

Abstract
Extensive protein cross-linking and aggregation are some of the most common molecular events in the pathogenesis of Alzheimer's disease (AD). Both β-amyloid (Aβ) plaques and neurofibrillary tangles, which are extracellular and intracellular proteinaceous aggregates, respectively, contribute to neuronal death and progressive cognitive decline. Although protein cross-linking has been recognized and extensively studied for many years, the underlying mechanisms are largely unknown. Recent data indicates that tissue transglutaminase (tTG), which catalyzes the cross-linking of a wide spectrum of proteins including Aβ, tau, α-synuclein and neurofilament proteins, may be involved in protein aggregation in AD. Many AD risk factors, such as trauma, inflammation, ischemia and stress, up-regulate tTG protein and activity levels. In this review, we summarize the evidence that tTG plays a role in AD, especially in cross-linking of Aβ, tau, α-synuclein and neurofilament proteins. An experimentally testable hypothesis is that tTG may play a central role in AD pathogenesis and that it provides a conceptual link between sporadic and familial AD through a shared pathogenic pathway.
Keywords: Tissue transglutaminase (tTG, TG2); Alzheimer's disease; β-amyloid (Aβ); tau; α-synuclein; neurofilament proteins; protein cross-linking
Introduction
Alzheimer's disease (AD) affects millions of people worldwide with, unfortunately, ever increasing incidence. Currently there is no cure for this devastating disease, and even symptomatic relief remains modestly effective. Underlying the behavioral and cognitive decline of AD is the progressive neuronal dysfunction and ultimately cell death by processes that are not fully understood.
Grossly, the brain of AD usually shows atrophy with reduced volume and weight due to extensive loss of neurons in the neocortex. Histologically, the most remarkable and consistent morphological features are the neuritic senile plaques and neurofibrillary tangles (NFTs) [1]. The major proteinaceous component of the plaques is the extensively cross-linked β-amyloid (Aβ) with non-amyloid components comprising the core of the plaques [2, 3]. Mature NFTs are composed of aggregates of hyperphosphorylated tau [4, 5] and many other proteins, such as ubiquitin [6–8] and neurofilaments [9–11]. The mechanism underlying the extensive protein cross-linking in AD is still unknown, but tissue transglutaminase (tTG) has been implicated in this process [12, 13]. In this review, we will focus on the potential biological significance of tTG in the pathogenesis of AD.
Tissue Transglutaminase
Tissue transglutaminase (also known as TG2, EC 2.3.2.13) is a member of the Ca++-dependent transglutaminase (TG) family that catalyzes protein cross-linking [12, 14, 15] (Figure 1). The γ-glutamyl-ε-lysine isopeptide bond formed by the action of these enzymes produces highly insoluble protein complexes that are extremely stable, showing resistance to 2% SDS and 8M urea or enzymatic degradation [14, 16]. These protein scaffolds may stabilize the structural integrity of the dying cells before their clearance by phagocytosis, thus preventing the nonspecific release of harmful intracellular components such as lysosomal enzymes, nucleic acid, and the resulting inflammatory responses.
Figure 1.
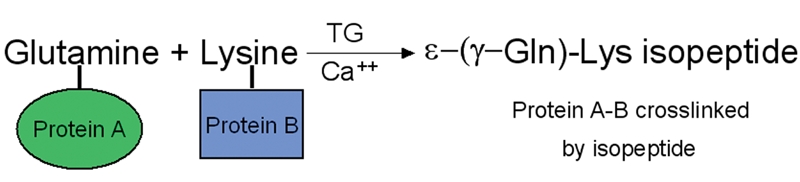
Simplified scheme of tTG-catalyzed isopeptide formation between glutamine and lysin in a calcium-dependent manner. Glutamyl residue in one protein molecule serves as acyl donor or amine acceptor, and lysyl residue in another protein serves acyl acceptor or amine donor. With calcium, tTG catalyzes a covalent cross-linking between the proteins by forming γ-glutamyl-ε-lysine isopeptide bond. Modified from Greenberg CS et al [12].
Nine different TGs have been identified in mammals and human [17–19] including TG C [20, 21], K [22], E [23], P [24], X [18], factor XIII [14, 17] and Band 4.2 protein [25, 26]. These enzymes are subject to various post-translational modifications such as phosphorylation, fatty acylation and proteolytic cleavage which regulate the activity and subcellular distribution of the enzyme under different biological conditions [27, 28]. The tTG gene encodes a monomeric protein composed of 685-691 amino acids in human and other vertebrates [29–33] with a calculated molecular weight of about 80 kDa, although a shorter form of tTG might also exist [34]. The human tTG gene has been mapped to chromosome 20 and includes 13 exons and 12 introns [35, 36]. General features of members of the TG family and detailed biochemistry of tTG have been summarized in several recent reviews [37, 38].
The x-ray crystal structure of human tTG complexed with GDP at 2.8-Å resolution showed that the monomer has four distinct domains that are quite similar to Factor XIII [39–41]. These include an N-terminal β-sandwich domain, a transamidation catalytic core, and two C-terminal barrels (Figure 2). These features suggest a structural basis for the negative regulation of transamidation activity by the bound nucleotide, and positive regulation of transamidation by Ca++ [41]. With truncated tTG-GST fusion protein, it was found that the N-terminal β-sandwich domain and the catalytic domain are required for tTG enzymatic activity, while the C-terminal barrels are not [42].
Figure 2.
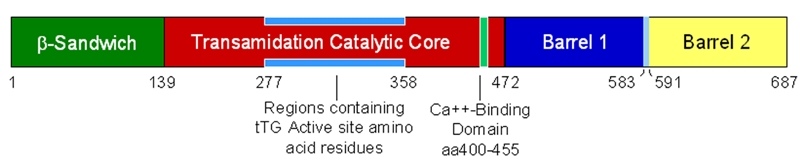
Schematic representation of the structural domains of transglutaminase, amino acid residue distribution region of the catalytic core and Ca++-binding domain. The scheme was drawn based on the data from Liu S. et al [42] with reference to [37, 38].
Tissue TG is particularly interesting due to its wide spread expression in many tissues including brain. It is expressed in both central and peripheral nervous systems [43–47]. In brains, tTG is localized mostly in the cytoplasmic compartment of neurons [43, 48, 49], although it can also be found in nuclei and extracellular matrix [19]. Growing data suggests that tTG is involved not only in some physiological processes such as differentiation and apoptosis but also in multiple pathological processes such as wound healing and neurodegenerative diseases by producing protein conjugates [50–58]. Among all members of the TG family, tTG is one of the most extensively studied and has been implicated in multiple human diseases including AD [59].
Many AD Risk Factors Induce Expression of tTG
Since the majority of cases of AD are sporadic without a clear genetic cause, and an even a large percentage of familial cases cannot be explained by the overproduction of Aβ, multiple factors, especially environmental factors are likely involved in the pathogenesis of AD. In fact, traumatic brain injury [60, 61], aging [62–64], inflammation [65, 66], ischemic damage (infarcts and ischemia) [67–71] and brain stress [72–75] have all be shown to increase the risk of AD. Many of them overly induce tTG expression and/or activity.
Tissue TG is Increased in Brain after Trauma
For many years, traumatic brain injury (TBI) has been associated with enhanced AD risk [76–78]. Epidemiological evidence and retrospective clinical studies implicated TBI as a common preceding event prior to AD [79, 80], especially in those without ApoE4, a known genetic risk factor for AD [81–83]. Dementia pugilistica (DP) is a progressive memory disorder that occurs after repeated head trauma in professional boxers. It is characterized by NFTs that are composed of hyperphosphorylated tau protein indistinguishable from NFTs in AD brains. Animal studies have shown that TBI induces cognitive impairment [84–86] and at an ultrastructural level increases deposition of Aβ [87]. Abnormal tau proteins isolated from DP brains were indistinguishable from the six abnormally phosphorylated brain tau isoforms in AD brains [88]. These data supports the notion that TBI increases susceptibility to AD [89, 90]. There are a variety of other similarities between TBI and AD including highly aggregated Aβ that is typically resistant to proteolytic degradation [91]. These aggregated Aβ species found in AD [92] can also be found in various TBI animal models [87, 93, 94]. In a study on human subjects, significantly more Aβ immunoreactive neurons were observed after head injury than controls [95]. The levels of Aβ was increased in the cerebrospinal fluid of patients after severe brain injury and remained elevated for some time after the initial event [78]. Extensively aggregated and phosphorylated tau is detected in rat brain after traumatic injury. In this model, normal-looking neurons in the telencephalon and brainstem were immunoreactive for phosphorylated tau six months after injury. Cortical neuronal counts gradually decreased, with up to 42% decrease at 6 months after injury [94]. These data suggests that recurrent TBI may cause DP through pathological mechanisms similar to those seen in AD. A single TBI may increase susceptibility to sporadic AD decades after the event.
