

Abstract
Mutations in the breast cancer susceptibility gene 2 (BRCA2) are commonly found in familial pancreatic cancer. Recently, EMSY (11q13.5) has been described as a BRCA2 interacting protein capable of binding and inactivating the protein domain encoded by exon 3 of the BRCA2 gene. Amplification of EMSY occurs in 13% of sporadic breast cancers and is directly linked to increased expression. Here we investigate the amplification status of this new potential oncogene in 59 sporadic pancreatic cancers using fluorescence in situ hybridization (FISH) and tissue microarray (TMA). Real-time quantitative RT-PCR was performed on 20 pancreatic cancer cell lines and overexpression was calculated using the delta-delta-Ct-method. Amplification of EMSY was found in 8/59 cases (13.6%). 9/20 (45%) cell line samples showed overexpression of EMSY. In conclusion, sporadic pancreatic cancer shows amplification of EMSY at prevalence similar to that found in other cancers.
Keywords: Pancreatic cancer, EMSY, BRCA2, FISH, amplification
Introduction
Pancreatic cancer is the deadliest of the common solid cancers and the fourth leading cause of cancer-related death in the United States [1]. Due to the late development of clinical symptoms, 80% of patients with pancreatic cancer have metastasis at the time of initial diagnosis [2] such that 1-year and 5-year survival is 19% and 4%, respectively [3]. Surgical resection remains the best option for cure, but only about 20% of patients are eligible for surgery [4]. Numerous genetic (K-ras, p16, p53, SMAD4 [5–7], telomere shortening) and epigenetic (e.g. SPARC, RELN, TFPI-2 [8–11] and others [12, 13]) alterations occur during the development of pancreatic adenocarcinoma. In addition, germline mutations of the BRCA2 gene occur in 5–15% of individuals with familial pancreatic cancer [14–16] and in about 5% of patients with clinically sporadic pancreatic cancer [17]. A polymorphic stop codon near the 3’ end of BRCA2 is more common in patients with familial pancreatic cancer than in controls [18].
BRCA2 is a cancer-susceptibility gene that functions in DNA repair pathways which are essential for the maintenance of genetic integrity [19, 20]. BRCA2 promotes highly accurate homologous recombination (HR) mediated double strand break (DSB) repair through direct interaction with RAD51 [21]. Cancer in BRCA2 mutation carriers is associated with loss of heterozygosity (LOH) of the wild-type allele, resulting in a functional deficiency [22]. Even though functional deficiency of BRCA2 strongly reduces HR-mediated DSB repair, other error-prone DSB repair systems such as nonhomologous end joining (NHEJ) and single-strand annealing (SSA) remain intact [21, 23]. Rerouting of DSB processing through these repair systems may cause the accumulation of structural chromosomal aberrations as seen in BRCA2-deficient human cancer cells [24]. BRCA2-deficient cells exhibit an impaired DSB repair mechanism resulting in chromosomal instability and carcinogenesis [25]. Their inability to successfully repair DSB renders BRCA2-null cells especially vulnerable to DNA damaging agents such as mitomycin C (MMC) [26, 27] and poly(ADP-Ribose) polymerase (PARP1) [28–35]. These findings have led to a clinical trial testing the utility of using MMC and gemcitabine in patients with BRCA2 germline mutations [36].
Although inherited mutations of BRCA2 are well known to predispose to breast and ovarian cancer, somatic BRCA2 mutations are rarely seen in sporadic cancer cases [37, 38]. In addition, in response to DNA damage the Fanconi anemia (FA) pathway functioning upstream of BRCA1 and BRCA2 forms a Fanconi protein complex that mono-ubiquinates FANCD2 which then interacts with BRCA1/BRCA2. Mostly somatic mutations in the Fanconi anemia genes, FANCC and FANCG occur in 5–10% of pancreatic cancers [39–42]. Fanconi anemia gene-deficient cells are also hypersensitive to DNA damaging agents such as MMC [27, 43, 44].
Other proteins that interact with the BRCA2 pathway are genetically altered in certain cancers. Recently, a novel gene, EMSY, has been described providing a new mechanism possibly linking BRCA2 to sporadic breast and ovarian cancer [45]. EMSY is amplified in 13% of sporadic breast cancer and 17% of high grade ovarian cancer [46]. EMSY (11q13.5) maps to the same amplicon as GARP, glycoprotein A repetitions predominant, a gene not expressed in breast cancer [47], presenting EMSY as the gene of interest within this amplicon. Amplification of the 11q13 locus is common in several tumor types. Investigation of this gene dense region has led to several candidate targets of amplification [46, 48]. Cyclin D1 (CCD1) and EMS1 (cortactin) – located on separate amplicons at 11q13.3 (CCD1 mapping 0.8 Mb proximal to EMS1) – are considered strong candidate oncogenes within this region and are frequently co-amplified in breast cancer [49]. Other genes in this region include RSF-1, a gene that was recently described as amplified in a subset of ovarian carcinomas [50]. Amplification of EMSY occurs independently of adjacent CCD1, and correlates directly with increased levels of mRNA [45, 51–53]. The amino terminal of EMSY (ENT-domain) binds to the independent activation domain of BRCA2 encoded by exon 3 of the BRCA2 gene. Deletion of this specific region is known to be the sole mutation in a Swedish breast/ovarian cancer family [54]. Overexpression of EMSY interferes with the activation potential domain of BRCA2 encoded by exon 3 resulting in decreased BRCA2 activity, mimicking the genomic instability phenotype as seen in BRCA2 deficient cells [45]. The importance of EMSY in tumorigenesis remains uncertain although EMSY amplification was associated with a poor outcome in one study [53].
