

Abstract
Pancreatic intraepithelial neoplasia (PanIN) is a histologically well-defined precursor to invasive ductal adenocarcinoma of the pancreas. PanINs are remarkably common lesions, particularly in the elderly population. Molecular studies have helped establish the progression of PanIN to invasive cancer, and recently genetically engineered mouse models have been generated that recapitulate the entire spectrum of lesions from precursor to invasive pancreatic cancer. Some PanIN lesions produce lobulocentric atrophy of the pancreatic parenchyma, and, when multifocal, this lobulocentric atrophy may be detectable using currently available imaging techniques such as endoscopic ultrasound. The association of acinar-ductal metaplasia with PanIN lesions has led some to hypothesize that PanINs develop from acinar cells that undergo acinar-ductal metaplasia.
Keywords: Pancreatic cancer, intraepithelial neoplasm, metaplasia, genetics, epigenetics
Introduction
Invasive adenocarcinoma of the pancreas is one of the, if not the, most lethal of all of the solid malignancies [1] It is usually not diagnosed until after the cancer has metastasized, and metastatic pancreatic cancer is a heterogeneous, complex and extremely challenging disease to treat. It will be difficult to make significant progress against pancreatic cancer if we focus our efforts solely on treating metastatic disease. Experience in the treatment of breast and colorectal neoplasms has demonstrated that the detection and treatment of early, non-invasive, disease have a major impact on cancer mortality [2–5]. We believe that the detection and treatment of early, non-invasive, pancreatic neoplasia will have a major impact on pancreatic cancer mortality.
Three morphologic forms of non-invasive pancreatic neoplasia have been defined. These are: 1) pancreatic intraepithelial neoplasia (PanIN), 2) mucinous cystic neoplasm (MCN), and 3) intraductal papillary mucinous neoplasm (IPMN). PanINs are by far the most common of these lesions. This review will focus on pancreatic intraepithelial neoplasia.
Clinical
A number of studies have examined the prevalence of PanINs in surgically resected pancreata and in autopsy material [6–8]. Like invasive pancreatic cancer, the prevalence of PanINs increases with age [9], and in most reports, PanINs are more common in the head of the gland than in the tail [6–8]. PanINs, and particularly higher grade PanINs (PanIN-3), are more common in pancreata with an invasive cancer than they are in pancreata with chronic pancreatitis, and PanINs are more common in pancreata with chronic pancreatitis than they are in controls. For example, Andea et al found a progressive increase in the number of PanINs and in the grade of PanINs when control pancreata were compared to pancreata with chronic pancreatitis and to pancreata with ductal adenocarcinoma [6]. PanINs were identified in 16% of the controls (none of which had PanIN-3), in 60% of the cases of chronic pancreatitis (4% had PanIN-3), and in 82% of pancreata with pancreatic cancer (40% harbored a PanIN-3) [9]. These clinical studies demonstrate that PanINs are very common lesions, and the clinical features of PanINs parallel those of invasive pancreatic cancer.
There have only been a handful of isolated case reports of patients with PanINs who later developed an invasive pancreatic cancer [10, 11]. The frequency and speed at which PanINs progress to invasive pancreatic cancer remain two important unanswered clinical questions. The only estimate of the progression of PanINs that we are aware of is a “back of the envelope” calculation reported by Terhune et al in a footnote to a paper on KRAS2 gene mutations [12]. Based on estimates of the prevalence of PanINs and the known prevalence of invasive pancreatic cancer, they estimated a 1% probability of a single PanIN lesion progressing to invasive cancer [12].
Morphology
Lesions which we now call PanINs have been recognized for over a century, but their significance was not recognized until recently [13–15]. As a result, these lesions were called by a wide variety of names including duct hyperplasia, hypertrophy, metaplasia, dysplasia, and, in German, zwishenformen [13–15]. PanINs arise in the smaller pancreatic ducts, they measure less than 0.5 cm, and they are classified morphologically into three grades. PanIN-1 lesions are composed of columnar epithelial cells with basally oriented uniform and round nuclei (Figure 1) [13, 14]. PanIN-1 lesions can be flat (PanIN-1A) or papillary (PanIN-1B) [13, 14]. PanIN-2 lesions are architecturally slightly more complex than PanIN-1 lesions, and they have more nuclear changes including loss of nuclear polarity, nuclear crowding, variation in nuclear size (pleomorphism), nuclear hyperchromasia, and nuclear pseudostratification [13, 14]. PanIN-3 lesions show the greatest degree of dysplasia. These lesions are architecturally complex, forming papillae and cribriform structures, and in some instances clusters of cells bud off of the epithelium into the lumen of the duct [13, 14]. Cytologically, the nuclei in PanIN-3 lesions are enlarged, pleomorphic, and poorly oriented [13, 14]. Nucleoli are often prominent and mitotic figures, and even abnormal mitoses, can be seen [13, 14].
Figure 1.
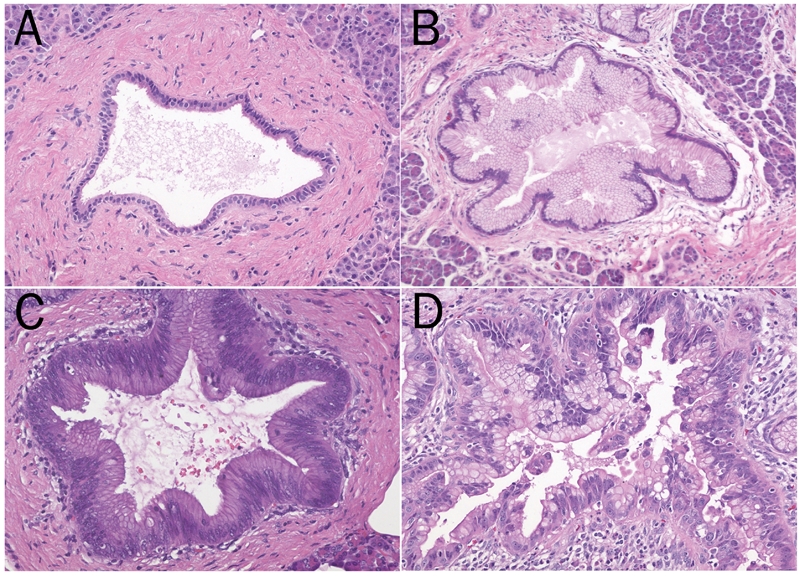
The progression from normal ducts (A) to pancreatic intraepithelial neoplasia (PanIN) (B-D) is associated with both architectural and cytological changes. In PanIN-1 the nuclei are uniform and basally oriented (B). The nuclei in PanIN-2 are slightly larger, more basophilic and there is some loss of nuclear polarity (C). The nuclear pleomorphism in PanIN-3 can be significant (D).