Tissue TG is usually up-regulated in injured tissues, which suggests that it plays a role in wound repair [96–98]. In model of spinal cord ischemia, overall TG activity increased transiently and then declined to control levels after one week [99]. After injury of superior cervical ganglion or vagus nerve, TG activity was also increased [100, 101]. Recent studies have shown that both tTG mRNA and protein are up-regulated after TBI [102]. While increased tTG synthesis and activation under such circumstances is part of the normal protective cellular response contributing to tissue homeostasis by stabilization of the extracellular matrix and cellular integrity, pathologic protein cross-linking may also occur as seen in AD.
Tissue TG is Up-Regulated in Brain with Ischemia, Inflammation and Other Cell Stresses
In addition to TBI, other AD risk factors such as ischemia, inflammation and cell stress [64, 65, 68, 72, 73, 103–105] induce tTG expression or activity. Focal brain infarct elicits inflammation in the lesion and the surrounding brain tissues with a rapid up-regulation of pro-inflammatory cytokines such as TN F-α and IL-1β [106]. Both TNF-α and IL-1β can induce tTG expression in cultured cells [107]. After global cerebral ischemia in gerbils, tTG activity was followed by incorporation of [3H]-putrescine into dimethyl-casein throughout the 48 hours of reperfusion following a 3 minute occlusion. In experimental animals, significant increases were found in the ischemic hippocampus at 24 hours of reperfusion, while minor changes were observed in the cortex. Both RT-PCR and western-blot demonstrated a substantial up-regulation of tTG in the ischemic hippo-campus, suggesting that tTG is part of the tissue stress response after global brain ischemia/reperfusion [108]. Increased expression of tTG at both mRNA and protein levels was also seen following middle cerebral artery occlusion in rats [109]. Tissue TG mRNA level peaked on day 5 after injury in the ipsilateral cortex. However, in the ipsilateral hippocampus, tTG induction peaked 1 day after injury and to a lesser extent than observed in the ipsilateral cortex. Western blot analysis demonstrated that tTG protein expression progressively increased from day 1 to day 7 after ischemia, with greater expression in cortex than hippocampus. These results demonstrate that tTG mRNA and protein expression increases significantly after ischemic injury. The temporal profile of tTG induction after ischemia was similar to that observed in TBI animal model [102], suggesting a similar role of tTG in both pathological conditions [109].
Tissue TG can also be induced by cerebral inflammation [106] and brain stress induced by glutamate excitotoxicity, calcium influx, oxidative stress, inflammatory cytokines and UV exposure [110]. These data suggests that Aβ can also induce tTG expression, possibly through effects on cellular redox status or calcium flux. There are indications that activated tTG redistributed to the plasma membrane [110]. At this location, tTG may play an active role in excitotoxic neuronal cell death, a likely component of acute central nerve system (CNS) injury and chronic CNS neurodegenerative disease [111, 112].
Tissue TG Catalyzes the Cross-Linking of Critical Proteins of AD Pathology
The most characteristic pathological structures of AD pathology are senile plaques and NFTs [1] (Figure 3A and B). The major components in senile plaques are Aβ1-40 and Aβ1-42. Some senile plaques have a condensed core that contains truncated α-synuclein fragments [2, 113]. Small amounts of neurofilaments can also be found in plaques [114]. The dominant component of NFTs is the hyperphosphorylated tau, a microtubule binding protein [115–117]. Recently, α-synuclein had also been found in NFTs [118–120]. So far, all of those major components found in senile plaques and NFTs have been shown to be substrates of tTG.
Figure 3.
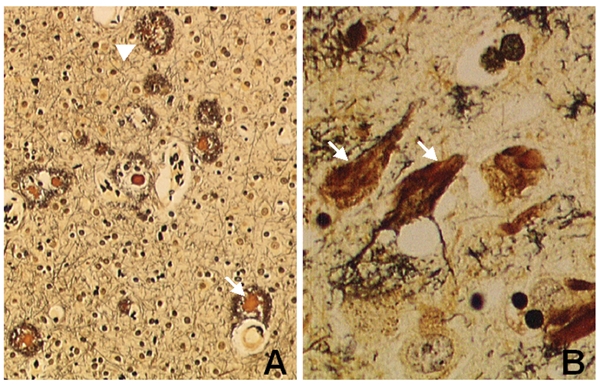
Characteristic structures of AD brain. (A) Senile plaque (arrowhead) and senile plaque with a condensed core (arrow) and (B) neurofibrillary tangles (arrows) (silver staining × 400).
The first suggestion that tTG may play a role in AD was made by Selkoe and colleagues [121] when they showed tTG can covalently cross-link neurofilament proteins into insoluble polymers in vitro by forming γ-glutamyl-ε-lysine intermolecular bridges. Later studies indicated that tTG can catalyze cross-linking of Aβ [122], amyloid precursor protein (APP) [123–127], tau [128–135] and α-synuclein [134, 136, 137] in addition to neurofilament proteins.
Tissue TG Cross-Links Aβ and APP
Several years after a potential link between tTG and AD was suggested [121], Ikura and colleagues reported that tTG could cross-link synthetic Aβ1-28 in vitro exclusively through Lys16 [123]. This finding was quickly extended to the Aβ1-42 [124] and APP [138] by independent groups. Using the incorporation of site-specific probes followed by enzymatic digestion and sequencing of tracer-containing fractions, Lys16, Lys28 and Gln15 in Aβ were all susceptible to cross-linking by tTG [139]. Aβ cross-linking catalyzed by tTG could be inhibited by specific inhibitors (e.g., dansylcadaverine and spermine) and non-steroidal anti-inflammatory drugs (e.g., indomethacin, meclofenamic acid, diflunisal and salicylic acid) [140]. Immunochemical demonstration of tTG in amyloid plaques in AD brains suggests a role in plaque formation by cross-linking Aβ or other components [141]. The in vivo data demonstrating a direct link between tTG and cross-linking of Aβ are still missing.
Tissue TG Cross-Links tau
Tau protein is an excellent substrate of TG and tTG both in vitro and in vivo [126]. Dudek and colleagues showed that in the presence of TG tau formed macromolecular complexes that were insoluble in ionic detergent, β-mercaptoethanol, guanidine-HCl and urea. Furthermore, they demonstrated that the filamentous tau aggregates had increased immunoreactivity to the monoclonal antibody Alz-50 [126]. To determine which domains of tau were modified by tTG, [3H]-putrescine-labeled tau was digested with chymotrypsin. Mass spectrometric analysis demonstrated that tau was modified at only one or a few discrete sites, primarily in the carboxyl half of the molecule. Thus, cross-linking is selective for only a subset of the many glutamine residues in tau. Furthermore, a tau deletion construct (T264) containing a portion of the microtubule binding domain, which is normally a substrate of TG, cannot be cross-linked. This provides evidence that the cross-linking may be conformation-dependent [142].
Similar observations were reported by another group who used purified tTG from guinea pig liver to cross-link recombinant human tau protein [143]. Cross-linking site analysis of human tau (tau23 and tau40) showed that eight glutamines can function as amine acceptor residues, with two major sites at Gln351 and Gln424. In addition, 10 lysine residues were identified as amine donors, most of which are clustered adjacent to the microtubule binding repeats of tau in regions known to be solvent accessible in filamentous tau [144]. When over-expressed in cultured SH-SY5Y cells, tTG was co-immunoprecipitated with tau [145]. Recently, tau protein cross-linking catalyzed by TG was further confirmed in P301L tau transgenic mice [135]. Studies on human specimens indicate that tTG may be involved in cross-linking of tau pathology seen in AD brains. A study performed on frozen prefrontal cortex of 9 AD and 9 age- and postmortem interval-matched controls showed that total TG activity was significantly higher in AD compared to controls. Tissue TG protein levels determined by quantitative immunoblotting were elevated approximately 3-fold in AD compared to controls. Interestingly, there were no significant differences in TG activity or tTG protein levels in the cerebellum from the same panel of samples between control and AD cases [146]. Furthermore, the level of isopeptide bonds, the catalytic product of tTG, was increased in AD brains compared to controls [147, 148]. In one study, a statistically significant (45%) elevation in ε-(γ-glutamyl)-lysine cross-links was found in AD when compared to control cortex [147]. Using single- and double-label immunofluorescence confocal microscopy and immunoaffinity purification and immunoblotting, another study found isopeptide bonds in NFTs and paired helical filament tau early in AD. The number of neurons that are immunoreactive with the antibody against ε-(γ-glutamyl)-lysine bonds was significantly higher in AD cortex compared with age-matched controls and schizophrenics [148].
TG activity, including tTG, may also play a role in NFTs seen in progressive supranuclear palsy (PSP) brains [149]. We double-stained AD brains with anti-tau and anti-isopeptide antibodies which showed that tau and isopeptide co-localized in many, although not all, tangle-bearing neurons (Figure 4A, Wang DS et al: unpublished data). Some amyloid plaques also showed colocalization of isopeptide and Aβ, but the intensities of isopeptide immunostaining were relatively weaker (Figure 4B). This may reflect further protein degradation in plaques, epitope masking or partially stronger immunostaining of very abundant Aβ. Together, the available data suggests that TG, especially tTG, could be a contributing factor in NFT formation.
