

Abstract
Alzheimer's disease and Fragile X syndrome both display synaptic phenotypes, and based on recent studies, likely share dendritic over expression of amyloid precursor protein (APP) and β-amyloid (Aβ). In order to create a mouse model to specifically study the effects of APP and Aβ at synapses, we crossed Tg2576, which over-express human APP with the Swedish mutation (hAPPsw), with fmr-1 KO mice. The progeny, named FRAXAD, displayed increased mortality (23% by 30 days of age) compared to Tg2576 (3%) and WT and fmr-1 KO littermate controls (0%) consistent with a developmental defect. By 60 days of age, both the Tg2576 and FRAXAD mice approached a 40% mortality rate compared to 0% for WT and fmr-1 KO littermates. To understand the mechanism underlying increased mortality in APP over-expressing mice, we assessed seizure thresholds in response to pentylenetetrazol (PTZ). Both the Tg2576 and FRAXAD mice had a lower threshold to PTZ-induced seizures (average seizure score of ≥4.0) in comparison to nontransgenic littermates (average seizure score 1.9–2.9). Seizures are a major phenotype of AD, FXS, Down syndrome, autism and epilepsy, and these data suggested that developmental over-expression of dendritic APP or Aβ increased seizure susceptibility.
Keywords: amyloid, amyloid precursor protein, Fragile X mental retardation protein, FRAXAD, seizure, synapse
Introduction
Beta-amyloid (Aβ), which is generated by β- and γ-secretase cleavage of amyloid precursor protein (APP), is the predominant protein found in senile plaques in Alzheimer's disease (AD). We have previously demonstrated that APP, Aβ40 and Aβ42 are up-regulated in a mouse model for Fragile X syndrome (FXS) [1]. Fragile X mental retardation protein (FMRP), which is absent in FXS and in fmr-1 KO mice, binds to and represses the dendritic translation of APP mRNA. Activation of group 1 metabotropic glutamate receptors (mGluRs) dislodges FMRP from APP mRNA thereby activating its translation and increasing APP accumulation. Excess APP is processed to Aβ40/42 with fmr-1 KO brain having 1.6–2.5-fold more than WT. Increasing evidence in the literature suggests that APP plays an important role in synapse formation while Aβ contributes to synaptic demise.
While Aβ toxicity likely contributes to neurodegeneration in AD and Down syndrome (DS), reciprocally reduced soluble APP (sAPP) may also play a role. sAPP is normally neuroprotective and its loss disrupts calcium homeostasis and increases neuronal excitability [2]. sAPP metabolites regulate cell proliferation, proteases, blood coagulation, cell adhesion, neuronal survival, glutamate responses, neurite outgrowth, intracellular calcium levels and cytokine release as well as protect against exocitotoxic insults [2]. There are decreased levels of sAPP in the cerebrospinal fluid (CSF) of sporadic AD patients [3] as well as in AD patients with the inherited Swedish APP670/671 mutation [4].
Consistent with these in vitro data, patients with AD and DS have a high prevalence of seizures, 10% and 8% respectively, compared to 1% for the general population [5–7]. In FXS, patients exhibit hyperarousal, hyperactivity, autism, aggression, anxiety, increased sensitivity to sensory stimuli as well as seizures [8]. We hypothesized that a common pathway involving APP, or one of its metabolites, was responsible for the lower seizure thresholds found in these neurological disorders.
There are several AD mouse models that exhibit enhanced seizure activity or facilitated seizure induction in response to chemical convulsants. APP23 mice over-express hAPP751sw under the control of the neuron-specific Thy1 promoter and exhibit myoclonic jumping and seizure activity [9]. CRND8 transgenic mice over-express the Swedish and Indiana familial AD mutations in APP695 (K670N/M671L and V717F). These mutations are located near the β- and γ-secretase sites resulting in elevated levels of Aβ and amyloid plaques and both pre- and post-plaque mice exhibit a lower threshold to PTZ-induced seizures [10]. Transgenic mice that over-express a double mutation in the α-secretase recognition site [R609D and K612E] in the Aβ region of mouse APP695 are hyperactive, exhibit seizures and die prematurely [11]. There was a 69% mortality rate by one year of age with the first death occurring at 30 days. Mice homozygous for a targeted mutation in exon 2 of the APP gene (β-APPΔ/Δ), as well as those having deletion of the entire gene, exhibit enhanced kainate-induced seizures indicating that deficiency of sAPP facilitates seizure activity [12]. BACE1−/−PDAPP− and BACE1−/−/PDAPP+ mice display spontaneous tonic-clonic seizures [13]. Thus, mice that over- and under-express APP and its metabolites display increased incidence or susceptibility to seizures.
Tg2576 mice over-express human APP695 (hAPPsw) with the Swedish familial AD mutation and exhibit exacerbated Aβ production and deposition [14], however, there are no previous reports that this strain displays spontaneous seizures or an increased susceptibility to chemically-induced seizures. Herein, we report that Tg2576 mice displayed spontaneous seizures, increased mortality and lower thresholds to PTZ-induced seizures. We have also generated a novel AD mouse that over-expresses hAPPsw in an fmr-1 background, FRAXAD mice. FRAXAD mice displayed similar phenotypes as the Tg2576 albeit they have increased mortality at juvenile ages indicative of neurodevelopmental defect. Our data supports a model whereby over-expression of APP or one of its proteolytic products contributes to seizure activity in neurodegenerative disorders.
Materials and Methods
Mouse Husbandry
The WT, fmr-1 KO, Tg2576 and FRAXAD mice were housed 1–4 per microisolator cage on a 6am-6pm light cycle with ad libitum access to food (Purina 5015 mouse diet) and water. The cages contained seeds and a neslet as the only sources of environmental enrichment and were changed once per week. All husbandry and euthanasia procedures were performed in accordance with NIH and an approved University of Wisconsin Madison animal care protocol through the Research Animal Resources Center. Genotypes were determined by PCR analysis of DNA extracted from tail biopsies taken at the time of weaning (postnatal day 18–21).