This study examines the prevalence of EMSY amplification in pancreatic adenocarcinomas using fluorescence in situ hybridization (FISH) and EMSY mRNA quantification in pancreatic cancer cell lines. We also determined if pancreatic cancers overexpressed the nearby gene, RSF-1.
Materials and Methods
Patients and Samples
The pancreatic cancer samples were chosen from tissue microarrays (TMAs) developed on patients with resectable infiltrating adenocarcinoma of the pancreas that underwent pancreatico-duodenectomy at Johns Hopkins Hospital, Baltimore, MD, between 1/2/1998 and 7/25/2003 [55]. Because the natural history for variant pancreatic neoplasms differs from usual pancreatic ductal adenocarcinoma, patients with IPMN’s, mucinous cystic adenocarcinomas and medullary adenocarcinomas were excluded. Patients were also excluded if gross metastatic or unresectable disease beyond the Whipple margins was found at the time of surgery.
All clinical and pathologic patient information is maintained in a regularly updated clinical database. The primary outcome of the study was overall survival as determined from date of Whipple resection to the time of death or last follow-up. Patients, their family or their primary physicians are contacted by postcard at least annually to confirm patient status with the last observation recorded in February 2007. This study was conducted as part of a Johns Hopkins Hospital IRB approved protocol.
Fluorescence In Situ Hybridization
DNA from EMSY PAC clones DJ824N10, DJ855A11 and DJ180012 provided by Dr. Carlos Caldas, University of Cambridge Department of Oncology, was extracted and purified and directly labeled with Spectrum Green by NICK translation. A Spectrum aqua-labeled centromeric probe, CEP11 was used to stain centromere-11. FISH was performed on 4 TMAs of formalin fixed, paraffin embedded tissues [55]. The slides were heat treated overnight at 56°C, deparaffinized (Xylene, 100%, 95% and 75% ethanol), demasked using 50TE2 pH 9.0 for 30min at 95°C, washed in 2XSSC and RNAse treated for 1 hr at 37°C (0.1mg/μL in 2 × SSC), after which they were washed again in 2XSSC, digested in a pepsin solution (0.1% in dH2O, pH 2.0) for 10min at 37°C, washed in PBS and dehydrated in ethanol (75%, 95% and 100%). Probes and target DNA were denatured simultaneously for 12 min at 80°C. Hybridization was done for 72 hrs under coverslips at 37°C. Posthybridization washes were performed using NP-40 rapid wash procedure. Nuclei were counterstained with 4,6-diamidino 2-phenylindole (DAPI, 1μg/mL) diluted 1:5 in antifade solution. Slides were analyzed with a Leica DMRA2 fluorescence microscope at a 100X magnification using FITC, DAPI and Aqua filters. Signals were counted manually only in non-overlapping, intact nuclei. A total of 150 nuclei were counted per case, divided over two TMA cores, unless fewer representative nuclei were available. The average number of probe signals per nucleus was calculated for both EMSY and CEP11. The amplification ratio was determined as the ratio between the average EMSY copy number and the average centromere-11 copy number. EMSY amplification was found when the amplification ratio exceeded 1.5 [45]. If the average copy number of EMSY per nucleus was higher than 2.5 but the amplification ratio did not exceed 1.5 due to a concomitant increase in the centromere-11 signal these cases were labeled as having an increased EMSY copy number (ICN) without gene amplification. Documentation of the images was done using the Leica DC350 FX camera and Leica CW4000 FISH software.
Real-Time RT-PCR Expression Analysis
Quantitative SYBRGreen RT-PCR was performed on 20 cell line RNA samples to screen for over expression of the EMSY gene. Results were normalized to the housekeeping gene GAPDH and expression was compared to normal levels in the diploid pancreatic cell lines HPDE and HPNE using the delta-delta-Ct-method. Cell-lines with a ratio higher than 2.5 were considered as having EMSY overexpression [53]. Primers detecting the GAPDH gene were run in a separate reaction tube from the EMSY primers (EMSY-HF1 5’CCA CCC CAC ATG TCT CCT GTA 3’, EMSY-HR1 5’ TGA GCT TGG TGA TGT GGT GAC 3’). The PCR reaction mix was prepared according to the standard protocol with a final primer concentration of 0.3 μM. The samples were run using the Cepheid SmartCycler System. The protocol consisted of a 30 min, 50°C reverse transcriptase step followed by 15 min at 95°C for the initial activation. 45 cycles of denaturation (15 sec at 94°C), annealing (30 sec at 58°C) and extension (30 sec at 72°C) were performed after which a melting curve was used to confirm the specificity of the PCR products. Experiments were performed in triplicate.