PanINs need to be distinguished from the larger intraductal papillary mucinous neoplasms (IPMNs) [16]. IPMNs are larger than PanINs, and IPMNs tend to have longer and more mucinous papillae than PanINs [14, 16]. Most PanINs cannot be appreciated grossly and most are smaller than 0.5 cm [14, 16]. Most IPMNs are larger than 1 cm, and most are easily seen grossly and radiographically [14, 16]. The expression of the mucin MUC2 also favors the diagnosis of an IPMN [17]. The two lesions are easy to distinguish at the extremes, but it is becoming increasingly clear that a number of precursor lesions fall in between the two. These in between lesions not only pose diagnostic challenges, but they also suggest that in some instances small lesions which we currently classify as PanINs may progress to larger lesions which we would classify as an IPMN. Additional studies are needed to establish this possible continuum.
Three new observations on the morphology of PanIN lesions deserve special note. First, Brune et al and Detlefsen et al have reported that distinctive lobulocentric atrophy is associated with many PanIN lesions, even low-grade PanIN lesions [18, 19]. This lobulocentric atrophy raises the possibility that PanIN lesions hamper the flow of acinar secretions producing a localized form of chronic pancreatitis [18, 19]. This may initiate a vicious cycle in which an early PanIN lesion causes localized duct obstruction, inflammation, and epithelial injury and regeneration which conspire to promote the progression of the PanIN lesion [18]. In addition, the foci of lobulocentric atrophy are larger than the PanIN lesions themselves, and, as will be discussed in greater detail later, these larger lesions may be clinically detectable [18, 20].
Second, PanINs, and particularly PanINs in individuals with a strong family history of pancreatic cancer, can be multifocal [18, 20, 21]. Brune et al reported eight patients with a strong family history of pancreatic cancer who underwent partial pancreatectomy because a screening endoscopic ultrasound revealed an early pancreatic lesion, and, remarkably, 11% of the duct profiles in these eight pancreata harbored a PanIN lesion (range 1.0% to 27.3%) [18]. This multifocality of disease has profound implications for patient management [20, 22]. If a patient with a strong family history of pancreatic cancer undergoes partial pancreatectomy and is found to have multi-focal PanINs, they are likely to have additional PanIN lesions in the unresected portion of their gland. On the plus side, the combination of lobulocentric atrophy and multifocality may produce a distinctive change in the pancreas that is detectable using currently available imaging technologies. For example, Canto et al screened seventy-eight high-risk patients (72 from familial pancreatic cancer kindreds, 6 individuals with the Peutz-Jeghers syndrome) and found that multifocal lobulocentric atrophy caused by multifocal PanIN lesions produced endoscopic ultrasound changes similar to those seen with chronic pancreatitis [18, 20]. This major advance establishes that early pancreatic neoplasia, in this case multifocal PanIN lesions, can be detected in asymptomatic individuals before an invasive cancer develops.
Third, a growing number of observations have highlighted acinar to ductal metaplasia in association with PanIN lesions [18, 23]. The acini in the lobules of lobulocentric atrophy described earlier are often characterized by prominent acinar-ductal metaplasia. As will be discussed in greater detail later, the association of acinar-ductal metaplasia with PanIN lesions has led investigators working with genetically engineered mouse models of pancreatic cancer to suggest that PanINs develop from acinar cells that undergo acinar-ductal metaplasia [23, 24].
Genetics
The genetic changes in PanIN lesions have been determined using a variety of techniques. Activating point mutations in the KRAS2 gene have been described, as has bi-allelic inactivation of the MAD4/DPC4, TP53 and p16/CDKN2A genes [12, 25–35]. Recently, telomere shortening has been shown to be an early event in PanIN development [36]. As one would expect for a precursor lesion, the genes mutated in PanINs are many of the same genes mutated in invasive pancreatic cancer.
Two important observations have been made in the genetic analyses of different grades of PanIN. First, although the mutations do not occur in a specific well-regulated order, some mutations usually occur before others. KRAS2 gene mutations and telomere shortening tend to appear first (in PanIN-1 lesions), followed by the inactivation of p16/CDKN2A (usually in PanIN-2), and finally the inactivation of TP53 and MAD4/DPC4 (typically in PanIN-3 lesions) (Figure 2) [25–35, 37]. Second, the prevalence of mutations increases in parallel with the degree of dysplasia. For example, Lohr et al conducted a meta-analysis of the studies published between 1988 and 2003 that provided information on KRAS2 gene mutations in PanIN lesions from pancreata of patients with pancreatic cancer [37]. As has been reported for p16/CDKN2A, they found a stepwise increase in KRAS2 gene mutations with the grade of dysplasia of the PanIN lesion [32, 37]. KRAS2 gene mutations were found in 36%, 44%, and 87% of PanIN-1A, 1B, and 2–3 lesions, respectively (trend statistic P <0.001) [37]. Thus, in PanIN lesions, we see a progression from apparently harmless precursors to deadly invasive cancers.
Figure 2.
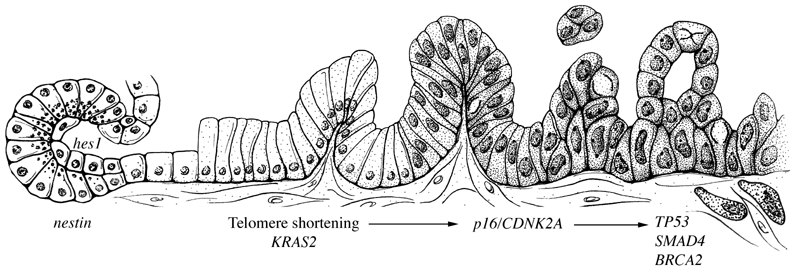
Model for the histological and genetic progression from normal cells (far left) through PanIN lesions (center), to invasive pancreatic cancer (far right). (Reprinted, with permission, from Annual Review of Pathology: Mechanisms of Disease. 2008;3:157–188 © by Annual Reviews. www.annualreviews.org).
