, even when examined well into their 90s, and therefore other genetic and nongenetic factors undoubtedly contribute to disease onset and progression in many cases (8).
Apolipoprotein E protein (apoE) is a 34-kDa very low-density lipoprotein synthesized primarily by the liver, which functions in the periphery as a mediator of lipoprotein metabolism and lipid clearance through binding of apoE-containing lipoprotein particles to the low-density lipoprotein receptor-related protein (2, 9, 10). In the central nervous system, apoE is synthesized and secreted primarily by astrocytes and microglia, and its importance is underscored by the low abundance of other apolipoproteins (11–13). Additionally, apoE is believed to play a pivotal role in the redistribution of lipid and cholesterol during membrane repair and has been postulated to be important for maintaining synaptic plasticity, especially after neuronal injury (14–16). In humans, APOE is a single gene located on chromosome 19q 13.2 with three major allelic variants (ɛ2, ɛ3, and ɛ4) encoding three protein isoforms (2). Rodents, however, have only a single gene, which differs from the human amino acid sequence by 30% at the C terminus, a region that is intimately associated with the core of neuritic plaques (17–19). Functionally, it is not clear how similar rodent apoE is to the human isoform(s). Targeted replacement studies demonstrate that when mouse Apoe is replaced with human APOE3, the resulting E3/E3 mice are more susceptible to diet-induced hyperlipidemia, suggesting that human apolipoproteins may not be good ligands for mouse receptors (20).
How do the three APOE alleles and their encoded protein isoforms alter the risk of developing AD? Several hypotheses have been proposed, including differential isoform-specific neurotrophic (21–24) and neurotoxic properties (25), antioxidative activity (26), and amyloidogenic effects (27–29). The latter hypothesis is supported by postmortem neuropathological findings from several laboratories, which have consistently demonstrated increased amyloid burden in APOE4 carriers (30–32). Although in vitro studies have demonstrated avid binding of amyloid β-peptide (Aβ) to the three apoE isoforms, forming in some cases rather stable complexes, the isoform-dependent specificity of this binding is controversial (34), and its relationship to amyloid clearance, deposition, and/or fibrillogenesis is still poorly understood.
To investigate the role of apoE on amyloid deposition in vivo, we crossed Apoe knockout mice (35) with transgenic mice overexpressing a mutant (valine to phenylalanine at position 717; V717F) β amyloid precursor protein (APP) gene (36, 37). We have previously reported the absence of amyloid deposits in 6-mo-old TG mice that are homozygous for APPV717F as well as being Apoe deficient (37). We hypothesized that the lack of amyloid deposition in these Apoe-deficient TG mice would be influenced by age, and that older mice would eventually develop amyloid deposits like their wild-type counterparts. We now report that heterozygous APPV717F+/− TG mice that lack Apoe do not develop amyloid deposits even when examined at 21–22 mo of age. Furthermore, the area occupied by Aβ immunoreactive deposits is dramatically decreased in the hippocampus and completely absent in the cerebral cortex of these mice. The marked astrogliosis and microgliosis observed in APPV717FApoe+/+ TG mice are also greatly reduced in Apoe-deficient TG mice. Our findings strongly suggest that apoE expression is critical for Aβ deposition and for its subsequent fibrillization into protease-resistant amyloid deposits.
Materials and Methods
Mice Deficient in Apoe Containing the APP V717F Transgene.
Mice (both male and female) that were homozygous for the APPV717F transgene (36) were crossed to an Apoe knockout mouse (35). The resulting hemizygous progeny (APPV717F+/− Apoe+/−) were subsequently bred, and the following genotypes were selected for this study: APPV717F+/− Apoe−/−; APPV717F+/− Apoe+/−; APPV717F+/− Apoe+/+. Age-matched littermates with the following genotypes: APPV717F−/− Apoe+/+ and/or APPV717F−/− Apoe−/− were used as controls. Three to six animals from each of the above gentoypes at 9, 15, and 21/22 mo of age were perfused and the brains processed for paraffin sectioning (see below).
Tissue Preparation and Immunohistochemistry.
Animals were heparinized (500 U. S. Pharmacopeia units i.p.), anesthetized, and perfused transcardially with 60 ml of saline followed by 120 ml of 4% paraformaldehyde in 0.1M phosphate buffer (pH 7.2). Brains were postfixed in the same fixative, dehydrated in graded ethanols, cleared in xylene, and embedded in paraffin. Serial coronal sections of 10-μm thickness were cut using a Leica RM 2135 microtome and placed on poly-l-lysine-coated slides (two sections per slide). Every tenth pair of sections was stained for thioflavine-S histochemistry (39). Additional sets of adjacent sections were immunoreacted with one of the following monoclonal antibodies: 10D5 (Aβ1–28, 1:100) or 21F12 (Aβ33–42, 1:1,000) (38) or an Aβ1–40-specific antibody (no. 44–348; Quality Control Biochemicals, Hopkington, MA). ApoE antiserum (1:1,500; Chemicon) was used to detect apoE. Antibodies were visualized by using goat anti-mouse IgG by the peroxidase-antiperoxidase method utilizing 3,3′diaminobenzidine (DAB) as the chromogen. For immunofluorescence, secondary antibodies were FITC-conjugated anti-mouse IgG and rhodamine red-conjugated anti-goat (1:100; Jackson ImmunoResearch). Six sections from each animal of each genotype were immunolabeled with antibodies specific for Aβ1–42 and Aβ1–40. An additional six sections per animal per genotype were immunolabeled with glial fibrillary acidic protein antisera (GFAP, 1:1,000; Boehringer Mannheim). Both Aβ, as well as GFAP, immunoreactivity was visualized by using DAB. The specificity of Aβ immunoreactivity was confirmed by preadsorption with the appropriate peptides as well as lack of signal when the primary antibody was omitted. Activated microglia were visualized by using tomato lectin histochemistry on paraffin-embedded sections, as previously described (40).
