

Abstract
Hepatocyte growth factor (HGF) is known to promote renal epithelial cell survival by dual mechanisms involving Bad phosphorylation and Bcl-xL induction. However, it remains elusive as to the relative contributions of these two events to HGF-mediated cytoprotection. Here we investigated the role and mechanism of HGF in protecting renal epithelial cells from death induced by oxidant stress both in vitro and in vivo. Simultaneous incubation of human kidney proximal tubular epithelial cells (HKC-8) with HGF failed to protect them from oxidant stress-induced cell death, even though it was capable of inducing endogenous Akt and Bad phosphorylation. However, pre-incubation of HKC-8 cells with HGF for 48 hours dramatically promoted their survival and prevented caspase-3 cleavage and activation induced by H2O2. A close association between Bcl-xL induction and effective cytoprotection by HGF was observed in HKC-8 cells after H2O2 treatment. Furthermore, ectopic expression of exogenous Bcl-xL via adenoviral vector prevented H2O2-triggered caspase-3 activation. In a mouse model of acute kidney injury induced by ischemia/reperfusion, pre-administration of HGF expression vector drastically prevented apoptosis and largely preserved kidney function, whereas much less protective effect was observed when HGF gene was given immediately after ischemic injury. These results suggest that Bcl-xL induction plays an imperative role in mediating HGF cytoprotection of renal epithelial cells after death challenge.
Keywords: HGF, c-met, Bcl-xL, apoptosis, Bad, ischemia/reperfusion, acute kidney injury
Introduction
Hepatocyte growth factor (HGF) and its c-met receptor consist of a signaling system that plays an essential role in mammalian development, tumorigenesis and organ regeneration [1–3]. In addition to the well-described mitogenic, motogenic and morphogenic activities, HGF has been demonstrated to be a potent survival factor capable of preventing diverse types of cells from undergoing apoptosis both in vitro and in vivo [4–6]. Studies indicate that HGF prevents MLP-29 liver progenitor cells from apoptotic death and inhibits hepatocyte apoptosis in a mouse model of Fas-induced fulminant hepatic failure [5, 7]. Expression of constitutively activated c-met tyrosine kinase in the liver of transgenic mice prevents their hepatocytes from apoptosis and permits immortalization of these cells [8]. HGF also protects various carcinoma cells from apoptosis induced by different DNA-damaging agents [9, 10]. Despite several studies suggesting the potential involvement of the members of the Bcl-2 gene family such as BAG-1 and Bcl-xL [7, 11, 12], the signaling and mediators that lead to HGF inhibition of apoptosis remain incompletely understood.
Earlier studies from our laboratory suggest that HGF may protect renal epithelial cell survival by dual mechanisms involving two distinct members of Bcl-2 family proteins [13]. HGF triggers pro-apoptotic protein Bad phosphorylation and subsequent inactivation through phosphoinositide (PI) 3-kinase/Akt pathway [14], while it induces pro-survival protein Bcl-xL expression [12, 15]. Such dual mechanisms of HGF are found to be shared by other growth factors including brain-derived neutrotrophic factor (BDNF) in promoting cerebellar granule neuron survival [16], suggesting that a dual action of HGF on both pro-death and pro-survival proteins in a coordinated fashion may be a general mechanism for growth factor to promote survival in diverse types of the cells in vivo.
Bad phosphorylation and Bcl-xL induction triggered by HGF in renal epithelial cells are kinetically distinct events [13]. While the signal pathway leading to Bad phosphorylation involves a cascade of sequential activation of kinases, it is completed via several posttranslational modifications that occur within a short period of time ranging from minutes to hours following HGF treatment. On the other hand, HGF-induced Bcl-xL expression is a delayed process that requires prolonged incubation ranging from hours to days [13]. Although it is postulated that the dual actions of HGF on renal epithelial cells may be necessary for promoting cell survival following severe death challenges, the functionality and biological significance of Bcl-xL induction in HGF-mediated cytoprotection remain to be established.
In this study, we investigated HGF protection against kidney tubular epithelial cell death induced by oxidant stress. Our results demonstrated that induction of Bcl-xL expression through pre-incubation is required for HGF to exert effective cytoprotection after treatment with H2O2. These studies reveal a critical role of Bcl-xL in HGF-mediated renal epithelial cell survival after injury.
Materials and Methods
Cell Culture and Treatment
Human proximal tubular epithelial cell line (HKC-8) were obtained from Dr. L. Racusen of the Johns Hopkins University and maintained in DMEM/F12 medium supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA), as described previously [17, 18]. HKC-8 cells were seeded on 6 or 12-well culture plates in the complete medium containing 10% FBS for 16 hours, and then changed to serum-free medium after washing twice with the medium. Recombinant human HGF (provided by the Genentech, Inc., South San Francisco, CA) was added to the culture to a final concentration of 20 ng per mL except otherwise indicated. At the same time, the cells were treated with or without H2O2 (Sigma, St. Louis, MO) at the final concentration of 0.5 mM to induce cell death, except when indicated otherwise. Following 16 hours of incubation, cells were harvested and subjected to various assays to determine the cell viability and cell death (see below). For some experiments, the cells were pre-incubated with 20 ng/mL of HGF or phosphate-buffered saline (PBS) vehicle for various periods of time as indicated, and then treated with or without 0.5 mM H2O2 for an additional 16 hours.
Cell Survival Assay
Cell viability was assessed by counting the number of cells that remained adherent to the cell monolayer and excluded trypan blue, as previously reported [13]. Cells were seeded in 6 or 12-well plates and incubated for a fixed time as indicated. Following various treatments, non-adherent cells were removed by two washes with PBS solution. Adherent cells were harvested by trypsin-EDTA digestion and stained with 0.04% trypan blue for 5 minutes. The number of cells excluding trypan blue was determined by counting in a hemacytometer. Cell survival rate following various treatments was expressed as a percentage of viable cells in the treatment groups relative to the controls.