These findings have a number of important implications. First, the finding that the same genes are targeted in PanINs and in invasive pancreatic cancer lends strong support to the hypothesis that PanINs are a precursor to invasive pancreatic cancer. Second, the finding that the prevalence of mutations increases with the degree of dysplasia validates the histological grading system that has been developed for PanIN lesions [13]. Third, knowledge of the relative order in which genes are mutated in PanINs has helped guide the creation of genetically engineered mouse (GEM) models of pancreatic cancer [38–40]. As discussed in greater detail later in this review, the major breakthrough in the creation of GEM models that mimic human disease came when Hingorani et al introduced endogenous KRAS gene mutations into the embryonic mouse pancreas [39]. Finally, an understanding of the relative order at which genetic alterations occur in PanINs forms an essential foundation for the development of early detection tests based on genetic changes. For example, KRAS2 gene mutations are a natural target for gene-based screening tests because these mutations occur relatively early in the development of pancreatic neoplasia [41, 42].
Epigenetic Alterations
Gene expression can be controlled by promoter methylation, and a number of genes have been shown to be abnormally methylated in pancreatic cancer [43–45]. For example, the p16/CDKN2A gene is genetically inactivated in ∼80% of invasive pancreatic cancers, and in another 15–20% it is inactivated by promoter hypermethylation [46]. As is true for genetic mutations, the prevalence of these abnormally methylated genes increases with the grade of the PanIN lesion [47, 48]. For example, Fukushima et al reported that the p16/CDKN2A gene is hypermethylated in 7% of PanIN-1 lesions and in 21% of PanIN-3 lesions [47]. A number of tests have been developed to detect abnormally methylated genes in a variety sample types, and this abnormal methylation can therefore potentially form the basis of a detection test for early, non-invasive, disease [49, 50].
Protein Expression
As one would expect for a precursor lesion, many of the same proteins expressed in invasive pancreatic cancer are also expressed in PanIN lesions [51–55]. Global analyses of gene expression have shown that a number of markers of gastric epithelial differentiation, such as pepsinogen C, MUC6, KLF4 and TFF1, are expressed in PanINs [55]. In addition, several cell adhesion molecules, such as claudin 18, and proteins that play a role in calcium regulation, such as the S-100 proteins, are expressed in both PanINs and in invasive pancreatic cancers [51–55]. Some of these proteins are expressed in early lesions (PanIN-1 lesions), while other proteins aren't expressed until late, in PanIN-3 lesions [51, 52]. For example, Bhanot et al reported an almost exponential increase in survivin transcript levels from low-grade PanINs (PanIN-1) to high-grade PanINs (PanINs-2 and 3) to invasive cancer [51]. The patterns of protein expression in PanIN lesions are important because the proteins expressed in low-grade PanINs may be reasonable chemoprevention targets, while those expressed late (in PanIN-3 lesions) are potential markers for the early detection of pancreatic neoplasia. For example, Maitra et al have shown that cyclooxygenase-2 (COX-2) is expressed in human PanINs and Funahashi et al have shown that COX-2 inhibitors delay the progression of PanINs to invasive cancer in a mouse model of pancreatic neoplasia [52, 56]. S100A11 is a protein expressed in PanINs that may serve as a good marker for the early detection of pancreatic neoplasia [53].
A number of mucins are also expressed in PanIN lesions [17, 57–60]. PanINs, particularly high-grade PanINs, express MUC1, MUC4, MUC5AC, and MUC6 [14, 17, 58–60]. These mucins can be used to distinguish PanINs from IPMNs, because PanINs, in contrast to IPMNs with intestinal differentiation, do not express MUC2 [17]. These mucins are also potentially detectable by imaging [61] and they may be useful for screening and as therapeutic targets for the treatment of precursor lesions [62, 63].
Mouse Models
An understanding of the genetics of human PanINs and invasive pancreatic cancer was critical to the development of genetically engineered mouse models of pancreatic cancer [64]. The first model to successfully recapitulate the human disease was developed by Hingorani et al [39]. They directed the endogenous expression of mutant KRAS(G12D) to progenitor cells of the mouse pancreas using the PDX1 and PTF1 promoters [39]. KRAS was chosen because, as discussed earlier, KRAS2 gene mutations occur early in human PanIN lesions and are almost ubiquitous in invasive pancreatic cancer [39]. These genetically engineered mice first developed lesions morphologically very similar to human PanINs and months later they developed invasive ductal adenocarcinomas [39]. The fact that these animals developed PanIN lesions before they developed invasive cancer has helped to validate the hypothesis that PanINs can progress to invasive cancer.
Once a KRAS gene-based genetically engineered mouse model was developed, the next natural step was to add additional genetic alterations to define the role of these additional genetic changes in the development of pancreatic neoplasia [64]. The genetic alterations that have been combined with KRAS mutations, based on the genes targeted in human pancreatic cancer, include p16(ink4a) mutations, TP53 mutations and MAD4/DPC4 [40, 65–68]. Invasive pancreatic cancer developed faster with the addition of p16/CDKN2A and TP53 mutations, and, of interest, cystic neoplasms developed with the addition of MAD4/DPC4 mutations [40, 65–68]. These experiments are complex and sometimes produce varying results. Nonetheless, they do help define the role of these genes in the progression of pancreatic neoplasia.
Mouse models can also be used to examine the role of other medical conditions and environmental factors in the development of pancreatic cancer [38, 69]. For example, Guerra et al demonstrated that when KRAS mutations are created in adult mice these genetically engineered mice do not develop lesions, but when pancreatitis is induced these same mice develop PanINs and pancreatic cancer [38]. Studies such as this one, when integrated in with observations made in humans, can help define the contribution of genetic, medical and environmental factors in the development of pancreatic cancer [38, 70].
