

Abstract
Polymorphic regions consisting of a variable number of tandem repeats within intron 2 of the gene coding for the serotonin transporter protein 5-HTT have been associated with susceptibility to affective disorders. We have cloned two of these intronic polymorphisms, Stin2.10 and Stin2.12, into an expression vector containing a heterologous minimal promoter and the bacterial LacZ reporter gene. These constructs were then used to produce transgenic mice. In embryonic day 10.5 embryos, both Stin2.10 and Stin2.12 produced consistent β-galactosidase expression in the embryonic midbrain, hindbrain, and spinal cord floor plate. However, we observed that the levels of β-galactosidase expression produced by both the Stin2.10 and Stin2.12 within the rostral hindbrain differed significantly at embryonic day 10.5. Our data suggest that these polymorphic variable number of tandem repeats regions act as transcriptional regulators and have allele-dependent differential enhancer-like properties within an area of the hindbrain where the 5-HTT gene is known to be transcribed at this stage of development.
Keywords: transcriptional regulation, promoter, transgenic mice, LacZ gene, rostral hindbrain
In addition to its accepted role as a neurotransmitter, several studies have suggested that serotonin (5-HT) acts as a morphogen during embryogenesis. These studies have suggested that 5-HT plays an important role in the development of the central nervous system (CNS; refs. 1–4). For example, 5-HT induces neurogenesis and neuronal differentiation (1–4), affects migration of cranial neural crest cells from the vicinity of the early hindbrain (5), and inhibits growth cone mobility in both mammals and invertebrates (6, 7). It has also been shown that 5-HT and many of its receptors are present in the embryo CNS during development (4, 8–12)
In the adult brain, the 5-HT transporter protein (5-HTT) limits the action of 5-HT on its receptors by acting as a sodium-dependent plasma membrane transporter recycling 5-HT into the presynaptic terminal (13). 5-HTT seems to be the major determinant in regulating extracellular 5-HT levels, and as a result, several studies have also suggested that 5-HTT may also play a role in morphogenesis (10, 11). Recent in situ hybridization analyses have further shown that 5-HTT transcripts are present in the CNS and many other areas of the early developing embryo (10, 11), such as the presumptive embryonic day (E)11 mouse rostral hindbrain (14).
5-HTT has been shown to be the major site of action of many antidepressants (15), and as a result, much interest has focused on the possible roles played by 5-HTT in the generation of affective disorders, psychosis, and anxiety symptoms. Consequently, the association of polymorphic regions to susceptibility to these problems has recently come under scrutiny. Linkage studies of polymorphisms within the 5-HTT gene have indicated that a possible relationship exists between certain polymorphisms and susceptibility to depressive disorders (12, 16–24). Much interest has focused on a polymorphism within intron 2, which consists of a variable number of tandem repeats (VNTR) containing between 9 (Stin2.9), 10 (Stin2.10), and 12 (Stin2.12) copies of a 17-bp element (16–20). The difference in the number of copies of the repeated 17-bp element has been correlated with susceptibility to bipolar and unipolar disorder (16–20). However, several other studies have produced data that seem to indicate that no link exists between 5-HTT intron 2 polymorphic regions and susceptibility to affective disorders (25, 26). Therefore, a possible role for these polymorphisms in the generation of affective disorders remains moot.
We have taken a different approach in studying these polymorphisms and have explored the possibility that 5-HTT intron 2 VNTR polymorphic regions may act as transcriptional regulatory sequences during embryonic development. We introduced expression constructs containing the Stin2.10 and Stin2.12 polymorphisms upstream of a heterologous promoter driving the LacZ marker gene into mouse embryos by pronuclear injection. Between E9 and E11.5, strong and consistent β-galactosidase (βgal) expression in specific areas of the developing CNS was produced by both constructs, particularly in the midbrain, hindbrain, and neural tube floor plate (FP). We can therefore show that the Stin2.10 and Stin2.12 polymorphisms permit marker gene expression in vivo in a manner consistent with their possible roles as putative CNS enhancers. Furthermore, we show that a significant difference exists between the ability of both the polymorphisms studied in allowing marker gene expression in the rostral hindbrain, an area associated with native 5-HTT mRNA expression and the development of rostral serotoninergic cell clusters.