Quantification of Aβ Deposits.
Immunoreactive Aβ deposits and thioflavine-S-fluorescent Aβ deposits were quantified semiautomatically by using a Quantimet 570 (Leica) image processing and analysis system fitted to a Leitz Aristoplan microscope. Control of the image analysis system and the automated microscope was achieved by using the standard Quantimet 570 QUIC and qbasic software written in the qbasic language. Aβ (10D5 immunoreactivity), and thioflavine-S-reactive deposits were measured by using a ×16 objective in a binary plane and Quantimet's color detection function and, when necessary, image editing. The number of deposits and the area of their cross-sectional profiles were obtained automatically on every set of 20th sections and accumulated separately for cortex and hippocampus. Data collected for areas were converted to square microns.
Analysis of immunoreactive deposits for apoE alone and Aβ colocalized with apoE were performed on a Macintosh computer by using the public domain NIH image program (http://rsb.info.nih.gov/nih-image) by defining a region of interest, measuring the area, and counting immunopositive deposits. Two regions were analyzed: an area of parietal cortex comprising layers I–VI (approximately 650 μm2) and an area of the hippocampal CA1 region comprising layers oriens, pyramidal, and stratum radiatum (approximately 350 μm2).
Reverse Transcriptase–PCR and Western Blot Analysis.
Total RNA (2.5 μg), extracted from the cortices of 15-mo-old APPV717F+/−Apoe−/− or APPV717F+/− Apoe+/+ TG mice, was reverse transcribed by using Superscript (GIBCO/BRL). One microliter of a 1:10 dilution of reverse-transcribed cDNA was used as the PCR template. Primers and cycling conditions were identical to those previously published (36, 38). Total protein was extracted from hippocampi of the above mice by homogenization in buffer containing 1% Nonidet P-40, 0.1% SDS, 50 mM Tris (pH 8.0), 50 mM NaCl, 0.05% deoxycholate, and protease inhibitors (Boehringer Mannheim). Samples were size fractionated on a 4–12% polyacrylamide gradient gel (NOVEX, San Diego) and transferred onto nitrocellulose (Hybond N, Amersham). The nitrocellulose filter was then probed sequentially with a monoclonal antibody specific for the human amyloid precursor protein (antibody 8E5) (36–38) and a polyclonal antibody specific for mouse apoE (BioDesign, Kennebunk, ME). The specific signal was then visualized by using enhanced chemiluminescence (Amersham).
Aβ ELISA Measurements.
Hippocampi and cortices from 10 2-mo-old TG mice from each of the following genotypes: APPV717F+/− Apoe−/− and APPV717F+/− Apoe+/+ were microdissected and quickly homogenized in 5.5 M guanidine buffer. Homogenates were diluted 1:10 with cold casein buffer (0.25% casein/0.05% sodium azide) followed by centrifugation for 20 min at 4°C at 10,000 × g. Total Aβ and Aβ1–42 were measured by sandwich ELISA, as previously described (37).
Statistical Evaluation.
Inferences made in this work are based on P values from t tests and ANOVA contrasts. Because these methods assume that the data are normally or Gaussian distributed, the data were power transformed to better meet this assumption. The optimal power for the transformation was determined to be 0.2 following the method of Box and Cox (41). The areas of both thioflavine-S-positive staining and Aβ immunoreactivity were thus transformed, and the transformed data were tested for normality. Because the sample sizes were small, a test was used that adjusts for bias in small samples. The Brown–Hettmansperger tests for normality (42) indicated that the transformed data are approximately normally distributed. A series of ANOVA contrasts on the transformed data were then preformed.
Results
Apoe-Deficient APPV717F+/− TG Mice Fail to Develop Amyloid Deposits.
Transgenic mice (APPV717F+/−) with the three Apoe genotypes (Apoe−/−; Apoe+/−; Apoe+/+) were studied at 9, 15, and 21/22 mo of age. Age-matched littermates without the APPV717F transgene served as controls. No amyloid deposits were observed in any brain region examined in APPV717F+/− Apoe−/− mice at any of the ages examined (9, 15, or 21/22 mo; Figs. 1 and 2). There was an age-dependent increase in the area occupied by amyloid deposits in both the hippocampus and cortex of APPV717F+/− Apoe+/+ and APPV717F+/− Apoe+/− TG mice (Fig. 3 A and B). However, a dramatic decrease in amyloid deposits was observed in Apoe hemizygous TG mice (compared with Apoe wild-type TG mice) at all time points examined. For example, at 21/22 mo of age, the amount of amyloid in Apoe+/− TG mice was ≤30% of that observed in Apoe+/+ TG mice (P < 0.01 compared with Apoe+/+ TG mice) (Figs. 2 and 3). Similar results were observed in the hippocampus and cerebral cortex (Fig. 2 A and B). No amyloid deposits were seen in age-matched mice lacking the APPV717F transgene (data not shown).
Figure 1.

