

Abstract
The re-expression of multiple cell cycle markers representing various cell cycle phases in postmitotic pyramidal neurons suggests that neurons in Alzheimer disease (AD) attempt to re-enter the cell cycle. Entry into the cell cycle requires activation of G1 to S phase cell cycle proteins, among which retinoblastoma protein (pRb) is a key regulator. pRb inhibits the transcription of cell cycle proteins in the nucleus of healthy cells by interaction and consequent blocking of the active site of E2F, dependent upon the phosphate stoichiometry and combination of the locations of their 16 potential phosphorylation sites on pRb. Therefore, to determine whether pRb is involved in the aberrant cell cycle phenotype in AD neurons, a systematic immunocytochemical evaluation of the phosphorylation status of pRb protein using antibodies specific for multiple phosphorylation sites (i.e., pSpT249/252, pS612, pS795, pS807, pS811 and pT821) was carried out in the hippocampal regions of brains from AD patients. Increased levels of phospho-pRb (ppRb) for all these phosphorylation sites were noted in the brains of AD patients as compared to control cases. More importantly, redistribution of ppRb from the nucleus to the cytoplasm of susceptible neurons, with significant localization in neurofibrillary tangles and neuritic plaques, was observed. Additional studies revealed extensive co-localization between phospho-p38 and ppRb, implicating that p38 activation may contribute to cell cycle abnormalities through pRb phosphorylation. Taken together, these data supports the concept of neuronal cell cycle re-entry in AD and indicates a crucial role for pRb in this process.
Keywords: Alzheimer disease, cell cycle, hyperphosphorylation, p38, retinoblastoma protein
Introduction
Alzheimer disease (AD) is a progressive and fatal neurodegenerative disease that is characterized by deterioration in memory and cognitive abilities, disturbances in behavior and gradual loss of the ability to communicate and carry out activities of daily living. The disease affects an estimated 4–5 million Americans and more than 30 million people worldwide [1]. Despite decades of extensive research, the cause of AD is not yet fully understood. Nonetheless, recent findings support the notion of mechanistic parallels between neurogenesis during normal development and neurodegeneration during AD [2, 3]. Although adult neurons are generally thought of as being in a quiescent, non-proliferative state, it is becoming increasingly apparent that degenerating neurons in AD brains exhibit activation in cell cycle apparatus and are characteristic of cells re-entering the cell division process [4]. Evidence of this aberrant re-entry into the cell cycle in AD includes the abnormal reexpression of various cyclins, cyclin-dependent kinases (CDKs), and cyclin dependant kinase inhibitors (CDKIs) that potentially lead to the active expression of growth promoting genes [5–21].
pRb, the tumor suppressor product of the retinoblastoma susceptibility gene, is involved in a number of cellular functions, including cell division, differentiation, senescence, and apoptosis [22]. It was the first of the three tumor suppressor proteins to be identified in the Retinoblastoma family [23]. Additional studies have revealed that pRb acts together with two other “pocket” proteins, p107 and p130, to inhibit proliferation in most, if not all cell types [24]. pRb and other pocket proteins exert their inhibitory effects in part by binding to and inhibiting the activity of critical regulatory proteins and transcription factors, including those of the E2F family, whose activation is necessary for the G1-S transition [25]. The pRb/E2F complex inhibits transcription of cell cycle proteins in the nucleus of healthy cells by blocking the active site of E2F promoters [26], and recent data suggests that this complex also participates in cell death via transcription-dependent or independent mechanisms [27]. pRb is a nuclear phospho-protein containing 16 potential phosphorylation sites spanning the entire sequence. The binding and interacting ability of pRb with E2F and other transcription factors is governed by its phosphorylation state which fluctuates as the cell passes through different phases of the cell division cycle: pRb is found in a hypophosphorylated state (1–2 mol of phosphate per mol of pRb) in G0 and much of G1, is converted into a hyperphosphorylated state (about 10 mol of phosphate per mol of pRb) as the cell approaches the end of G1 phase and persists in this state through the remainder of the cell cycle, finally it is converted back to hypophosphorylated state during late M phase [28]. Hypophosphorylated pRb is the active form of pRb which binds to E2F and other transcription factors and inhibits cell cycle progression [29]. Hyperphosphorylation of pRb, presumably by the sequential action of various cyclin-CDK pairs and potentially other kinases such as p38 or ERK [30], causes it to dissociate from E2F, leading to the expression of E2F target genes [31] which is critical for cell cycle progression through the restriction point. Since pRb remains in its hyperphosphorylated state throughout the remainder of the cycle [32], it may also play a role in guiding the cell through S, G2, and M. During late M phase, the active hypophosphorylated pRb is reconstituted by dephosphorylation catalyzed by protein phosphatase 1.
Prior studies demonstrated higher levels of pRb phosphorylated at Ser795 [ppRb(S795)] in AD compared to controls with ppRb(S795) predominantly located in the nucleus of cells in proximity to neurofibrillary tangles (NFT) or plaques but rarely colocalized with NFT markers [33]. It was also absent in the dystrophic neurites around plaques. Indeed, amyloid-β (Aβ) induced phosphorylation of pRb at serine 795 in the nucleus in vitro [34]. However, given that ppRb(S795) is also present in the hypophosphorylated form of pRb [35] and phosphorylation at this site alone does not necessarily release E2F [36], it would be premature to conclude, based on these studies, that pRb becomes hyperphosphorylated and cell cycle repression is lifted without examining the phosphorylation status of other phosphorylation sites. Therefore, in the present study, we carried out a systematic examination of the phosphorylation state of pRb using a battery of pRb antibodies specific for multiple phosphorylation sites (i.e., pSpT249/252, pS612, pS795, pS807, pS811 and pT821) in the hippocampal regions in AD brains. Consistent to the previous findings, we found an increased level of ppRb immunoreactivity in AD brains compared to control cases. More importantly, we noticed redistribution of ppRb to the cytoplasm of susceptible neurons with significant localization in NFTs and neuritic plaques, which suggests not only elevated hyperphosphorylation but also aberrant distribution of ppRb contributes to the abnormal neuronal re-entry into cell cycle in AD.