Methods
Plasmid Construction.
Stin2.10 and Stin2.12 polymorphisms were obtained from the DNA of nonaffected and bipolar individuals, respectively, by PCR by using the following oligonucleotides (27, 28): 5′-GTCAGTATCACAGGCTGCGAG-3′ and 5′-TGTTCCTAGTCTTACGCCAGTG-3′ (MWG Biotec, Ebersberg, Germany). The PCR products were cloned into pCR2.1 (Invitrogen), and several clones of each construct were sequenced on a LI-COR 4000L DNA sequencer (MWG Biotec) to ascertain PCR fidelity. Correct sequences were then subcloned into the NotI–SpeI site of the heterologous promoter (human β-globin promoter) LacZ expression vector pGZ40 (which has been shown not to express LacZ in embryos on its own) to produce p29Stin2.10 and p29Stin2.12 (Fig. 1A; ref. 29). Before oocyte injection, both constructs were removed from their plasmid backbones by restriction digestion with NotI/SalI.
Figure 1.

(A) A limited restriction map of the p29Stin2.10 and p29Stin2.12 constructs used in this study. hβg, human β-globin minimal promoter sequence; LacZ, βgal gene; pA, simian virus 40 polyadenylation sequence. (B–J) Expression patterns of the LacZ gene in p29Stin2.10 transgenic embryos at E9 (B–D), E10.5 (E–G), E11.5 (H), and E12.5 (I and J). B, E, H, and I represent side views of the embryos; C, F, and J are viewed ventrally; D and G are viewed caudally. Fb, forebrain; Mb, midbrain; Hb, hindbrain. Sc, spinal cord; Fp, FP; Ov, otic vesicle; r1–5, rhombomeres 1–5; H, heart.
Transgenic Mice.
Transgenic mice were generated as described (30). DNA constructs were injected at a concentration of 2 ng/μl into the pronuclei of one-cell embryos derived from superovulated (CBA × C57BL/6)F1 females. Injected eggs were transferred into pseudopregnant CD1 females. Transgenic embryos were harvested at E8, E9, E10.5, E11.5, E12.5, and E13.5 and assayed for βgal activity (30).
PCR Screening of Transgenic Lines.
Tail-tip DNA was extracted by incubating mouse tail-tip biopsy material overnight in 500-μl tail-tip buffer [200 μg/ml−1 proteinase K (Roche Molecular Biochemicals)/300 mM sodium acetate, pH 7/1% SDS/l0 mM Tris, pH 8/1 mM EDTA, pH 8]. After incubation, biopsy samples were frozen and then centrifuged at 13,000 × g at 4°C for 10 min. After the removal of 1 μl of the supernatant and its dilution ×10, PCR analysis was carried out with the following PCR primers: 5′-CGCTGATTTCTGTAGTCGGTT-3′ and 5′-GAATTATTTTTGATGGCGTT-3′ (MWG Biotec).
Vibratome Sections.
Vibratome sections were produced from embryos stained for LacZ to ascertain the exact locations of internal staining sites.
LacZ-stained embryos were washed twice in PBS and left overnight in 4% (vol/vol) sucrose/PBS. Embryos were then incubated for 6 h in 20% (vol/vol) sucrose/PBS then transferred into BSG solution [14% (vol/vol) BSA/20% (vol/vol) sucrose/0.5% gelatin in PBS] and incubated overnight with gentle agitation. On the next day, the embryos were embedded in fresh BSG solution mixed with 2.5% (vol/vol) glutaraldehyde, and 100-μm thick sections were cut with a Vibratome series 1000.
Results
Transient Transgenic and Transgenic Line Analysis of the p29Stin2.10 Construct.