Generation of the FRAXAD Strain, Breeding Strategy and Official Jackson Laboratory Nomenclature
Tg2576 males in a C57BL/6J background were crossed with fmr-1 KO females in a C57BL/6 background to produce first generation FRAXAD male mice. For subsequent backcrosses, both male and female Tg2576 and FRAXAD mice were bred with WT and fmr-1 KO C57BL/6 mice, respectively.
The Tg2576 and FRAXAD males are aggressive resulting in fighting and injury to cagemates. For breeding purposes, the males were co-housed with mates soon after genotyping to circumvent the aggressive behavior. When multiple males were co-housed, there was a high incidence of anal injury to the non-transgenic littermates. The official nomenclature for the FRAXAD mice as recommended by Jackson Laboratories is: B6.Cg.Fmr1<tm1Cgr>Tg(APPSWE)2576Kahs/Cjw.
APP/APPα and Aβ ELISA Analyses
APP/APPα and Aβ40/42 levels in 14–16 day old Tg2576 and FRAXAD whole brain homogenates and synaptoneurosomes (SNs) were determined by sandwich ELISAs per BioSource recommendations http://www.invitrogen.com (catalog #KHB0051 for APP/APPα, catalog #KHB3482 for Aβ40 and catalog # KHB3442 for Aβ42). Whole homogenates and SNs were prepared as previously described [1]. Protein concentrations were determined by BCA assay (Pierce, http://www.piercnet.com). Whole homogenates were solubilized in 6.4M GnHCl for 4hr at room temperature with mixing, diluted 80-fold with ELISA diluent buffer containing protease inhibitor cocktail, and spun at 16,000 g for 20 min at 4°C. This dilution reduces the GnHCl concentration below 0.1M to avoid interference with the ELISA assay. The cleared GnHCl lysates were assayed at a 1:800 dilution for APP/APPα and 1:500 for Aβ40/42. SNs were frozen/thawed and assayed at a 1:10 dilution for APP/APPα or undiluted for Aβ40/42. APP/APPα and Aβ40/42 levels were quantified based upon standard curves run on the same ELISA plates. APP/APPα levels were expressed as ng/mL APP/APPα per μg of brain homogenate or SN analyzed. Aβ40 levels were expressed as pg/mL Aβ40 per μg of brain homogenate or SN analyzed.
Seizure Induction and Scoring
Mice (8–10 weeks old) were acclimated to the testing room, weighed and administered 50 mg/kg PTZ [Sigma catalog #P6500] via intraperitoneal (IP) injection. Latency time to seizure was assessed by two observers and based on a five-point scale (Supplementary Table 1). Mice were monitored for 60 min and those that did not die from the seizure induction were culled.
Results
We crossed Tg2576 mice, which over-express hAPPsw, with fmr-1 KO mice, which lack FMRP. Our goal was to generate a novel mouse model for AD research that specifically over-expressed hAPPsw and Aβ at synapses. The progeny, named FRAXAD, generated 23% more Aβ40 compared to Tg2576 in GnHCl-solublized, whole brain lysates prepared from 14–16 day-old mice (Figure 1A; p<0.03) and equivalent Aβ40 in SN samples (Figure 1B). APP/APPα levels appeared slightly higher in Tg2576 mice in both whole homogenates and SNs, but were not statistically different from the FRAXAD (Figure 1C, D). Aβ42 levels were below/at the detection limits of the ELISA assay (30 pg/mL). Thus, this data demonstrates that FRAXAD mice, which over-express hAPPsw in an fmr-1 KO background, produce significantly more Aβ40 by 2 weeks of age than Tg2576.
Figure 1.

FRAXAD mice generated more Aβ40 than Tg2576. GnHCl-soluble brain lysates (A and C) and SN (B and D) from 14–16 day-old Tg2576 and FRAXAD mice were analyzed by ELISA for Aβ40 (A and B) and APP/APPα (C and D). Each cohort (n=6) from backcrosses n7-n9 consisted of 3 female and 3 male mice. The difference between FRAXAD (13,571 ± 666 pg/mL/μg) and Tg2576 (11,060 ± 696 pg/mL/μg) Aβ40 levels in brain homogenates was statistically significant, Student T-test: p<0.03. The difference between FRAXAD (3.63 ± 0.24 pg/mL/μg) and Tg2576 (3.40 ± 0.39 pg/mL/μg) Aβ40 levels in SNs was not statistically significant, Student T-test: p=0.6. The difference between FRAXAD (25,897 ± 2047 ng/mL/μg) and Tg2576 (28,962 ± 2875 ng/mL/μg) APP/APPα levels in brain homogenate was not statistically significant, Student T-test: p=0.4. The difference between FRAXAD (80.64 ± 11.14 ng/mL/μg) and Tg2576 (97.45 ± 10.86 ng/mL/μg) APP/APPα levels in SNs was not statistically significant, Student T-test: p=0.3.
FRAXAD mice were viable and fertile although we did observe increased juvenile mortality. Longevity studies revealed a 23% death rate in the FRAXAD mice by 30 days of age compared to 3% for the Tg2576 (Figure 2). By 60 days of age, both strains approached a 40% death rate. The WT and fmr-1 KO littermates had a 100% survival during this timeframe. The survival of the Tg2576 and FRAXAD was similar from 54 days out to one year (Supplementary Figure 1), and overt signs of deteriorated health did not precede death. Necropsy analysis of FRAXAD mice (1–3 months old) indicated no gross morphological changes in the organs compared with WT and KO controls.
Figure 2.