Immunohistochemistry
Immunohistochemistry (IHC) for RSF-1 was performed as previously described [50].
Statistics
Simple descriptive statistics were used to determine the prevalence of clinico-pathological differences between pancreatic cancer with EMSY amplification and those without.
Results
Fluorescence In Situ Hybridization
In total 4 TMAs were stained with 18 cases per TMA using a FISH-array for EMSY and centromere 11. A total of 59 cases of pancreatic cancer were successfully scored. Each case consisted of four cores on the TMA, two of which tumor tissue and two controls. Approximately 150 random intact nuclei were scored per case. No gain in copy number was found in any of the control tissue (Figure 1).
Figure 1.
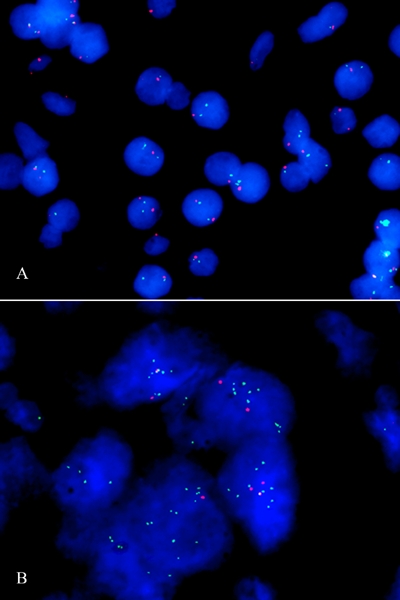
FISH analysis of EMSY amplification, conducted on paraffin embedded pancreatic tissue. The EMSY probe is colored green, while the control signal, the centromere-11 probe is red. A. No EMSY amplification in normal pancreatic tissue. B. EMSY amplification in a cluster of pancreatic tumor cells.
As can be seen in Table 1, low-level amplification of EMSY (EMSY/CEP11 copy number ratio >1.5) was found in eight of 59 cases (13.6%). An increased copy number (ICN) of EMSY (>2.5 EMSY probe signals on average per nucleus) was found in 12 of 59 cases (20.3%) including the 8 with EMSY amplification. But in 4 of these cases, the centromeric probe signal was also increased suggesting that in these cases the increased EMSY signal was due to aneuploidy. There were no obvious clinicopathologic differences (age, gender, tumor size, differentiation, stage) between the eight cases with EMSY amplification and the remaining cases without such amplification, although our sample size was not powered to identify such differences.
Table 1.
Summary of FISH analysis of 59 cases of sporadic pancreatic cancer
| Average # of signals per nucleus | ||||
|---|---|---|---|---|
| case # | # of nuclei scored | EMSY | C11 | EMSY/C11 ratio |
| 1 | 133 | 1.86 | 1.56 | 1.19 |
| 2 | 128 | 1.69 | 1.67 | 1.01 |
| 3 | 123 | 1.51 | 1.49 | 1.01 |
| 4 | 115 | 1.46 | 1.42 | 1.03 |
| 5 | 135 | 2.55 | 2.02 | 1.26 |
| 6 | 129 | 2.58 | 2.35 | 1.1 |
| 7 | 79 | 1.72 | 1.78 | 0.97 |
| 8 | 122 | 2.02 | 1.91 | 1.06 |
| 9 | 103 | 1.75 | 1.63 | 1.07 |
| 10 | 118 | 1.52 | 1.47 | 1.03 |
| 11 | 122 | 1.83 | 1.73 | 1.06 |
| 12 | 169 | 3.93 | 2.39 | 1.64 |
| 13 | 126 | 1.57 | 1.45 | 1.08 |
| 14 | 109 | 2.99 | 1.87 | 1.6 |
| 15 | 121 | 2.11 | 1.75 | 1.21 |
| 16 | 86 | 1.76 | 1.55 | 1.14 |
| 17 | 152 | 3.31 | 1.42 | 2.33 |
| 18 | 131 | 1.81 | 1.54 | 1.18 |
| 19 | 135 | 1.44 | 1.37 | 1.05 |
| 20 | 104 | 1.55 | 1.5 | 1.03 |
| 21 | 45 | 1.66 | 1.44 | 1.15 |
| 22 | 153 | 2.2 | 1.39 | 1.58 |
| 23 | 126 | 1.54 | 1.58 | 0.97 |
| 24 | 73 | 2.47 | 2.44 | 1.01 |
| 25 | 135 | 1.62 | 1.51 | 1.07 |
| 26 | 138 | 2.47 | 1.49 | 1.66 |
| 27 | 67 | 1.49 | 1.43 | 1.04 |
| 28 | 99 | 1.59 | 1.58 | 1.01 |
| 29 | 128 | 1.94 | 1.68 | 1.15 |
| 30 | 111 | 1.62 | 1.48 | 1.09 |
| 31 | 123 | 1.57 | 1.49 | 1.05 |
| 32 | 110 | 2 | 1.66 | 1.2 |
| 33 | 171 | 2.61 | 1.49 | 1.75 |
| 34 | 62 | 2.69 | 2.36 | 1.14 |
| 35 | 117 | 1.83 | 1.69 | 1.08 |
| 36 | 54 | 2.98 | 2.11 | 1.41 |
| 37 | 127 | 2.02 | 1.91 | 1.06 |
| 38 | 134 | 2.19 | 2.06 | 1.06 |
| 39 | 140 | 3.42 | 2.26 | 1.51 |
| 40 | 145 | 4.01 | 2.99 | 1.34 |
| 41 | 137 | 1.47 | 1.53 | 0.96 |
| 42 | 160 | 1.67 | 1.54 | 1.08 |
| 43 | 198 | 2.96 | 2.27 | 1.3 |
| 44 | 141 | 2.27 | 1.91 | 1.19 |