Mouse models are potentially useful tools for the discovery of markers of early pancreatic neoplasia. One of the limitations in studying human disease is that it is rare to identify patients with documented PanINs who do not also have an invasive neoplasm. By contrast, it is relatively easy to obtain biosamples from genetically engineered mice with PanINs but no invasive cancer. Indeed, Bardeesy et al have recently reported that they were able to identify serum markers of human pancreatic cancer through proteomic analysis of biosamples from genetically engineered mouse models of PanIN and pancreatic cancer (presented at the 2007 SPORE Meeting, Baltimore, MD).
Finally, the issue of “cell of origin” has been examined in genetically engineered mouse models using different promoters to target the expression of mutant KRAS in different cell populations [24, 38, 71–73]. While the early models targeted pancreatic progenitor cells using the PDX1 and PTF1 promoters, the expression of mutant KRAS in acinar and centroacinar cells as directed by the nestin and even the elastase promoters produces PanIN and invasive pancreatic cancer [38, 71]. Surprisingly, the expression of mutant KRAS in mature ductal cells as driven by the cytokeratin 19 promoter did not induce PanINs nor did it produce invasive cancer [74]. Careful examination of the pathologic changes in these models suggests that acinar-ductal metaplasia and centroacinar cell expansion play a role in the formation of PanINs, at least in mice (Figure 2) [39, 72, 73].
Integrating Mouse Models and Human Disease
Although they are often performed in isolation, it is helpful to integrate the observations made with human disease with the results produced by genetically engineered mouse models. First, both the experience in humans and the observations in mice support the importance of genetic alterations, including mutations in the KRAS2, p16/CDKN2A, TP53 and MAD4/DPC4 genes, in the development of pancreatic neoplasia [39, 40, 46, 65–67, 68, 75, 76]. These mutations are common in human disease and can drive the formation and/or progression of disease in mouse models. Second, both the experience in humans and the observations in mice support the importance of non-genetic factors, such as chronic pancreatitis, contributing to disease [38, 69, 70]. Third, both the experience in humans and the observations in mice support the hypothesis that invasive pancreatic cancers can evolve from PanIN lesions [13]. Finally, observations made in the mouse models and then translated to observations in humans, suggest that PanINs may not be the earliest precursor lesions; that centroacinar cells and acinar-ductal metaplasia may play a role in some instances [39, 73].
Early Detection
It is our belief that the early detection of precursor lesions, such as PanINs, offers the greatest hope of saving lives that would otherwise be lost to pancreatic cancer [2]. A number of potential targets for the early detection of PanIN lesions have been discussed, including mutant genes, aberrantly methylated genes, and over expressed proteins. While these are all exciting markers for the future, we need to do something now. This need is felt greatest in individuals with a strong family history of pancreatic cancer because they appear to be the most at-risk for developing pancreatic cancer [77]. Patients with three or more first-degree relatives with pancreatic cancer have a 14 to 32-fold increased risk of developing pancreatic cancer, and this risk is significant [77]. For example, as of September 1, 2007, 56 incident pancreatic cancers have developed in at-risk family members in the National Familial Pancreas Tumor Registry and almost all of these patients presented with metastatic disease [77].
Endoscopic ultrasound (EUS) is one of the best available technologies to image the pancreas and M. Canto et al and Brentnall et al have used EUS to screen asymptomatic, apparently healthy, members of families in which there have already been several pancreatic cancers [20, 22, 78]. Close to 10% of the individuals screened by Canto et al were found to have a significant precursor lesion in their pancreas [20, 78]. Most of the lesions detected were intraductal papillary mucinous neoplasms [78]. In addition, as discussed earlier in the section on the morphology of PanINs, it was noted that multifocal PanINs, because they produce multifocal lobulocentric atrophy, can be detected by EUS because they give the pancreas an appearance similar to that of chronic pancreatitis [18]. Based on these early successes, a multicenter study, called “CAPS 3”, examining the efficacy of EUS and other imaging modalities in the early detection of pancreatic neoplasia is underway.
Unanswered Questions
While the last two decades have witnessed an explosive growth in our understanding of PanIN lesions and the critical role they play as precursors to invasive pancreatic cancer, there remain several important unanswered questions. First, long-standing pancreatitis, and familial pancreatitis in particular, has been shown to significantly increase the risk of pancreatic cancer in humans [70]. Similarly, the induction of pancreatitis in genetically engineered mouse models can promote pancreatic neoplasia [38]. Yet, most patients with pancreatic cancer don't have a clinical history of pancreatitis. This leads to the question: is subclinical pancreatitis common in the population? Second, if subclinical pancreatitis is common in the population what is the mechanism by which pancreatitis promotes the development of pancreatic cancer? Is it the result of inflammation, injury and repair, or does pancreatitis induce acinar to ductal metaplasia and thereby promote pancreatic neoplasia? The answer to this question may guide intervention strategies. Should efforts to prevent the progression of normal cells to PanINs to cancer focus on preventing acinar to ductal metaplasia or should they focus on reducing inflammation [53, 62, 79]? Third, what is the natural history of untreated PanIN lesions [10, 80]? What percentage of PanINs progress and how fast do the various PanIN lesions progress to invasive carcinoma? Finally, does a PanIN arising in an individual with a strong family history of pancreatic cancer behave the same as PanIN arising as a sporadic lesion [18]? This is a critical issue for screening efforts directed at the high-risk population with a family history of pancreatic cancer.
Conclusion
Pancreatic intraepithelial neoplasia is a precursor to one of the deadliest of all of the solid malignancies. Efforts to prevent, to detect, and to treat these lesions have the potential to save many lives.