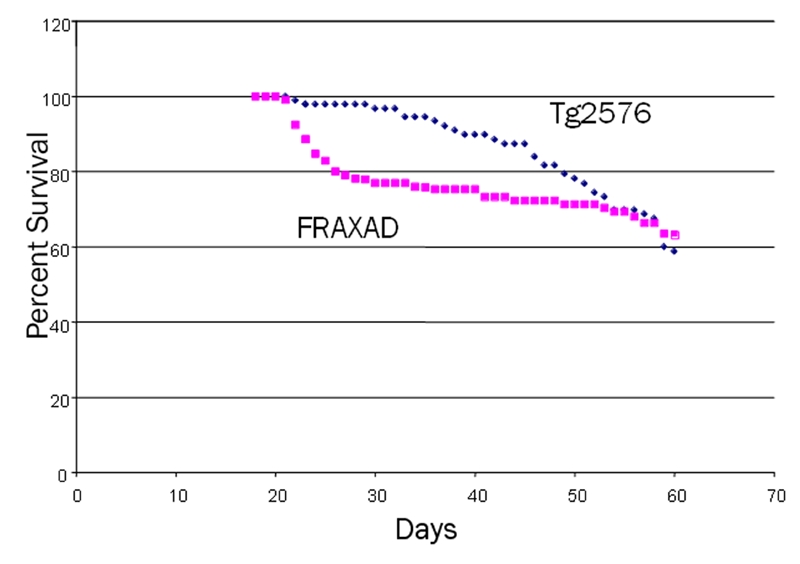
FRAXAD mice exhibited increased juvenile mortality compared to Tg2576. Deaths for Tg2576 and FRAXAD mice were monitored over a 378-day period (backcrosses 1–6) and the mouse longevity in days was plotted against the percent of mice surviving (60 days shown, see Supplementary Figure 1 for the 378 day period). As a few mice were removed from the colony for use in experiments, the number of mice decreased during the survival time course. At the beginning of the time course (18 days), survival was based on 105 FRAXAD mice and 91 Tg2576 mice, and by the end of the time course (60 days), survival was based on 93 FRAXAD and 80 Tg2576 mice.
We observed spontaneous, repetitive behaviors in approximately 3% of the FRAXAD and Tg2576 mice. These behaviors started at about 6 months of age and were manifested as wild running or myoclonic jumping. We also observed home-cage seizure activity in a small percentage of Tg2576 and FRAXAD mice. Several transgenic mouse lines, which over-express familial mutation of APP, display tonic-clonic seizures in their home-cages, which have not been previously reported for Tg2576 mice. Thus, we surmised that a lower threshold to seizure induction could account for the increased mortality in the Tg2576 and FRAXAD mice compared to non-transgenic littermate controls. Therefore, we assessed seizure threshold after pentylenetetrazole (PTZ) injection in 8–10 week old mice. Seizures were graded on a five-point scale (Supplementary Table 1 and Supplementary Videos 1–5).
To establish a baseline for seizure scores, we assessed seizures in WT and fmr-1 KO mice in a pure C57BL/6 background (Figure 3 and Tables 1 and 2). Mice were injected with 50 mg PTZ per kg body weight intraperitoneally, an established dose that generates grade 3 seizures in WT male mice, and seizure activity was monitored for 60 min. We observed an average seizure grade of 2.2 in WT female, 3.13 in WT male, 2.67 in fmr-1 KO female and 2.9 in fmr-1 KO male mice in a pure C57BL/6 background (Figure 3, Table 1). The 0.93 increase in seizure grade between the WT male and female mice was statistically significant (p=0.015). The male and female fmr-1 KO mice had average seizure scores intermediate between the male and female WT mice and were not statistically different. In general, the female mice exhibited lower grade seizures as well as longer latency times to reach grades 1–3, albeit the latency times were not statistically different from the males (Table 2). There was a higher tendency in the female mice to “walk off” the seizures resulting in stronger, lower stage seizures, while the males tended to “freeze-up” and enter higher stage seizures. While the female mice had less severe seizures, they had longer recovery times than the males (Table 2). Overall, PTZ (50 mg/kg) induced convulsive grade 3 seizures in 80% of WT male mice and 40–50% of WT female and fmr-1 KO male and female mice without causing any deaths (Table 2).
Figure 3.
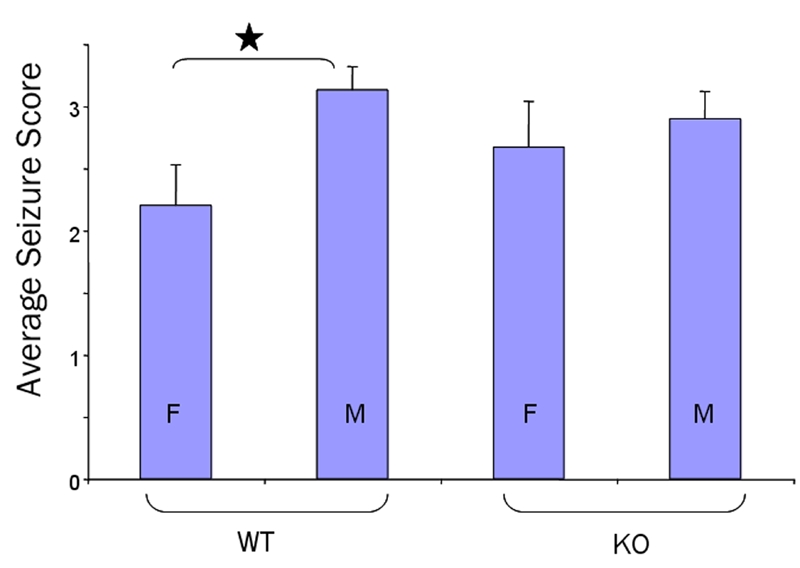
PTZ-induced seizures in male and female WT and fmr-1 KO mice in a pure C57BL/6 background. C57BL/6 mice (8–10 weeks old) were injected intraperitoneally with 50 mg PTZ per kg body weight and seizure activity was monitored for 60 min. Average seizure scores were 2.2±0.33 (WT females, n=10), 3.13±0.19 (WT males, n=15), 2.67±0.37 (fmr-1 KO females, n=9) and 2.9±0.22 (fmr-1 KO males, n=20). The 0.93 difference in seizure grade between the WT male and female mice was statistically significant (p=0.015).
Table 1.