Cell Death Assay
A simple double staining with acridine orange plus ethidium bromide was utilized to determine cell death after oxidative stress, as described elsewhere [19]. Briefly, HKC-8 cells after various treatments were harvested and suspended in PBS. An aliquot of cell suspension (25 µL) was incubated with 1 µL of the staining mixture containing acridine orange (100 µg/mL) and ethidium bromide (100 µg/mL) for 2 min. Cell suspension was then placed on glass slide and examined under a Nikon Eclipse E600 Epi-fluorescence microscope equipped with a digital camera (Melville, NY). Viable cells are identified by a bright green nucleus, whereas apoptotic cells show nuclear orange staining with condensed chromatin structure. Necrotic cells display an orange nucleus without condensed chromatin.
LDH Activity Assay
The measurement of lactate dehydrogenase (LDH) activity released from the cytosol of damaged cells into the supernatant was performed by using a Cytotoxicity Detection Kit (Boehringer Mannheim GmbH, Germany) [20]. This colorimetric assay for the quantification of cell death and cell lysis was based on the released LDH activity in the supernatant upon damage of the plasma membrane. LDH activity was determined in the supernatants of HKC-8 cells after various treatments according to the procedures specified by the manufacturer.
Western Blot Analysis
HKC-8 cells were lysed with SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, and 0.1% bromophenol blue). For assessing caspase-3 activation, cells were lysed into CHAPS buffer (Cell Signaling Technology) supplemented with protease and phosphatase inhibitor cocktails (Sigma). After centrifuging to remove cell debris, supernatant was mixed with SDS sample buffer. Samples were then heated at 100°C for 5–10 minutes, separated on 10% or 15% SDS-polyacrylamide gels and subjected to Western blot as previously described [21]. The pro- and cleaved caspase-3 antibodies, phospho-specific Akt (Ser473) and Bad (Ser112) antibodies that detect Akt and Bad only when phosphorylated at specific sites, and the antibodies that detect total Akt and Bad as well as Bcl-xL were obtained from the Cell Signaling Technology, Inc. (Danvers, MA). Anti-actin antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Caspase-3 Activity Assay
The analysis of caspase-3 activity was performed according to the procedures reported elsewhere [22]. Briefly, HKC-8 cells were washed with PBS, and suspended in a lysis buffer containing 50 mM Tris HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 0.1% SDS, 1 mM EDTA, 50 mM NaF, 0.5% sodium deoxycholate and 1% protease inhibitor cocktail (Sigma). Cell lysate was centrifuged at 10,000 rpm for 10 minutes at 4°C, and the supernatant was used for the enzymatic assay. Twenty µg of protein extracts were incubated at 37°C for 16 hours with 20 µM site-specific tetrapeptide caspase-3 substrate conjugated to aminotrifluromethyl-coumarin (Ac-DEVD-AFC, Calbiochem. La Jolla, CA) in a caspase assay buffer (20 mM PIPES, 100 mM NaCl, 10 mM DTT, 1 mM EDTA, 0.1% CHAPS, 10% sucrose, pH 7.2). The release of the fluorogenic group AFC was measured by a fluorescence spectrometer (PerkinElmer Life Sciences LS50-B), at the excitation/emission wave-lengths of 405/500 nm.
Adenovirus Infection
The recombinant adenoviral vectors containing cDNAs encoding for β-galactosidase (Ad.LacZ) and Bcl-xL (Ad.Bcl-xL) were provided by Dr. A. Gambotto of the Vector Core Facility at the University of Pittsburgh. HKC-8 cells were seeded on 6-well plates, incubated for 24 hours, and then infected with increasing amount of adenoviral vectors (0.25, 0.5, 1.0, 2.5 and 5.0 × 107 particles per mL) in serum-free medium. Infected cells were incubated for 4 hours, and then restored to complete medium. After 24 hours, infected cells were treated with or without 0.5 mM H2O2 in the absence or presence of HGF as indicated. Caspase-3 activation was determined by the procedures described above.
Animals and HGF Plasmid Injection
Male CD-1 mice that weighed between 20 and 24 g were obtained from Harlan Sprague-Dawley (Indianapolis, IN). They were housed in the animal facilities of the University of Pittsburgh Medical Center with free access to food and water. Recombinant human HGF expression plasmid (pCMV-HGF) or empty vector pcDNA3 (Invitrogen) was administrated into mice at 1 mg/kg body weight by a hydrodynamic-based gene transfer technique via rapid injection of a large volume of DNA solution through the tail vein, as previously described [15, 23]. Mice were randomly assigned to five groups: 1) sham control group; 2) ischemia/reperfusion (I/R) control group with pre-administration of pcDNA3 24 h prior to I/R; 3) I/R with pre-administration of pCMV-HGF 24 h prior to I/R; 4) I/R control group with simultaneous administration of pcDNA3 immediately after I/R; and 5) I/R with simultaneous administration of pCMV-HGF immediately after I/R. Animals were anesthetized with pentobarbital sodium and body temperature was maintained at 36–37.5°C. Mice underwent bilateral renal ischemia by clamping both renal pedicles with nontraumatic clamps for 30 minutes. Sham-operated mice had their kidneys exposed, manipulated, but occlusion of the renal pedicles was omitted. The incisions were temporarily closed during ischemia or sham operation. After 30 minutes, the clamps were removed, and reperfusion of the kidneys was visually confirmed. Mice were sacrificed at 48 hours after I/R, and serum was collected. Kidney cryosections were prepared and used for terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) staining.
TUNEL Staining
Cryosections were prepared at 5 µm thick in a cryostat by routine procedures [15]. In situ detection of DNA fragmentation was performed using TUNEL staining with Apoptosis Detection System (Promega, Madison, WI). TUNEL staining was carried out according to the specific protocol suggested by the manufacturer, as described previously [15]. Slides were observed on Nikon Eclipse E600 Epi-fluorescence microscope.