References
- 1.American Cancer Society. Cancer. New York, New York: American Cancer Society; 2007. Cancer Facts and Figures 2007; pp. 1–52. [Google Scholar]
- 2.Berry DA, Cronin KA, Plevritis SK, Fryback DG, Clarke L, Zelen M, Mandelblatt JS, Yakovlev AY, Habbema JD, Feuer EJ. Effect of screening and adjuvant therapy on mortality from breast cancer. N Engl J Med. 2005;353:1784–1792. doi: 10.1056/NEJMoa050518. [DOI] [PubMed] [Google Scholar]
- 3.Faivre J, Dancourt V, Lejeune C, Tazi MA, Lamour J, Gerard D, Dassonville F, Bonithon-Kopp C. Reduction in colorectal cancer mortality by fecal occult blood screening in a French controlled study. Gastroenterology. 2004;126:1674–1680. doi: 10.1053/j.gastro.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 4.Frazier AL, Colditz GA, Fuchs CS, Kuntz KM. Cost-effectiveness of screening for colorectal cancer in the general population. JAMA. 2000;284:1954–1961. doi: 10.1001/jama.284.15.1954. [DOI] [PubMed] [Google Scholar]
- 5.Newcomb PA, Storer BE, Morimoto LM, Templeton A, Potter JD. Long-term efficacy of sigmoidoscopy in the reduction of colorectal cancer incidence. J Natl Cancer Inst. 2003;95:622–625. doi: 10.1093/jnci/95.8.622. [DOI] [PubMed] [Google Scholar]
- 6.Andea A, Sarkar F, Adsay NV. Clinicopathological correlates of pancreatic intraepithelial neoplasia: a comparative analysis of 82 cases with and 152 cases without pancreatic ductal adenocarcinoma. Mod Pathol. 2003;16:996–1006. doi: 10.1097/01.MP.0000087422.24733.62. [DOI] [PubMed] [Google Scholar]
- 7.Cubilla AL, Fitzgerald PJ. Morphological lesions associated with human primary invasive nonendocrine pancreas cancer. Cancer Res. 1976;36:2690–2698. [PubMed] [Google Scholar]
- 8.Kozuka S, Sassa R, Taki T, Masamoto K, Nagasawa S, Saga S, Hasegawa K, Takeuchi M. Relation of pancreatic duct hyperplasia to carcinoma. Cancer. 1979;43:1418–1428. doi: 10.1002/1097-0142(197904)43:4<1418::aid-cncr2820430431>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz AM, Henson DE. Familial and sporadic pancreatic carcinoma, epidemiologic concordance. Am J Surg Pathol. 2007;31:645–646. doi: 10.1097/PAS.0b013e31802d6d42. [DOI] [PubMed] [Google Scholar]
- 10.Brat DJ, Lillemoe KD, Yeo CJ, Warfield PB, Hruban RH. Progression of pancreatic intraductal neoplasias to infiltrating adenocarcinoma of the pancreas. Am J Surg Pathol. 1998;22:163–169. doi: 10.1097/00000478-199802000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Brockie E, Anand A, Albores-Saavedra J. Progression of atypical ductal hyperplasia/carcinoma in situ of the pancreas to invasive adenocarcinoma. Ann Diagn Pathol. 1998;2:286–292. doi: 10.1016/s1092-9134(98)80020-8. [DOI] [PubMed] [Google Scholar]
- 12.Terhune PG, Phifer DM, Tosteson TD, Longnecker DS. K-ras mutation in focal proliferative lesions of human pancreas. Cancer Epidemiol Biomarkers Prev. 1998;7:515–521. [PubMed] [Google Scholar]
- 13.Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett E, Goodman SN, Kern SE, Klimstra DS, Klöppel G, Longnecker DS, Lüttges J, Offerhaus GJ. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579–586. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 14.Hruban RH, Pitman MB, Klimstra DS, editors. Tumors of the pancreas. Atlas of tumor pathology. 4th series. Washington, DC: American Registry of Pathology and Armed Forces Institute of Pathology; 2007. [Google Scholar]
- 15.Hulst SPL. Zur kenntnis der Genese des Adenokarzinoms und Karzinoms des Pankreas. Virchows Arch (B) 1905;180:288–316. [Google Scholar]
- 16.Hruban RH, Takaori K, Klimstra DS, Adsay NV, Albores-Saavedra J, Biankin AV, Biankin SA, Compton C, Fukushima N, Furukawa T, Goggins M, Kato Y, Klöppel G, Longnecker DS, Lüttges J, Maitra A, Offerhaus GJ, Shimizu M, Yonezawa S. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2004;28:977–987. doi: 10.1097/01.pas.0000126675.59108.80. [DOI] [PubMed] [Google Scholar]
- 17.Adsay NV, Merati K, Andea A, Sarkar F, Hruban RH, Wilentz RE, Goggins M, Iocobuzio-Donahue C, Longnecker DS, Klimstra DS. The dichotomy in the preinvasive neoplasia to invasive carcinoma sequence in the pancreas: differential expression of MUC1 and MUC2 supports the existence of two separate pathways of carcinogenesis. Mod Pathol. 2002;15:1087–1095. doi: 10.1097/01.MP.0000028647.98725.8B. [DOI] [PubMed] [Google Scholar]
- 18.Brune KA, Abe T, Canto MI, O'Malley L, Klein AP, Maitra A, Adsay NV, Fishman EK, Cameron JL, Yeo CJ, Kern SE, Goggins M, Hruban RH. Multifocal neoplastic precursor lesions associated with lobular atrophy of the pancreas in patients having a strong family history of pancreatic cancer. Am J Surg Pathol. 2006;30:1067–1076. [PMC free article] [PubMed] [Google Scholar]
- 19.Detlefsen S, Sipos B, Feyerabend B, Kloppel G. Pancreatic fibrosis associated with age and ductal papillary hyperplasia. Virchows Arch. 2005;447:800–805. doi: 10.1007/s00428-005-0032-1. [DOI] [PubMed] [Google Scholar]