Lack of apoE reduces both Aβ immunoreactive as well as amyloid deposits in a transgenic mouse model of AD. In 21/22-mo-old APPV717+/− TG mice wild-type for Apoe (APPV717F+/− Apoe+/+), numerous thioflavine-S-fluorescent (A) and Aβ immunoreactive deposits (B and C) are evident in both the hippocampus and the cerebral cortex. By contrast, in 21/22-mo-old APPV717F+/− Apoe−/− mice, no thioflavine-S-fluorescent Aβ deposits are observed in any brain region. Furthermore, Aβ-immunoreactive deposits are confined to the hippocampus. APPV717F+/− Apoe+/− mice had an essentially intermediate level of both thioflavine-S-fluorescent and Aβ immunoreactive deposits. Neither thioflavine-S-fluorescent nor Aβ-immunoreactive deposits were found in APPV717F−/− Apoe+/+ mice. The photomicrographs are from representative sections of each of the genotypes indicated. (Original magnification, A and B ×9, C ×35.)
Figure 2.

ApoE expression is required for amyloid deposition in the APPV717F TG mouse. APPV717F+/− TG mice with various apoE alleles (Apoe+/+; +/−;−/−) were studied at 9, 15, and 21/22 mo of age. Serial coronal sections were stained with thioflavine-S, and the area occupied by fluorescence was quantified for both the hippocampus and the cerebral cortex (see text for details). Note the complete absence of amyloid deposits in APPV717F+/− TG mice in the absence of Apoe and the modest increase with age in Apoe+/− mice. Amyloid deposition in APPV717F+/− Apoe+/− mice never exceeds 20–30% of that observed in APPV717F+/− Apoe+/+ mice despite significant Aβ immunoreactive deposits in the hippocampus (see Fig. 3). All data were log transformed and tested for normality before statistical analysis (see Materials and Methods). ***, P < 0.001; **, P < 0.01; P = not significant (ns) compared with APPV717F−/− Apoe+/+ mice. #, P < 0.01 compared with APPV717F+/− Apoe+/+ mice.
Figure 3.

Effect of apoE on Aβ immunoreactivity in the hippocampus and cerebral cortex of APPV717F+/− TG mice with various Apoe alleles. Serial coronal sections were stained for total Aβ (10D5, 1:1,000), and the area occupied by immunoreactive deposits was quantified (see Materials and Methods). The area occupied by Aβ immunoreactivity increased significantly from 9 to 21/22 mo in APPV717F+/− Apoe+/+ and APPV717F+/− Apoe+/− mice. A reduction in the area occupied by Aβ immunoreactivity was seen in the hippocampus (A) of APPV717F+/− Apoe−/− mice at all ages examined. In contrast to the hippocampus, no Aβ immunoreactive deposits were observed in the cortex (B) of Apoe-deficient APPV717F+/− mice. Note that by 21/22 mo, Apoe hemizygous mice have a similar amount of Aβ immunoreactivity as Apoe homozygous mice, whereas the amount of amyloid deposition is significantly decreased to ≤30% of homozygous mice (Fig. 2). All data were log transformed and tested for normality before statistical analysis (see Materials and Methods). ***, P < 0.001; **, P < 0.01; P = ns compared with APPV717F−/− Apoe+/+ TG mice. #, P < 0.01 compared with APPV717F+/− Apoe+/+ TG mice.
The Deposition and Distribution of Aβ Immunoreactivity Is Markedly Influenced by apoE.
The area occupied by Aβ immunoreactivity in the hippocampus of APPV717F+/− Apoe+/+ TG mice increases dramatically with age (Fig. 3A). The age-associated increase in hippocampal Aβ immunoreactivity observed in APP V717F+/− Apoe+/− TG mice was similar but delayed when compared with that found in APPV717F+/− Apoe+/+ TG mice. For example, the overall area of hippocampal Aβ immunoreactive deposits was significantly lower at 9 mo in Apoe+/− TG mice (P < 0.001 compared with Apoe+/+ TG mice), but reached levels similar to Apoe+/+ TG mice by 21/22 mo of age (P = not significant vs. Apoe+/+ TG mice) (Fig. 3A). In the cerebral cortex of APPV717F+/− Apoe+/− TG mice, the increase in Aβ immunoreactivity eventually equaled the area occupied by Aβ immunoreactivity in the hippocampus of APPV717F+/− Apoe+/+ TG mice (i.e., by 21/22 mo of age). Like the hippocampus, however, the increase in Aβ immunoreactivity was delayed in Apoe+/− TG mice and was significantly reduced compared with Apoe+/+ TG mice at 9 and 15 mo of age (P < 0.01) (Fig. 3B). Strikingly, and in contrast to the hippocampus, no Aβ immunoreactive deposits were observed in the cortex of Apoe-deficient APPV717F+/− TG mice at any age examined (Fig. 3B).
The regional distribution of Aβ immunoreactive deposits was similar in APPV717F+/− TG mice with one or two Apoe alleles. Aβ deposits varied from small single to large multifocal deposits reaching up to 100 μm in diameter and affecting all regions of the cerebral cortex as well as the hippocampus. However, the polymorph layer of the dentate gyrus of the hippocampus did not show Aβ immunoreactivity in Apoe+/+ or Apoe+/− TG mice. The amount and regional distribution of Aβ immunoreactivity differed dramatically in APPV717F+/− Apoe−/− TG mice as compared with Apoe+/+ or Apoe+/− TG mice. Of significance was the reduction in the area occupied by Aβ immunoreactivity in the hippocampus (Figs. 1, 2, and 3A) and the complete lack of Aβ immunoreactivity in the cerebral cortex of APPV717F+/−Apoe−/− mice at any of the ages examined (Fig. 3B). In the hippocampus of APPV717F+/− Apoe−/− TG mice, nearly all Aβ immunoreactivity was Aβ1–42 (Fig. 4). Furthermore, the distribution of Aβ immunoreactivity in the hippocampus was similar to the other genotypes examined, with the distinct exception of the dentate gyrus, where the polymorph layer was preferentially affected (Figs. 1 and 4).