Seizure scores in WT and fmr-1 KO mice in a pure C57BL/6 background
| Mouse | N | Avg Age (days) | Ave Weight (g) | Seizure Score |
|---|---|---|---|---|
| WT female | 10 | 65.0 ± 2.11 | 20.38 ± 0.51 | 2.20 ± 0.33 |
| WT male | 15 | 68.8 ± 1.77 | 26.01 ± 0.72 | 3.13 ± 0.19 |
| KO female | 9 | 64.9 ± 1.03 | 21.02 ± 0.68 | 2.67 ± 0.37 |
| KO male | 20 | 63.0 ± 0.53 | 26.13 ± 0.28 | 2.90 ± 0.22 |
Table 2.
Latency time to seizure in WT and fmr-1 KO in a pure C57BL/6 background
| Mouse | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Recovery (min) | % Mice Grade 3 | % Mice Grade 4 |
|---|---|---|---|---|---|---|---|
| WT Female | 124.7 ± 12.3 | 550.1 ± 86.7 | 669.0 ± 108.8 | 662.0 ± 0 | 40 ± 5 | 40 | 10 |
| n=10 | n=7 | n=4 | n=1 | n=10 | |||
| WT Male | 105.3 ± 10.5 | 445.1 ± 52.4 | 598.3 ± 62.6 | 687.8 ± 189.6 | 28 ± 4 | 80 | 33 |
| n=15 | n=15 | n=12 | n=5 | n=15 | |||
| KO Female | 129.6 ± 23.4 | 540.1 ± 198.8 | 583.5 ± 89.0 | 595.0 ± 124.5 | 43 ± 5 | 44 | 33 |
| n=9 | n=8 | n=4 | n=3 | n=8 | |||
| KO Male | 117.2 ± 9.1 | 534.7 ± 90.5 | 474.2 ± 58.3 | 510.3 ± 65.1 | 33 ± 3 | 50 | 40 |
| n=20 | n=20 | n=10 | n=8 | n=20 |
With our seizure baseline established, we assessed PTZ-induced seizures in Tg2576, FRAXAD and littermate controls. These mice were from n3-n6 backcrosses (87.5–98.4% C57BL/6). In the non-transgenic littermate controls, the average seizure scores ranged from 1.89–2.85 with trends for slightly reduced seizure thresholds in males compared to females and in fmr-1 KO compared to WT mice, albeit not statistically significant differences (WT female 1.89, WT male 2.42, KO female 2.55 and KO male 2.85) (Figure 4A, Table 1). Thus, in a mixed background, average seizure scores were slightly lower than in the pure C57BL/6 background but maintained a trend whereby male mice were somewhat more sensitive to seizure induction than females.
Figure 4.
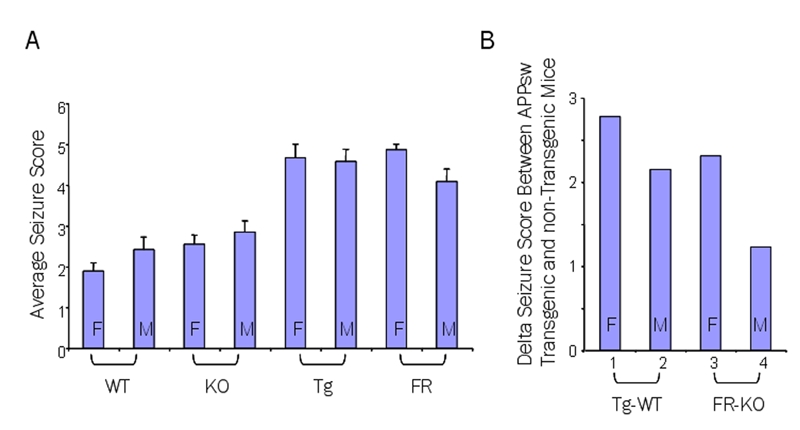
Male and female Tg2576 and FRAXAD mice had a lower threshold to PTZ-induced seizures. Mice from the n3-n6 backcrosses (87.5–98.4% C57BL/6) were injected with 50 mg/kg and (A) mouse strain was plotted against the average seizure score: 1.89±0.20 (WT female, n=9), 2.42±0.31 (WT male, n=12), 2.55±0.22 (frm-1 KO female, n=20), 2.85±0.27 (frm-1 KO male, n=13), 4.67±0.33 (Tg2576 female, n=6), 4.57±0.30 (Tg2576 male, n=7), 4.86±0.14 (FRAXAD female, n=7) and 4.08±0.31 (FRAXAD male, n=12). The difference between FRAXAD males and females was statistically significant, p=0.04, as was the difference between each hAPPsw transgenic mouse and its nontransgenic littermate control, p<0.007. The delta seizure scores (B) were 2.78 (WT and Tg2576 females), 2.15 (WT and Tg2576 males), 2.31 (fmr-1 KO and FRAXAD females) and 1.23 (fmr-1 KO and FRAXAD males).
In the Tg2576 and FRAXAD mice, PTZ induced an average seizure score greater than or equal to 4 for males and females of both strains (Tg2576 female 4.67, Tg2576 male 4.57, FRAXAD female 4.86 and FRAXAD male 4.08) (Figure 4A, Table 3). Comparison of the hAPPsw-over-expressing mice (average seizure scores 4.08–4.86) to nontransgenic littermates (1.89–2.85) was statistically significant in all cases. Therefore, mice over-expressing hAPPsw, and likely proteolytic products of hAPPsw, show elevated rates of spontaneous and PTZ-induced seizures.
Table 3.