| 45 | 123 | 1.56 | 1.53 | 1.02 |
| 46 | 158 | 2.16 | 1.95 | 1.11 |
| 47 | 84 | 1.49 | 1.57 | 0.95 |
| 48 | 146 | 1.64 | 1.62 | 1.01 |
| 49 | 166 | 1.8 | 1.53 | 1.18 |
| 50 | 119 | 1.88 | 1.67 | 1.13 |
| 51 | 72 | 2.27 | 1.99 | 1.14 |
| 52 | 164 | 2.68 | 1.74 | 1.54 |
| 53 | 132 | 1.99 | 1.65 | 1.21 |
| 54 | 102 | 1.59 | 1.55 | 1.03 |
| 55 | 74 | 1.54 | 1.47 | 1.05 |
| 56 | 170 | 1.63 | 1.67 | 0.98 |
| 57 | 168 | 1.58 | 1.56 | 1.01 |
| 58 | 166 | 1.93 | 1.71 | 1.13 |
| 59 | 191 | 1.58 | 1.54 | 1.03 |
| 37.3% had an increased copy number (>2.0 signals on average per nucleus) | 13.6% had EMSY amplification (ratio >1.5) | |||
 EMSY amplification
EMSY amplification
 Increased EMSY copy number
Increased EMSY copy number
Real-Time RT-PCR Expression Analysis
EMSY amplification is associated with an increase in RNA levels. We determined the RNA expression levels in 20 pancreatic cell lines. Nine of 20 cell lines tested (45%) had an EMSY expression ratio >2.5 relative to the mean expression ratio in 2 non-neoplastic pancreas cell lines, but none of the pancreatic cancer cell lines showed high levels (>5-fold above reference normal levels) of EMSY overexpression. A subset of cell lines have been previously studied with regard to BRCA2 mutation status [17], defects in FANCD2 monoubiquitination and hypersensitivity to MMC due to FA or BRCA2 deficiency [44]. Results have been incorporated in Table 2.
Table 2.
Pancreatic cancer cell lines and corresponding EMSY expression level
| Cell lines | EMSY mRNA expression ratio | BRCA2 mutation status [17] | Fancd2 mono-ubiquitination [44] | Hypersensitivity to MMC [44] |
|---|---|---|---|---|
| Colo357 | 2.1 | pos | NA | |
| PL14 | 2.2 | NA | NA | |
| Hs766T | 1 | neg | pos | |
| CFPAC1 | 2 | LOH | pos | NA |
| BxPC3 | 1.5 | LOH | pos | neg |
| PL4 | 3.1 | NA | NA | |
| PaCa2 | 0.9 | NA | NA | |
| PL11 | 1.2 | neg | pos | |
| Su86.86 | 2.7 | LOH | pos | neg |
| CAPAN2 | 2.3 | pos | NA | |
| PL3 | 3 | pos | NA | |
| PL9 | 2.5 | NA | NA | |
| PL6 | 2 | pos | NA | |
| PL1 | 1.1 | NA | NA | |
| PL12 | 2.6 | NA | NA | |
| Aspe1 | 2 | NA | NA | |
| CAPAN1 | 2.6 | 6174delT/LOH | pos | pos |
| MiaPaCa | 3.4 | NA | NA | |
| AsPC1 | 3.1 | LOH | pos | NA |
| Panc-1 | 4.1 | LOH | pos | NA |
LOH, loss of heterozygosity; del, deletion; pos, positive; neg, negative; NA, not applicable
Immunohistochemistry
Expression of the nearby gene RSF-1 recently identified as overexpressed in ovarian cancers was evaluated in pancreatic ductal adenocarcinomas by immunohistochemistry of pancreatic cancer TMAs. RSF-1 protein was similarly expressed in pancreatic cancer and normal epithelium of the pancreatic duct.
Discussion
Given the poor survival of patients with pancreatic adenocarcinoma, there is a tremendous need to characterize the molecular profiles of these cancers in the hopes that molecular profiles can guide early diagnosis and the optimal use of novel therapies. EMSY has recently been proposed as a potential oncogene in breast and ovarian cancer through its interaction with BRCA2. The involvement of the BRCA2 pathway in pancreatic cancer led us to believe EMSY may also play a role in pancreatic carcinogenesis. EMSY is exclusively nuclear and, like BRCA2, re-localizes to DSB repair sites following DNA damage. Furthermore, the ENT-domain of EMSY interacts with a number of chromatin-regulator proteins, including HP1β and BS69, suggesting a role in chromatin remodeling [45, 56–58].