Figure 4.
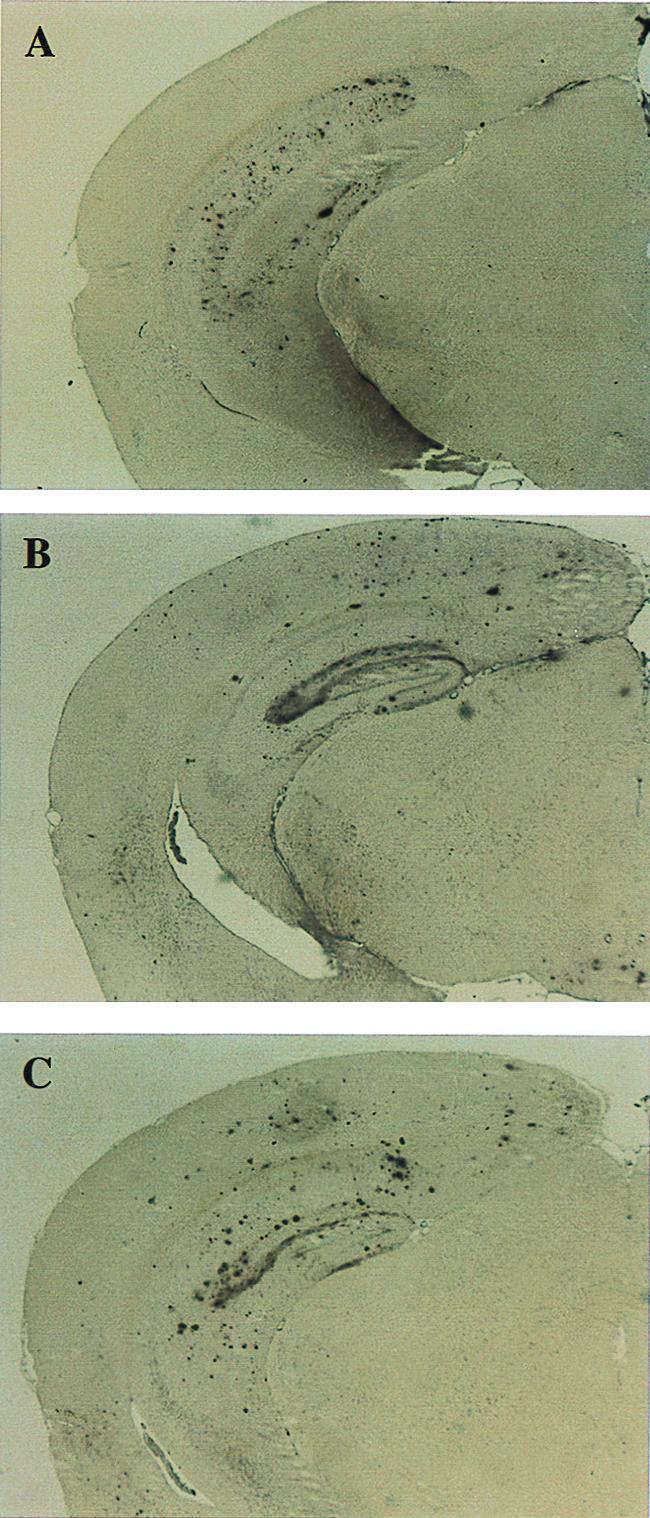
Aβ immunoreactivity in APPV717F+/− TG mice with the three Apoe genotypes: (A) Apoe−/−; (B) Apoe+/−; and (C) Apoe+/+ at 21/22 mo of age. Sections were stained with an antibody specific for Aβ1–42 (21F12; see Materials and Methods for details). There is a decrease in the area occupied by Aβ immunoreactivity in the hippocampus of APPV717F+/− Apoe−/− mice and a complete absence of Aβ immunoreactivity (including total Aβ; see also Fig. 1) in the cerebral cortex (A). The distribution of Aβ1–42 immunoreactivity was similar in all genotypes, with the exception of the dentate gyrus of the hippocampus where the polymorph layer was preferentially affected in APPV717F+/− Apoe−/− mice. Note the absence of Aβ deposits in the outer molecular layer of the dentate gyrus in Apoe−/− compared with Apoe+/+ mice (A vs. B). (Original magnification ×5.)
Glial Activation Associated with Aβ Deposition Is Markedly Reduced in Mice Lacking apoE.