Material and Methods
Brain tissue
Paraffin embedded tissue sections of hippocampus and neocortex were obtained from 12 AD cases and nine non-demented age-matched controls and fixed with either routine formalin or in methacarn (methanol; chloroform; acetic acid; 6:3:1). All human tissues were obtained from the Alzheimer Disease Research Center at Case Western Reserve University with appropriate Institutional Review Board approval. AD cases were confirmed pathologically and met the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria for neuropathologic diagnosis of AD [37, 38]. The average age at death was 85 years for AD cases (ages range from 79–91) and 69 years for controls (ages range from 57–81 years). The average post-mortem interval was 29 hours for AD cases (range of 3–55 hours) and 25 hours for control cases (range of 4–46 hours). Control cases were also assigned by CERAD criteria and, in some cases, showed only age-related pathological structures identified with phosphorylated tau (AT8).
Immunocytochemistry
Immunocytochemistry was performed on the tissue sections using the peroxidase-anti-peroxidase protocol as described previously [39]. Briefly, the slides were immersed in xylene to remove the paraffin, and then hydrated through graded ethanol solutions and the endogenous peroxidase activity removed with 3% H2O2 for 30 minutes. The sections were then incubated with 10% normal goat serum (NGS) in TBS at room temperature for 30 min to prevent non-specific binding, then overnight at 4°C with primary antibodies. Antibodies used were mouse monoclonal anti-retinoblastoma (unphosphorylated) KAM-CP124 (Stressgen Biotechnology, Ann Arbor, MI at 1:40 dilution); rabbit polyclonal antibodies against phosphoretinoblastoma protein (ppRb) pS807, pS811, pS612, pSpT 249/252 and pT821 (BioSource International, Camarillo, CA at 1:100 dilution); monoclonal antiserum to phosphorylated tau (AT8, Pierce Endogen at 1:1000 dilution; PHF1, gift of Sharon Greenberg at 1:1000 dilution; 12E8, gift of Elan Pharmaceuticals at 1:1000 dilution); and rabbit polyclonal antibodies against phospho-p38 (pp38) (Cell Signaling, Danvers, MA, at 1:100 dilution). Following incubation with species specific secondary antibodies and peroxidase-anti-peroxidase complexes, the antibodies were detected with 3, 3'-diaminobenzidine (DAB) as the chromogen. Some sections were pretreated with trypsin (400μg/mL for 10 minutes at room temperature) prior to application of primary antibodies to visualize nuclear localization.
The specificity of ppRb(S807) antibody was confirmed following adsorption with the specific antigen (Abgent, 50μg/mL). The immunocytochemical procedure was repeated with absorbed antibody prepared by the overnight incubation of anti-phosphorylated retinoblastoma primary antibody with its immunizing peptide.
Double staining was performed on some sections to directly compare the localization of ppRb(S807) and phosphorylated tau (12E8 or PHF-1), as previously described [40]. Briefly, the ppRb was detected with DAB using the peroxidase anti-peroxidase method, and the 12E8 or PHF-1 was detected on the same section using the Fast Blue as chromogen following the alkaline phosphatase anti-alkaline phosphatase method. Sections were mounted with crystal mount.
Results
To systematically evaluate the phosphorylation status of pRb protein in AD, a battery of pRb antibodies specific for seven phosphorylation sites (i.e., pSpT249/252, pS612, pS795, pS807, pS811 and pT821) were used. Consistent with a previous report [33], antisera specific for ppRb(S795) labeled nuclei of pyramidal neurons and granular cells in dentate gyrus in both control cases (Figure 1A and inset) and some AD cases (Figure 1B and inset) following trypsin pretreatment. However, in other AD cases, the cytoplasm of pyramidal neurons in the hippocampus (Figure 1C) showed some ppRb(S795) immunoreactivity, while in all control cases the cytoplasm remained unstained. Most notably, although ppRb(S795) was rarely associated with neurofibrillary pathology in AD [33], antibodies specific for other pRb phosphorylation sites demonstrated strong labeling of pathological structures (a schematic of phosphorylation sites and overview of staining patterns is shown in Figure 2). Immunolabeling with anti-phosphoretinoblastoma protein S807 [ppRb(S807)] antibody revealed intense staining of pathology in all cases of AD examined, specifically both intracellular and extracellular NFT in pyramidal neurons, neuropil threads, dystrophic neurites accumulating around amyloid plaques, and granulovacuolar degeneration (GVD) (Figure 3A), while non-diseased aged patients without any pathology showed little immunoreactivity (Figure 3B). To demonstrate the specificity of ppRb(S807) antibody, adsorption with its immunizing peptide, greatly reduced the immunostaining (Figure 3D), compared to the non-adsorbed antibody on adjacent sections (Figure 3C). During the process of optimizing detection condition, there was no effect of different fixative (i.e., formalin or methacarn) on staining pattern, while, as anticipated, pretreatment with trypsin greatly enhanced the nuclear staining.
Figure 1.
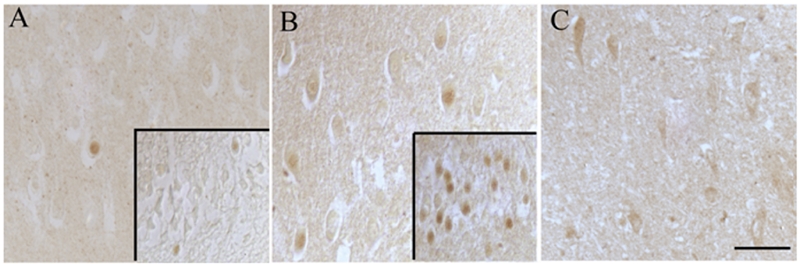
Localization of ppRb(S795) recognizes nuclei in both pyramidal neurons and granule neurons in control cases (A and inset, respectively) as well as in more nuclei in both pyramidal neurons and granule neurons in AD cases (B and inset, respectively). In some AD cases, neuronal cytoplasm also contains ppRb(S795) (C), yet never in control cytoplasm. Scale bar = 50 μm.
Figure 2.

A. Schematic representation of the pRb protein showing known phosphorylation sites. Those epitopes examined in this study are denoted with an (*). Epitopes found to be present in AD pathology in the present study are denoted in the dashed box. B. An overview of the pathological structures revealed by antisera specific for different phosphorylation sites spanning pRb.
Figure 3.