Seizure scores in Tg2576, FRAXAD and littermate controls
| Mouse | N | Avg Age (days) | Avg Weight (g) | Seizure Score |
|---|---|---|---|---|
| WT Female | 9 | 59.4 | 20.55 ± 0.71 | 1.89 ± 0.20 |
| WT Male | 12 | 60.8 | 26.80 ± 0.65 | 2.42 ± 0.31 |
| KO Female | 20 | 62.0 | 21.05 ± 0.35 | 2.55 ± 0.22 |
| KO Male | 13 | 62.8 | 27.61 ± 0.42 | 2.85 ± 0.27 |
| Tg2576 Female | 6 | 59.5 | 19.49 ± 0.59 | 4.67 ± 0.33 |
| Tg2576 Male | 7 | 61.3 | 25.98 ± 1.39 | 4.57 ± 0.30 |
| FRAXAD Female | 7 | 59.3 | 21.16 ± 0.76 | 4.86 ± 0.14 |
| FRAXAD Male | 12 | 62.2 | 25.90 ± 0.70 | 4.08 ± 0.31 |
The fmr-1 KO male mice had the highest average seizure score (2.85) of the nontransgenic controls and the FRAXAD males had the lowest average seizure score (4.08) of the hAPPsw transgenic strains. Thus, while the other hAPPsw over-expressing strains were 2+ seizure grades higher than their nontransgenic counterparts (WT and Tg2576 females Δ seizure score = 2.78, males = 2.15, fmr-1 KO and FRAXAD females = 2.31), the delta seizure score for the FRAXAD and fmr-1 KO male mice was 1.23 (Figure 4B). The percentage of mice exhibiting grades 3, 4 and 5 seizures versus strain and gender is given in Supplementary Figures 2A, 2B and 2C, respectively. The lower average seizure score for the FRAXAD males can be attributed to a substantial decrease in the number of mice exhibiting grade 5 seizures (42%) compared to the female and male Tg2576 and female FRAXAD, 83, 71 and 86%, respectively, as well as to a small decrease in the number of grade 3 seizures. However, while the FRAXAD males exhibited the lowest percentage of grade 5 seizures of the hAPPsw over-expressing mice, the mice in this group that did reach grade 5 displayed the shortest latency time to death (335 sec ± 82) (Table 4).
Table 4.
Latency time to seizure in Tg2576, FRAXAD and littermate controls
| Mouse | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Grade 5 | Recovery Time (min) | Time of Death (sec) | Mice Dead (%) |
|---|---|---|---|---|---|---|---|---|
| WT (F)** | 154.25 ± 19.39* | 660.83 ± 24.52 | 20.00 | N/A | N/A | 43 ± 4 | N/A | 0 |
| n=8 | n=6 | n=1 | n=0 | n=0 | n=9 | n=0 | ||
| WT (M) | 107.50 ± 10.81* | 399.89 ± 77.39 | 599.67 ± 60.73 | 588.50 ± 161.50 | N/A | 55 ± 8 | N/A | 0 |
| n=12 | n=9 | n=6 | n=2 | n=0 | n=12 | n=0 | ||
| KO (F) | 144.74 ± 8.63 | 340.24 ± 55.63 | 405.75 ± 53.44 | 370.80 ± 73.21 | N/A | 62 ± 4 | N/A | 0 |
| n=19 | n=17 | n=8 | n=5 | n=0 | n=20 | n=0 | ||
| KO (M) | 127.17 ± 18.51 | 345.17 ± 58.76 | 550.80 ± 97.28 | 574.40 ± 80.81 | N/A | 52 ± 5 | N/A | 0 |
| n=12 | n=12 | n=5 | n=5 | n=0 | n=13 | n=0 | ||
| Tg2576 (F) | 215.67 ± 41.57 | 363.83 ± 178.24 | 562.80 ± 235.51 | 690.60 ± 226.87 | 1022.00 ± 425.79 | 66.00 | 1077.60 ± 426.84 | 83 |
| n=3 | n=6 | n=5 | n=5 | n=5 | n=1 | n=5 | ||
| Tg2576 (M) | 174.50 ± 31.76 | 197.14 ± 31.43 | 337.29 ± 62.41 | 443.17 ± 84.07 | 430.00 ± 97.60 | 29 ± 4 | 469.60 ± 105.03 | 71 |
| n=4 | n=7 | n=7 | n=6 | n=5 | n=2 | n=5 | ||
| FRAXAD (F) | 139.25 ± 15.87 | 208.50 ± 25.59 | 336.57 ± 49.70 | 360.71 ± 58.35 | 621.50 ± 166.41* | 36.00 | 669.67 ± 155.88 | 86 |
| n=4 | n=6 | n=7 | n=7 | n=6 | n=1 | n=6 | ||
| FRAXAD (M) | 156.13 ± 15.02 | 221.80 ± 29.38 | 278.11 ± 63.83 | 313.43 ± 63.46 | 212.40 ± 69.39* | 45 ± 5 | 334.80 ± 81.51 | 42 |
| n=8 | n=10 | n=9 | n=10 | n=5 | n=7 | n=5 |
denotes statistically significant difference between the two samples.
M: male; F: female
Of note, seizure threshold was assessed at 8–10 weeks of age when the mice are sexually mature and survival of both strains is equivalent, 68–70% survival rate (8 weeks old) and 47–53% survival rate (10 weeks old). It would be difficult to assign seizure grades in mice at 29 days of age, which coincides with the maximum difference in mortality rate (19.9%) between the strains, due to the smaller body size and increased activity of juvenile mice. Hence, the mice injected with PTZ for these seizure studies were the healthiest ones in that they survived to age 8–10 weeks of age. As there was a large gender-based difference in average seizure scores between the male and female FRAXAD mice (females: 4.86 versus males: 4.08, p=0.04), we graphed the longevity data from Figure 2 based on gender (Figure 5). At 29 days of age, the death rates are: 18% (FRAXAD males), 28% (FRAXAD females) and 2% Tg2576 males and females. The FRAXAD males, which exhibit a higher tolerance to PTZ-induced seizures, also have an increased survival rate from weaning to postnatal week 10 compared with FRAXAD females.
Figure 5.