Serum Creatinine Assay
Serum was collected from animals at 48 h after I/R. Serum creatinine level was determined using a creatinine assay kit according to the protocols specified by the manufacturer (Sigma, St. Louis, MO) [15]. The level of creatinine was expressed as milligram per 100 milliliter (dL).
Statistical Analysis
Statistical analysis of the data was performed using SigmaStat software (Jandel Scientific Software, San Rafael, CA). Comparison between multiple groups was made using one-way ANOVA followed by the Student-Newman-Keuls test or Student t test between two groups. A P value of less than 0.05 was considered significant.
Results
Oxidant Stress Induces Renal Epithelial Cell Death via Different Mechanisms
We first examined the responses of renal epithelial cells to death challenges such as oxidant stress in vitro. Human kidney proximal tubular epithelial cells (HKC-8) were treated with different concentrations of H2O2 for 16 hours in the absence of serum. As shown in Figure 1A, H2O2 induced marked cell death in a dose-dependent manner, leading to substantial decline in cell survival. To define the mode of cell death after oxidant stress, we examined the cleavage and activation of pro-caspase-3, a marker for apoptosis. As illustrated in Figure 1B, a dose-dependent activation of caspase-3 was observed in HKC-8 cells when H2O2 was used at the concentrations less than 0.5 mM. When the concentrations of H2O2 increased beyond this level, however, less cleaved caspase-3 was detected in HKC-8 cells. Similar results were obtained when active casapse-3 was determined quantitatively by measuring the enzymatic activity (Figure 1C). We also assessed the activity of lactate dehydrogenase (LDH), an enzyme released from the cytosol into the supernatant upon damage of the plasma membrane when cells undergo necrosis. As shown in Figure 1D, when H2O2 concentrations reached 0.5 mM or higher, significant LDH activity was detected in the supernatant of HKC-8 cells. These results suggest that oxidant stress, dependent on its dosages, may induce renal epithelial cell death through both apoptotic and necrotic pathways.
Figure 1.
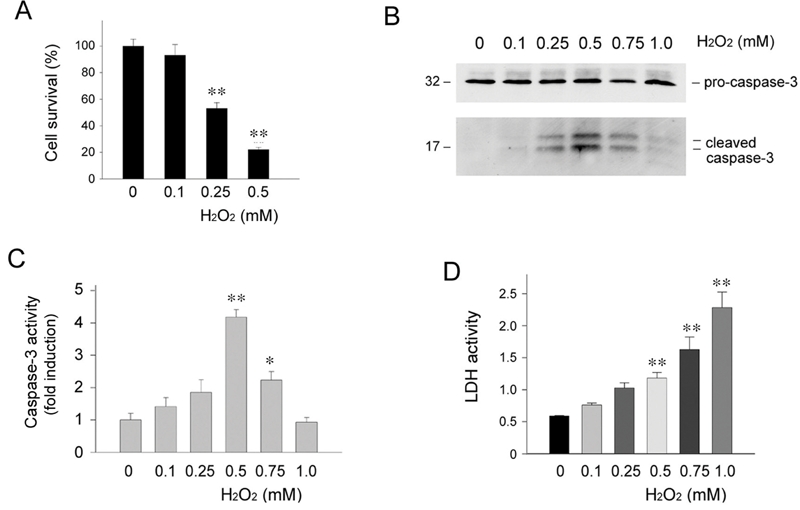
Oxidant stress induces kidney proximal tubular epithelial cell death through different mechanisms. Human kidney proximal tubular epithelial cells (HKC-8) were treated with different concentrations of H2O2 as indicated for 16 hours in the absence of serum. A. Cell survival rate. B. Western blot analyses demonstrated the cleavage and activation of pro-caspase-3 in HKC-8 cells after treatment with different concentrations of H2O2 as indicated. C. Quantitative determination of active caspase-3 activity after H2O2 treatment. Data are presented as means ± SEM of three experiments. ** P < 0.01; * P < 0.05 verse controls. D. LDH activity in the supernatants of HKC-8 cells after H2O2 treatment. Data are presented as means ± SEM of three experiments. ** P < 0.01 verse controls.
Simultaneous Incubation with HGF Fails to Promote Renal Epithelial Cell Survival
To determine whether HGF can prevent renal epithelial cell death induced by oxidant stress, we examined the effects of exogenous HGF on HKC-8 cell survival following H2O2 treatment. As shown in Figure 2A, simultaneous addition of HGF did not significantly improve HKC-8 cell survival after incubation with 0.5 mM H2O2. This observation was confirmed independently by a cell death assay using double staining with acridine orange and ethidium bromide (Figures 2B and C). Furthermore, HGF also failed to prevent caspase-3 cleavage and activation in HKC-8 cells after oxidative stress (Figure 2D). Thus, simultaneous incubation with exogenous HGF does not protect renal epithelial cells against oxidant stress-induced cell death.
Figure 2.

Simultaneous incubation with HGF fails to protect renal epithelial cells against oxidant stress-induced cell death. HKC-8 cells were treated simultaneously with or without 20 ng/mL HGF and 0.5 mM H2O2 for 16 hours in the absence of serum. A. HGF failed to promote cell survival after oxidant stress. B and C. HGF did not significantly protect HKC-8 cells from oxidant stress-induced cell death. Cell death was assessed by double staining with acridine orange and ethidium bromide. D. Simultaneous incubation of HKC-8 cells with HGF did not affect H2O2 -induced caspase-3 activation.