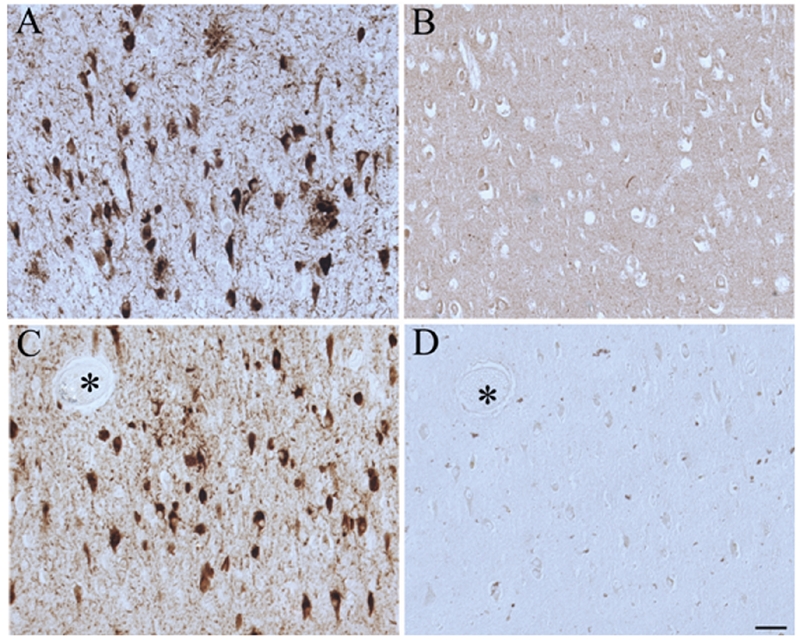
Pathological structures in AD hippocampus, namely NFT and neuritic plaques, accumulate phosphorylated pRb(S807) (A), while control cases show little staining (B). Adjacent serial sections of AD hippocampus stained with antibodies against ppRb(S807) adsorbed with its immunizing peptide (D) almost completely eliminated the immunostaining compared with that without adsorption (C). Asterisks indicate landmark vessels. Scale bar = 50 μm.
The localization of ppRb(S807) to pathological structures is confirmed by the colocalization with 12E8 and PHF-1 positive structures as revealed by double immunocytochemistry or by comparison of adjacent serial sections. In the double staining experiment, comparison of ppRb(S807) with 12E8 (Figure 4B), which is specific for tau phosphorylated at Ser262 and is a relatively early marker for tau pathology, revealed some overlap, yet many cells contained either only ppRb(S807) or only 12E8. Comparison of ppRb(S807) with PHF-1 (Figure 4A), which is specific for tau phosphorylated at Ser396 and Ser404 and is a relatively late marker for tau pathology, revealed significantly more overlap with only a few neurons containing only ppRb(S807) or PHF-1. This was also confirmed by immunostaining in adjacent serial sections both in cases of AD (Figure 4C, D) and in aged control cases with some pathology (Figure 4E, F).
Figure 4.
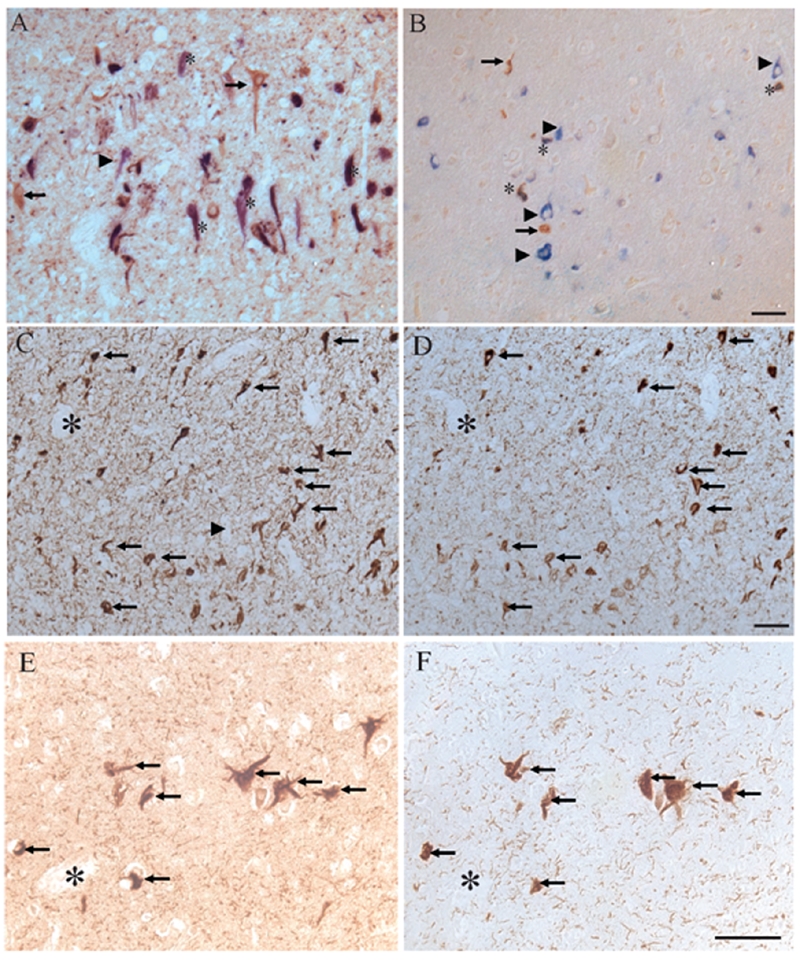
Using double-label immunocytochemistry, localization of ppRb(S807), stained brown, was compared with PHF-1 (blue, A) and 12E8 (B). Neurons containing both phosphorylated tau and ppRb(S807) were marked by *. Neurons containing only ppRb(S807) (arrows), or only phosphorylated tau (arrowheads) were also marked. In adjacent sections of other hippocampal regions in AD, there is striking overlap of ppRb(S807) with PHF-1 (C, D respectively) as well as with the small number of NFT in an aged control case (E, F). Scale bar = 50 μm.
Similar to ppRb(S807), antibodies specific for other five sites of phosphorylation along the pRb molecule examined in this study also showed distinct and strong labeling of pathological structures. A series of adjacent serial sections demonstrates the pattern of localization and the comparison of different epitopes within the same structures. Antisera specific for the C-terminal phosphorylation sites S811 (Figure 5A) and T821 (Figure 5B) localize to NFT, dystrophic neurites, neuropil threads and GVD. Most of the same neurons, marked by arrows, contain pRb phosphorylated at both S811 and T821. In comparison, while the ppRb(S249T252) epitope (Figure 5C) is present in some NFT and dystrophic neurites, the amount of overlap within the same cells as epitope ppRb(S811) on adjacent serial sections (Figure 5D), is not as apparent. Consistently, ppRb(S807) was found within many more pathological structures than these other epitopes. Adjacent sections show virtually all NFT stained for ppRb(S811) (Figure 5F) also contain ppRb(S807) (Figure 5E), with a greater number of NFT and neuropil threads containing ppRb(S807) only.