- 20.Canto MI, Goggins M, Hruban RH, Fishman EK, Axilbund JE, Griffin CA, Ali SZ, Richman J, Jagannath S, Kantsevoy SV, Petersen GM, Giardiello FM, Kalloo AN. Screening for early pancreatic neoplasia in high-risk individuals: a prospective controlled study. Clin Gastroenterol Hepatol. 2006;4:766–781. doi: 10.1016/j.cgh.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 21.Meckler KA, Brentnall TA, Haggitt RC, Crispin D, Byrd DR, Kimmey MB, Bronner MP. Familial fibrocystic pancreatic atrophy with endocrine cell hyperplasia and pancreatic carcinoma. Am J Surg Pathol. 2001;25:1047–1053. doi: 10.1097/00000478-200108000-00009. [DOI] [PubMed] [Google Scholar]
- 22.Brentnall TA, Bronner MP, Byrd DR, Haggitt RC, Kimmey MB. Early diagnosis and treatment of pancreatic dysplasia in patients with a family history of pancreatic cancer. Ann Intern Med. 1999;131:247–255. doi: 10.7326/0003-4819-131-4-199908170-00003. [DOI] [PubMed] [Google Scholar]
- 23.Zhu L, Shi G, Schmidt CM, Hruban RH, Konieczny SF. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am J Pathol. 2007;171:263–273. doi: 10.2353/ajpath.2007.061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murtaugh LC, Leach SD. A case of mistaken identity? Nonductal origins of pancreatic “ductal” cancers. Cancer Cell. 2007;11:211–213. doi: 10.1016/j.ccr.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 25.Goggins M, Hruban RH, Kern SE. BRCA2 is inactivated late in the development of pancreatic intraepithelial neoplasia: evidence and implications. Am J Pathol. 2000;156:1767–1771. doi: 10.1016/S0002-9440(10)65047-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinmöller E, Dietmaier W, Zirngibl H, Heinmöller P, Scaringe W, Jauch K-W, Hofstädter F, Rüschoff J. Molecular analysis of microdissected tumors and preneoplastic intraductal lesions in pancreatic carcinoma. Am J Pathol. 2000;157:83–92. doi: 10.1016/S0002-9440(10)64520-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lüttges J, Schlehe B, Menke MA, Vogel I, Henne-Bruns D, Klöppel G. The K-ras mutation pattern in pancreatic ductal adenocarcinoma usually is identical to that in associated normal, hyperplastic, and metaplastic ductal epithelium. Cancer. 1999;85:1703–1710. [PubMed] [Google Scholar]
- 28.Lüttges J, Galehdari H, Brocker V, Schwarte-Waldhoff I, Henne-Bruns D, Klöppel G, Schmiegel W, Hahn SA. Allelic loss is often the first hit in the biallelic inactivation of the p53 and DPC4 genes during pancreatic carcinogenesis. Am J Pathol. 2001;158:1677–1683. doi: 10.1016/S0002-9440(10)64123-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moskaluk CA, Hruban RH, Kern SE. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997;57:2140–2143. [PubMed] [Google Scholar]
- 30.Rosty C, Geradts J, Sato N, Wilentz RE, Roberts H, Sohn T, Cameron JL, Yeo CJ, Hruban RH, Goggins M. p16 Inactivation in pancreatic intraepithelial neoplasias (PanINs) arising in patients with chronic pancreatitis. Am J Surg Pathol. 2003;27:1495–1501. doi: 10.1097/00000478-200312000-00001. [DOI] [PubMed] [Google Scholar]
- 31.Tada M, Ohashi M, Shiratori Y, Okudaira T, Komatsu Y, Kawabe T, Yoshida H, Machinami R, Kishi K, Omata M. Analysis of K-ras gene mutation in hyperplastic duct cells of the pancreas without pancreatic disease. Gastroenterology. 1996;110:227–231. doi: 10.1053/gast.1996.v110.pm8536861. [DOI] [PubMed] [Google Scholar]
- 32.Wilentz RE, Geradts J, Maynard R, Offerhaus GJ, Kang M, Goggins M, Yeo CJ, Kern SE, Hruban RH. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res. 1998;58:4740–4744. [PubMed] [Google Scholar]
- 33.Wilentz RE, Iacobuzio-Donahue CA, Argani P, McCarthy DM, Parsons JL, Yeo CJ, Kern SE, Hruban RH. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000;60:2002–2006. [PubMed] [Google Scholar]
- 34.Yamano M, Fujii H, Takagaki T, Kadowaki N, Watanabe H, Shirai T. Genetic progression and divergence in pancreatic carcinoma. Am J Pathol. 2000;156:2123–2133. doi: 10.1016/S0002-9440(10)65083-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yanagisawa A, Ohtake K, Ohashi K, Hori M, Kitagawa T, Sugano H, Kato Y. Frequent c-Ki-ras oncogene activation in mucous cell hyperplasias of pancreas suffering from chronic inflammation. Cancer Res. 1993;53:953–956. [PubMed] [Google Scholar]
- 36.van Heek NT, Meeker AK, Kern SE, Yeo CJ, Lillemoe KD, Cameron JL, Offerhaus GJ, Hicks JL, Wilentz RE, Goggins M, De Marzo AM, Hruban RH, Maitra A. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am J Pathol. 2002;161:1541–1547. doi: 10.1016/S0002-9440(10)64432-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lohr M, Kloppel G, Maisonneuve P, Lowenfels AB, Luttges J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia. 2005;7:17–23. doi: 10.1593/neo.04445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by k-ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 39.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, Tuveson DA. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 40.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 41.Caldas C, Hahn SA, Hruban RH, Redston MS, Yeo CJ, Kern SE. Detection of K-ras mutations in the stool of patients with pancreatic adenocarcinoma and pancreatic ductal hyperplasia. Cancer Res. 1994;54:3568–3573. [PubMed] [Google Scholar]