HGF Induces Bad Phosphorylation in the Presence of Oxidant Stress
Earlier studies showed that HGF prevents apoptosis induced by serum deprivation through dual mechanisms involving Bad phosphorylation and Bcl-xL induction [13]. To exclude the possibility that the failure of HGF cytoprotection is due to its inability to induce Bad phosphorylation in the presence of death stimuli such as H2O2, we examined the phosphorylation status of Akt and Bad proteins by simultaneous incubation of HKC-8 cells with HGF and H2O2. As shown in Figure 3, in the presence of H2O2, HGF was capable of inducing both Akt and Bad phosphorylation in HKC-8 cells, suggesting that death stimuli did not significantly abrogate HGF-induced Bad phosphorylation and resultant inactivation. Moreover, H2O2 also did not eradicate the pre-existed phosphorylated Akt and Bad induced by pre-incubation of HKC-8 cells with HGF for short periods of time ranging from 10 to 30 minutes (Figure 2). These results suggest that even in the presence of death stimuli, HGF is able to induce the phosphorylation and subsequent inactivation of pro-apoptotic Bad protein. Therefore, it appears that endogenous Bad phosphorylation alone may not be sufficient for mediating HGF cytoprotection after oxidant stress.
Figure 3.
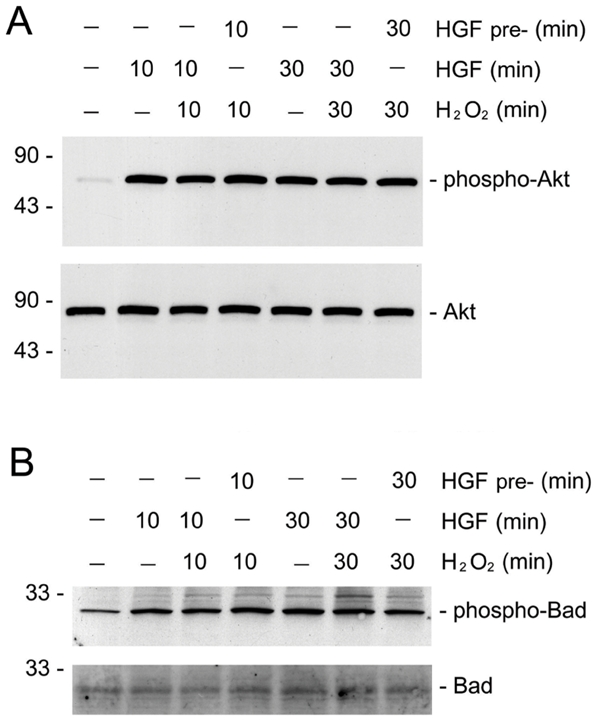
HGF induces endogenous Akt and Bad phosphorylation in renal epithelial cells in the presence of oxidant stress. HKC-8 cells were treated with 20 ng/mL HGF for various periods of time as indicated, either simultaneously with or prior to addition of 0.5 mM H2O2. The cell lysates were immunoblotted with antibodies against phospho-Akt and phospho-Bad, or against total Akt and Bad protein, respectively. A. phospho-Akt (Ser473) and total Akt. B. phospho-Bad (Ser112) and total Bad.
HGF Promotion of Renal Epithelial Cell Survival Requires Preincubation
In addition to triggering Bad phosphorylation, HGF is known to induce Bcl-xL expression in renal epithelial cells in a delayed fashion that requires prolonged incubation for 36 – 48 hours [13]. It is thus conceivable that long-term pre-incubation with HGF could be required to pre-condition renal epithelial cells to prevail during a death challenge. To test this hypothesis, we pre-incubated HKC-8 cells with exogenous HGF for various periods of time prior to treatment with H2O2. As shown in Figure 4A, pre-incubation with HGF for 48 hours dramatically protected HKC-8 cells from death induced by 0.5 mM H2O2. The survival rate of HKC-8 cells was dependent on the duration of pre-incubation, and marked cytoprotection was only observed in the cultures pre-incubated with HGF for longer than 36 hours (Figure 4A), consistent with the time points when significant Bcl-xL induction was evident [13]. Similar results were obtained when cell death was assessed (Figures 4D-F). Pre-incubation with HGF also reduced caspase-3 activation in HKC-8 cells after oxidant stress (Figure 4G). This cytoprotective effect of HGF was also dose-dependent (Figure 4H). Together, these observations demonstrate that pre-incubation is required for HGF to protect renal epithelial cells against cell death induced by oxidant stress.
Figure 4.
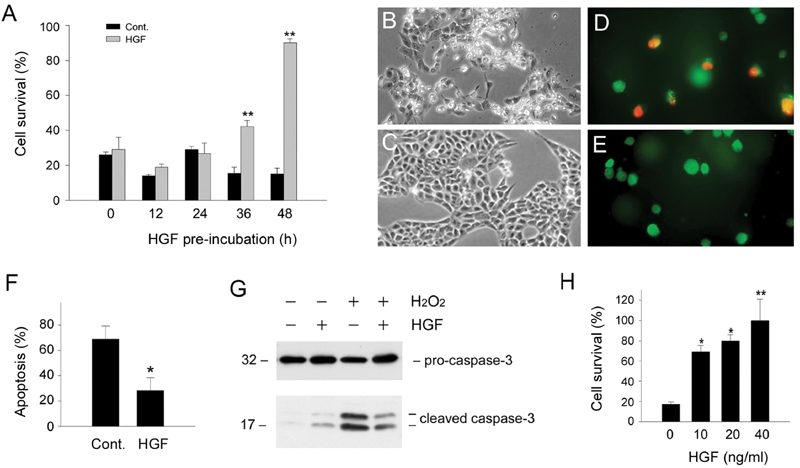
Pre-incubation with HGF protects renal epithelial cells from apoptosis induced by oxidant stress. A. HKC-8 cells were pre-incubated with HGF (20 ng/mL) in serum-free medium for various periods of time as indicated, followed by treatment with 0.5 mM H2O2 for 16 hours. Significant cell survival was only observed in HKC-8 cells pre-incubated with HGF for longer than 36 hours. **P < 0.01. B and C. Representative phase-contrast morphology of HKC-8 cells pre-incubated without (B) or with HGF (C) for 48 hours, followed by incubation with 0.5 mM H2O2 for 16 hours. D and E. Representative micrographs showed apoptosis assessed by double staining of HKC-8 cells pre-incubated without (D) or with HGF (E) for 48 hours, followed by incubation with 0.5 mM H2O2 for 16 hours. F. Quantitative determination of apoptosis in HKC-8 cells pre-incubated without or with HGF. *P < 0.05. G. Western blot analyses demonstrated that HGF pre-incubation reduced casapse-3 activation in HKC-8 cells after H2O2 treatment. H. HGF pre-incubation promoted HKC-8 cell survival in a dose-dependent manner. *P < 0.05. **P < 0.01.