Figure 4.
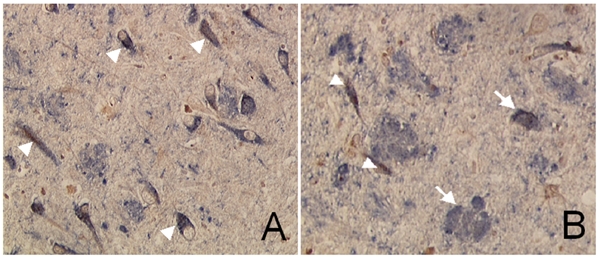
A. Isopeptides and tau protein co-localized in neurofibrillary tangles. The section was stained with mouse monoclonal anti-isopeptide antibody manually first with HRP-DAB. Then the section was treated with DAKO double staining kit followed by CP13 antibody with AP-NACP for color-development. Brown is isopeptide and blue is tau. Arrows indicate tangle-bearing neurons stained by both anti-isopeptide and anti-tau antibodies. B. Isopeptide and Aβ double-staining show colocalization of both proteins in the plaques (arrows) and NFTs (arrow head). The intensities of isopeptide in plaques are relatively weaker that are potentially due to further protein degradation in this type of lesions during the relative lengthy morphogenesis compared to structures like NFT. Magnification: 200 ×.
Tissue TG Cross-Links α-Synuclein
Alpha-Synuclein, an intracellular neuronal protein and a synaptic marker, is also a common substrate of tTG. A 35-residue peptide derived from α-synuclein is a major, non-Aβ component (NAC) of plaques [3, 150, 151]. It is usually localized to the condensed core of the amyloid plaques [152]. TG catalyzes the formation of covalently linked NAC polymers in vitro as well as polymers with Aβ. The tTG-reactive amino acid residues in NAC are Gln79 and Lys80. Lys80 is localized in a consensus motif Lys-Thr-Lys-Glu-Gly-Val, which is conserved in the synuclein gene family [134]. Purified tTG catalyzed α-synuclein cross-linking, leading to the formation of high molecular weight aggregates in vitro. Over-expression of tTG resulted in formation of detergent-insoluble α-synuclein aggregates in cellular models. Immunohistochemical studies on postmortem brain tissue confirmed the presence of TG-catalyzed ε-(γ-glutamyl)-lysine isopeptide in the halo of Lewy bodies in Parkinson's disease and dementia with Lewy bodies, co-localizing with α-synuclein [136]. Furthermore, both tTG protein and isopeptide coimmunoprecipitated with α-synuclein in extracts of PD substantia nigra. The isopeptide was detected in both α-synuclein monomer and its higher molecular weight oligomers, indicating this modification was early in Lewy body formation [137]. Interestingly, we and others found α-synuclein and tau co-exist in many NFTs or Lewy body-like structures, usually with an α-synuclein core surrounded by tau-positive periphery [119, 120, 153]. This indicates α-synuclein cross-linking and aggregation may be an initial event which precedes tau aggregation in the morphogenesis of Lewy bodies and NFTs [120].
Tissue TG and Isopeptide are Increased in AD Compared to the Age-Matched Controls
Recently we showed that tTG and tTG activity are elevated in AD brains compared to controls [37, 48, 154]. As discussed above, isopeptides have been found in plaques [141] and tangles [148]. Our recent study showed that levels of tTG, tTG activity and isopeptide immunoreactivity in brain homogenates correlate inversely with neuropsychological test scores reflective of overall cognitive function (Wang DS et al: manuscript in preparation). In these brains, tTG and tTG activity were increased compared to age-matched normal controls. Isopeptide levels showed a more robust inverse correlation with clinical cognitive measures than tTG or tTG activity. This suggests that although tTG and its activity are increased during AD pathogenesis, this increase may be limited or may reach a steady-state level, but that isopeptide immunoreactivity, the product of tTG activity, may continue to increase and accumulate during the disease process. The results suggest that accumulation of cross-linked protein gradually results in neuronal dysfunction and cognitive decline.
Although insoluble and 70% formic acid-extractable isopeptide correlated with both neuropathological and neuropsycho-logical data, total isopeptide levels in crude homogenate only showed significant correlation with some neuropsychological, but not neuropathologic measures (Wang DS et al: unpublished data). It is likely that formic acid-extractable isopeptides are derived from insoluble end-stage structures such as neuritic plaques and NFTs. Thus, one would expect a robust correlation with neuropathological measures (e.g., plaque and tangles counts) as well as clinical cognitive data. On the other hand, total isopeptide immunoreactivity includes isopeptides from other proteins that may not be present in insoluble lesions. It is tempting to speculate that these soluble isopeptide-containing proteins may contribute to neuronal dysfunction independent of plaques and tangles.
Hypothetical Role of tTG in AD Pathogenesis
Based on the above studies, we propose a hypothetical mechanism for the role of tTG in AD pathogenesis (Figure 5). Increased brain tTG induced by environmental factors such as brain trauma, inflammation and ischemic injury will lead to cross-linked proteins, such as α-synuclein [119, 120, 155], Aβ [124, 125] and tau [147, 148]. Increased production of Aβ due to trauma, inflammation and ischemia in sporadic AD or overproduction of Aβ in familial AD due to mutations in presenilin or APP will increase the stress in brain and further up-regulate tTG, causing a feed-forward response. Aggregated Aβ may serve as a long-term chronic stimulant for tTG and perpetuate the pathogenic process [157]. During this chronic process, neuronal cells are gradually lost, which leads to progressive cognitive decline. Reversal or attenuation of this protein cross-linking and aggregation may help slow cognitive decline and neurodegeneration in AD. Future research is needed to establish the sequence of events after initiating factors are no longer present. In familial AD, the stress due to the excessive production of Aβ alone may be sufficient to increase brain tTG and initiate AD pathogenesis, with or without additional factors needed to initiate the pathogenic cascade in sporadic AD.
Figure 5.
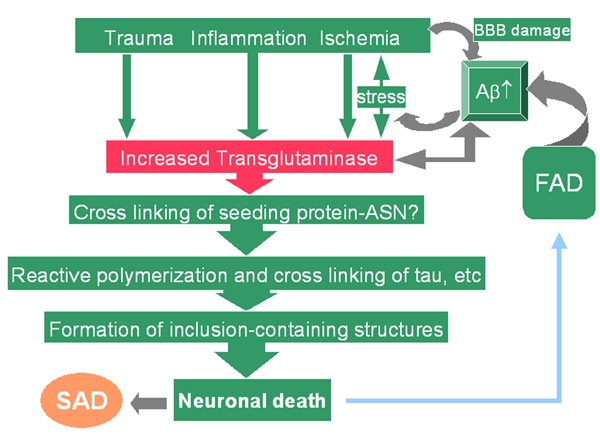
Hypothetical mechanisms for the role of tTG in AD pathogenesis. Increased brain tTG induced by multiple factors such as trauma, inflammation and ischemic injury will cross-link protein like α-synuclein (ASN), Aβ and tau. In sporadic AD, increased Aβ due to trauma, inflammation and ischemia will further increase tTG levels. Aggregated Aβ may serve as a long-term chronic stimulant for the tTG and keep the pathogenic process going even after the initial factors no longer present. In familial AD (FAD), excessive production Aβ may be sufficient to increase tTG and initiate AD pathogenesis, with or without additional factors seen in sporadic AD. FAD: familial Alzheimer's disease; SAD: sporadic Alzheimer's disease.
Summary
Extensive protein cross-linking and aggregation involving a variety of proteins are commonly occurring molecular processes during the pathogenesis of AD [156]. The initiating factors are likely to be environmental insults (e.g., trauma, inflammation or ischemic damage) that lead to increased tTG activity and increased cross-linking for tau, Aβ and other molecules, which leads to functional impairment, structural lesions characteristic of AD (e.g., plaques and tangles) and eventually neuronal death. If the relationship between increased tTG and deleterious cross-linking of proteins such as α-synuclein, tau and Aβ are critical to AD pathogenesis, therapeutic measures should be developed to manipulate tTG protein and activity levels.
Acknowledgments
The authors thank the financial support from the Research Fund of the Department of Pathology and Laboratory Medicine, University of Wisconsin School of Medicine, Madison and NIH Grant AG25722 to DSW, NIH Grants R01-AG10675 and P30-HD63352 to JSM, NIH Grant R01-AG14449 to DWD.