The expression of the marker gene throughout several developmental stages was studied in whole mount (Figs. 1 and 2). Embryo staining patterns between the transgenic line and the transient individuals generated showed a high degree of consistency between E9.5 and E10.5. For this reason, the data will be presented together.
Figure 2.
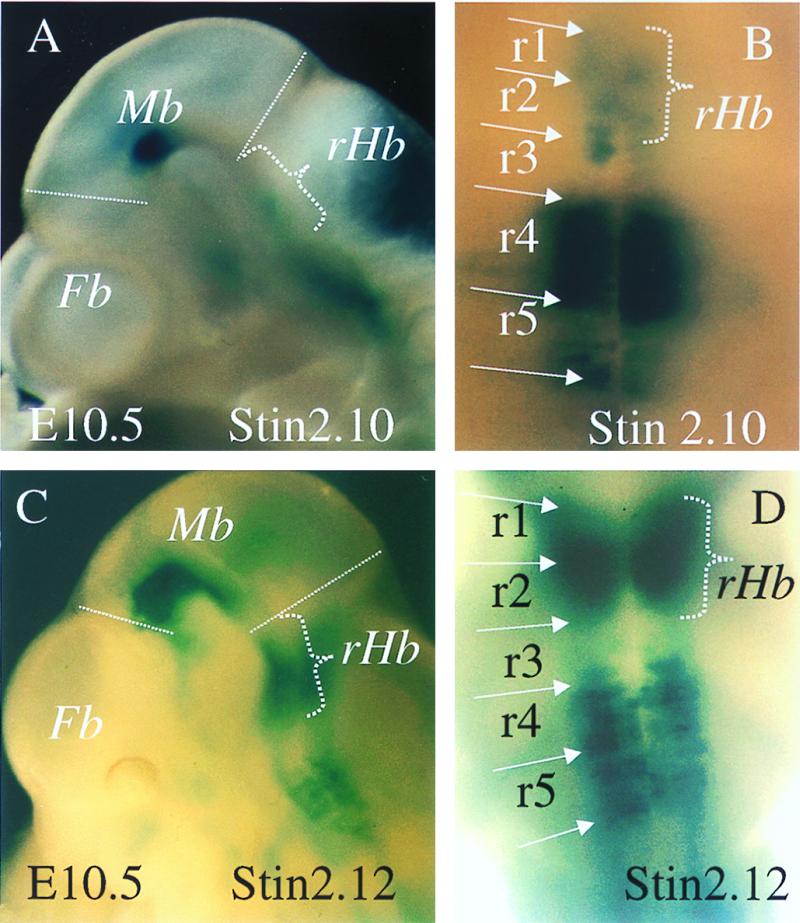
A comparison of the hindbrain LacZ expression patterns shown by E10.5 embryos transgenic for p29Stin2.10 (A and B) and p29Stin2.12 (C and D). A and C represent side views of the embryo head, and B and D represent a caudal view of the E10.5 embryo hindbrain. rHb, rostral hindbrain FP; other abbreviations are defined in Fig. 1.
E8.
(Transgenic line only.) No LacZ expression was detectable in any of the individuals from the transgenic line examined.
E9.
(Two transients generated.) Strong and consistent expression of the marker gene could now be detected in both the transgenic line and transient embryos (Fig. 1 B–D). LacZ staining was seen in two discrete spots flanking the midbrain midline (Fig. 1 C and D), in the FP of the hindbrain (but with a much higher intensity in rhombomeres 4 and 5; Fig. 1C), and throughout the spinal cord FP (Fig. 1 B and C).
E10.5.
(Four transient individuals generated.) Consistent expression was seen in the same regions described for E9 with the addition of small spots of somite expression (Figs. 1 E–G). At this stage, however, rostral hindbrain FP expression had become less defined. To determine in which specific cell layers the LacZ reporter gene was expressed within the hindbrain, Vibratome sections of several transgenic embryos were examined. These sections confirmed whole-mount observations of LacZ expression in the neural tube FP (data not shown).