HGF-mediated Cytoprotection Correlates with Bcl-xL Induction
To test whether HGF-mediated cytoprotection after pre-incubation is associated with its induction of Bcl-xL, we examined the Bcl-xL expression in the absence or presence of oxidant stress. As shown in Figure 5A, pre-incubation of HKC-8 cells with HGF for 48 hours induced endogenous Bcl-xL expression. Although H2O2 inhibited Bcl-xL at basal conditions, HGF was still able to upregulate Bcl-xL protein in the presence of H2O2. Given that HGF pre-incubation promotes HKC-8 cell survival, these studies suggest that the cytoprotection elicited by HGF is closely associated with the up-regulation of endogenous Bcl-xL expression.
Figure 5.
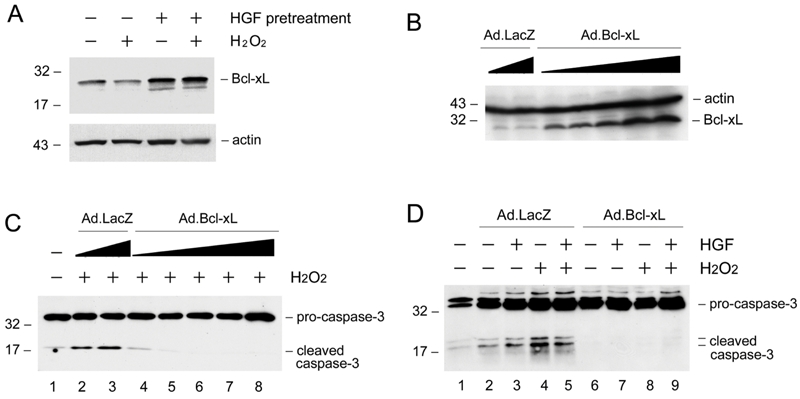
HGF induces Bcl-xL expression and ectopic expression of Bcl-xL protects renal epithelial cells from apoptosis. A. HGF induces Bcl-xL expression in the absence or presence of H2O2. HKC-8 cells were incubated with or without HGF (20 ng/mL) for 48 hours, and then treated with 0.5 mM H2O2 for 16 hours. Cell lysates were immunoblotted with specific antibodies against Bcl-xL and actin, respectively. B. Ectopic expression of Bcl-xL in HKC-8 cells after infection with adenoviral vector. HKC-8 cells were infected with increasing amounts of adenovirus harboring Bcl-xL gene (Ad.Bcl-xL) or control adenovirus (Ad.LacZ). Cell lysates were immunoblotted with antibodies against Bcl-xL and actin, respectively. C. Expression of exogenous Bcl-xL protected HKC-8 cells from H2O2-induced caspase-3 activation in a dose-dependent manner. D. Over-expression of Bcl-xL abolished caspase-3 activation in HKC-8 cells after H2O2 treatment.
To evaluate the role of Bcl-xL induction in mediating renal epithelial cell survival, we examined the impact of ectopic expression of exogenous Bcl-xL on oxidant stress-induced cell death. As shown in Figure 5B, infection of HKC-8 cells with adenovirus harboring Bcl-xL gene resulted in significant Bcl-xL expression in a dose-dependent manner. Comparing with the controls, ectopic expression of Bcl-xL abolished the activation of caspase-3 after H2O2 treatment (Figures 5C and D).
Preadministration of HGF Prevents Apoptosis and Ameliorates Ischemic Acute Kidney Injury In Vivo
To assess the cytoprotective potential of HGF in vivo, we investigated the effect of exogenous HGF on cell survival in acute kidney injury induced by ischemia/reperfusion (I/R), a model characterized by prominent tubular epithelial cell death. As shown in Figure 6, a dramatic increase in apoptosis was found in the kidney at 2 days after I/R, consistent with other reports [24, 25]. However, pre-administration of HGF expression plasmid largely prevented apoptotic cell death. This cytoprotective effect of HGF was also dependent on the timing of administration. When HGF plasmid was given immediately after I/R, little cytoprotection was observed (Figure 6).
Figure 6.
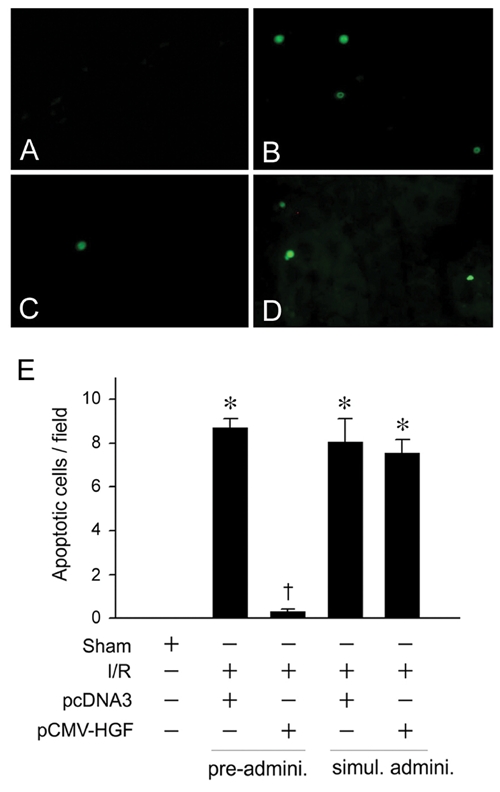
Pre-administration of HGF gene prevents apoptosis after acute kidney injury induced by ischemia/reperfusion. Mice were administrated HGF expression plasmid (pCMV-HGF) or control vector (pcDNA3). Apoptosis was assessed in kidney cyrosections at day 2 after ischemia/reperfusion (I/R) by TUNEL staining. A-D. Representative micrographs demonstrated TUNEL-positive cells in various groups. E. Quantitative determination of apoptosis in the kidney after various treatments. *P < 0.05 verse sham controls. †P < 0.05 verse I/R controls (n = 4). Pre-admini., HGF gene was given 24 hours prior to I/R. Simul. Admini., HGF gene was given at the same time with I/R.