Figure 5.
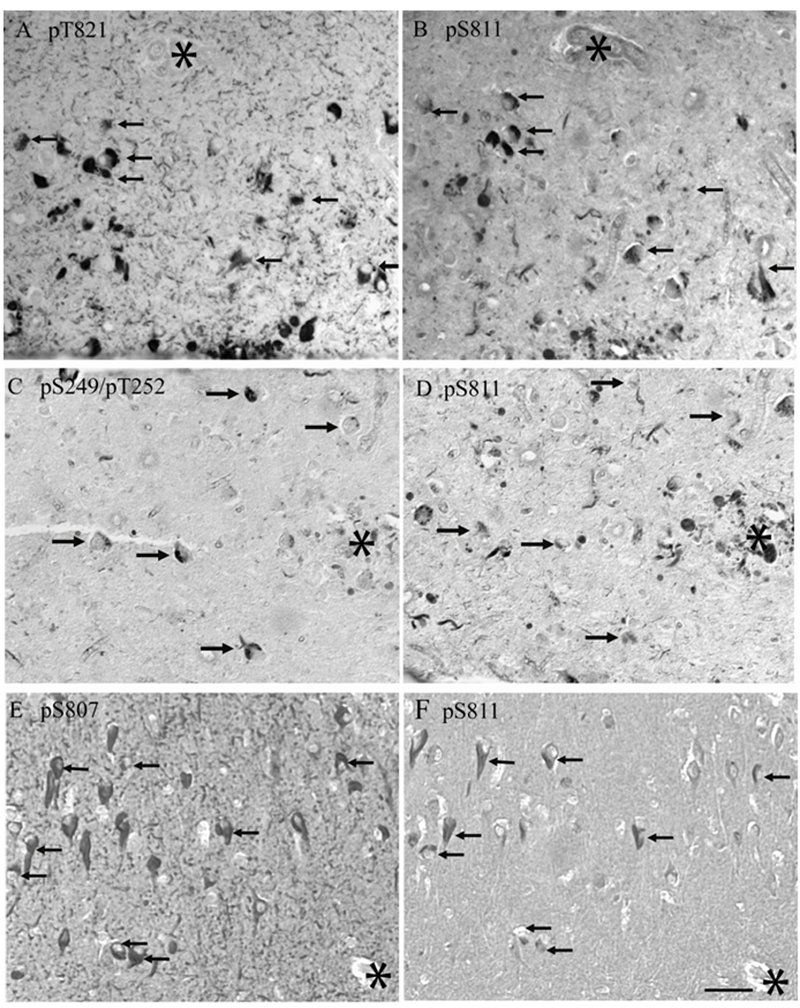
Adjacent serial sections from different cases of AD show the relationship among various phosphorylation sites within the same pathological structures. A, B. Antisera specific for ppRb(S811) and ppRb(T821) recognized pathology in most of the same neurons, arrows. In another case, an antibody recognizing ppRb(S249T 252) (C) is present in NFT which also contain ppRb(S811) (arrows), while additional structures contain ppRb(S811) only (D). In a third case of AD, ppRb(S807) (E) is present in all, and many other NFT, recognized by anti-ppRb(S811) (F). Scale bar = 50 μM.
To visualize the pRb epitopes in nuclei more readily, some tissue sections were pretreated with trypsin. Neuronal nuclear localization was seen in both control and AD cases with antisera specific for ppRb(S807) (shown in Figure 6), ppRb(S249T252) and ppRb(T821) (not shown). Further distinguishing the AD and control cases was the pattern of nuclear localization in the CA1- CA4 areas of the hippocampus and entorhinal cortex as shown in Figure 6, which shows representative areas for the CA1, CA2, and CA3 area from the same tissue sections. Consistently, in some control cases with some pathology, many pyramidal neurons throughout the CA1-CA4 readily demonstrated nuclear staining for ppRb(S807) (Figure 6A, B, C). However, in most cases of AD, the nuclear localization predominantly seen in the CA3 (Figure 6D), was gradually replaced by in a systematic way by examining seven of the sixteen potential phosphorylation sites and found that all these phosphorylation sites demonstrated disease-specific alterations: 1) NFT localization in the CA2 (Figure 6E), and finally, in the CA1 (Figure 6F), only NFTs were positive, while the nuclei were left virtually unstained. Nuclear membranes were prominently labeled in some cases of AD by antisera specific for ppRb(S807) and ppRb(T821) (Figure 7A). Also, in addition to NFT, dystrophic neurites and neuropil threads, GVD were readily detected by antisera against ppRb(S612) (Figure 7B) and ppRb(S811) (Figure 7A and inset).
Figure 6.
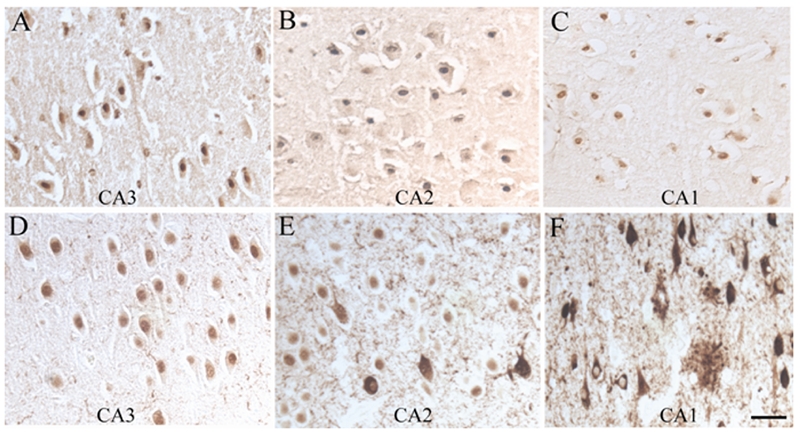
Nuclear localization of ppRb is readily detected in sections of AD and control following pretreatment of the tissue sections with trypsin. In this figure, antisera to ppRb(S807) recognize nuclei in pyramidal neurons in a section from a control case, throughout the CA3, CA2 and CA1 regions (A, B, C). Conversely, in a single section from a representative AD case, pyramidal neuronal nuclei are positive in the CA3 area (D), while nuclei and a few NFT stain in the CA2 (E), and pathological structures are positive in the CA1, while the nuclei remain unstained (F). Scale bar = 50 μm.
Figure 7.