E11.5.
(Transgenic line only.) Expression was still to be seen in the midbrain and spinal cord FP but to a lesser intensity. No expression could be seen in the hindbrain (Fig. 1H).
E12.5.
(Transgenic line only.) The only detectable expression was in the nasal pits (Fig. 1 I and J).
E13.5.
(Two transients generated.) No βgal expression was detected in any of the transgenic embryos recovered.
No expression was detected in any p29Stin2.10 transgenic individuals after this developmental time point or in any of the neonate mice or adults examined.
Transient Analysis of the p29Stin2.12 Construct.
Three transient transgenics were generated by using the p29Stin2.12 construct and were recovered at E10.5. The patterns produced were comparable to those generated by using the p29Stin2.10 construct in that midbrain, hindbrain, and FP expression was present. However, strong expression was seen in the rostral hindbrain, in a region that corresponded to rhombomeres 1 and 2, of all three p29Stin2.12 embryos recovered with an intensity similar to that seen in rhombomeres 4 and 5 (Fig. 2 C and D). This pattern is significantly different to all of the p29Stin2.10 individuals examined in this study, in which only low levels of βgal expression were seen in rhombomeres 1 and 2 (Fig. 2 A and B).
Discussion
Transgene Expression in the Embryonic Hindbrain.
We have shown in this study that two polymorphisms of the 5-HTT intron 2 VNTR region, Stin2.10 and Stin2.12, which are correlated with susceptibility to depressive disorders, act as strong and consistent positive transcriptional regulatory elements. Moreover, they were found to differ in the strength of their transcriptional inducing abilities within the developing rostral hindbrain at E10.5, where Stin2.12 seemed to be significantly stronger than the Stin2.10 polymorphism. These findings are supported by in vitro data that show that Stin2.12 was found to act as a significantly more potent positive regulator of marker gene expression than the Stin2.10 polymorphism when transformed into embryonic stem cells deprived of leukemia inhibitory factor (31). Several lines of research may serve to put these findings into context.
One study used in situ hybridization to show that, at E11, transcripts of the 5-HTT gene are also present in the rostral hindbrain FP just caudal of the mesencephalic flexure (14). We suggest that the Stin2.10/12 transcriptional regulator elements within the 5-HTT gene contribute to its transcriptional regulation within the rostral hindbrain. One reason that has been suggested to explain why 5-HTT is expressed in the hindbrain comes from the observation that little or no 5-HT is produced in the young rodent embryo (10, 11, 14), a situation that may also exist in the human embryo. Therefore, one role for the 5-HTT protein in development could be the concentration of 5-HT in the rostral hindbrain from maternal sources where it can then act as a morphogen (1–4, 6, 7). Several other studies have shown that 5-HT neurons first develop in the rostral hindbrain in a cluster designated B5-9 (32–35).
It is intriguing that, in addition to our findings, two other groups have identified the rostral hindbrain as being significant in terms of the expression of 5-HTT mRNA and the eventual location of 5-HTT cell clusters. Therefore, we hypothesize that a link may exist between the expression patterns produced by the Stin2.10/12 transcriptional regulator elements, the expression of 5-HTT mRNA, and the eventual hindbrain localization of 5-HT neuron cluster B5-9. We suggest that the difference in levels of expression in the rostral hindbrain between the two polymorphisms used in this study might provide a clue as to how these polymorphisms may contribute to depressive illness. Several groups have provided data that show that inappropriate expression of certain genes such as epidermal growth factor, protein tyrosine phosphorylase, and interleukin-6 (36–38) within the brains of transgenic animals results in abnormal CNS development. Thus, inappropriate levels of 5-HTT expression in the rostral hindbrain during development may be produced if the gene is driven by a stronger or weaker than normal positive transcriptional regulator. This situation may have the effect of increasing/decreasing local morphogenic 5-HT levels and leading to possible aberrant 5-HT neuron development. Because of the association of 5-HT neurons in the generation of different behavioral states (13), the situation described in this study may have repercussions for the emotional well being of an individual in later life. It is intriguing in this context that the rostral raphe nuclei, derived from rostral hindbrain, have been shown to send axons into the forebrain and cerebral cortex where they have been suggested to have a function in emotional modulation (34).