- 42.Shi C, Eshleman SH, Jones D, Fukushima N, Hua L, Parker AR, Yeo CJ, Hruban RH, Goggins M, Eshleman JR. LigAmp for sensitive detection of single-nucleotide differences. Nat Methods. 2004;1:141–147. doi: 10.1038/nmeth713. [DOI] [PubMed] [Google Scholar]
- 43.Sato N, Fukushima N, Maitra A, Matsubayashi H, Yeo CJ, Cameron JL, Hruban RH, Goggins M. Discovery of novel targets for aberrant methylation in pancreatic carcinoma using high-throughput microarrays. Cancer Res. 2003;63:3735–3742. [PubMed] [Google Scholar]
- 44.Sato N, Goggins M. The role of epigenetic alterations in pancreatic cancer. J Hepatobiliary Pancreat Surg. 2006;13:286–295. doi: 10.1007/s00534-005-1057-1. [DOI] [PubMed] [Google Scholar]
- 45.Ueki T, Toyota M, Sohn T, Yeo CJ, Issa JP, Hruban RH, Goggins M. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000;60:1835–1839. [PubMed] [Google Scholar]
- 46.Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, Moskaluk CA, Hahn SA, Schwarte-Waldhoff I, Schmiegel W, Baylin SB, Kern SE, Herman JG. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57:3126–3130. [PubMed] [Google Scholar]
- 47.Fukushima N, Sato N, Ueki T, Rosty C, Walter KM, Wilentz RE, Yeo CJ, Hruban RH, Goggins M. Aberrant methylation of preproenkephalin and p16 genes in pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am J Pathol. 2002;160:1573–1581. doi: 10.1016/S0002-9440(10)61104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jansen M, Fukushima N, Rosty C, Walter K, Altink R, Heek TV, Hruban RH, Offerhaus GJ, Goggins M. Aberrant methylation of the 5' CpG island of TSLC1 is common in pancreatic ductal adenocarcinoma and is first manifest in high-grade PanlNs. Cancer Biol Ther. 2002;1:293–296. doi: 10.4161/cbt.84. [DOI] [PubMed] [Google Scholar]
- 49.Fukushima N, Walter KM, Uek T, Sato N, Matsubayashi H, Cameron JL, Hruban RH, Canto M, Yeo CJ, Goggins M. Diagnosing pancreatic cancer using methylation specific PCR analysis of pancreatic juice. Cancer Biol Ther. 2003;2:78–83. doi: 10.4161/cbt.183. [DOI] [PubMed] [Google Scholar]
- 50.Matsubayashi H, Canto M, Sato N, Klein AP, Abe T, Yamashita K, Yeo CJ, Kalloo AN, Hruban RH, Goggins M. DNA methylation alterations in the pancreatic juice of patients with suspected pancreatic disease. Cancer Res. 2005;66:1208–1217. doi: 10.1158/0008-5472.CAN-05-2664. [DOI] [PubMed] [Google Scholar]
- 51.Bhanot U, Heydrich R, Moller P, Hasel C. Survivin expression in pancreatic intraepithelial neoplasia (PanIN): steady increase along the developmental stages of pancreatic ductal adenocarcinoma. Am J Surg Pathol. 2006;30:754–759. doi: 10.1097/00000478-200606000-00013. [DOI] [PubMed] [Google Scholar]
- 52.Maitra A, Ashfaq R, Gunn CR, Rahman A, Yeo CJ, Sohn TA, Cameron JL, Hruban RH, Wilentz RE. Cyclooxygenase 2 expression in pancreatic adenocarcinoma and pancreatic intraepithelial neoplasia: an immunohistochemical analysis with automated cellular imaging. Am J Clin Pathol. 2002;118:194–201. doi: 10.1309/TPG4-CK1C-9V8V-8AWC. [DOI] [PubMed] [Google Scholar]
- 53.Ohuchida K, Mizumoto K, Ohhashi S, Yamaguchi H, Konomi H, Nagai E, Yamaguchi K, Tsuneyoshi M, Tanaka M. S100A11, a putative tumor suppressor gene, is over-expressed in pancreatic carcinogenesis. Clin Cancer Res. 2006;12:5417–5422. doi: 10.1158/1078-0432.CCR-06-0222. [DOI] [PubMed] [Google Scholar]
- 54.Ohuchida K, Mizumoto K, Egami T, Yamaguchi H, Fujii K, Konomi H, Nagai E, Yamaguchi K, Tsuneyoshi M, Tanaka M. S100P is an early developmental marker of pancreatic carcinogenesis. Clin Cancer Res. 2006;12:5411–5416. doi: 10.1158/1078-0432.CCR-06-0298. [DOI] [PubMed] [Google Scholar]
- 55.Prasad N, Biankin AV, Fukushima N, Maitra A, Elkahloun AG, Hruban RH, Goggins M, Leach SD. Gene expression profiles in pancreatic intraepithelial neoplasia reflect the effects of Hedgehog signaling on pancreatic ductal epithelial cells. Cancer Res. 2004;65:1619–1626. doi: 10.1158/0008-5472.CAN-04-1413. [DOI] [PubMed] [Google Scholar]
- 56.Funahashi H, Satake M, Dawson D, Huynh NA, Reber HA, Hines OJ, Eibl G. Delayed progression of pancreatic intraepithelial neoplasm in a conditional Kras (G12D) mouse model by a selective cyclooxygenase-2 inhibitor. Cancer Res. 2007;67:7068–7071. doi: 10.1158/0008-5472.CAN-07-0970. [DOI] [PubMed] [Google Scholar]
- 57.Kim GE, Bae HI, Park HU, Kuan SF, Crawley SC, Ho JJ, Kim YS. Aberrant expression of MUC5AC and MUC6 gastric mucins and sialyl Tn antigen in intraepithelial neoplasms of the pancreas. Gastroenterology. 2002;123:1052–1060. doi: 10.1053/gast.2002.36018. [DOI] [PubMed] [Google Scholar]
- 58.Nagata K, Horinouchi M, Saitou M, Higashi M, Nomoto M, Goto M, Yonezawa S. Mucin expression profile in pancreatic cancer and the precursor lesions. J Hepatobiliary Pancreat Surg. 2007;14:243–254. doi: 10.1007/s00534-006-1169-2. [DOI] [PubMed] [Google Scholar]
- 59.Park HU, Kim JW, Kim GE, Bae HI, Crawley SC, Yang SC, Gum JR, Jr, Batra SK, Rousseau K, Swallow DM, Sleisenger MH, Kim YS. Aberrant expression of MUC3 and MUC4 membrane-associated mucins and sialyl Le(x) antigen in pancreatic intraepithelial neoplasia. Pancreas. 2003;26:e48–e54. doi: 10.1097/00006676-200304000-00022. [DOI] [PubMed] [Google Scholar]