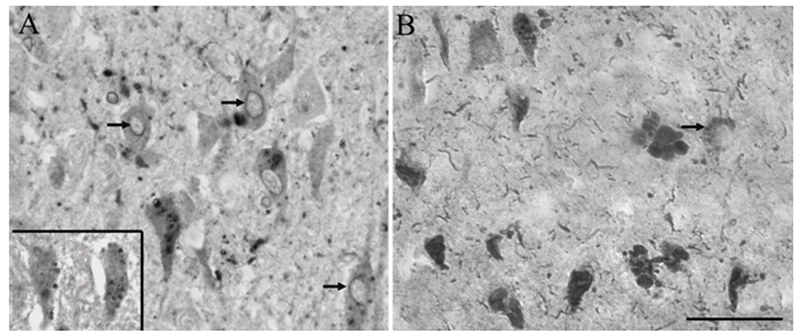
Nuclear membranes are often found to contain both ppRb(S807) (A, arrows) and ppRb(T821) (not shown). Also, GVD are also found to contain ppRb, most readily detected with antisera to ppRb(S612) (B, arrows) and ppRb(S811) (A, inset). Scale bar = 50 μm.
Since p38 can phosphorylate pRb and a prior study demonstrated that pp38 does not colocalize with ppRb(S795) in AD [41], we sought to determine whether pp38 colocalized with other phospho-pRb epitopes. Indeed, immunostaining of adjacent sections with pp38 and ppRb(S807) revealed significant colocalization of the two proteins in the same neurons (Figure 8 A and B respectively). Specifically, some neurons positive for ppRb(S807) showed pp38 in the NFT or in GVD structures. Following trypsin pretreatment, some nuclei also showed pp38 immunoreactivity.
Figure 8.
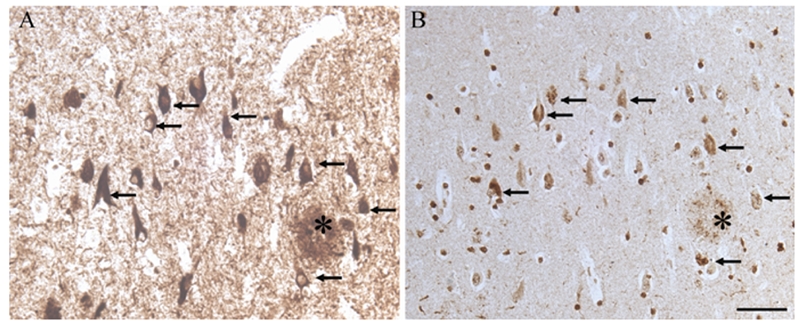
On adjacent serial sections, many NFT with ppRb(S807) (A) also contain phospho-p38, in either the NFT or GVD structures (B) (arrows). Scale bar = 50 μm.
Discussion
Prior studies demonstrated that Aβ induces pRb phosphorylation at Ser795 in neuronal cells and ppRb(S795) showed increased nuclear immunoreactivity in AD neurons [34, 33]. However, pRb contains multiple phosphorylation sites and its binding to E2F and regulation of cell cycle progression is regulated by phosphorylation of these sites in a cumulative manner [42]. To substantiate the potential involvement of pRb in neuronal cell cycle abnormalities in AD, it is essential to evaluate whether pRb is indeed hyperphosphorylated, i.e., phosphorylation at multiple sites, in AD neurons. In this study, we determined the phosphorylation status of pRb phosphorylation at all of these sites is increased in AD; 2) increased phosphorylation at six sites examined (i.e., pSpT249/252, pS612, pS807, pS811 and pT821) spanning the majority of the sequence is associated with AD pathologies; 3) increased phosphorylation at multiple sites is present in the same pyramidal neuron; 4) although neuronal nuclei staining is noted for five of these sites (i.e., pSpT249/252, pS795, pS807, and pT821) in both AD and control cases, there is a disease specific re-distribution of these phosphoepitopes in AD cases with a gradual replacement by cytoplasmic/pathological staining in the order of CA3-CA2-CA1. Based on these findings, we concluded that pRb is likely hyperphosphorylated and redistributed from the nucleus to cytoplasm in vulnerable neurons in AD. Since these neurons also demonstrate abnormal cell cycle, it is likely that aberrant pRb signaling plays an important role in this process in AD.
As a key G1/S regulator, the ability of pRb to control the cell cycle is largely attributed to repression of E2F transcription factors. A/B pocket and additional C-terminal sequences of pRb protein (amino acids 379–870, termed the ‘larger pocket’) are required in binding of E2F and to confer growth suppression through pRb/E2F interaction [43]. Phosphorylation/dephosphorylation at the 16 potential phosphorylation sites plays a specific regulatory role of pRb binding to E2F: hypophosphorylated pRb binds and repress E2F activity while hyperphosphorylated pRb releases E2F and enables cell cycle progression. Interestingly, two-dimensional phospho-peptide analysis of in vivo hypophosphorylated pRb showed that 13 of the 16 phosphorylation sites are occupied, suggesting that hypophosphorylated pRb is comprised of a complex mixture of multiple phospho-isoforms containing a limited number of phosphates per molecule of pRB [35, 29]. Nevertheless, this same study also suggested the nuclear-bound hypophosphorylated pRb lacks phosphorylation at three specific sites (pT252, pT356 and pT373) compared to hyperphosphorylated pRb [35]. Another group demonstrated the binding of pRb to free E2F is regulated by dual mechanisms involving phosphorylation either at a number of C-terminal sites or at two serines in the spacer domain of pRb [36]. Overall, these phosphorylation sites are used to both activate pRb by hypophosphorylation and inactivate it by hyperphosphorylation, dependent upon the phosphate stoichiometry and combination of locations. Therefore, it is insufficient to evaluate pRb signaling by examining the phosphorylation status of only one phosphorylation site. Thus, we determined the phosphorylation status of pRb at seven of the sixteen potential phosphorylation sites: two in the N-terminus (i.e., pS249, pT252), one in the spacer region (pS612) and four in the C-terminus (pS795, pS807, pS811 and pS821), combination of most of these sites are implicated in pRb binding to E2F in prior studies [35, 36].