Here we looked at EMSY copy number aberrations in 59 cases of sporadic pancreatic carcinoma and expression of EMSY in 20 pancreatic cancer cell lines. We identified eight of 59 (13.6%) cases of low-level EMSY amplification (EMSY/CEP11 copy number ratio >1.5) in sporadic pancreatic carcinomas. EMSY overexpression was found in 9 of 20 pancreatic cancer cell lines, but the level of overexpression was modest with no cell lines showing expression more than five-fold above the reference level.
Twelve (20.3%) cases demonstrated increased copy number of EMSY (>2.5 EMSY probe signals on average per nucleus), with most cells appearing aneuploid containing also aberrant copy numbers of centromere 11. Nonetheless, six out of 12 cases do not show EMSY amplification due to an increase in centromere-11 copies. In all 12 cases increase in EMSY copy number was greater than that of centromere 11.
Cells containing increased copy numbers of EMSY appeared to be relatively large and clustered, surrounded by tumor cells expressing normal copy numbers, suggesting a late onset amplification with disease progression, rather than a mutation early in carcinogenesis. This may explain an earlier report that found a negative association of this mutation on disease outcome.
The inability to effectively repair DSB caused by DNA damaging agents results in the accumulation of chromatid breaks and aberrations leading to cell cycle arrest and eventually apoptosis. EMSY's inhibiting effect on the BRCA2 pathway suggests cells overexpressing EMSY may also express an increased sensitivity to DNA damaging agents. A subset of pancreatic cancer cell lines had been studied prior with regard to BRCA2 mutation status [17], defects in FANCD2 monoubiquitination and hypersensitivity to MMC due to FA or BRCA2 deficiency [44]. One cell line overexpressing EMSY was found not to be hypersensitive to MMC treatment. However, the level of EMSY amplification and RNA overexpression ratio of this particular cell line was not very high and it maybe that a higher level of EMSY expression is needed to be functionally relevant.
In summary, we find that pancreatic cancers display low-level amplification of EMSY at prevalence similar to what has been reported in other cancers. Further investigation is needed to determine if such low-level amplification of EMSY is sufficient to affect the pancreatic cancer sensitivity to DNA damaging agents such as MMC and PARP1.
Acknowledgments
This work was supported by the NCI grant (CA62924) and the Michael Rolfe Foundation.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 2.Jaffee EM, Hruban RH, Canto M, Kern SE. Focus on pancreas cancer. Cancer Cell. 2002;2:25–28. doi: 10.1016/s1535-6108(02)00093-4. [DOI] [PubMed] [Google Scholar]
- 3.Niederhuber JE, Brennan MF, Menck HR. The National Cancer Data Base report on pancreatic cancer. Cancer. 1995;76:1671–1677. doi: 10.1002/1097-0142(19951101)76:9<1671::aid-cncr2820760926>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 4.Yeo CJ, Sohn TA, Cameron JL, Hruban RH, Lillemoe KD, Pitt HA. Periampullary adenocarcinoma: analysis of 5-year survivors. Ann Surg. 1998;227:821–831. doi: 10.1097/00000658-199806000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biankin AV, Morey AL, Lee CS, Kench JG, Biankin SA, Hook HC, Head DR, Hugh TB, Sutherland RL, Henshall SM. DPC4/Smad4 expression and outcome in pancreatic ductal adenocarcinoma. J Clin Oncol. 2002;20:4531–4542. doi: 10.1200/JCO.2002.12.063. [DOI] [PubMed] [Google Scholar]
- 6.Infante JR, Matsubayashi H, Sato N, Tonascia J, Klein AP, Riall TA, Yeo C, Iacobuzio-Dohahue C, Goggins M. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J Clin Oncol. 2007;25:319–325. doi: 10.1200/JCO.2006.07.8824. [DOI] [PubMed] [Google Scholar]
- 7.Tascilar M, Skinner HG, Rosty C, Sohn T, Wilentz RE, Offerhaus GJ, Adsay V, Abrams RA, Cameron JL, Kern SE, Yeo CJ, Hruban RH, Goggins M. The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2001;7:4115–4121. [PubMed] [Google Scholar]
- 8.Ueki T, Toyota M, Sohn T, Yeo CJ, Issa JP, Hruban RH, Goggins M. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000;60:1835–1839. [PubMed] [Google Scholar]
- 9.Sato N, Parker AR, Fukushima N, Miyagi Y, Iacobuzio-Donahue CA, Eshleman JR, Goggins M. Epigenetic inactivation of TFPI-2 as a common mechanism associated with growth and invasion of pancreatic ductal adenocarcinoma. Oncogene. 2005;24:850–858. doi: 10.1038/sj.onc.1208050. [DOI] [PubMed] [Google Scholar]