References
- 1.Dickson DW. Neuropathological diagnosis of Alzheimer's disease: a perspective from longitudinal clinicopathological studies. Neurobiol Aging. 1997;18:S21–26. doi: 10.1016/s0197-4580(97)00065-1. [DOI] [PubMed] [Google Scholar]
- 2.Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brookes AJ, St Clair D. Synuclein proteins and Alzheimer's disease. Trends Neurosci. 1994;17:404–405. doi: 10.1016/0166-2236(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 4.Wood JG, Mirra SS, Pollock NJ, Binder LI. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau) Proc Natl Acad Sci USA. 1986;83:4040–4043. doi: 10.1073/pnas.83.11.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Wiche G, Seitelberger F, Grundke-Iqbal I, Iqbal K, Wisniewski HM. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res. 1989;477:90–99. doi: 10.1016/0006-8993(89)91396-6. [DOI] [PubMed] [Google Scholar]
- 6.Mori H, Kondo J, Ihara Y. Ubiquitin is a component of paired helical filaments in Alzheimer's disease. Science. 1987;235:1641–1644. doi: 10.1126/science.3029875. [DOI] [PubMed] [Google Scholar]
- 7.Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1992;13:179–189. doi: 10.1016/0197-4580(92)90027-u. [DOI] [PubMed] [Google Scholar]
- 8.Iqbal K, Alonso A, Gong C, Khatoon S, Kudo T, Singh T, Grundke-Iqbal I. Molecular pathology of Alzheimer neurofibrillary degeneration. Acta Neurobiol Exp (Wars) 1993;53:325–335. [PubMed] [Google Scholar]
- 9.Iqbal K, Grundke-Iqbal I, Wisniewski HM, Terry RD. Chemical relationship of the paired helical filaments of Alzheimer's dementia to normal human neurofilaments and neuro-tubules. Brain Res. 1978;142:321–332. doi: 10.1016/0006-8993(78)90638-8. [DOI] [PubMed] [Google Scholar]
- 10.Rasool CG, Selkoe DJ. Alzheimer's disease: exposure of neurofilament immunoreactivity in SDS-insoluble paired helical filaments. Brain Res. 1984;322:194–198. doi: 10.1016/0006-8993(84)91205-8. [DOI] [PubMed] [Google Scholar]
- 11.Rasool CG, Abraham C, Anderton BH, Haugh M, Kahn J, Selkoe DJ. Alzheimer's disease: immunoreactivity of neurofibrillary tangles with anti-neurofilament and anti-paired helical filament antibodies. Brain Res. 1984;310:249–260. doi: 10.1016/0006-8993(84)90148-3. [DOI] [PubMed] [Google Scholar]
- 12.Greenberg CS, Birckbichler PJ, Rice RH. Transglutaminases: multifunctional cross-linking enzymes that stabilize tissues. FASEB J. 1991;5:3071–3077. doi: 10.1096/fasebj.5.15.1683845. [DOI] [PubMed] [Google Scholar]
- 13.Cooper AJ, Jeitner TM, Blass JP. The role of transglutaminases in neurodegenerative diseases: overview. Neurochem Int. 2002;40:1–5. doi: 10.1016/s0197-0186(01)00055-9. [DOI] [PubMed] [Google Scholar]
- 14.Lorand L, Conrad SM. Transglutaminases. Mol Cell Biochem. 1984;58:9–35. doi: 10.1007/BF00240602. [DOI] [PubMed] [Google Scholar]
- 15.Aeschlimann D, Mosher D, Paulsson M. Tissue transglutaminase and factor XIII in cartilage and bone remodeling. Semin Thromb Hemost. 1996;22:437–443. doi: 10.1055/s-2007-999043. [DOI] [PubMed] [Google Scholar]
- 16.Folk JE. The trimethylacetyl-transglutaminase complex. Methods Enzymol. 1982;87:36–42. doi: 10.1016/s0076-6879(82)87005-5. [DOI] [PubMed] [Google Scholar]
- 17.Aeschlimann D, Paulsson M. Transglutaminases: Protein crosslinking enzymes in tissues and body fluids. Thromb Haemostasis. 1994;71:402–415. [PubMed] [Google Scholar]
- 18.Aeschlimann D, Koeller MK, Allen-Hoffmann BL, Mosher DF. Isolation of a cDNA encoding a novel member of the transglutaminase gene family from human keratinocytes. Detection and identification of transglutaminase gene products based on reverse transcription-polymerase chain reaction with degenerate primers. J Biol Chem. 1998;273:3452–3460. doi: 10.1074/jbc.273.6.3452. [DOI] [PubMed] [Google Scholar]
- 19.Esposito C, Caputo I. Mammalian transglutaminases. FASEB J. 2005;272:615–631. doi: 10.1111/j.1742-4658.2004.04476.x. [DOI] [PubMed] [Google Scholar]
- 20.Thomazy V, Fesus L. Differential expression of tissue transglutaminase in human cells. An immunohistochemical study. Cell Tissue Res. 1989;255:215–224. doi: 10.1007/BF00229084. [DOI] [PubMed] [Google Scholar]
- 21.Muszbek L, Adany R, Mikkola H. Novel aspects of blood coagulation factor XIII. I. Structure, distribution, activation, and function. Crit Rev Clin Lab Sci. 1996;33:357–421. doi: 10.3109/10408369609084691. [DOI] [PubMed] [Google Scholar]
- 22.Kim SY, Chung SI, Steinert PM. Highly active soluble processed forms of the transglutaminase 1 enzyme in epidermal keratinocytes. J Biol Chem. 1995;270:18026–18035. doi: 10.1074/jbc.270.30.18026. [DOI] [PubMed] [Google Scholar]
- 23.Kim IG, Gorman JJ, Park SC, Chung SI, Steinert PM. The deduced sequence of the novel protransglutaminase E (TGase3) of human and mouse. J Biol Chem. 1993;268:12682–12690. [PubMed] [Google Scholar]
- 24.Dubbink HJ, Verkaik NS, Faber PW, Trapman J, Schroder FH, Romijn JC. Tissue specific and androgen-regulated expression of human prostate-specific transglutaminase. Biochem J. 1996;315:901–908. doi: 10.1042/bj3150901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen CM, Dotimas E, Korsgren C. Human erythrocyte membrane protein band 4.2. Semin Hematol. 1993;30:119–137. [PubMed] [Google Scholar]
- 26.Gwynn B, Korsgren C, Cohen CM, Ciciotte SL, Peters LL. The gene encoding protein 4.2 is distinct from the mouse platelet storage pool,deficiency mutation pallid. Genomics. 1997;42:532–535. doi: 10.1006/geno.1997.4764. [DOI] [PubMed] [Google Scholar]
- 27.Steinert PM, Kim SY, Chung SI, Marekov LN. The transglutaminase 1 enzyme is variably acylated by myristate and palmitate during differentiation in epidermal keratinocytes. J Biol Chem. 1996;271:26242–26250. doi: 10.1074/jbc.271.42.26242. [DOI] [PubMed] [Google Scholar]
- 28.Esposito C, Pucci P, Amoresano A, Marino G, Cozzolino A, Porta R. Transglutaminase from rat coagulating gland secretion. Post-translational modifications and activation by phosphatidic acids. J Biol Chem. 1996;271:27416–27423. doi: 10.1074/jbc.271.44.27416. [DOI] [PubMed] [Google Scholar]
- 29.Gentile V, Saydak M, Chiocca EA, Akande O, Birckbichler PJ, Lee KN, Stein JP, Davies PJ. Isolation and characterization of cDNA clones to mouse macrophage and human endothelial cell tissue transglutaminases. J Biol Chem. 1991;266:478–483. [PubMed] [Google Scholar]
- 30.Ikura K, Nasu T, Yokota H, Tsuchiya Y, Sasaki R, Chiba H. Amino acid sequence of guinea pig liver transglutaminase from its cDNA sequence. Biochemistry. 1988;27:2898–2905. doi: 10.1021/bi00408a035. [DOI] [PubMed] [Google Scholar]
- 31.Nakanishi K, Nara K, Hagiwara H, Aoyama Y, Ueno H, Hirose S. Cloning and sequence analysis of cDNA clones for bovine aortic-endothelial-cell transglutaminase. Eur J Biochem. 1991;202:15–21. doi: 10.1111/j.1432-1033.1991.tb16338.x. [DOI] [PubMed] [Google Scholar]
- 32.Weraarchakul-Boonmark N, Jeong JM, Murthy SN, Engel JD, Lorand L. Cloning and expression of chicken erythrocyte transglutaminase. Proc Natl Acad Sci USA. 1992;89:9804–9808. doi: 10.1073/pnas.89.20.9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwak SJ, Kim SY, Kim YS, Song KY, Kim IG, Park SC. Isolation and characterization of brain-specific transglutaminases from rat. Exp Mol Med. 1998;30:177–185. doi: 10.1038/emm.1998.26. [DOI] [PubMed] [Google Scholar]
- 34.Monsonego A, Shani Y, Friedmann I, Paas Y, Eizenberg O, Schwartz M. Expression of GTP-dependent and GTP-independent tissue-type transglutaminase in cytokine-treated rat brain astrocytes. J Biol Chem. 1997;272:3724–3732. doi: 10.1074/jbc.272.6.3724. [DOI] [PubMed] [Google Scholar]