In AD, deposition of Aβ in senile plaques is associated with astrocytic and microglial proliferation. Thus, we investigated whether the lack of amyloid observed in APPV717F+/−Apoe−/− TG mice would result in decreased astrogliosis and microgliosis. The astroglial and microglial response was evaluated in 15- and 21/22-mo-old APPV717F+/− Apoe+/+ and APPV717F+/− Apoe−/− TG mice. The degree of gliosis, as determined by GFAP immunolabeling, differed among genotypes, being more severe in the cerebral cortex and molecular layer of the hippocampus of APPV717F+/− Apoe+/+ TG mice (Fig. 5A). Qualitatively, clusters of intensely GFAP-positive astroglial cells were present in APPV717F+/− Apoe+/+ TG mice in both the hippocampus and frontal cortex (Fig. 5A and Inset). These clusters appeared as two or more GFAP immunoreactive astrocytes that were hypertrophied and had multiple thick cytoplasmic processes. These clusters were found mostly in the CA1 and CA3 regions and the dentate gyrus of the hippocampus, as well as the cerebral cortex. Fewer GFAP-positive astrocyte clusters were observed in both the hippocampus and cortex of APPV717F+/− Apoe−/− TG mice when compared with Apoe+/+ TG mice (Fig. 5 A and B). The intensity (and clustering) of tomato lectin staining for activated microglia was also greatly decreased in the cerebral cortex and hippocampus of APPV717F+/− Apoe−/− TG mice compared with APPV717F+/− Apoe+/+ TG mice (Fig. 5 C and D).
Figure 5.
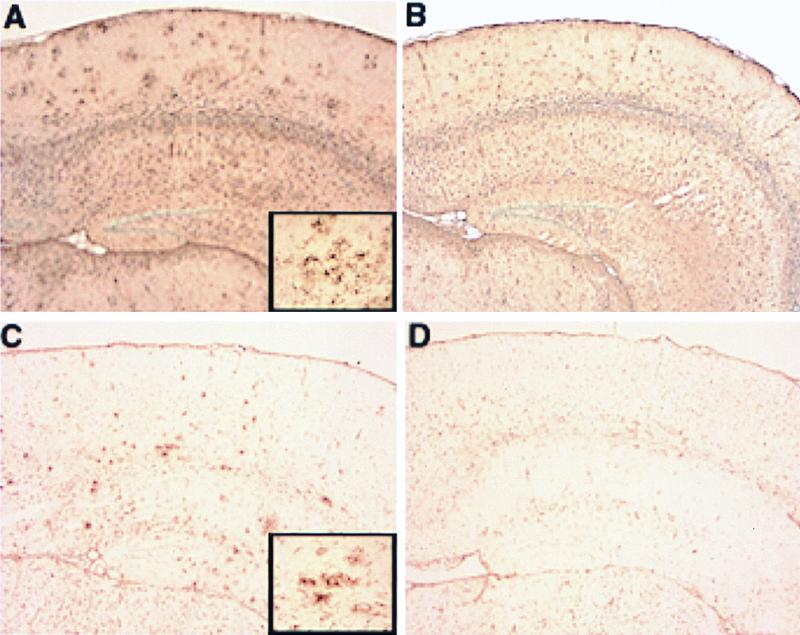
Reduced glial activation in APPV717F+/− Apoe−/− TG mice. Glial activation was evaluated by GFAP immunohistochemistry (A and B) and tomato lectin histochemistry (C and D) on paraffin-embedded brain sections from 21/22-mo-old APPV717F+/− Apoe+/+ and APPV717F+/− Apoe−/− mice (see Materials and Methods for details). A range of glial staining for each genotype analyzed was apparent with an increase in GFAP immunoreactivity and lectin staining in the superficial layers of the cerebral cortex and hippocampus in APPV717F+/− Apoe+/+ mice (A and C, respectively) compared with APPV717F+/− Apoe−/− mice (B and D, respectively). Note clusters of intensely stained astrocytes (A, Inset) and microglia (C, Inset), which are more prominent in APPV717F+/− Apoe+/+ than in APPV717F+/− Apoe−/− mice. (×5; Inset, ×40.)
ApoE Immunoreactivity Is Associated with a Subset of Aβ Immunoreactive Deposits and Virtually All Thioflavine-S-Fluorescent Aβ Deposits.
To determine whether apoE is associated with immunoreactive Aβ deposits and amyloid, we quantified them in serial sections of the cerebral cortex of APPV717F+/− Apoe+/+ TG mice at 21/22 mo of age. It is apparent from Fig. 6 that apoE immunoreactive Aβ deposits represent a subpopulation of the total Aβ deposits labeled with the 10D5 antibody (all Aβ species), because the number of apoE-positive deposits represents ≤50% of the total Aβ deposits quantified in alternate sections from the same brain region (Fig. 6D). As a control for nonspecific binding of the apoE antisera to Aβ deposits, we stained brain sections from APPV717F+/− Apoe−/− TG mice with the anti-apoE antisera and failed to detect any staining (data not shown). Furthermore, thioflavine-S-fluorescent Aβ deposits represent only a subset of the total apoE-positive Aβ deposits (Fig. 6). Nevertheless, by double staining it is apparent that virtually all (>99.9%) thioflavine-S-fluorescent Aβ deposits are apoE immunoreactive (Fig. 6 legend).
Figure 6.

ApoE immunoreactivity is associated with a subset of Aβ immunoreactive deposits. (A) Total Aβ immunoreactive deposits (10D5 antibody); (B) apoE immunoreactive deposits; (C) thioflavine-S-fluorescent deposits; and (D) quantitation of Aβ, apoE, and thioflavine-S fluorescent (see Materials and Methods for details). ApoE immunoreactive deposits represent a subpopulation of the total Aβ deposits, but virtually all (≥99%) thioflavine-S-positive Aβ deposits are immunoreactive for apoE. The latter was determined by counting thioflavine-S-fluorescent Aβ deposits in both the hippocampus (n = 273) and cortex (n = 256) and determining by double staining in the same sections (B and C) whether these were also apoE positive (≥99% of thioflavine-S-fluorescent Aβ deposits in each region were apoE positive) (data not shown).
Lack of Apoe Does Not Affect APPV717F Transgene Expression.