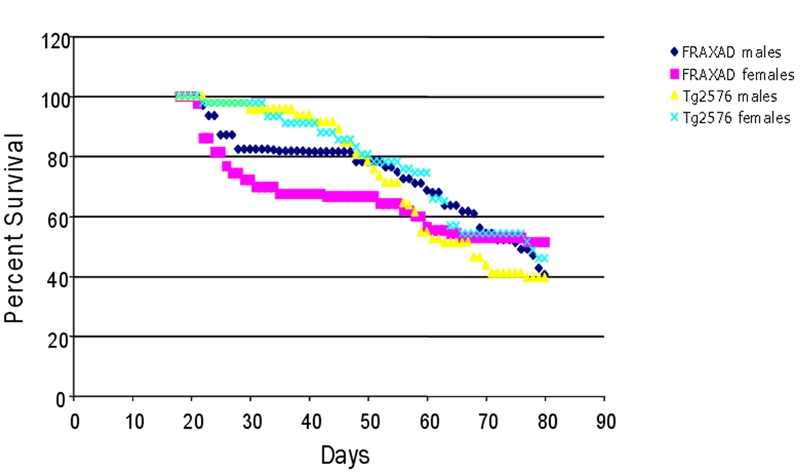
FRAXAD female mice had a higher juvenile mortality than FRAXAD males. The survival data for the mice included in Figure 1 was plotted separating the male and female mice. Tg2576 females (n=44), Tg2576 males (n=47), FRAXAD females (n=43) and FRAXAD males (n=62).
These mice were weaned at postnatal day 18. To test if the increased juvenile mortality in FRAXAD mice was due to a developmental delay that could be circumvented by extra time on the mother's milk, we also weaned mice at 21 days and assessed survival for the following 2 weeks (Table 5). Between 21 and 34 days of age, there was 0%, 14.3%, 4.5% and 9.1% mortality in FRAXAD males, FRAXAD females, Tg2576 males and Tg2576 females weaned at 21 days, respectively, compared to 18%, 27.9%, 4.3% and 6.8% for mice weaned at 18 days. Thus, three extra days on the mother's milk substantially reduced juvenile mortality in both male and female FRAXAD mice while not decreasing mortality in the Tg2576. These data suggest that (1) there is a developmental defect or delay in the FRAXAD, which is more pronounced in the female mice, and (2) the survival rate of juvenile mice weaned at 21 days is FRAXAD male > Tg2576 male > Tg2576 female > FRAXAD female, which inversely correlates with PTZ-induced seizure sensitivity in these strains (Table 3 and Figure 4A).
Table 5.
Tg2576 & FRAXAD mortality between days 21–34 after weaning
| Mouse | weaned at 18 days | weaned at 21 days |
|---|---|---|
| Tg2576 females | 6.80% | 9.10% |
| Tg2576 males | 4.30% | 4.50% |
| FRAXAD females | 27.90% | 14.30% |
| FRAXAD males | 18% | 0% |
Discussion
APP is ubiquitously expressed, but its normal physiological function is not well defined. Increasing evidence points to roles for APP in cell adhesion and synaptogenesis. APP and the related amyloid precursor-like protein 2 (APLP2) are transported to presynaptic terminals [15] while another homologue, APLP1, localizes to the postsynaptic density [16]. APP, APLP1 and APLP2 can homo- and hetero-dimerize and APP/APLP1 complexes are found in synaptic membrane preparations [17]. These data suggest that APP may act as a cell adhesion molecule (CAM) at the neuronal surface with APP and/or APLP2 on the presynaptic terminus binding to APLP1 on the postsynaptic membrane. The role of synaptic dysfunction has received increasing attention as a primary upstream lesion in early AD [18]. APP expression is increased during neuronal differentiation with maximal levels at synaptic connection completion [19–22]. APP promotes synapse differentiation at the neuromuscular junction in Drosophila [23] and increases the number of presynaptic terminals in transgenic mice [24]. siRNA targeted against APP decreased presynaptic APP/APLP2 levels and reduced synaptic activity in the rat superior colliculus [25]. Administration of anti-APP antibodies prevented memory formation in day-old chicks [26] and in rats [27]. These data suggest a critical physiological role for APP in synapse formation and maintenance.
APP presents a conundrum, as it is required early in development for synaptogenesis, yet, its over-expression in AD results in increased Aβ production and neuronal cell death. There is very limited data in the literature regarding APP mRNA and protein levels in FXS patients. There is a marked increase in APP mRNA in the cerebral cortex, hippocampus and cerebellar cortex in FXS mice [28] and a significant increase in APP at dendrites and total Aβ levels in brain [1]. APP over-expression in these mice could help explain the enhanced number of immature dendritic spines and dysregulation of spine pruning characteristic of FXS.
In order to assess the role of APP and its metabolites on synaptic function, we created a novel mouse model by crossing fmr-1 KO (mouse model for FXS) with Tg2576 mice (mouse model for AD). The progeny (named FRAXAD) produced 23% more Aβ40 by 14–16 days of age than Tg2576 and were viable and fertile with no gross morphological differences by necroscopy analyses. We observed increased juvenile mortality and a lower threshold to PTZ-induced seizures in both hAPPsw-over-expressing strains (Tg2576 and FRAXAD). At 29 days postnatal, mortality was highest in FRAXAD females (28%) compared to FRAXAD males (18%) and Tg2576 males and females (2%). We observed home-cage seizure activity in a small percentage of the Tg2576 and FRAXAD mice and surmised that a lower seizure threshold could account for the increased mortality. The PTZ seizure studies utilized mice at the n3-n6 backcross (87.5–98.4% C57BL/6). The C57BL mouse strain is more resistant to seizures and to drugs inducing seizures [29], yet, all of the hAPPsw-over-expressing mice exhibited a lower threshold to PTZ-induced seizures with an average seizure score of 4.0 or greater compared to 1.89–2.85 for nontransgenic littermates.
PTZ acts as a potent, competitive antagonist of GABAA receptors thereby inducing seizure activity [30]. Fmr-1 KO mice have a high seizure susceptibility to auditory stimulation [31–33], but a previous report indicated that PTZ elicited similar effects in WT and fmr-1 KO mice [32]. Likewise, we did not observe a statistically significant difference in PTZ-induced seizures between WT and fmr-1 KO mice; however, we observed a large and significant reduction in seizure threshold in both male and female Tg2576 and FRAXAD mice compared to nontransgenic mice. Seizures are a recurrent phenotype in many neurological disorders, including FXS and AD, as well as in several mouse models for AD and our data strongly supports a role for APP or an APP metabolite in seizure susceptibility.