Stin2.10 and 2.12 Transcriptional Regulation in the Midbrain and Neural Tube FP.
With an absence of data showing expression of 5-HTT mRNA in the embryonic midbrain, rhombomeres 4 and 5, and spinal cord FP, it is not easy to reconcile these expression patterns produced by both constructs in the embryo with any known developmental process. However, several studies have shown that the proximity of sequences such as chromatin-anchoring sites and DNA hypersensitivity sites often influence the normal function of transcriptional control elements (39–42). The expression patterns may therefore reflect the removal of regulatory influences on the Stin VNTR elements once isolated from their normal promoter environment.
We have shown that a VNTR within intron 2 of the 5-HTT gene produces expression in the rostral hindbrain, where serotoninergic neurons are known to differentiate and 5-HTT is known to be expressed (32–35, 43, 44). However, this VNTR also produces expression in the midbrain, where dopaminergic neurons derive (13, 32, 33). It is interesting in this context that a structurally similar VNTR region close to the human dopamine transporter (DAT1) gene has been identified (45). In addition, several studies have also discovered correlations with different polymorphisms of the DAT1 VNTR and a susceptibility to drug addiction, schizophrenia, attention deficiency disorder, and chronic depression (46–49). Therefore, expression of the marker gene driven by the 5-HTT VNTR regions in the midbrain might hint at the existence of an overlap in the regulation of expression of the 5-HTT gene and the determination of dopaminergic cell clusters where the DAT1 gene may play a role. In agreement with this hypothesis, the DNA sequence of the 5-HTT VNTR contains extensive homology to that found within the DAT1 VNTR (31). The relationship between these VNTR sequences and their ability to support gene expression warrants further study.
The FP expression described in this paper is interesting with respect to 5-HTT expression where it is known that exposure of chick embryos to 5-HTT inhibitors, such as cocaine and antidepressants, leads to abnormal development of the FP and the notochord (50). It may be significant therefore that Stin2.10/12, situated in intron 2 of the 5-HTT gene, should drive expression in the spinal cord FP. Although no FP expression of 5-HTT has been reported, one study reveals the expression of 5-HTT by immunohistochemistry in the spinal cord at E12 in the rat (14). If Stin2.10 was shown to drive 5-HTT expression in the FP, it would not be unreasonable to extrapolate the role 5-HTT might play within the neural tube in modulating morphogenic 5-HT levels and neural tube dorsoventral axis determination.
Acknowledgments
We thank San Bing Shen, Bob Hill, and Nick Hastie for critically reading this manuscript and Brendan Doe and the rest of the staff at the Medical Research Council Evans Building for excellent technical support. This work was funded by the Medical Research Council.