- 35.Gentile V, Davies PJ, Baldini A. The human tissue transglutaminase gene maps on chromosome 20q12 by in situ fluorescence hybridization. Genomics. 1994;20:295–297. doi: 10.1006/geno.1994.1170. [DOI] [PubMed] [Google Scholar]
- 36.Fraij BM, Gonzales RA. Organization and structure of the human tissue transglutaminase gene. Biochim Biophys Acta. 1997;1354:65–71. doi: 10.1016/s0167-4781(97)00132-2. [DOI] [PubMed] [Google Scholar]
- 37.Lesort M, Tucholski J, Miller ML, Johnson GV. Tissue transglutaminase: a possible role in neurodegenerative diseases. Prog Neurobiol. 2000;61:439–463. doi: 10.1016/s0301-0082(99)00052-0. [DOI] [PubMed] [Google Scholar]
- 38.Chen JS, Mehta K. Tissue transglutaminase: an enzyme with a split personality. Int J Biochem Cell Biol. 1999;31:817–836. doi: 10.1016/s1357-2725(99)00045-x. [DOI] [PubMed] [Google Scholar]
- 39.Lai TS, Slaughter TF, Koropchak CM, Haroon ZA, Greenberg CS. C-terminal deletion of human tissue transglutaminase enhances magnesium-dependent GTP/ATPase activity. J Biol Chem. 1996;271:31191–31195. doi: 10.1074/jbc.271.49.31191. [DOI] [PubMed] [Google Scholar]
- 40.Hwang KC, Gray CD, Sivasubramanian N, Im MJ. Interaction site of GTP binding Gh (transglutaminase II) with phospholipase C. J Biol Chem. 1995;270:27058–27062. doi: 10.1074/jbc.270.45.27058. [DOI] [PubMed] [Google Scholar]
- 41.Liu S, Cerione RA, Clardy J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc Natl Acad Sci USA. 2002;99:2743–2747. doi: 10.1073/pnas.042454899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iismaa SE, Chung L, Wu MJ, Teller DC, Yee VC, Graham RM. The core domain of the tissue transglutaminase Gh hydrolyzes GTP and ATP. Biochemistry. 1997;36:11655–11664. doi: 10.1021/bi970545e. [DOI] [PubMed] [Google Scholar]
- 43.Maggio N, Sellitti S, Capano CP, Papa M. Tissue-transglutaminase in rat and human brain: light and electron immunocytochemical analysis and in situ hybridization study. Brain Res Bull. 2001;56:173–182. doi: 10.1016/s0361-9230(01)00649-9. [DOI] [PubMed] [Google Scholar]
- 44.Gilad GM, Varon LE. Transglutaminase activity in rat brain: characterization, distribution, and changes with age. J Neurochem. 1985;45:1522–1526. doi: 10.1111/j.1471-4159.1985.tb07222.x. [DOI] [PubMed] [Google Scholar]
- 45.Hand D, Campoy FJ, Clark S, Fisher A, Haynes LW. Activity and distribution of tissue transglutaminase in association with nerve-muscle synapses. J Neurochem. 1993;61:1064–1072. doi: 10.1111/j.1471-4159.1993.tb03621.x. [DOI] [PubMed] [Google Scholar]
- 46.Perry MJ, Haynes LW. Localization and activity of transglutaminase, a retinoid-inducible protein, in developing rat spinal cord. Int J Dev Neurosci. 1993;11:325–337. doi: 10.1016/0736-5748(93)90004-w. [DOI] [PubMed] [Google Scholar]
- 47.Perry MJ, Mahoney SA, Haynes LW. Transglutaminase C in cerebellar granule neurons: regulation and localization of substrate cross-linking. Neuroscience. 1995;65:1063–1076. doi: 10.1016/0306-4522(94)00556-k. [DOI] [PubMed] [Google Scholar]
- 48.Kim SY, Grant P, Lee JH, Pant HC, Steinert PM. Differential expression of multiple transglutaminases in human brain. Increased expression and cross-linking by transglutaminases 1 and 2 in Alzheimer's disease. J Biol Chem. 1999;274:30715–30721. doi: 10.1074/jbc.274.43.30715. [DOI] [PubMed] [Google Scholar]
- 49.Lesort M, Chun W, Johnson GV, Ferrante RJ. Tissue transglutaminase is increased in Huntington's disease brain. J Neurochem. 1999;73:2018–2027. [PubMed] [Google Scholar]
- 50.Fesus L, Thomazy V. Searching for the function of tissue transglutaminase: its possible involvement in the biochemical pathway of programmed cell death. Adv Exp Med Biol. 1988;231:119–134. doi: 10.1007/978-1-4684-9042-8_10. [DOI] [PubMed] [Google Scholar]
- 51.Fukuda K, Kojiro M, Chiu JF. Differential regulation of tissue transglutaminase in rat hepatoma cell lines McA-RH7777 and McA-RH8994: relation to growth rate and cell death. J Cell Biochem. 1994;54:67–77. doi: 10.1002/jcb.240540108. [DOI] [PubMed] [Google Scholar]
- 52.Szegezdi E, Szondy Z, Nagy L, Nemes Z, Friis RR, Davies PJ, Fesus L. Apoptosis-linked in vivo regulation of the tissue transglutaminase gene promoter. Cell Death Differ. 2000;7:1225–1233. doi: 10.1038/sj.cdd.4400751. [DOI] [PubMed] [Google Scholar]
- 53.Zhang H, Yousem SA, Franklin WA, Elder E, Landreneau R, Ferson P, Keenan R, Whiteside T, Levitt ML. Differentiation and programmed cell death-related intermediate biomarkers for the development of non-small cell lung cancer: a pilot study. Hum Pathol. 1998;29:965–971. doi: 10.1016/s0046-8177(98)90202-7. [DOI] [PubMed] [Google Scholar]
- 54.Fabbi M, Marimpietri D, Martini S, Brancolini C, Amoresano A, Scaloni A, Bargellesi A, Cosulich E. Tissue transglutaminase is a caspase substrate during apoptosis. Cleavage causes loss of transamidating function and is a biochemical marker of caspase 3 activation. Cell Death Differ. 1999;6:992–1001. doi: 10.1038/sj.cdd.4400573. [DOI] [PubMed] [Google Scholar]
- 55.Piacentini M, Farrace MG, Hassan C, Serafini B, Autuori F. ‘Tissue’ transglutaminase release from apoptotic cells into extracellular matrix during human liver fibrogenesis. J Pathol. 1999;189:92–98. doi: 10.1002/(SICI)1096-9896(199909)189:1<92::AID-PATH386>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 56.Melino G, Annicchiarico-Petruzzelli M, Piredda L, Candi E, Gentile V, Davies PJ, Piacentini M. Tissue transglutaminase and apoptosis: sense and antisense transfection studies with human neuroblastoma cells. Mol Cell Biol. 1994;14:6584–6596. doi: 10.1128/mcb.14.10.6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Melino G, Draoui M, Bellincampi L, Bernassola F, Bernardini S, Piacentini M, Reichert U, Cohen P. Retinoic acid receptors alpha and gamma mediate the induction of “tissue” transglutaminase activity and apoptosis in human neuroblastoma cells. Exp Cell Res. 1997;235:55–61. doi: 10.1006/excr.1997.3656. [DOI] [PubMed] [Google Scholar]
- 58.Piredda L, Farrace MG, Lo Bello M, Malorni W, Melino G, Petruzzelli R, Piacentini M. Identification of ‘tissue’ transglutaminase binding proteins in neural cells committed to apoptosis. FASEB J. 1999;13:355–364. doi: 10.1096/fasebj.13.2.355. [DOI] [PubMed] [Google Scholar]
- 59.Kim SY, Jeitner TM, Steinert PM. Transglutaminases in disease. Neurochem Int. 2002;40:85–103. doi: 10.1016/s0197-0186(01)00064-x. [DOI] [PubMed] [Google Scholar]
- 60.Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry. 2003;74:857–862. doi: 10.1136/jnnp.74.7.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Szczygielski J, Mautes A, Steudel WI, Falkai P, Bayer TA, Wirths O. Traumatic brain injury: cause or risk of Alzheimer's disease? A review of experimental studies. J Neural Transm. 2005;112:1547–1564. doi: 10.1007/s00702-005-0326-0. [DOI] [PubMed] [Google Scholar]
- 62.Swaab DF, Dubelaar EJ, Hofman MA, Scherder EJ, van Someren EJ, Verwer RW. Brain aging and Alzheimer's disease; use it or lose it. Prog Brain Res. 2002;138:343–373. doi: 10.1016/S0079-6123(02)38086-5. [DOI] [PubMed] [Google Scholar]
- 63.Pike CJ, Rosario ER, Nguyen TV. Androgens, aging, and Alzheimer's disease. Endocrine. 2006;29:233–241. doi: 10.1385/ENDO:29:2:233. [DOI] [PubMed] [Google Scholar]