To investigate whether the lack of murine Apoe reduces the expression of the APPV717F transgene (either mRNA or protein processing), we analyzed APPV717F+/− TG mice at various ages with the three Apoe genotypes by using reverse transcription–PCR, Western blot analysis, and ELISA measurements of the Aβ peptides. No obvious decrease in the level of either mRNAs (APP 695, 751, 770) or protein was observed. In APPV717F+/− mice, brain-tissue Aβ levels measured by ELISA rise as a function of age, which parallels the dramatic increase in Aβ immunoreactivity detected immunohistochemically (36–38). We therefore measured hippocampal and cortical total Aβ and Aβ1–42 levels before Aβ deposition in young APPV717F+/− mice with and without Apoe. Total Aβ and Aβ1–42 levels in both the cerebral cortex and hippocampus were not significantly different at 2 mo of age (data not shown).
Discussion
The inheritance of one or two APOE4 alleles confers increased risk and an earlier age of onset of AD compared with individuals who inherit the more common APOE3 allele (1, 2). Moreover, APOE4 has been reported to be associated with increased plaque burden in patients with Down's syndrome, traumatic brain injury, amyloid angiopathy, normal aging, and, in some families, with autosomal dominant APP mutations that cause early-onset familial AD (43–49). Taken together, these genetic–epidemiological studies suggest that the three apoE isoforms differentially contribute to AD risk; however, the exact pathogenic mechanism(s) remains obscure.
An association between apoE and amyloid deposition is supported by postmortem studies demonstrating significantly more Aβ deposits and amyloid in APOE4 carriers compared with noncarrier AD patients (1). Numerous studies have shown avid, and in some cases isoform-specific, binding of Aβ peptide(s) to apoE (33, 34). However, both pro-(27, 28) and anti-amyloidogenic (29) actions of apoE have been reported and/or postulated based on in vitro studies. To determine whether apoE directly impacts amyloid deposition in vivo, we have crossed Apoe knockout mice with transgenic mice overexpressing a human mutant APP gene (V717F). The APPV717F TG mouse develops age- and region-dependent amyloid deposition in brain, which is accompanied by neuritic changes (dystrophic neurites) and glial activation (36, 38). Remarkably, in the absence of Apoe, these mice do not develop amyloid deposits when examined as old as 21/22 mo of age (Fig. 1). APPV717F TG mice that are hemizygous for Apoe have greatly reduced amyloid burden that increases with age but is still ≤30% of that observed in Apoe+/+ TG mice at 21/22 mo of age. Despite the fact that we observed substantial immunoreactive Aβ deposits in the hippocampus of APPV717F Apoe-deficient TG mice at both the 15- and 21/22-mo timepoints, no amyloid was ever detected. These data suggest a critical (perhaps necessary) role for apoE in the process of Aβ fibrillogenesis.
As previously reported (37), the absence of Apoe affects neither the transcription nor translation of the APPV717F transgene nor its processing to Aβ peptides. In the absence of Apoe, therefore, Aβ peptide synthesis in the APPV717F TG mouse is unaffected and, although brain Aβ levels are quite high relative to wild-type (non-TG) mice, there is apparently sufficient clearance of the peptide to prevent substantial extracellular deposition. The presence of murine apoE eventually impedes Aβ clearance (see below), perhaps by sequestering the peptide in a form and/or cellular compartment that promotes its deposition, aggregation, and subsequent fibrillization.
Our data suggest that apoE plays at least two critical and possibly distinct roles to facilitate Aβ deposition. Clearly, apoE has a fibrillogenic action and may be required for the maturation of diffuse Aβ deposits to fibrillar Aβ deposits (senile plaques). However, apoE also appears to affect the amount and anatomical distribution of diffuse Aβ deposits and may be required for Aβ deposition in certain brain regions (e.g., the cerebral cortex). We and others have postulated that apoE may alter the clearance of Aβ in brain tissue, and over time it appears that apoE (probably in an isoform-specific manner) reduces clearance, thus promoting deposition and eventually fibrillogenesis. Recently, we have observed that human apoE3 and apoE4 expression reduces Aβ deposition (compared with murine apoE or no apoE) in young APPV717F+/− TG mice (51). However, as these mice age, they eventually display both nonfibrillar and fibrillar Aβ deposits like their murine apoE-expressing counterparts and to a greater extent than that observed in Apoe-deficient mice (unpublished data). Thus, it appears that in young APPV717F TG mice expressing mouse or human apoE, Aβ clearance may be enhanced or facilitated. However, as APPV717F TG mice age, apoE expression somehow impedes Aβ clearance, facilitating both nonfibrillar and fibrillar Aβ deposition. Importantly, we have recently observed a 10-fold greater amount of amyloid in 15-mo-old apoE4 compared with apoE3-expressing APPV717F+/− TG mice (unpublished data), confirming that the effects of apoE on amyloid deposition are both qualitative (isoform specific) and quantitative (dependent on level of expression) in nature. Finally, our findings suggest that treatments that decrease glial apoE expression should reduce amyloid plaque burden and conceivably prevent or slow disease progression.
Acknowledgments
We thank Rosemarie Richardson, Brenda Dupree, and Constance Alyea, for their technical assistance and Pamela J. Edmonds for editorial assistance. We also acknowledge the following grants: Public Health Service, P30 AG 10133; Alzheimer's Association, TLL-97-027; and Lilly Research Laboratories to B.G.