- 60.Swartz MJ, Batra SK, Varshney GC, Hollingsworth MA, Yeo CJ, Cameron JL, Wilentz RE, Hruban RH, Argani P. MUC4 expression increases progressively in pancreatic intraepithelial neoplasia. Am J Clin Pathol. 2002;117:791–796. doi: 10.1309/7Y7N-M1WM-R0YK-M2VA. [DOI] [PubMed] [Google Scholar]
- 61.Foss CA, Fox JJ, Feldmann G, Maitra A, Iacobuzio-Donohue C, Kern SE, Hruban RH, Pomper MG. Radiolabeled anti-claudin 4 and anti-prostate stem cell antigen: initial imaging in experimental models of pancreatic cancer. Mol Imaging. 2007;6:131–139. [PubMed] [Google Scholar]
- 62.Ohuchida K, Mizumoto K, Yamada D, Fujii K, Ishikawa N, Konomi H, Nagai E, Yamaguchi K, Tsuneyoshi M, Tanaka M. Quantitative analysis of MUC1 and MUC5AC mRNA in pancreatic juice for preoperative diagnosis of pancreatic cancer. Int J Cancer. 2006;118:405–411. doi: 10.1002/ijc.21317. [DOI] [PubMed] [Google Scholar]
- 63.Singh AP, Chaturvedi P, Batra SK. Emerging roles of MUC4 in cancer: a novel target for diagnosis and therapy. Cancer Res. 2007;67:433–436. doi: 10.1158/0008-5472.CAN-06-3114. [DOI] [PubMed] [Google Scholar]
- 64.Hruban RH, Adsay NV, Albores-Saavedra J, Anver MR, Biankin AV, Boivin GP, Furth EE, Furukawa T, Klein AP, Klimstra DS, Kloppel G, Lauwers GY, Longnecker DS, Luttges J, Maitra A, Offerhaus GJ, Perez-Gallego L, Redston M, Tuveson DA. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res. 2006;66:95–106. doi: 10.1158/0008-5472.CAN-05-2168. [DOI] [PubMed] [Google Scholar]
- 65.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bardeesy N, Morgan J, Sinha M, Signoretti S, Srivastava S, Loda M, Merlino G, DePinho RA. Obligate roles for p16(Ink4a) and p19(Arf)-p53 in the suppression of murine pancreatic neoplasia. Mol Cell Biol. 2002;22:635–643. doi: 10.1128/MCB.22.2.635-643.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D, DePinho RA. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006;20:3130–3146. doi: 10.1101/gad.1478706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Izeradjene K, Combs C, Best M, Gopinathan A, Wagner A, Grady WM, Deng CX, Hruban RH, Adsay NV, Tuveson DA, Hingorani SR. Kras(G12D) and Smad4/Dpc4 haplo-insufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell. 2007;11:229–243. doi: 10.1016/j.ccr.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 69.Archer H, Jura N, Keller J, Jacobson M, Bar-Sagi D. A mouse model of hereditary pancreatitis generated by transgenic expression of R122H trypsinogen. Gastroenterology. 2006;131:1844–1855. doi: 10.1053/j.gastro.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 70.Lowenfels AB, Maisonneuve EP, Dimagno YE, Gates LK, Perrault J, Whitcomb DC, International Hereditary Pancreatitis Study Group Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1997;89:442–446. doi: 10.1093/jnci/89.6.442. [DOI] [PubMed] [Google Scholar]
- 71.Carriere C, Seeley ES, Goetze T, Longnecker DS, Korc M. The Nestin progenitor lineage is the compartment of origin for pancreatic intraepithelial neoplasia. Proc Natl Acad Sci USA. 2007;104:4437–4442. doi: 10.1073/pnas.0701117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stanger BZ, Stiles B, Lauwers GY, Bardeesy N, Mendoza M, Wang Y, Greenwood A, Cheng KH, McLaughlin M, Brown D, DePinho RA, Wu H, Melton DA, Dor Y. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell. 2005;8:185–195. doi: 10.1016/j.ccr.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 73.Tuveson DA, Zhu L, Gopinathan A, Willis NA, Kachatrian L, Grochow R, Pin CL, Mitin NY, Taparowsky EJ, Gimotty PA, Hruban RH, Jacks T, Konieczny SF. Mist1-KrasG12D knock-in mice develop mixed differentiation metastatic exocrine pancreatic carcinoma and hepato-cellular carcinoma. Cancer Res. 2006;66:242–247. doi: 10.1158/0008-5472.CAN-05-2305. [DOI] [PubMed] [Google Scholar]
- 74.Brembeck FH, Schreiber FS, Deramaudt TB, Craig L, Rhoades B, Swain G, Grippo P, Stoffers DA, Silberg DG, Rustgi AK. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003;63:2005–2009. [PubMed] [Google Scholar]
- 75.Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ, Kern SE. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8:27–32. doi: 10.1038/ng0994-27. [DOI] [PubMed] [Google Scholar]
- 76.Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 77.Klein AP, Brune KA, Petersen GM, Goggins M, Tersmette AC, Offerhaus GJ, Griffin C, Cameron JL, Yeo CJ, Kern SE, Hruban RH. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004;64:2634–2638. doi: 10.1158/0008-5472.can-03-3823. [DOI] [PubMed] [Google Scholar]
- 78.Canto MI, Goggins M, Yeo CJ, Griffin C, Axilbund JE, Brune KA, Ali SZ, Jagannath S, Petersen GM, Fishman EK, Piantadosi S, Giardiello FM, Hruban RH. Screening for pancreatic neoplasia in high-risk individuals: An EUS-based approach. Clin Gastroenterol Hepatol. 2004;2:606–621. doi: 10.1016/s1542-3565(04)00244-7. [DOI] [PubMed] [Google Scholar]
- 79.Maitra A, Adsay NV, Argani P, Iacobuzio-Donahue CA, De Marzo A, Cameron JL, Yeo CJ, Hruban RH. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol. 2003;16:902–912. doi: 10.1097/01.MP.0000086072.56290.FB. [DOI] [PubMed] [Google Scholar]
- 80.Hruban RH, Goggins M, Parsons JL, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6:2969–2972. [PubMed] [Google Scholar]