Consistent with renal cytoprotection, we found that pre-administration of HGF plasmid also prevented kidney dysfunction and renal failure, as assessed by serum creatinine levels (Figure 7). However, much less amelioration of kidney dysfunction was noticed when HGF plasmid was given immediately after I/R (Figure 7). These results suggest that HGF only elicits a full cytoprotection in vivo when it is given prior to ischemic injury.
Figure 7.
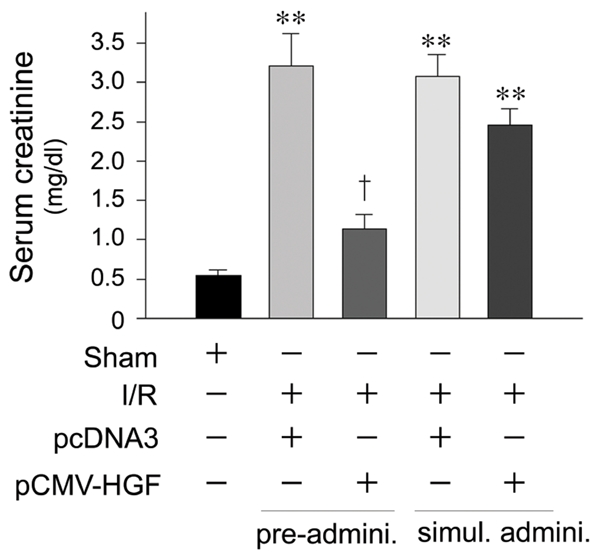
Pre-administration of HGF gene prevents acute renal failure after ischemia/reperfusion injury. Mice were administrated HGF expression plasmid (pCMV-HGF) or control vector (pcDNA3). Serum creatinine levels were determined at day 2 after ischemia/reperfusion (I/R). **P < 0.01 verse sham controls. †P < 0.01 verse I/R controls (n = 4). Pre-admini., HGF gene was given 24 hours prior to I/R. Simul. Admini., HGF gene was given at the same time with I/R.
Discussion
HGF has been demonstrated to induce pro-death Bad phosphorylation and pro-survival Bcl-xL expression [13]; however, the relative importance of these events in HGF-mediated cytoprotection remains ambiguous. In this study, by taking advantage of the fact that Bad phosphorylation and Bcl-xL induction triggered by HGF are two kinetically distinct events, we have investigated their roles in promoting renal epithelial cell survival after oxidant stress. Our results indicate that Bad phosphorylation by HGF alone is not sufficient for protection of renal epithelial cells against cell death induced by H2O2. Instead, Bcl-xL induction via pre-incubation with HGF produces profound effects favoring cell survival, thereby playing the decisive role protecting renal epithelial cells after injury.
In the presence of death stimuli, HGF is able to stimulate the signal pathway leading to the phosphorylation of Akt and Bad proteins in renal epithelial cells, and it fails to protect against cell death. However, this by no means suggests that Bad phosphorylation and subsequent inactivation are not necessary for HGF-mediated cell survival. In fact, overexpression of Bad protein alone causes HKC-8 cell death, and HGF abolishes Bad-induced apoptosis by phosphorylating Bad protein at both Ser112 and Ser136 sites in renal epithelial cells as demonstrated previously [13]. The inability of Bad phosphorylation to protect cells following severe death challenges such as oxidant stress is probably due to the fact that the endogenous Bad level is extremely low in these cells, as shown by Western blot analysis. Therefore, inactivation of pro-apoptotic Bad alone would have a limited impact on the ratio of anti- vs pro-apoptotic proteins inside the cells, even if the pro-death activity of endogenous cellular Bad could be completely abolished via phosphorylation by HGF.
The results of the present study clearly illustrate that Bcl-xL protein level dictates the fate of renal epithelial cells after injury. In this regard, it is conceivable that, although HGF regulates multiple survival pathways [26, 27], one of the apoptosis regulatory proteins such as Bcl-xL may play a predominant role in controlling the cell survival/death. Consistent with this notion, ectopic expression of exogenous Bcl-xL is sufficient for protecting renal epithelial cells from death induced by H2O2. It should be noted that the experimental manipulations such as adenovirus infection may lead to a high level of Bcl-xL expression that is probably neither achievable nor realistic physiologically in vivo. Therefore, in normal physiologic conditions, the possibility exists that both Bad phosphorylation and Bcl-xL induction are required for HGF-mediated protection of renal epithelial cells from death after injury.
Although HGF-induced Bcl-xL expression is known to be a delayed process, the mechanism underlying the Bcl-xL induction by HGF remains largely unknown. There are potentially two pathways that could lead to Bcl-xL expression in HGF-treated renal epithelial cells. One probably involves Ras-mitogen-activated protein kinase (Ras-MAKP) signaling pathway [16]. Studies show that one of the downstream kinases of HGF-activated Ras-MAPK signaling, p90RSK, phosphorylates and activates the transcription factor cAMP response element-binding protein (CREB) at serine 133 [28], which in turn promotes pro-survival genes such as Bcl-xL expression. Another potential mechanism involves the signaling of Akt and transcription factor NFκB [29]. As established in previous studies [13, 28, 30], HGF rapidly and markedly phosphorylates and activates Akt kinase in a PI 3-kinase-dependent fashion. Activated Akt may directly activate IκB kinases which in turn phosphorylates IκB, the endogenous inhibitor of transcription factor NFκB, resulting in its proteasome-mediated degradation and subsequent activation of NFκB, thereby promoting the transcription of pro-survival genes [31, 32].