As previously reported [33], ppRb(S795) demonstrated increased nuclear staining in some AD cases compared to age-matched control cases. However, we also noted that more than half of the AD cases only demonstrated elevated cytoplasmic staining of ppRb(S795). In fact, increased cytoplasmic staining in AD neurons was also noted in the pRb phosphorylated at the other four sites (i.e., pSpT249/252, pS807, and pT821), suggesting that this is a general phenomenon or these sites are phosphorylated on the same molecules. Importantly, although the prior study only found a minor portion of ppRb(S795) was associated with NFTs which was also confirmed in this study, we found that pRb phosphorylated at the other six sites (i.e., pSpT249/252, pS612, pS807, and pT821) also demonstrated increased immunoreactivity in AD neurons and more significantly, they are associated with various neurofibrillary pathologies. Given that these seven phosphorylation sites spread throughout the entire sequence, our finding indicates that increased phosphorylation occurs throughout the entire sequence of pRb protein in AD neurons. Moreover, we also demonstrated that pRb phosphorylated at multiple sites is present in the same pyramidal neurons. Although the possibility that these observations are due to the increase in hypophosphorylated pRb mixture of multiple phospho-isoforms containing phosphates on each of these sites can not be ruled out, it is more likely that these findings suggested pRb is hyperphosphorylated in susceptible neurons in AD because 1) hypophosphorylated pRb lacks phosphorylation at three specific sites including Thr252 [35]. Our study suggested increased phosphorylation at Thr252 in AD neurons; 2) hypophosphorylated pRb is tightly bound to the nucleus while hyperphosphorylated form loosely binds to nucleus [44]. Indeed, changes in the subcellular distribution of pRb from nucleus to cytoplasm accompanied by alterations in its phosphorylation state as well as by cytoplasmic re-localization of E2F was previously reported in neurons in encephalitic midfrontal cortex [45]. Therefore, the redistribution of various ppRbs from the nucleus to the cytoplasm in our study suggests that these ppRbs are likely hyperphosphorylated forms. Because pRb hyperphosphorylation easily translates into E2F release and cell cycle progression in normal cells, our findings suggest aberrant pRb signaling likely contributes to the abnormal cell cycle re-entry and progression in susceptible neurons in AD.
Our study demonstrated increased pRb phosphorylation at all sites examined. It is generally accepted that pRb becomes sequentially phosphorylated by the actions of cyclin D-CDK4/6 and cyclin E-CDK2 during the G1 phase of the cell cycle [25]. Additional phosphorylation occurs by cyclin A-CDK2 during S-phase and cyclin B-cdc2 during G2/M phases [46]. Notably, increased aberrant expression of various CDKs (i.e., CdDK, CDK2, and cdc2) and cyclins (i.e., cyclin D, cyclin E and cyclin B1) were found in AD neurons, many of which are also associated with neurofibrillary pathologies [6–10, 47]. Therefore, hyperphosphorylated pRb in AD neurons may be generated by activated cyclin D-CDK4, cyclin E-CDK2 and/or cyclin B-cdc2. One key question is whether pRb becomes hyperphosphorylated in the nucleus or in the cytoplasm in these neurons. Given the presence of many nuclearly-stained neurons for multiple phospho-pRb epitope and their gradual replacement by cytoplasmic/pathology-stained neurons in the order of CA3-2-1, it is likely that pRb becomes hyperphosphorylated in the nucleus. Then the next relevant question is how ppRb translocates to the cytoplasm in these neurons. Since pRb is released from its tight nuclear tethering once hyperphosphorylated by cyclin/CDKs [44] and it also binds to various cyclins [46], the presence of these cyclin-CDKs in the cytoplasm of these neurons or neurofibrillary pathologies implicates that the phosphorylation by and binding to cyclin-CDKs may play a role in the redistribution of pRb. Some CDKs phosphorylate tau in vitro and may be involved in tau phosphorylation and neurofibrillary tangles formation in vivo [48–50]. Phosphorylation of pRb at some sites (i.e., pS807) and/or its translocation may be a relative later event compared to tangle formation since it shares more complete overlap with PHF-1, a late marker, than 12E8, an early marker for neurofibrillary pathology. Nevertheless, these likely roles of cyclin-CDKs in aberrant pRb signaling do not exclude the potential involvement of other kinases that are known to phosphorylate pRb such as ERK or p38. In fact, ERK and p38 are activated in vulnerable neurons in AD [51, 40, 52, 39, 53] and our data suggests that p38 also demonstrated significant overlap with hyperphosphorylated pRb. Given that pRb hyperphosphorylation during cell cycle is a gradual progressive process which may be even more so during the aberrant cell cycle events in AD neurons, it is very possible that p38 contributes to the phosphorylation of at least some of the phosphorylation sites. One interesting aspect of our findings is the difference in the intensity and structures immunolabeled by different phospho-pRb antibodies, especially that of ppRb(S795) compared to other ppRbs. Given our incomplete understanding of how pRb get hyperphosphorylated except for the known fact that it is a progressive process, by inference, this difference may suggest some sites get phosphorylated by specific kinases or at an earlier time point than other sites.
In conclusion, by systematic examination of phosphorylation status of pRb at seven potential phosphorylation sites, we demonstrated increased phosphorylation at all these sites and a translocation of ppRb from the nucleus to the cytoplasm in susceptible neurons in AD. ppRb demonstrated an extensive association with various neurofibrillary pathologies and pRb phosphorylated at multiple sites is present within the same AD neurons. Overall, these findings suggest that hyperphosphorylated and aberrantly translocated pRb likely contributes to the cell cycle abnormalities in AD neurons.
Acknowledgments
Work in the authors' laboratories is supported by the National Institutes of Health (AG024028), and Philip Morris USA Inc. and Philip Morris International.