Abbreviations
- apoE
apolipoprotein E protein
- APP
amyloid precursor protein
- Aβ
amyloid β-peptide
- AD
Alzheimer's disease
- TG
transgenic
- GFAP
glial fibrillary acidic protein
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Strittmatter W J, Saunders A M, Schmechel D, Pericak-Vance M A, Enghild J, Salvesen G S, Roses A D. Proc Natl Acad Sci USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weisgraber K H, Mahley R W. FASEB J. 1996;10:1485–1494. doi: 10.1096/fasebj.10.13.8940294. [DOI] [PubMed] [Google Scholar]
- 3.Corder E H, Saunders A M, Risch N J, Strittmatter W J, Schmechel D E, Gaskell P C, Jr, Rimmler J B, Locke P A, Conneally P M, Schmader K E, et al. Nat Genet. 1994;7:180–184. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- 4.Lambert J-C, Berr C, Pasquier F, Delacourte A, Frigard B, Cottel D, Pérez-Tur J, Mouroux V, Mohr M, Cécyre D, et al. Hum Mol Genet. 1998;7:1511–1516. doi: 10.1093/hmg/7.9.1511. [DOI] [PubMed] [Google Scholar]
- 5.Bullido M J, Artiga M J, Recuero M, Sastre I, Garcia M A, Aldudo J, Lendon C, Han S W, Morris J C, Frank A, et al. Nat Genet. 1998;18:69–71. doi: 10.1038/ng0198-69. [DOI] [PubMed] [Google Scholar]
- 6.Artiga M J, Bullido M J, Frank A, Sastre I, Recuero M, García M A, Lendon C L, Han S W, Morris J C, Vázquez J, et al. Hum Mol Genet. 1998;7:1887–1892. doi: 10.1093/hmg/7.12.1887. [DOI] [PubMed] [Google Scholar]
- 7.Farrer L A, Cupples L A, Haines J L, Hyman B, Kukull W A, Mayeux R, Myers R H, Pericak-Vance M A, Risch N, van Duijn C M. J Am Med Assoc. 1997;278:1349–1356. [PubMed] [Google Scholar]
- 8.Meyer M R, Tschanz J T, Norton M C, Welsh-Bohmer K A, Steffens D C, Wyse B W, Breitner J C S. Nat Genet. 1998;19:321–322. doi: 10.1038/1206. [DOI] [PubMed] [Google Scholar]
- 9.Wolf B B, Lopes M B S, VandenBerg S R, Gonias S L. Am J Pathol. 1992;141:37–42. [PMC free article] [PubMed] [Google Scholar]
- 10.Rebeck W G, Harr S D, Strickland D K, Hyman B T. Ann Neurol. 1995;37:211–217. doi: 10.1002/ana.410370212. [DOI] [PubMed] [Google Scholar]
- 11.Newman T C, Dawson P A, Rudel L L, Williams D L. J Biol Chem. 1985;260:2452–2457. [PubMed] [Google Scholar]
- 12.Boyles J K, Pitas R E, Wilson E, Mahley R W, Taylor J M. J Clin Invest. 1985;76:1501–1513. doi: 10.1172/JCI112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakai M, Kawamata T, Maeda K, Tanaka C. Neurosci Lett. 1996;211:41–44. doi: 10.1016/0304-3940(96)12716-6. [DOI] [PubMed] [Google Scholar]
- 14.Boyles J K, Zoellner C D, Anderson L J, Kosick L M, Pitas R E, Hui D Y, Mahley R W, Gebicke-Haerter P J, Ignatius M J, Shooter E M. J Clin Invest. 1989;83:1015–1031. doi: 10.1172/JCI113943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poirier J, Baccichet A, Dea D, Gauthier S. Neuroscience. 1993;55:81–90. doi: 10.1016/0306-4522(93)90456-p. [DOI] [PubMed] [Google Scholar]
- 16.Guillaume D, Bertrand P, Dea D, Davignon J, Poirier J. J Neurochem. 1996;66:2410–2418. doi: 10.1046/j.1471-4159.1996.66062410.x. [DOI] [PubMed] [Google Scholar]
- 17.Weisgraber K H. Adv Protein Chem. 1994;45:249–302. doi: 10.1016/s0065-3233(08)60642-7. [DOI] [PubMed] [Google Scholar]
- 18.Näslund J, Thyberg J, Tjernberg L O, Wernstedt C, Kariström A R, Bogdanovic N, Gandy S E, Lannfelt L, Terenius L, Nordstedt C. Neuron. 1995;15:219–228. doi: 10.1016/0896-6273(95)90079-9. [DOI] [PubMed] [Google Scholar]
- 19.Wisniewski T, Ghiso J, Frangione B. Neurobiol Dis. 1997;4:313–328. doi: 10.1006/nbdi.1997.0147. [DOI] [PubMed] [Google Scholar]
- 20.Sullivan P M, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick R L, Quarfordt S H, Maeda N. J Biol Chem. 1997;272:17972–17980. doi: 10.1074/jbc.272.29.17972. [DOI] [PubMed] [Google Scholar]
- 21.Holtzman D M, Pitas R E, Kilbridge J, Nathan B, Mahley R W, Bu G, Schwartz A L. Proc Natl Acad Sci USA. 1995;92:9480–9484. doi: 10.1073/pnas.92.21.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeMattos R B, Curtiss L K, Williams D L. J Biol Chem. 1998;273:4206–4212. doi: 10.1074/jbc.273.7.4206. [DOI] [PubMed] [Google Scholar]
- 23.Sun Y, Wu S, Bu G, Onifade M K, Patel S N, LaDu M J, Fagan A M, Holtzman D M. J Neurosci. 1998;18:3261–3272. doi: 10.1523/JNEUROSCI.18-09-03261.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nathan B P, Chang K-C, Bellosta S, Brisch E, Ge N, Mahley R W, Pitas R E. J Biol Chem. 1995;270:19797–19799. doi: 10.1074/jbc.270.34.19791. [DOI] [PubMed] [Google Scholar]