- 10.Sato N, Fukushima N, Maehara N, Matsubayashi H, Koopmann J, Su GH, Hruban RH, Goggins M. SPARC/osteonectin is a frequent target for aberrant methylation in pancreatic adenocarcinoma and a mediator of tumor-stromal interactions. Oncogene. 2003;22:5021–5030. doi: 10.1038/sj.onc.1206807. [DOI] [PubMed] [Google Scholar]
- 11.Sato N, Fukushima N, Chang R, Matsubayashi H, Goggins M. Differential and epigenetic gene expression profiling identifies frequent disruption of the RELN pathway in pancreatic cancers. Gastroenterology. 2006;130:548–565. doi: 10.1053/j.gastro.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 12.Sato N, Goggins M. Epigenetic alterations in intraductal papillary mucinous neoplasms of the pancreas. J Hepatobiliary Pancreat Surg. 2006;13:280–285. doi: 10.1007/s00534-005-1056-2. [DOI] [PubMed] [Google Scholar]
- 13.Sato N, Goggins M. The role of epigenetic alterations in pancreatic cancer. J Hepatobiliary Pancreat Surg. 2006;13:286–295. doi: 10.1007/s00534-005-1057-1. [DOI] [PubMed] [Google Scholar]
- 14.Couch FJ, Johnson MR, Rabe KG, Brune K, de Andrade M, Goggins M, Rothenmund H, Gallinger S, Klein A, Petersen GM, Hruban RH. The prevalence of BRCA2 mutations in familial pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:342–346. doi: 10.1158/1055-9965.EPI-06-0783. [DOI] [PubMed] [Google Scholar]
- 15.Hahn SA, Greenhalf B, Ellis I, Sina-Frey M, Rieder H, Korte B, Gerdes B, Kress R, Ziegler A, Raeburn JA, Campra D, Grutzmann R, Rehder H, Rothmund M, Schmiegel W, Neoptolemos JP, Bartsch DK. BRCA2 germline mutations in familial pancreatic carcinoma. J Natl Cancer Inst. 2003;95:214–221. doi: 10.1093/jnci/95.3.214. [DOI] [PubMed] [Google Scholar]
- 16.Murphy KM, Brune KA, Griffin C, Sollenberger JE, Petersen GM, Bansal R, Hruban RH, Kern SE. Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: deleterious BRCA2 mutations in 17% Cancer Res. 2002;62:3789–3793. [PubMed] [Google Scholar]
- 17.Goggins M, Schutte M, Lu J, Moskaluk CA, Weinstein CL, Petersen GM, Yeo CJ, Jackson CE, Lynch HT, Kruban RH, Kern SE. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996;56:5360–5364. [PubMed] [Google Scholar]
- 18.Martin ST, Matsubayashi H, Rogers CD, Philips J, Couch FJ, Brune K, Yeo CJ, Kern SE, Hruban RH, Goggins M. Increased prevalence of the BRCA2 polymorphic stop codon K3326X among individuals with familial pancreatic cancer. Oncogene. 2005;24:3652–3656. doi: 10.1038/sj.onc.1208411. [DOI] [PubMed] [Google Scholar]
- 19.Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, Sands A, Eichele G, Hasty P, Bradley A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 1997;386:804–810. doi: 10.1038/386804a0. [DOI] [PubMed] [Google Scholar]
- 20.Patel KJ, Yu VP, Lee H, Corcoran A, Thistlethwaite FC, Evans MJ, Colledge WH, Friedman LS, Ponder BA, Venkitaraman AR. Involvement of Brca2 in DNA repair. Mol Cell. 1998;1:347–357. doi: 10.1016/s1097-2765(00)80035-0. [DOI] [PubMed] [Google Scholar]
- 21.Xia F, Taghian DG, DeFrank JS, Zeng ZC, Willers H, Iliakis G, Powell SN. Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal non-homologous end joining. Proc Natl Acad Sci USA. 2001;98:8644–9864. doi: 10.1073/pnas.151253498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goggins M, Hruban RH, Kern SE. BRCA2 is inactivated late in the development of pancreatic intraepithelial neoplasia: evidence and implications. Am J Pathol. 2000;156:1767–1771. doi: 10.1016/S0002-9440(10)65047-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tutt A, Bertwistle D, Valentine J, Gabriel A, Swift S, Ross G, Griffin C, Thacker J, Ashworth A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001;20:4704–4716. doi: 10.1093/emboj/20.17.4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–182. doi: 10.1016/s0092-8674(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 25.Abaji C, Cousineau I, Belmaaza A. BRCA2 regulates homologous recombination in response to DNA damage: implications for genome stability and carcinogenesis. Cancer Res. 2005;65:4117–4125. doi: 10.1158/0008-5472.CAN-04-3071. [DOI] [PubMed] [Google Scholar]
- 26.Abbott DW, Freeman ML, Holt JT. Double-strand break repair deficiency and radiation sensitivity in BRCA2 mutant cancer cells. J Natl Cancer Inst. 1998;90:978–985. doi: 10.1093/jnci/90.13.978. [DOI] [PubMed] [Google Scholar]