In conclusion, over-expression of hAPPsw in an fmr-1 KO background (FRAXAD) increased mortality in juvenile mice. The threshold for PTZ-induced seizures in male and female Tg2576 and FRAXAD mice was significantly lower than non-transgenic littermates. Male FRAXAD mice had a better survival rate at juvenile ages and were less susceptible to PTZ-induced seizures than female FRAXAD, albeit both genders had decreased survival at juvenile ages compared to Tg2576. Our data suggests that over-production of hAPPsw, or one of its proteolytic products, contributes to altered synaptic plasticity leading to an increased incidence of seizures and death. Aβ as well as C-terminal fragments of APP are neurotoxic in vitro and in vivo as assessed by channel effects, blockage of LTP in the hippocampus, free radical generation and inflammation [34], whereas soluble APP is neuroprotective [2]. We did not observe statistically different levels of APP/APPα by ELISA in brain homogenates or SN from Tg2576 and FRAXAD mice. The ELISA assay detects full-length APP as well as soluble, neuroprotective APP cleaved by α-secretase (APPα), but does not detect soluble APP cleaved by β-secretase (APPβ). Thus, with this assay, it was not possible to discern if total APP levels were increased while APPα levels decreased in FRAXAD mice resulting in about an equivalent APP/APPα level as Tg2576. We did observe increased brain hAβ40 in FRAXAD mice compared to Tg2576 (14–16 days-old). Therefore, an increase in neurotoxic Aβ40, and possibly a decrease in neuroprotective sAPPα, during postnatal developmental likely caused the increased mortality in FRAXAD mice following weaning.
Supplementary Material
Acknowledgments
We thank Drs. Jessica Crain and Jeff Johnson in the Pharmacology Department at the UW Madison for Tg2576 mice, Dr. Wayne Frankel of Jackson Laboratories for advice on testing seizure threshold in mice, Dr. Beverly Richards-Smith, the scientific curator at Jackson Laboratories, for determining the official FRAXAD nomenclature, Dr. Craig Atwood in the Department of Medicine at the UW Madison for his suggestion to test survival after a 21 day wean time and Dr. Ruth Sullivan, a veterinary pathologist at the Waisman Center, for necroscopy analyses. This work was supported by National Institutes of Health Grants R01 AG10675 (to J.S.M.), P30 HD03352 (to the Waisman Center), the Wisconsin Comprehensive Memory Program (WCMP) and a private donation by Bill and Doris Willis.
References
- 1.Westmark CJ, Malter JS. FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 2007;5:e52. doi: 10.1371/journal.pbio.0050052. (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mattson MP, Barger SW, Cheng B, Lieberburg I, Smith-Swintosky VL, Rydel RE. Beta-amyloid precursor protein metabolites and loss of neuronal Ca2+ homeostasis in Alzheimer's disease. Trends Neurosci. 1993;16:409–414. doi: 10.1016/0166-2236(93)90009-b. [DOI] [PubMed] [Google Scholar]
- 3.Van Nostrand WE, Wagner SL, Shankle WR, Farrow JS, Dick M, Rozemuller JM, Juiper MA, Wolters EC, Zimmerman J, Cotman CW, et al. Decreased levels of soluble amyloid beta-protein precursor in cerebrospinal fluid of live Alzheimer disease patients. Proc Natl Acad Sci USA. 1992;89:2551–2555. doi: 10.1073/pnas.89.7.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lannfelt L, Basun H, Wahlund LO, Rowe BA, Wagner SL. Decreased alpha-secretase-cleaved amyloid precursor protein as a diagnostic marker for Alzheimer's disease. Nat Med. 1995;1:829–832. doi: 10.1038/nm0895-829. [DOI] [PubMed] [Google Scholar]
- 5.Velez L, Selwa LM. Seizure disorders in the elderly. Am Fam Physician. 2003;67:325–332. [PubMed] [Google Scholar]
- 6.Risse SC, Lampe TH, Bird TD, Nochlin D, Sumi SM, Keenan T, Cubberley L, Peskind E, Raskind MA. Myoclonus, seizures, and paratonia in Alzheimer disease. Alzheimer Dis Assoc Disord. 1990;4:217–225. doi: 10.1097/00002093-199040400-00003. [DOI] [PubMed] [Google Scholar]
- 7.Menendez M. Down syndrome, Alzheimer's disease and seizures. Brain Dev. 2005;27:246–252. doi: 10.1016/j.braindev.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 8.Hagerman RJ, Hagerman PJ. Physical and behavioral phenotype. Baltimore, MD: The Johns Hopkins University Press; 2002. pp. 3–109. [Google Scholar]
- 9.Lalonde R, Dumont M, Staufenbiel M, Strazielle C. Neurobehavioral characterization of APP23 transgenic mice with the SHIRPA primary screen. Behav Brain Res. 2005;157:91–98. doi: 10.1016/j.bbr.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 10.Del Vecchio RA, Gold LH, Novick SJ, Wong G, Hyde LA. Increased seizure threshold and severity in young transgenic CRND8 mice. Neurosci Lett. 2004;367:164–167. doi: 10.1016/j.neulet.2004.05.107. [DOI] [PubMed] [Google Scholar]
- 11.Moechars D, Lorent K, De Strooper B, Dewachter I, Van Leuven F. Expression in brain of amyloid precursor protein mutated in the alpha-secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. EMBO J. 1996;15:1265–1274. [PMC free article] [PubMed] [Google Scholar]