- 64.Candore G, Balistreri CR, Grimaldi MP, Vasto S, Listi F, Chiappelli M, Licastro F, Lio D, Caruso C. Age-related inflammatory diseases: role of genetics and gender in the pathophysiology of Alzheimer's disease. Ann N Y Acad Sci. 2006;1089:472–486. doi: 10.1196/annals.1386.008. [DOI] [PubMed] [Google Scholar]
- 65.Sastre M, Klockgether T, Heneka MT. Contribution of inflammatory processes to Alzheimer's disease: molecular mechanisms. Int J Dev Neurosci. 2006;24:167–176. doi: 10.1016/j.ijdevneu.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 66.Rogers JT, Lahiri DK. Metal and inflammatory targets for Alzheimer's disease. Curr Drug Targets. 2004;5:535–551. doi: 10.2174/1389450043345272. [DOI] [PubMed] [Google Scholar]
- 67.Rosano C, Newman AB. Cardiovascular disease and risk of Alzheimer's disease. Neurol Res. 2006;28:612–620. doi: 10.1179/016164106X130407. [DOI] [PubMed] [Google Scholar]
- 68.Koistinaho M, Koistinaho J. Interactions between Alzheimer's disease and cerebral ischemia–focus on inflammation. Brain Res Rev. 2005;48:240–250. doi: 10.1016/j.brainresrev.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 69.Bailey TL, Rivara CB, Rocher AB, Hof PR. The nature and effects of cortical microvascular pathology in aging and Alzheimer's disease. Neurol Res. 2004;26:573–578. doi: 10.1179/016164104225016272. [DOI] [PubMed] [Google Scholar]
- 70.Pansari K, Gupta A, Thomas P. Alzheimer's disease and vascular factors: facts and theories. Int J Clin Pract. 2002;56:197–203. [PubMed] [Google Scholar]
- 71.Kudo T, Imaizumi K, Tanimukai H, Katayama T, Sato N, Nakamura Y, Tanaka T, Kashiwagi Y, Jinno Y, Tohyama M, Takeda M. Are cerebrovascular factors involved in Alzheimer's disease? Neurobiol Aging. 2000;21:215–224. doi: 10.1016/s0197-4580(00)00129-9. [DOI] [PubMed] [Google Scholar]
- 72.Polidori MC. Oxidative stress and risk factors for Alzheimer's disease: clues to prevention and therapy. J Alzheimers Dis. 2004;6:185–191. doi: 10.3233/jad-2004-6211. [DOI] [PubMed] [Google Scholar]
- 73.Butterfield DA, Griffin S, Munch G, Pasinetti GM. Amyloid beta-peptide and amyloid pathology are central to the oxidative stress and inflammatory cascades under which Alzheimer's disease brain exists. J Alzheimers Dis. 2002;4:193–201. doi: 10.3233/jad-2002-4309. [DOI] [PubMed] [Google Scholar]
- 74.Veurink G, Fuller SJ, Atwood CS, Martins RN. Genetics, lifestyle and the roles of amyloid beta and oxidative stress in Alzheimer's disease. Ann Hum Biol. 2003;30:639–667. doi: 10.1080/03014460310001620144. [DOI] [PubMed] [Google Scholar]
- 75.Ohta S, Ohsawa I. Dysfunction of mitochondria and oxidative stress in the pathogenesis of Alzheimer's disease: on defects in the cytochrome c oxidase complex and aldehyde detoxification. J Alzheimers Dis. 2006;9:155–166. doi: 10.3233/jad-2006-9208. [DOI] [PubMed] [Google Scholar]
- 76.Jellinger KA, Paulus W, Wrocklage C, Litvan I. Traumatic brain injury as a risk factor for Alzheimer disease. Comparison of two retrospective autopsy cohorts with evaluation of ApoE genotype. BMC Neurol. 2001;1:3. doi: 10.1186/1471-2377-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lye TC, Shores EA. Traumatic brain injury as a risk factor for Alzheimer's disease: a review. Neuropsychol Rev. 2000;10:115–129. doi: 10.1023/a:1009068804787. [DOI] [PubMed] [Google Scholar]
- 78.Emmerling MR, Morganti-Kossmann MC, Kossmann T, Stahel PF, Watson MD, Evans LM, Mehta PD, Spiegel K, Kuo YM, Roher AE, Raby CA. Traumatic brain injury elevates the Alzheimer's amyloid peptide A beta 42 in human CSF. A possible role for nerve cell injury. Ann N Y Acad Sci. 2000;903:118–122. doi: 10.1111/j.1749-6632.2000.tb06357.x. [DOI] [PubMed] [Google Scholar]
- 79.Rocca WA, Amaducci LA, Schoenberg BS. Epidemiology of clinically diagnosed Alzheimer's disease. Ann Neurol. 1986;19:415–424. doi: 10.1002/ana.410190502. [DOI] [PubMed] [Google Scholar]
- 80.Rasmusson DX, Brandt J, Martin DB, Folstein MF. Head injury as a risk factor in Alzheimer's disease. Brain Inj. 1995;9:213–219. doi: 10.3109/02699059509008194. [DOI] [PubMed] [Google Scholar]
- 81.Guo Z CL, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD, Duara R, DeCarli C, Johnson K, Go RC, Growdon JH, Haines JL, Kukull WA, Farrer LA. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000;54:1316–1323. doi: 10.1212/wnl.54.6.1316. [DOI] [PubMed] [Google Scholar]
- 82.Luukinen H VP, Koski K, Laippala P, Kivela SL. Head injuries and cognitive decline among older adults: a population-based study. Neurology. 1999;52:557–562. doi: 10.1212/wnl.52.3.557. [DOI] [PubMed] [Google Scholar]
- 83.Plassman BL HR, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JC. Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology. 2000;55:1158–1166. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- 84.Fox GB, Faden AI. Traumatic brain injury causes delayed motor and cognitive impairment in a mutant mouse strain known to exhibit delayed Wallerian degeneration. J Neurosci Res. 1998;53:718–727. doi: 10.1002/(SICI)1097-4547(19980915)53:6<718::AID-JNR9>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 85.Zohar O, Schreiber S, Getslev V, Schwartz JP, Mullins PG, Pick CG. Closed-head minimal traumatic brain injury produces long-term cognitive deficits in mice. Neuroscience. 2003;118:949–955. doi: 10.1016/s0306-4522(03)00048-4. [DOI] [PubMed] [Google Scholar]
- 86.DeFord SM, Wilson MS, Rice AC, Clausen T, Rice LK, Barabnova A, Bullock R, Hamm RJ. Repeated mild brain injuries result in cognitive impairment in B6C3F1 mice. J Neurotrauma. 2002;19:427–438. doi: 10.1089/08977150252932389. [DOI] [PubMed] [Google Scholar]
- 87.Uryu K, Laurer H, McIntosh T, Pratico D, Martinez D, Leight S, Lee VM, Trojanowski JQ. Repetitive mild brain trauma accelerates Abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J Neurosci. 2002;22:446–454. doi: 10.1523/JNEUROSCI.22-02-00446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schmidt ML, Zhukareva V, Newell KL, Lee VM, Trojanowski JQ. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer's disease. Acta Neuropathol (Berl) 2001;101:518–524. doi: 10.1007/s004010000330. [DOI] [PubMed] [Google Scholar]
- 89.Cribbs DH, Chen LS, Cotman CW, LaFerla FM. Injury induces presenilin-1 gene expression in mouse brain. Neuroreport. 1996;7:1773–1776. doi: 10.1097/00001756-199607290-00016. [DOI] [PubMed] [Google Scholar]
- 90.Nakagawa Y, Reed L, Nakamura M, McIntosh TK, Smith DH, Saatman KE, Raghupathi R, Clemens J, Saido TC, Lee VM, Trojanowski JQ. Brain trauma in aged transgenic mice induces regression of established abeta deposits. Exp Neurol. 2000;163:244–252. doi: 10.1006/exnr.2000.7375. [DOI] [PubMed] [Google Scholar]
- 91.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 92.Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA. Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett. 2001;306:116–120. doi: 10.1016/s0304-3940(01)01876-6. [DOI] [PubMed] [Google Scholar]
- 93.Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, Leoni MJ, Xu BN, Wolf JA, Meaney DF. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:982–992. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- 94.Hoshino S, Tamaoka A, Takahashi M, Kobayashi S, Furukawa T, Oaki Y, Mori O, Matsuno S, Shoji S, Inomata M, Teramoto A. Emergence of immunoreactivities for phosphorylated tau and amyloid-beta protein in chronic stage of fluid percussion injury in rat brain. Neuroreport. 1998;9:1879–1883. doi: 10.1097/00001756-199806010-00039. [DOI] [PubMed] [Google Scholar]