The finding that Bcl-xL induction is required for HGF-mediated protection against renal epithelial cell death presented in this study could have important implications. HGF is widely considered to be a promising therapeutic agent that protects against organ failure and promotes tissue regeneration after acute injury [2, 33–35]. Similar to in vitro studies, pre-administration of HGF gene prior to acute kidney injury has profound effects on renal epithelial cell survival and preservation of kidney function after ischemic injury, whereas injection of HGF gene after acute insult displays much less protective effect in vivo (Figures 6 and 7). Notably, HGF is shown to induce Bcl-xL expression in the kidney after acute injury [15]. Therefore, pre-administration of HGF would allow the cells to precondition themselves via Bcl-xL induction and to prepare for severe death challenges. In this regard, our studies have provided significant mechanistic insights into understanding why pretreatment is required for HGF to exert maximal cytoprotection in vivo.
Acknowledgments
We thank Dr. Andrea Gambotto for kindly providing adenoviral vectors and Dr. Lawrence Kiss for critically reading this manuscript. This work was supported by the National Institutes of Health grants DK054922, DK061408 and DK064005.
References
- 1.Liu Y. Hepatocyte growth factor in kidney fibrosis: therapeutic potential and mechanisms of action. Am J Physiol Renal Physiol. 2004;287:F7–F16. doi: 10.1152/ajprenal.00451.2003. [DOI] [PubMed] [Google Scholar]
- 2.Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 3.Sattler M, Salgia R. c-Met and hepatocyte growth factor: potential as novel targets in cancer therapy. Curr Oncol Rep. 2007;9:102–108. doi: 10.1007/s11912-007-0005-4. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, Sun AM, Dworkin LD. Hepatocyte growth factor protects renal epithelial cells from apoptotic cell death. Biochem Biophys Res Commun. 1998;246:821–826. doi: 10.1006/bbrc.1998.8676. [DOI] [PubMed] [Google Scholar]
- 5.Kosai K, Matsumoto K, Nagata S, Tsujimoto Y, Nakamura T. Abrogation of Fas-induced fulminant hepatic failure in mice by hepatocyte growth factor. Biochem Biophys Res Commun. 1998;244:683–690. doi: 10.1006/bbrc.1998.8293. [DOI] [PubMed] [Google Scholar]
- 6.Fornoni A, Li H, Foschi A, Striker GE, Striker LJ. Hepatocyte growth factor, but not insulin-like growth factor I, protects podocytes against cyclosporin A-induced apoptosis. Am J Pathol. 2001;158:275–280. doi: 10.1016/S0002-9440(10)63966-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bardelli A, Longati P, Albero D, Goruppi S, Schneider C, Ponzetto C, Comoglio PM. HGF receptor associates with the anti-apoptotic protein BAG-1 and prevents cell death. EMBO J. 1996;15:6205–6212. [PMC free article] [PubMed] [Google Scholar]
- 8.Amicone L, Spagnoli FM, Spath G, Giordano S, Tommasini C, Bernardini S, De Luca V, Della Rocca C, Weiss MC, Comoglio PM, Tripodi M. Transgenic expression in the liver of truncated Met blocks apoptosis and permits immortalization of hepatocytes. EMBO J. 1997;16:495–503. doi: 10.1093/emboj/16.3.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fan S, Wang JA, Yuan RQ, Rockwell S, Andres J, Zlatapolskiy A, Goldberg ID, Rosen EM. Scatter factor protects epithelial and carcinoma cells against apoptosis induced by DNA-damaging agents. Oncogene. 1998;17:131–141. doi: 10.1038/sj.onc.1201943. [DOI] [PubMed] [Google Scholar]
- 10.Fan S, Ma YX, Wang JA, Yuan RQ, Meng Q, Cao Y, Laterra JJ, Goldberg ID, Rosen EM. The cytokine hepatocyte growth factor/scatter factor inhibits apoptosis and enhances DNA repair by a common mechanism involving signaling through phosphatidyl inositol 3' kinase. Oncogene. 2000;19:2212–2223. doi: 10.1038/sj.onc.1203566. [DOI] [PubMed] [Google Scholar]
- 11.Li Z, Mizuno S, Nakamura T. Antinecrotic and antiapoptotic effects of hepatocyte growth factor on cholestatic hepatitis in a mouse model of bile-obstructive diseases. Am J Physiol Gastrointest Liver Physiol. 2007;292:G639–646. doi: 10.1152/ajpgi.00292.2006. [DOI] [PubMed] [Google Scholar]
- 12.Dai C, Li Y, Yang J, Liu Y. Hepatocyte growth factor preserves beta cell mass and mitigates hyperglycemia in streptozotocin-induced diabetic mice. J Biol Chem. 2003;278:27080–27087. doi: 10.1074/jbc.M211947200. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y. Hepatocyte growth factor promotes renal epithelial cell survival by dual mechanisms. Am J Physiol. 1999;277:F624–633. doi: 10.1152/ajprenal.1999.277.4.F624. [DOI] [PubMed] [Google Scholar]
- 14.Kakazu A, Chandrasekher G, Bazan HE. HGF protects corneal epithelial cells from apoptosis by the PI-3K/Akt-1/Bad- but not the ERK1/2-mediated signaling pathway. Invest Ophthalmol Vis Sci. 2004;45:3485–3492. doi: 10.1167/iovs.04-0372. [DOI] [PubMed] [Google Scholar]