References
- 1.Smith MA. Alzheimer disease. Int Rev Neurobiol. 1998;42:1–54. doi: 10.1016/s0074-7742(08)60607-8. [DOI] [PubMed] [Google Scholar]
- 2.Casadesus G, Smith MA, Zhu X, Aliev G, Cash AD, Honda K, Petersen RB, Perry G. Alzheimer disease: evidence for a central pathogenic role of iron-mediated reactive oxygen species. J Alzheimers Dis. 2004;6:165–169. doi: 10.3233/jad-2004-6208. [DOI] [PubMed] [Google Scholar]
- 3.Zhu X, Lee HG, Perry G, Smith MA. Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta. 2007;1772:494–502. doi: 10.1016/j.bbadis.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 4.Bowser R, Smith MA. Cell cycle proteins in Alzheimer's disease: plenty of wheels but no cycle. J Alzheimers Dis. 2002;4:249–254. doi: 10.3233/jad-2002-4316. [DOI] [PubMed] [Google Scholar]
- 5.Arendt T, Rodel L, Gartner U, Holzer M. Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer's disease. Neuroreport. 1996;7:3047–3049. doi: 10.1097/00001756-199611250-00050. [DOI] [PubMed] [Google Scholar]
- 6.Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer's disease? J Cell Biol. 1996;132:413–425. doi: 10.1083/jcb.132.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol. 1997;150:1933–1939. [PMC free article] [PubMed] [Google Scholar]
- 8.Nagy Z, Esiri MM, Cato AM, Smith AD. Cell cycle markers in the hippocampus in Alzheimer's disease. Acta Neuropathol (Berl) 1997;94:6–15. doi: 10.1007/s004010050665. [DOI] [PubMed] [Google Scholar]
- 9.Nagy Z, Esiri MM, Smith AD. Expression of cell division markers in the hippocampus in Alzheimer's disease and other neuro-degenerative conditions. Acta Neuropathol (Berl) 1997;93:294–300. doi: 10.1007/s004010050617. [DOI] [PubMed] [Google Scholar]
- 10.Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci. 1997;17:3588–3598. doi: 10.1523/JNEUROSCI.17-10-03588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagy Z, Esiri MM, Smith AD. The cell division cycle and the pathophysiology of Alzheimer's disease. Neuroscience. 1998;87:731–739. doi: 10.1016/s0306-4522(98)00293-0. [DOI] [PubMed] [Google Scholar]
- 12.McShea A, Wahl AF, Smith MA. Re-entry into the cell cycle: a mechanism for neuro-degeneration in Alzheimer disease. Med Hypotheses. 1999;52:525–527. doi: 10.1054/mehy.1997.0680. [DOI] [PubMed] [Google Scholar]
- 13.Raina AK, Monteiro MJ, McShea A, Smith MA. The role of cell cycle-mediated events in Alzheimer's disease. Int J Exp Pathol. 1999;80:71–76. doi: 10.1046/j.1365-2613.1999.00106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris PL, Zhu X, Pamies C, Rottkamp CA, Ghanbari HA, McShea A, Feng Y, Ferris DK, Smith MA. Neuronal polo-like kinase in Alzheimer disease indicates cell cycle changes. Neurobiol Aging. 2000;21:837–841. doi: 10.1016/s0197-4580(00)00218-9. [DOI] [PubMed] [Google Scholar]
- 15.Zhu X, Rottkamp CA, Raina AK, Brewer GJ, Ghanbari HA, Boux H, Smith MA. Neuronal CDK7 in hippocampus is related to aging and Alzheimer disease. Neurobiol Aging. 2000;21:807–813. doi: 10.1016/s0197-4580(00)00217-7. [DOI] [PubMed] [Google Scholar]
- 16.Ogawa O, Lee HG, Zhu X, Raina A, Harris PL, Castellani RJ, Perry G, Smith MA. Increased p27, an essential component of cell cycle control, in Alzheimer's disease. Aging Cell. 2003;2:105–110. doi: 10.1046/j.1474-9728.2003.00042.x. [DOI] [PubMed] [Google Scholar]
- 17.Ogawa O, Zhu X, Lee HG, Raina A, Obrenovich ME, Bowser R, Ghanbari HA, Castellani RJ, Perry G, Smith MA. Ectopic localization of phosphorylated histone H3 in Alzheimer's disease: a mitotic catastrophe? Acta Neuropathol (Berl) 2003;105:524–528. doi: 10.1007/s00401-003-0684-3. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J Neurosci. 2003;23:2557–2563. doi: 10.1523/JNEUROSCI.23-07-02557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marlatt M, Lee HG, Perry G, Smith MA, Zhu X. Sources and mechanisms of cytoplasmic oxidative damage in Alzheimer's disease. Acta Neurobiol Exp (Wars) 2004;64:81–87. doi: 10.55782/ane-2004-1493. [DOI] [PubMed] [Google Scholar]
- 20.Evans TA, Raina AK, Delacourte A, Aprelikova O, Lee HG, Zhu X, Perry G, Smith MA. BRCA1 may modulate neuronal cell cycle re-entry in Alzheimer disease. Int J Med Sci. 2007;4:140–145. doi: 10.7150/ijms.4.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Previll LA, Crosby ME, Castellani RJ, Bowser R, Perry G, Smith MA, Zhu X. Increased expression of p130 in Alzheimer disease. Neurochem Res. 2007;32:639–644. doi: 10.1007/s11064-006-9146-3. [DOI] [PubMed] [Google Scholar]
- 22.Ma D, Zhou P, Harbour JW. Distinct mechanisms for regulating the tumor suppressor and antiapoptotic functions of Rb. J Biol Chem. 2003;278:19358–19366. doi: 10.1074/jbc.M301761200. [DOI] [PubMed] [Google Scholar]
- 23.Harbour JW, Dean DC. Rb function in cell-cycle regulation and apoptosis. Nat Cell Biol. 2000;2:E65–67. doi: 10.1038/35008695. [DOI] [PubMed] [Google Scholar]
- 24.Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24:2796–2809. doi: 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- 25.Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998;18:753–761. doi: 10.1128/mcb.18.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu L, van den Heuvel S, Helin K, Fattaey A, Ewen M, Livingston D, Dyson N, Harlow E. Inhibition of cell proliferation by p107, a relative of the retinoblastoma protein. Genes Dev. 1993;7:1111–1125. doi: 10.1101/gad.7.7a.1111. [DOI] [PubMed] [Google Scholar]
- 27.Ranganathan S, Scudiere S, Bowser R. Hyperphosphorylation of the retinoblastoma gene product and altered subcellular distribution of E2F-1 during Alzheimer's disease and amyotrophic lateral sclerosis. J Alzheimers Dis. 2001;3:377–385. doi: 10.3233/jad-2001-3403. [DOI] [PubMed] [Google Scholar]
- 28.Mittnacht S. Control of pRB phosphorylation. Curr Opin Genet Dev. 1998;8:21–27. doi: 10.1016/s0959-437x(98)80057-9. [DOI] [PubMed] [Google Scholar]