- 95.McKenzie JE, Gentleman SM, Roberts GW, Graham DI, Royston MC. Increased numbers of beta APP-immunoreactive neurones in the entorhinal cortex after head injury. Neuroreport. 1994;6:161–164. doi: 10.1097/00001756-199412300-00041. [DOI] [PubMed] [Google Scholar]
- 96.Haroon ZA, Hettasch JM, Lai TS, Dewhirst MW, Greenberg CS. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999;13:1787–1795. doi: 10.1096/fasebj.13.13.1787. [DOI] [PubMed] [Google Scholar]
- 97.Griffin M, Casadio R, Bergamini CM. Transglutaminases: nature's biological glues. Biochem J. 2002;368:377–396. doi: 10.1042/BJ20021234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Aeschlimann D, Thomazy V. Protein crosslinking in assembly and remodelling of extracellular matrices: the role of transglutaminases. Connect Tissue Res. 2000;41:1–27. doi: 10.3109/03008200009005638. [DOI] [PubMed] [Google Scholar]
- 99.Fujita K, Ando M, Yamauchi M, Nagata Y, Honda M. Alteration of transglutaminase activity in rat and human spinal cord after neuronal degeneration. Neurochem Res. 1995;20:1195–1201. doi: 10.1007/BF00995383. [DOI] [PubMed] [Google Scholar]
- 100.Ando M, Kunii S, Tatematsu T, Nagata Y. Rapid and transient alterations in transglutaminase activity in rat superior cervical ganglia following denervation or axotomy. Neurosci Res. 1993;17:47–52. doi: 10.1016/0168-0102(93)90028-o. [DOI] [PubMed] [Google Scholar]
- 101.Tetzlaff W, Gilad VH, Leonard C, Bisby MA, Gilad GM. Retrograde changes in transglutaminase activity after peripheral nerve injuries. Brain Res. 1988;445:142–146. doi: 10.1016/0006-8993(88)91083-9. [DOI] [PubMed] [Google Scholar]
- 102.Tolentino PJ, DeFord SM, Notterpek L, Glenn CC, Pike BR, Wang KK, Hayes RL. Up-regulation of tissue-type transglutaminase after traumatic brain injury. J Neurochem. 2002;80:579–588. doi: 10.1046/j.0022-3042.2001.00726.x. [DOI] [PubMed] [Google Scholar]
- 103.Balin BJ, Appelt DM. Role of infection in Alzheimer's disease. J Am Osteopath Assoc. 2001;101:S1–6. [PubMed] [Google Scholar]
- 104.Kalaria RN. The role of cerebral ischemia in Alzheimer's disease. Neurobiol Aging. 2000;21:321–330. doi: 10.1016/s0197-4580(00)00125-1. [DOI] [PubMed] [Google Scholar]
- 105.Pratico D. Alzheimer's disease and oxygen radicals: new insights. Biochem Pharmacol. 2002;63:563–567. doi: 10.1016/s0006-2952(01)00919-4. [DOI] [PubMed] [Google Scholar]
- 106.Kim SY. Transglutaminase 2 in inflammation. Front Biosci. 2006;11:3026–3035. doi: 10.2741/2030. [DOI] [PubMed] [Google Scholar]
- 107.Dvorak CM, Hardstedt M, Xie H, Wang M, Papas KK, Hering BJ, Murtaugh MP, Fahrenkrug SC. Transcriptional profiling of stress response in cultured porcine islets. Biochem Biophys Res Commun. 2007;357:118–125. doi: 10.1016/j.bbrc.2007.03.101. [DOI] [PubMed] [Google Scholar]
- 108.Ientile R, Caccamo D, Marciano MC, Curro M, Mannucci C, Campisi A, Calapai G. Transglutaminase activity and transglutaminase mRNA transcripts in gerbil brain ischemia. Neurosci Lett. 2004;363:173–177. doi: 10.1016/j.neulet.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 109.Tolentino PJ, Waghray A, Wang KK, Hayes RL. Increased expression of tissue-type transglutaminase following middle cerebral artery occlusion in rats. J Neurochem. 2004;89:1301–1307. doi: 10.1111/j.1471-4159.2004.02436.x. [DOI] [PubMed] [Google Scholar]
- 110.Ientile R, Caccamo D, Griffin M. Tissue transglutaminase and the stress response. Amino Acids. 2007;33:385–394. doi: 10.1007/s00726-007-0517-0. [DOI] [PubMed] [Google Scholar]
- 111.Tucholski J, Roth KA, Johnson GV. Tissue transglutaminase overexpression in the brain potentiates calcium-induced hippocampal damage. J Neurochem. 2006;97:582–594. doi: 10.1111/j.1471-4159.2006.03780.x. [DOI] [PubMed] [Google Scholar]
- 112.Caccamo D, Campisi A, Curro M, Li Volti G, Vanella A, Ientile R. Excitotoxic and post-ischemic neurodegeneration: Involvement of transglutaminases. Amino Acids. 2004;27:373–379. doi: 10.1007/s00726-004-0117-1. [DOI] [PubMed] [Google Scholar]
- 113.Iwai A, Yoshimoto M, Masliah E, Saitoh T. Non-A beta component of Alzheimer's disease amyloid (NAC) is amyloidogenic. Biochemistry. 1995;34:10139–10145. doi: 10.1021/bi00032a006. [DOI] [PubMed] [Google Scholar]
- 114.Arai H, Lee VM, Otvos L, Jr, Greenberg BD, Lowery DE, Sharma SK, Schmidt ML, Trojanowski JQ. Defined neurofilament, tau, and beta-amyloid precursor protein epitopes distinguish Alzheimer from non-Alzheimer senile plaques. Proc Natl Acad Sci USA. 1990;87:2249–2253. doi: 10.1073/pnas.87.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dickson DW, Ksiezak-Reding H, Davies P, Yen SH. A monoclonal antibody that recognizes a phosphorylated epitope in Alzheimer neurofibrillary tangles, neuro-filaments and tau proteins immunostains granulovacuolar degeneration. Acta Neuropathol (Berl) 1987;73:254–258. doi: 10.1007/BF00686619. [DOI] [PubMed] [Google Scholar]
- 116.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ihara Y, Nukina N, Miura R, Ogawara M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer's disease. J Biochem (Tokyo) 1986;99:1807–1810. doi: 10.1093/oxfordjournals.jbchem.a135662. [DOI] [PubMed] [Google Scholar]
- 118.Iwai A, Masliah E, Sundsmo MP, DeTeresa R, Mallory M, Salmon DP, Saitoh T. The synaptic protein NACP is abnormally expressed during the progression of Alzheimer's disease. Brain Res. 1996;720:230–234. doi: 10.1016/0006-8993(96)00014-5. [DOI] [PubMed] [Google Scholar]
- 119.Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol. 2003;62:389–397. doi: 10.1093/jnen/62.4.389. [DOI] [PubMed] [Google Scholar]
- 120.Wang DS. From Lewy body disease to Alzheimer's disease: hypothesis and evidence. Front Biosci. 2003;8:S223–227. doi: 10.2741/1029. [DOI] [PubMed] [Google Scholar]
- 121.Selkoe DJ, Abraham C, Ihara Y. Brain transglutaminase: in vitro crosslinking of human neurofilament proteins into insoluble polymers. Proc Natl Acad Sci USA. 1982;79:6070–6074. doi: 10.1073/pnas.79.19.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang W, Johnson BR, Suri DE, Martinez J, Bjornsson TD. Immunohistochemical demonstration of tissue transglutaminase in amyloid plaques. Acta Neuropathol (Berl) 1998;96:395–400. doi: 10.1007/s004010050910. [DOI] [PubMed] [Google Scholar]
- 123.Ikura K, Takahata K, Sasaki R. Cross-linking of a synthetic partial-length (1–28) peptide of the Alzheimer beta/A4 amyloid protein by transglutaminase. FEBS Lett. 1993;326:109–111. doi: 10.1016/0014-5793(93)81772-r. [DOI] [PubMed] [Google Scholar]
- 124.Dudek SM, Johnson GV. Transglutaminase facilitates the formation of polymers of the beta-amyloid peptide. Brain Res. 1994;651:129–133. doi: 10.1016/0006-8993(94)90688-2. [DOI] [PubMed] [Google Scholar]
- 125.Boros S, Kamps B, Wunderink L, de Bruijn W, de Jong WW, Boelens WC. Transglutaminase catalyzes differential crosslinking of small heat shock proteins and amyloid-beta. FEBS Lett. 2004;576:57–62. doi: 10.1016/j.febslet.2004.08.062. [DOI] [PubMed] [Google Scholar]
- 126.Dudek SM, Johnson GV. Transglutaminase catalyzes the formation of sodium dodecyl sulfate-insoluble, Alz-50-reactive polymers of tau. J Neurochem. 1993;61:1159–1162. doi: 10.1111/j.1471-4159.1993.tb03636.x. [DOI] [PubMed] [Google Scholar]
- 127.Ho GJ, Gregory EJ, Smirnova IV, Zoubine MN, Festoff BW. Cross-linking of beta-amyloid protein precursor catalyzed by tissue transglutaminase. FEBS Lett. 1994;349:151–154. doi: 10.1016/0014-5793(94)00663-6. [DOI] [PubMed] [Google Scholar]