- 15.Dai C, Yang J, Liu Y. Single injection of naked plasmid encoding hepatocyte growth factor prevents cell death and ameliorates acute renal failure in mice. J Am Soc Nephrol. 2002;13:411–422. doi: 10.1681/ASN.V132411. [DOI] [PubMed] [Google Scholar]
- 16.Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- 17.Yang J, Liu Y. Blockage of tubular epithelial to myofibroblast transition by hepatocyte growth factor prevents renal interstitial fibrosis. J Am Soc Nephrol. 2002;13:96–107. doi: 10.1681/ASN.V13196. [DOI] [PubMed] [Google Scholar]
- 18.Racusen LC, Monteil C, Sgrignoli A, Lucskay M, Marouillat S, Rhim JG, Morin JP. Cell lines with extended in vitro growth potential from human renal proximal tubule: characterization, response to inducers, and comparison with established cell lines. J Lab Clin Med. 1997;129:318–329. doi: 10.1016/s0022-2143(97)90180-3. [DOI] [PubMed] [Google Scholar]
- 19.Arany I, Megyesi JK, Kaneto H, Tanaka S, Safirstein RL. Activation of ERK or inhibition of JNK ameliorates H(2)O(2) cytotoxicity in mouse renal proximal tubule cells. Kidney Int. 2004;65:1231–1239. doi: 10.1111/j.1523-1755.2004.00500.x. [DOI] [PubMed] [Google Scholar]
- 20.Wen X, Li Y, Hu K, Dai C, Liu Y. Hepatocyte growth factor receptor signaling mediates the anti-fibrotic action of 9-cis-retinoic acid in glomerular mesangial cells. Am J Pathol. 2005;167:947–957. doi: 10.1016/S0002-9440(10)61185-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai C, Yang J, Liu Y. Transforming growth factor-beta1 potentiates renal tubular epithelial cell death by a mechanism independent of Smad signaling. J Biol Chem. 2003;278:12537–12545. doi: 10.1074/jbc.M300777200. [DOI] [PubMed] [Google Scholar]
- 22.Yin XM, Luo Y, Cao G, Bai L, Pei W, Kuharsky DK, Chen J. Bid-mediated mitochondrial pathway is critical to ischemic neuronal apoptosis and focal cerebral ischemia. J Biol Chem. 2002;277:42074–42081. doi: 10.1074/jbc.M204991200. [DOI] [PubMed] [Google Scholar]
- 23.Yang J, Dai C, Liu Y. Hepatocyte growth factor gene therapy and angiotensin II blockade synergistically attenuate renal interstitial fibrosis in mice. J Am Soc Nephrol. 2002;13:2464–2477. doi: 10.1097/01.asn.0000031827.16102.c1. [DOI] [PubMed] [Google Scholar]
- 24.Sheridan AM, Bonventre JV. Cell biology and molecular mechanisms of injury in ischemic acute renal failure. Curr Opin Nephrol Hypertens. 2000;9:427–434. doi: 10.1097/00041552-200007000-00015. [DOI] [PubMed] [Google Scholar]
- 25.Wei Q, Yin XM, Wang MH, Dong Z. Bid deficiency ameliorates ischemic renal failure and delays animal death in C57BL/6 mice. Am J Physiol Renal Physiol. 2006;290:F35–42. doi: 10.1152/ajprenal.00184.2005. [DOI] [PubMed] [Google Scholar]
- 26.Moumen A, Ieraci A, Patane S, Sole C, Comella JX, Dono R, Maina F. Met signals hepatocyte survival by preventing Fas-triggered FLIP degradation in a PI3k-Akt-dependent manner. Hepatology. 2007;45:1210–1217. doi: 10.1002/hep.21604. [DOI] [PubMed] [Google Scholar]
- 27.Fassetta M, D'Alessandro L, Coltella N, Di Renzo MF, Rasola A. Hepatocyte growth factor installs a survival platform for colorectal cancer cell invasive growth and overcomes p38 MAPK-mediated apoptosis. Cell Signal. 2006;18:1967–1976. doi: 10.1016/j.cellsig.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 28.Tan R, Zhang X, Yang J, Li Y, Liu Y. Molecular basis for the cell type specific induction of SnoN expression by hepatocyte growth factor. J Am Soc Nephrol. 2007;18:2340–2349. doi: 10.1681/ASN.2007010128. [DOI] [PubMed] [Google Scholar]
- 29.Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 30.Okada M, Sugita K, Inukai T, Goi K, Kagami K, Kawasaki K, Nakazawa S. Hepatocyte growth factor protects small airway epithelial cells from apoptosis induced by tumor necrosis factor-alpha or oxidative stress. Pediatr Res. 2004;56:336–344. doi: 10.1203/01.PDR.0000134255.58638.59. [DOI] [PubMed] [Google Scholar]
- 31.Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol. 2001;11:297–305. doi: 10.1016/s0959-4388(00)00211-7. [DOI] [PubMed] [Google Scholar]
- 32.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 33.Conway K, Price P, Harding KG, Jiang WG. The molecular and clinical impact of hepatocyte growth factor, its receptor, activators, and inhibitors in wound healing. Wound Repair Regen. 2006;14:2–10. doi: 10.1111/j.1743-6109.2005.00081.x. [DOI] [PubMed] [Google Scholar]
- 34.Matsumoto K, Nakamura T. Hepatocyte growth factor: renotropic role and potential therapeutics for renal diseases. Kidney Int. 2001;59:2023–2038. doi: 10.1046/j.1523-1755.2001.00717.x. [DOI] [PubMed] [Google Scholar]
- 35.Franquesa M, Alperovich G, Herrero-Fresneda I, Lloberas N, Bolanos N, Fillat C, Rama I, Cruzado JM, Grinyo JM, Torras J. Direct electrotransfer of hHGF gene into kidney ameliorates ischemic acute renal failure. Gene Ther. 2005;12:1551–1558. doi: 10.1038/sj.gt.3302569. [DOI] [PubMed] [Google Scholar]