- 25.Tolar M, Keller J N, Chan S, Matteson M P, Margnes M A, Crutcher K A. J Neurosci. 1999;15:7100–7110. doi: 10.1523/JNEUROSCI.19-16-07100.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyata M, Smith J D. Nat Genet. 1996;14:55–61. doi: 10.1038/ng0996-55. [DOI] [PubMed] [Google Scholar]
- 27.Wisniewski T, Castaño E M, Golabek A, Vogel T, Frangione B. Am J Pathol. 1994;145:1030–1035. [PMC free article] [PubMed] [Google Scholar]
- 28.Ma J, Yee A, Brewer H B, Das S, Potter H. Nature (London) 1994;372:92–94. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- 29.Evans K C, Berger E P, Cho C-G, Weisgraber K H, Lansbury P T. Proc Natl Acad Sci USA. 1994;92:763–767. doi: 10.1073/pnas.92.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmechel D E, Saunders A M, Strittmatter W J, Crain B J, Hulette C M, Joo S H, Pericak-Vance M A, Goldgaber D, Roses A D. Proc Natl Acad Sci USA. 1993;90:9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wisniewski T, Frangione B. Neurosci Lett. 1992;135:235–238. doi: 10.1016/0304-3940(92)90444-c. [DOI] [PubMed] [Google Scholar]
- 32.Rebeck G W, Reiter J S, Strickland D K, Hyman B T. Neuron. 1993;11:575–580. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 33.Strittmatter W J, Weisgraber K H, Huang D Y, Dong L-M, Salvesen G S, Pericak-Vance M, Schmechel D, Saunders A M, Goldgaber D, Roses A D. Proc Natl Acad Sci USA. 1993;90:8098–8102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.LaDu M J, Falduto M T, Manelli A M, Reardon C A, Getz G S, Frail D E. J Biol Chem. 1994;269:23403–23406. [PubMed] [Google Scholar]
- 35.Zhang S H, Reddick R L, Piedrahita J A, Maeda N. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 36.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Nature (London) 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 37.Bales K R, Verina T, Dodel R C, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone E M, Little S P, Cummins D J, et al. Nat Genet. 1997;17:263–264. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- 38.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, et al. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bancroft J D, Stevens A. Theory and Practice of Histological Techniques. 4th Ed. New York: Churchill Livingstone; 1996. p. 365. [Google Scholar]
- 40.Acarin L, Vela J M, Gonzalez B, Castellano B. J Histochem Cytochem. 1994;42:1033–1041. doi: 10.1177/42.8.8027523. [DOI] [PubMed] [Google Scholar]
- 41.Box G E P, Cox D R. J R Stat Soc B. 1964;26:211–243. [Google Scholar]
- 42.Brown B H, Hettmansperger T P. J Am Stat Assoc. 1996;91:1668–1675. [Google Scholar]
- 43.Lemere C A, Blusztajn J K, Yamaguchi H, Wisniewski T, Saido T C, Selkoe D J. Neurobiol Dis. 1996;3:16–32. doi: 10.1006/nbdi.1996.0003. [DOI] [PubMed] [Google Scholar]
- 44.Gearing M, Mori H, Mirra S S. Ann Neurol. 1996;39:395–399. doi: 10.1002/ana.410390320. [DOI] [PubMed] [Google Scholar]
- 45.Hyman B T, West H L, Rebeck G W, Buldyrev S V, Mantegna R N, Ukleja M, Havlin S, Stanley H E. Proc Natl Acad Sci USA. 1995;92:3586–3590. doi: 10.1073/pnas.92.8.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Greenberg S M, Rebeck G W, Vonsattel J P G, Gomez-Isla T, Hyman B T. Ann Neurol. 1995;38:254–259. doi: 10.1002/ana.410380219. [DOI] [PubMed] [Google Scholar]
- 47.Berr C, Hauw J-J, Delaere P, Duyckaerts C, Amouyel P. Neurosci Lett. 1994;178:221–224. doi: 10.1016/0304-3940(94)90763-3. [DOI] [PubMed] [Google Scholar]
- 48.Nicoll J A R, Roberts G W, Graham D I. Nat Med. 1995;1:135–137. doi: 10.1038/nm0295-135. [DOI] [PubMed] [Google Scholar]
- 49.Houlden H, Crook R, Backhovens H, Prihar G, Baker M, Hutton M, Rossor M, Martin J J, Van Broeckhoven C, Hardy J. Am J Med Genet. 1998;81:117–121. doi: 10.1002/(sici)1096-8628(19980207)81:1<117::aid-ajmg19>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 50.Wood S J, Chan W, Wetzel R. Biochemistry. 1996;35:12623–12628. doi: 10.1021/bi961074j. [DOI] [PubMed] [Google Scholar]
- 51.Holtzman D M, Bales K R, Wu S, Bhat P, Parsadanian M, Fagan A M, Chang L K, Sun Y, Paul S M. J Clin Invest. 1999;103:R15–R21. doi: 10.1172/JCI6179. [DOI] [PMC free article] [PubMed] [Google Scholar]