- 27.van der Heijden MS, Brody JR, Dezentje DA, Gallmeier E, Cunningham SC, Swartz MJ, DeMarzo AM, Offerhaus GJ, Isacoff WH, Hruban RH, Kern SE. In vivo therapeutic responses contingent on Fanconi anemia/BRCA2 status of the tumor. Clin Cancer Res. 2005;11:7508–7515. doi: 10.1158/1078-0432.CCR-05-1048. [DOI] [PubMed] [Google Scholar]
- 28.Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, Bontcheva-Diaz VD, Cox BF, DeWeese TL, Dillehay LE, Ferguson DC, Ghoreishi-Haack NS, Grimm DR, Guan R, Han EK, Holley-Shanks RR, Hristov B, Idler KB, Jarvis K, Johnson EF, Kleinberg LR, Klinghofer V, Lasko LM, Liu X, Marsh KC, McGonigal TP, Meulbroek JA, Olson AM, Palma JP, Rodriguez LE, Shi Y, Stavropoulos JA, Tsurutani AC, Zhu GD, Rosenberg SH, Giranda VL, Frost DJ. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13:2728–2737. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Liu L, Montagna C, Ried T, Deng CX. Haploinsufficiency of Parp1 accelerates Brca1-associated centrosome amplification, telomere shortening, genetic instability, apoptosis, and embryonic lethality. Cell Death Differ. 2007;14:924–931. doi: 10.1038/sj.cdd.4402105. [DOI] [PubMed] [Google Scholar]
- 30.McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O'connor MJ, Tutt AN, Zdzienicka MZ, Smith GC, Ashworth A. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-Ribose) polymerase Inhibition. Cancer Res. 2006;66:8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 31.Hay T, Jenkins H, Sansom OJ, Martin NM, Smith GC, Clarke AR. Efficient deletion of normal Brca2-deficient intestinal epithelium by poly(ADP-ribose) polymerase inhibition models potential prophylactic therapy. Cancer Res. 2005;65:10145–10148. doi: 10.1158/0008-5472.CAN-05-1186. [DOI] [PubMed] [Google Scholar]
- 32.McCabe N, Lord CJ, Tutt AN, Martin NM, Smith GC, Ashworth A. BRCA2-deficient CAPAN-1 cells are extremely sensitive to the inhibition of poly(ADP-Ribose) polymerase: an issue of potency. Cancer Biol Ther. 2005;4:934–936. doi: 10.4161/cbt.4.9.2141. [DOI] [PubMed] [Google Scholar]
- 33.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 34.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 35.Gallmeier E, Kern SE. Absence of specific cell killing of the BRCA2-deficient human cancer cell line CAPAN1 by poly(ADP-ribose) polymerase inhibition. Cancer Biol Ther. 2005;4:703–706. doi: 10.4161/cbt.4.7.1909. [DOI] [PubMed] [Google Scholar]
- 36. http://www.myriad.com/news/release/920082
- 37.Breast Cancer Linkage Consortium. Pathology of familial breast cancer: differences between breast cancers in carriers of BRCA1 or BRCA2 mutations and sporadic cases. Lancet. 1997;349:1505–1510. [PubMed] [Google Scholar]
- 38.Schutte M, da Costa LT, Hahn SA, Moskaluk C, Hoque AT, Rozenblum E, Weinstein CL, Bittner M, Meltzer PS, Trent JM. Identification by representational difference analysis of a homozygous deletion in pancreatic carcinoma that lies within the BRCA2 region. Proc Natl Acad Sci USA. 1995;92:5950–5954. doi: 10.1073/pnas.92.13.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Couch FJ, Johnson MR, Rabe K, Boardman L, McWilliams R, de Andrade M, Petersen G. Germ line Fanconi anemia complementation group C mutations and pancreatic cancer. Cancer Res. 2005;65:383–386. [PubMed] [Google Scholar]
- 40.Rogers CD, van der Heijden MS, Brune K, Yeo CJ, Hruban RH, Kern SE, Goggins M. The genetics of FANCC and FANCG in familial pancreatic cancer. Cancer Biol Ther. 2004;3:167–169. doi: 10.4161/cbt.3.2.609. [DOI] [PubMed] [Google Scholar]
- 41.Rogers CD, Couch FJ, Brune K, Martin ST, Philips J, Murphy KM, Petersen G, Yeo CJ, Hruban RH, Goggins M. Genetics of the FANCA gene in familial pancreatic cancer. J Med Genet. 2004;41:e126. doi: 10.1136/jmg.2004.024851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Heijden MS, Yeo CJ, Hruban RH, Kern SE. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res. 2003;63:2585–2588. [PubMed] [Google Scholar]
- 43.Gallmeier E, Kern SE. Targeting Fanconi anemia/BRCA2 pathway defects in cancer: the significance of preclinical pharmacogenomic models. Clin Cancer Res. 2007;13:4–10. doi: 10.1158/1078-0432.CCR-06-1637. [DOI] [PubMed] [Google Scholar]