- 12.Steinbach JP, Muller U, Leist M, Li ZW, Nicotera P, Aquzzi A. Hypersensitivity to seizures in beta-amyloid precursor protein deficient mice. Cell Death Differ. 1998;5:858–866. doi: 10.1038/sj.cdd.4400391. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi D, Zeller M, Cole T, Buttini M, McConlogue L, Sinha S, Freedman S, Morris RG, Chen KS. BACE1 gene deletion: Impact on behavioral function in a model of Alzheimer's disease. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 15.Lyckman AW, Confaloni AM, Thinakaran G, Sisodia SS, Moya KL. Post-translational processing and turnover kinetics of pre-synaptically targeted amyloid precursor super-family proteins in the central nervous system. J Biol Chem. 1998;273:11100–11106. doi: 10.1074/jbc.273.18.11100. [DOI] [PubMed] [Google Scholar]
- 16.Kim TW, Wu K, Xu JL, McAuliffe G, Tanzi RE, Wasco W, Black IB. Selective localization of amyloid precursor-like protein 1 in the cerebral cortex postsynaptic density. Brain Res Mol Brain Res. 1995;32:36–44. doi: 10.1016/0169-328x(95)00328-p. [DOI] [PubMed] [Google Scholar]
- 17.Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, Kreger S, Lower A, Langer A, Merdes G, Paro R, Masters CL, Muller U, Kins S, Beyreuther K. Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 2005;24:3624–3634. doi: 10.1038/sj.emboj.7600824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coleman P, Federoff H, Kurlan R. A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology. 2004;63:1155–1162. doi: 10.1212/01.wnl.0000140626.48118.0a. [DOI] [PubMed] [Google Scholar]
- 19.Hung AY, Koo EH, Haass C, Selkoe DJ. Increased expression of beta-amyloid precursor protein during neuronal differentiation is not accompanied by secretory cleavage. Proc Natl Acad Sci USA. 1992;89:9439–9443. doi: 10.1073/pnas.89.20.9439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loffler J, Huber G. Beta-amyloid precursor protein isoforms in various rat brain regions and during brain development. J Neurochem. 1992;59:1316–1324. doi: 10.1111/j.1471-4159.1992.tb08443.x. [DOI] [PubMed] [Google Scholar]
- 21.Masliah E, Mallory M, Ge N, Saitoh T. Amyloid precursor protein is localized in growing neurites of neonatal rat brain. Brain Res. 1992;593:323–328. doi: 10.1016/0006-8993(92)91329-d. [DOI] [PubMed] [Google Scholar]
- 22.Moya KL, Benowitz LI, Schneider GE, Allinquant B. The amyloid precursor protein is developmentally regulated and correlated with synaptogenesis. Dev Biol. 1994;161:597–603. doi: 10.1006/dbio.1994.1055. [DOI] [PubMed] [Google Scholar]
- 23.Torroja L, Packard M, Gorczyca M, White K, Budnik V. The Drosophila beta-amyloid precursor protein homolog promotes synapse different-tiation at the neuromuscular junction. J Neurosci. 1999;19:7793–7803. doi: 10.1523/JNEUROSCI.19-18-07793.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mucke L, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, forss-Petter S, Pietropaolo M, Mallory M, Abraham CR. Synaptotrophic effects of human amyloid beta protein precursors in the cortex of transgenic mice. Brain Res. 1994;666:151–167. doi: 10.1016/0006-8993(94)90767-6. [DOI] [PubMed] [Google Scholar]
- 25.Herard AS, Besret L, Dubois A, Dauguet J, Delzescaux T, Hantraye P, Bonvento G, Moya KL. siRNA targeted against amyloid precursor protein impairs synaptic activity in vivo. Neurobiol Aging. 2005;27:1740–1750. doi: 10.1016/j.neurobiolaging.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 26.Mileusnic R, Lancashire CL, Rose SP. Amyloid precursor protein: From synaptic plasticity to Alzheimer's disease. Ann N Y Acad Sci. 2005;1048:149–165. doi: 10.1196/annals.1342.014. [DOI] [PubMed] [Google Scholar]
- 27.Doyle E, Bruce MT, Breen KC, Smith DC, Anderton B, Regan CM. Intraventricular infusions of antibodies to amyloid-beta-protein precursor impair the acquisition of a passive avoidance response in the rat. Neurosci Lett. 1990;115:97–102. doi: 10.1016/0304-3940(90)90524-d. [DOI] [PubMed] [Google Scholar]
- 28.D'Agata V, Warren ST, Zhao W, Torre ER, Alkon DL, Cavallaro S. Gene expression profiles in a transgenic animal model of fragile X syndrome. Neurobiol Dis. 2002;10:211–218. doi: 10.1006/nbdi.2002.0506. [DOI] [PubMed] [Google Scholar]
- 29.Kamatchi GL, Kofuji P, Wang JB, Fernando JC, Liu Z, Mathura JR, Burt DR. GABAA receptor beta 1, beta 2, and beta 3 subunits: Comparisons in DBA/2J and C57BL/6J mice. Biochim Biophys Acta. 1995;1261:134–142. doi: 10.1016/0167-4781(95)00009-6. [DOI] [PubMed] [Google Scholar]
- 30.Huang RQ, Bell-Horner CL, Dibas MI, Covey DF, Drewe JA, Dillon GH. Pentylene-tetrazole-induced inhibition of recombinant gamma-aminobutyric acid type A (GABA(A)) receptors: mechanism and site of action. J Pharmacol Exp Ther. 2001;298:986–995. [PubMed] [Google Scholar]
- 31.Peier AM, McIlwain KL, Kenneson A, Warren ST, Paylor R, Nelson DL. (Over)correction of FMR1 deficiency with YAC transgenics: Behavioral and physical features. Hum Mol Genet. 2000;9:1145–1159. doi: 10.1093/hmg/9.8.1145. [DOI] [PubMed] [Google Scholar]
- 32.Chen L, Toth M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience. 2001;103:1043–1050. doi: 10.1016/s0306-4522(01)00036-7. [DOI] [PubMed] [Google Scholar]
- 33.Musumeci SA, Bosco P, Calabrese G, Bakker C, De Sarro GB, Elia M, Ferri R, Oostra BA. Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia. 2000;41:19–23. doi: 10.1111/j.1528-1157.2000.tb01499.x. [DOI] [PubMed] [Google Scholar]
- 34.Chang KA, Suh YH. Pathophysiological roles of amyloidogenic carboxy-terminal fragments of the beta-amyloid precursor protein in Alzheimer's disease. J Pharmacol Sci. 2005;97:461–471. doi: 10.1254/jphs.cr0050014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.