- 128.Appelt DM, Balin BJ. The association of tissue transglutaminase with human recombinant tau results in the formation of insoluble filamentous structures. Brain Res. 1997;745:21–31. doi: 10.1016/s0006-8993(96)01121-3. [DOI] [PubMed] [Google Scholar]
- 129.Citron BA, SantaCruz KS, Davies PJ, Festoff BW. Intron-exon swapping of transglutaminase mRNA and neuronal Tau aggregation in Alzheimer's disease. J Biol Chem. 2001;276:3295–3301. doi: 10.1074/jbc.M004776200. [DOI] [PubMed] [Google Scholar]
- 130.Grierson AJ, Johnson GV, Miller CC. Three different human tau isoforms and rat neurofilament light, middle and heavy chain proteins are cellular substrates for transglutaminase. Neurosci Lett. 2001;298:9–12. doi: 10.1016/s0304-3940(00)01714-6. [DOI] [PubMed] [Google Scholar]
- 131.Miller ML, Johnson GV. Transglutaminase cross-linking of the tau protein. J Neurochem. 1995;65:1760–1770. doi: 10.1046/j.1471-4159.1995.65041760.x. [DOI] [PubMed] [Google Scholar]
- 132.Murthy SN, Wilson JH, Lukas TJ, Kuret J, Lorand L. Cross-linking sites of the human tau protein, probed by reactions with human transglutaminase. J Neurochem. 1998;71:2607–2614. doi: 10.1046/j.1471-4159.1998.71062607.x. [DOI] [PubMed] [Google Scholar]
- 133.Norlund MA, Lee JM, Zainelli GM, Muma NA. Elevated transglutaminase-induced bonds in PHF tau in Alzheimer's disease. Brain Res. 1999;851:154–163. doi: 10.1016/s0006-8993(99)02179-4. [DOI] [PubMed] [Google Scholar]
- 134.Jensen PH, Sorensen ES, Petersen TE, Gliemann J, Rasmussen LK. Residues in the synuclein consensus motif of the alpha-synuclein fragment, NAC, participate in transglutaminase-catalysed cross-linking to Alzheimer-disease amyloid beta A4 peptide. Biochem J. 1995;310:91–94. doi: 10.1042/bj3100091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Halverson RA, Lewis J, Frausto S, Hutton M, Muma NA. Tau protein is cross-linked by transglutaminase in P301L tau transgenic mice. J Neurosci. 2005;25:1226–1233. doi: 10.1523/JNEUROSCI.3263-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Junn E, Ronchetti RD, Quezado MM, Kim SY, Mouradian MM. Tissue transglutaminase-induced aggregation of alpha-synuclein: Implications for Lewy body formation in Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. 2003;100:2047–2052. doi: 10.1073/pnas.0438021100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Andringa G, Lam KY, Chegary M, Wang X, Chase TN, Bennett MC. Tissue transglutaminase catalyzes the formation of alpha-synuclein crosslinks in Parkinson's disease. FASEB J. 2004;18:932–934. doi: 10.1096/fj.03-0829fje. [DOI] [PubMed] [Google Scholar]
- 138.Ho GJ, Gregory EJ, Smirnova IV, Zoubine MN, Festoff BW. Cross-linking of beta-amyloid protein precursor catalyzed by tissue transglutaminase. FEBS Lett. 1994;349:151–154. doi: 10.1016/0014-5793(94)00663-6. [DOI] [PubMed] [Google Scholar]
- 139.Rasmussen LK, Sorensen ES, Petersen TE, Gliemann J, Jensen PH. Identification of glutamine and lysine residues in Alzheimer amyloid beta A4 peptide responsible for transglutaminase-catalysed homopolymerization and cross-linking to alpha 2M receptor. FEBS Lett. 1994;338:161–166. doi: 10.1016/0014-5793(94)80356-0. [DOI] [PubMed] [Google Scholar]
- 140.Zhang W, Johnson BR, Bjornsson TD. Pharmacologic inhibition of transglutaminase-induced cross-linking of Alzheimer's amyloid beta-peptide. Life Sci. 1997;60:2323–2332. doi: 10.1016/s0024-3205(97)00288-9. [DOI] [PubMed] [Google Scholar]
- 141.Zhang W, Johnson BR, Suri DE, Martinez J, Bjornsson TD. Immunohistochemical demonstration of tissue transglutaminase in amyloid plaques. Acta Neuropathol (Berl) 1998;96:395–400. doi: 10.1007/s004010050910. [DOI] [PubMed] [Google Scholar]
- 142.Miller ML, Johnson GV. Transglutaminase cross-linking of the tau protein. J Neurochem. 1995;65:1760–1770. doi: 10.1046/j.1471-4159.1995.65041760.x. [DOI] [PubMed] [Google Scholar]
- 143.Appelt DM, Balin BJ. The association of tissue transglutaminase with human recombinant tau results in the formation of insoluble filamentous structures. Brain Res. 1997;745:21–31. doi: 10.1016/s0006-8993(96)01121-3. [DOI] [PubMed] [Google Scholar]
- 144.Murthy SN, Wilson JH, Lukas TJ, Kuret J, Lorand L. Cross-linking sites of the human tau protein, probed by reactions with human transglutaminase. J Neurochem. 1998;71:2607–2614. doi: 10.1046/j.1471-4159.1998.71062607.x. [DOI] [PubMed] [Google Scholar]
- 145.Tucholski J, Kuret J, Johnson GV. Tau is modified by tissue transglutaminase in situ: possible functional and metabolic effects of polyamination. J Neurochem. 1999;73:1871–1880. [PubMed] [Google Scholar]
- 146.Johnson GV, Cox TM, Lockhart JP, Zinnerman MD, Miller ML, Powers RE. Transglutaminase activity is increased in Alzheimer's disease brain. Brain Res. 1997;751:323–329. doi: 10.1016/s0006-8993(96)01431-x. [DOI] [PubMed] [Google Scholar]
- 147.Norlund MA, Lee JM, Zainelli GM, Muma NA. Elevated transglutaminase-induced bonds in PHF tau in Alzheimer's disease. Brain Res. 1999;851:154–163. doi: 10.1016/s0006-8993(99)02179-4. [DOI] [PubMed] [Google Scholar]
- 148.Singer SM, Zainelli GM, Norlund MA, Lee JM, Muma NA. Transglutaminase bonds in neurofibrillary tangles and paired helical filament tau early in Alzheimer's disease. Neurochem Int. 2002;40:17–30. doi: 10.1016/s0197-0186(01)00061-4. [DOI] [PubMed] [Google Scholar]
- 149.Zemaitaitis MO, Kim SY, Halverson RA, Troncoso JC, Lee JM, Muma NA. Transglutaminase activity, protein, and mRNA expression are increased in progressive supranuclear palsy. J Neuropathol Exp Neurol. 2003;62:173–184. doi: 10.1093/jnen/62.2.173. [DOI] [PubMed] [Google Scholar]
- 150.Takeda A, Mallory M, Sundsmo M, Honer W, Hansen L, Masliah E. Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am J Pathol. 1998;152:367–372. [PMC free article] [PubMed] [Google Scholar]
- 151.Shibasaki Y, Baillie DA, St Clair D, Brookes AJ. High-resolution mapping of SNCA encoding alpha-synuclein, the non-A beta component of Alzheimer's disease amyloid precursor, to human chromosome 4q21.3–>q22 by fluorescence in situ hybridization. Cytogenet Cell Genet. 1995;71:54–55. doi: 10.1159/000134061. [DOI] [PubMed] [Google Scholar]
- 152.Dickson TC, Vickers JC. The morphological phenotype of beta-amyloid plaques and associated neuritic changes in Alzheimer's disease. Neuroscience. 2001;105:99–107. doi: 10.1016/s0306-4522(01)00169-5. [DOI] [PubMed] [Google Scholar]
- 153.Yokota O, Terada S, Ishizu H, Tsuchiya K, Kitamura Y, Ikeda K, Ueda K, Kuroda S. NACP/alpha-synuclein immunoreactivity in diffuse neurofibrillary tangles with calcification (DNTC) Acta Neuropathol (Berl) 2002;104:333–341. doi: 10.1007/s00401-002-0545-5. [DOI] [PubMed] [Google Scholar]
- 154.Johnson GV, Cox TM, Lockhart JP, Zinnerman MD, Miller ML, Powers RE. Transglutaminase activity is increased in Alzheimer's disease brain. Brain Res. 1997;751:323–329. doi: 10.1016/s0006-8993(96)01431-x. [DOI] [PubMed] [Google Scholar]
- 155.Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300:636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- 156.Citron BA, Suo Z, SantaCruz K, Davies PJ, Qin F, Festoff BW. Protein crosslinking, tissue transglutaminase, alternative splicing and neurodegeneration. Neurochem Int. 2002;40:69–78. doi: 10.1016/s0197-0186(01)00062-6. [DOI] [PubMed] [Google Scholar]
- 157.Dickson DW. Misfolded, protease-resistant proteins in animal models and human neurodegenerative disease. J Clin Invest. 2002;110:1403–1405. doi: 10.1172/JCI17164. [DOI] [PMC free article] [PubMed] [Google Scholar]