- 44.van der Heijden MS, Brody JR, Gallmeier E, Cunningham SC, Dezentje DA, Shen D, Hruban RH, Kern SE. Functional defects in the fanconi anemia pathway in pancreatic cancer cells. Am J Pathol. 2004;165:651–657. doi: 10.1016/S0002-9440(10)63329-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hughes-Davies L, Huntsman D, Ruas M, Fuks F, Bye J, Chin SF, Milner J, Brown LA, Hsu F, Gilks B, Nielsen T, Schulzer M, Chia S, Ragaz J, Cahn A, Linger L, Ozdag H, Cattaneo E, Jordanova ES, Schuuring E, Yu DS, Venkitaraman A, Ponder B, Doherty A, Aparicio S, Bentley D, Theillet C, Ponting CP, Caldas C, Kouzarides T. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell. 2003;115:523–535. doi: 10.1016/s0092-8674(03)00930-9. [DOI] [PubMed] [Google Scholar]
- 46.Karlseder J, Zeillinger R, Schneeberger C, Czerwenka K, Speiser P, Kubista E, Birnbaum D, Gaudray P, Theillet C. Patterns of DNA amplification at band q13 of chromosome 11 in human breast cancer. Genes Chromosomes Cancer. 1994;9:42–48. doi: 10.1002/gcc.2870090108. [DOI] [PubMed] [Google Scholar]
- 47.Bekri S, Adelaide J, Merscher S, Grosgeorge J, Caroli-Bosc F, Perucca-Lostanlen D, Kelley PM, Pebusque MJ, Theillet C, Birnbaum D, Gaudray P. Detailed map of a region commonly amplified at 11q13–>q14 in human breast carcinoma. Cytogenet Cell Genet. 1997;79:125–131. doi: 10.1159/000134699. [DOI] [PubMed] [Google Scholar]
- 48.Bashyam MD, Bair R, Kim YH, Wang P, Hernandez-Boussard T, Karikari CA, Tibshirani R, Maitra A, Pollack JR. Array-based comparative genomic hybridization identifies localized DNA amplifications and homozygous deletions in pancreatic cancer. Neoplasia. 2005;7:556–562. doi: 10.1593/neo.04586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hui R, Ball JR, Macmillan RD, Kenny FS, Prall OW, Campbell DH. EMS1 gene expression in primary breast cancer: relationship to cyclin D1 and oestrogen receptor expression and patient survival. Oncogene. 1998;17:1053–1059. doi: 10.1038/sj.onc.1202023. [DOI] [PubMed] [Google Scholar]
- 50.Shih IeM, Sheu JJ, Santillan A, Nakayama K, Yen MJ, Bristow RE, Vang R, Parmigiani G, Kurman RJ, Trope CG, Davidson B, Wang TL. Amplification of a chromatin remodeling gene, Rsf-1/HBXAP, in ovarian carcinoma. Proc Natl Acad Sci USA. 2005;102:14004–4009. doi: 10.1073/pnas.0504195102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown LA, Irving J, Parker R, Kim H, Press JZ, Longacre TA, Chia S, Magliocco A, Makretsov N, Gilks B, Pollack J, Huntsman D. Amplification of EMSY, a novel oncogene on 11q13, in high grade ovarian surface epithelial carcinomas. Gynecol Oncol. 2006;100:264–270. doi: 10.1016/j.ygyno.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 52.Raouf A, Brown L, Vrcelj N, To K, Kowk W, Huntsman D, Eaves CJ. Genomic instability of human mammary epithelial cells over-expressing a truncated form of EMSY. J Natl Cancer Inst. 2005;97:1302–1306. doi: 10.1093/jnci/dji254. [DOI] [PubMed] [Google Scholar]
- 53.Rodriguez C, Hughes-Davies L, Valles H, Orsetti B, Cuny M, Ursule L, Kouzarides T, Theillet C. Amplification of the BRCA2 pathway gene EMSY in sporadic breast cancer is related to negative outcome. Clin Cancer Res. 2004;10:5785–5791. doi: 10.1158/1078-0432.CCR-03-0410. [DOI] [PubMed] [Google Scholar]
- 54.Nordling M, Karlsson P, Wahlstrom J, Engwall Y, Wallgren A, Martinsson T. A large deletion disrupts the exon 3 transcription activation domain of the BRCA2 gene in a breast/ovarian cancer family. Cancer Res. 1998;58:1372–1375. [PubMed] [Google Scholar]
- 55.Infante JR, Matsubayashi H, Sato N, Tonascia J, Klein AP, Riall TA, Yeo C, Iacobuzio-Donahue C, Goggins M. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J Clin Oncol. 2007;25:319–25. doi: 10.1200/JCO.2006.07.8824. [DOI] [PubMed] [Google Scholar]
- 56.Ekblad CM, Chavali GB, Basu BP, Freund SM, Veprintsev D, Hughes-Davies L, Kouzarides T, Doherty AJ, Itzhaki LS. Binding of EMSY to HP1beta: implications for recruitment of HP1beta and BS69. EMBO Rep. 2005;6:675–680. doi: 10.1038/sj.embor.7400415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chavali GB, Ekblad CM, Basu BP, Brissett NC, Veprintsev D, Hughes-Davies L, Kouzarides T, Itzhaki LS, Doherty AJ. Crystal structure of the ENT domain of human EMSY. J Mol Biol. 2005;350:964–973. doi: 10.1016/j.jmb.2005.05.047. [DOI] [PubMed] [Google Scholar]
- 58.Huang Y, Myers MP, Xu RM. Crystal structure of the HP1-EMSY complex reveals an unusual mode of HP1 binding. Structure. 2006;14:703–712. doi: 10.1016/j.str.2006.01.007. [DOI] [PubMed] [Google Scholar]