- 29.Ezhevsky SA, Ho A, Becker-Hapak M, Davis PK, Dowdy SF. Differential regulation of retinoblastoma tumor suppressor protein by G(1) cyclin-dependent kinase complexes in vivo. Mol Cell Biol. 2001;21:4773–4784. doi: 10.1128/MCB.21.14.4773-4784.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo J, Sheng G, Warner BW. Epidermal growth factor-induced rapid retinoblastoma phosphorylation at Ser780 and Ser795 is mediated by ERK1/2 in small intestine epithelial cells. J Biol Chem. 2005;280:35992–35998. doi: 10.1074/jbc.M504583200. [DOI] [PubMed] [Google Scholar]
- 31.Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2F transcription factor is a cellular target for the RB protein. Cell. 1991;65:1053–1061. doi: 10.1016/0092-8674(91)90557-f. [DOI] [PubMed] [Google Scholar]
- 32.Israels ED, Israels LG. The cell cycle. Stem Cells. 2001;19:88–91. doi: 10.1634/stemcells.19-1-88. [DOI] [PubMed] [Google Scholar]
- 33.Jordan-Sciutto KL, Malaiyandi LM, Bowser R. Altered distribution of cell cycle transcriptional regulators during Alzheimer disease. J Neuropathol Exp Neurol. 2002;61:358–367. doi: 10.1093/jnen/61.4.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jordan-Sciutto K, Rhodes J, Bowser R. Altered subcellular distribution of transcriptional regulators in response to Abeta peptide and during Alzheimer's disease. Mech Ageing Dev. 2001;123:11–20. doi: 10.1016/s0047-6374(01)00334-7. [DOI] [PubMed] [Google Scholar]
- 35.Mittnacht S, Lees JA, Desai D, Harlow E, Morgan DO, Weinberg RA. Distinct sub-populations of the retinoblastoma protein show a distinct pattern of phosphorylation. EMBO J. 1994;13:118–127. doi: 10.1002/j.1460-2075.1994.tb06241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knudsen ES, Wang JY. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol Cell Biol. 1997;17:5771–5783. doi: 10.1128/mcb.17.10.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khachaturian ZS. Diagnosis of Alzheimer's disease. Arch Neurol. 1985;42:1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 38.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 39.Zhu X, Rottkamp CA, Hartzler A, Sun Z, Takeda A, Boux H, Shimohama S, Perry G, Smith MA. Activation of MKK6, an upstream activator of p38, in Alzheimer's disease. J Neurochem. 2001;79:311–318. doi: 10.1046/j.1471-4159.2001.00597.x. [DOI] [PubMed] [Google Scholar]
- 40.Zhu X, Rottkamp CA, Boux H, Takeda A, Perry G, Smith MA. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol. 2000;59:880–888. doi: 10.1093/jnen/59.10.880. [DOI] [PubMed] [Google Scholar]
- 41.Hoozemans JJ, Veerhuis R, Rozemuller AJ, Arendt T, Eikelenboom P. Neuronal COX-2 expression and phosphorylation of pRb precede p38 MAPK activation and neurofibrillary changes in AD temporal cortex. Neurobiol Dis. 2004;15:492–499. doi: 10.1016/j.nbd.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 42.Brown VD, Phillips RA, Gallie BL. Cumulative effect of phosphorylation of pRB on regulation of E2F activity. Mol Cell Biol. 1999;19:3246–3256. doi: 10.1128/mcb.19.5.3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hiebert SW. Regions of the retinoblastoma gene product required for its interaction with the E2F transcription factor are necessary for E2 promoter repression and pRb-mediated growth suppression. Mol Cell Biol. 1993;13:3384–3391. doi: 10.1128/mcb.13.6.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mittnacht S, Weinberg RA. G1/S phosphorylation of the retinoblastoma protein is associated with an altered affinity for the nuclear compartment. Cell. 1991;65:381–393. doi: 10.1016/0092-8674(91)90456-9. [DOI] [PubMed] [Google Scholar]
- 45.Jordan-Sciutto KL, Wang G, Murphy-Corb M, Wiley CA. Induction of cell-cycle regulators in simian immunodeficiency virus encephalitis. Am J Pathol. 2000;157:497–507. doi: 10.1016/S0002-9440(10)64561-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herwig S, Strauss M. The retinoblastoma protein: a master regulator of cell cycle, differentiation and apoptosis. Eur J Biochem. 1997;246:581–601. doi: 10.1111/j.1432-1033.1997.t01-2-00581.x. [DOI] [PubMed] [Google Scholar]
- 47.Zhu X, McShea A, Harris PL, Raina AK, Castellani RJ, Funk JO, Shah S, Atwood C, Bowen R, Bowser R, Morelli L, Perry G, Smith MA. Elevated expression of a regulator of the G2/M phase of the cell cycle, neuronal CIP-1-associated regulator of cyclin B, in Alzheimer's disease. J Neurosci Res. 2004;75:698–703. doi: 10.1002/jnr.20028. [DOI] [PubMed] [Google Scholar]
- 48.Paudel HK. Phosphorylation by neuronal cdc2-like protein kinase promotes dimerization of Tau protein in vitro. J Biol Chem. 1997;272:28328–28334. doi: 10.1074/jbc.272.45.28328. [DOI] [PubMed] [Google Scholar]
- 49.Lee KY, Clark AW, Rosales JL, Chapman K, Fung T, Johnston RN. Elevated neuronal Cdc2-like kinase activity in the Alzheimer disease brain. Neurosci Res. 1999;34:21–29. doi: 10.1016/s0168-0102(99)00026-7. [DOI] [PubMed] [Google Scholar]
- 50.Pei JJ, Braak H, Gong CX, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Up-regulation of cell division cycle (cdc) 2 kinase in neurons with early stage Alzheimer's disease neurofibrillary degeneration. Acta Neuropathol (Berl) 2002;104:369–376. doi: 10.1007/s00401-002-0565-1. [DOI] [PubMed] [Google Scholar]
- 51.Perry G, Roder H, Nunomura A, Takeda A, Friedlich AL, Zhu X, Raina AK, Holbrook N, Siedlak SL, Harris PL, Smith MA. Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. Neuroreport. 1999;10:2411–2415. doi: 10.1097/00001756-199908020-00035. [DOI] [PubMed] [Google Scholar]
- 52.Zhu X, Castellani RJ, Takeda A, Nunomura A, Atwood CS, Perry G, Smith MA. Differential activation of neuronal ERK, JNK/SAPK and p38 in Alzheimer disease: the ‘two hit’ hypothesis. Mech Ageing Dev. 2001;123:39–46. doi: 10.1016/s0047-6374(01)00342-6. [DOI] [PubMed] [Google Scholar]
- 53.Zhu X, Sun Z, Lee HG, Siedlak SL, Perry G, Smith MA. Distribution, levels, and activation of MEK1 in Alzheimer's disease. J Neurochem. 2003;86:136–142. doi: 10.1046/j.1471-4159.2003.01820.x. [DOI] [PubMed] [Google Scholar]