Abbreviation
- 5-HT
5-hydroxytryptamine or serotonin
- CNS
central nervous system
- En
embryonic day n
- VNTR
variable number of tandem repeats
- βgal
β-galactosidase
- FP
floor plate
References
- 1.Buznikov G A, Shmukler Y B, Lauder J M. Cell Mol Neurobiol. 1996;16:537–59. doi: 10.1007/BF02152056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chubakov A R, Gromova E A, Konovalov G V, Sarkisova E F, Chumasov E I. Brain Res. 1986;369:285–297. doi: 10.1016/0006-8993(86)90537-8. [DOI] [PubMed] [Google Scholar]
- 3.Chubakov A R, Gromova E A, Konovalov G V, Chumasov E I, Sarkisova E F. Neurosci Behav Physiol. 1986;16:490–497. doi: 10.1007/BF01191453. [DOI] [PubMed] [Google Scholar]
- 4.Whitaker-Azmitia P M, Azmitia E C. Brain Res. 1989;497:80–85. doi: 10.1016/0006-8993(89)90972-4. [DOI] [PubMed] [Google Scholar]
- 5.Moiseiwitsch J R, Lauder J M. Proc Natl Acad Sci USA. 1995;92:7182–7186. doi: 10.1073/pnas.92.16.7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haydon P G, McCobb D P, Kater S B. Science. 1984;226:561–564. doi: 10.1126/science.6093252. [DOI] [PubMed] [Google Scholar]
- 7.Ivgy-May N, Tamir H, Gershon M D. J Neurosci. 1994;14:1011–1029. doi: 10.1523/JNEUROSCI.14-03-01011.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hellendall R P, Schambra U B, Liu J P, Lauder J M. Exp Neurol. 1993;120:186–201. doi: 10.1006/exnr.1993.1054. [DOI] [PubMed] [Google Scholar]
- 9.Hellendall R P, Schambra U, Liu J, Breese G R, Millhorn D E, Lauder J M. J Chem Neuroanat. 1992;5:299–310. doi: 10.1016/0891-0618(92)90017-k. [DOI] [PubMed] [Google Scholar]
- 10.Hansson S R, Mezey E, Hoffman B J. Neuroscience. 1998;83:1185–1201. doi: 10.1016/s0306-4522(97)00444-2. [DOI] [PubMed] [Google Scholar]
- 11.Hansson S R, Mezey E, Hoffman B J. Neuroscience. 1999;89:243–265. doi: 10.1016/s0306-4522(98)00281-4. [DOI] [PubMed] [Google Scholar]
- 12.Hillion J, Catelon J, Raid M, Hamon M, De Vitry F. Brain Res Dev Brain Res. 1994;79:195–202. doi: 10.1016/0165-3806(94)90124-4. [DOI] [PubMed] [Google Scholar]
- 13.Kandel E R, Schwartz J M, Jessel T M. Principles of Neural Science. New York: Elsevier; 1991. [Google Scholar]
- 14.Schroeter S, Blakely R D. Ann NY Acad Sci. 1996;801:239–255. doi: 10.1111/j.1749-6632.1996.tb17446.x. [DOI] [PubMed] [Google Scholar]
- 15.Borowsky B, Hoffman B J. Int Rev Neurobiol. 1995;38:139–199. doi: 10.1016/s0074-7742(08)60526-7. [DOI] [PubMed] [Google Scholar]
- 16.Heils A, Mossner R, Lesch K P. J Neural Transm. 1997;104:1005–1014. doi: 10.1007/BF01273314. [DOI] [PubMed] [Google Scholar]
- 17.Collier D A, Arranz M J, Sham P, Battersby S, Vallada H, Gill P, Aitchison K J, Sodhi M, Li T, Roberts G W, et al. NeuroReport. 1996;7:1675–1679. doi: 10.1097/00001756-199607080-00030. [DOI] [PubMed] [Google Scholar]
- 18.Collier D A, Stober G, Li T, Heils A, Catalano M, Di Bella D, Arranz M J, Murray R M, Vallada H P, Bengel D, et al. Mol Psychiatr. 1996;1:453–460. [PubMed] [Google Scholar]
- 19.Ogilvie A D, Harmar A J. Mol Med. 1997;3:90–93. [PMC free article] [PubMed] [Google Scholar]
- 20.Ogilvie A D, Russell M B, Dhall P, Battersby S, Ulrich V, Smith C A, Goodwin G M, Harmar A J, Olesen J. Cephalalgia. 1998;18:23–26. doi: 10.1046/j.1468-2982.1998.1801023.x. [DOI] [PubMed] [Google Scholar]
- 21.Gelernter J, Cubells J F, Kidd J R, Pakstis A J, Kidd K K. Am J Med Genet. 1999;88:61–66. [PubMed] [Google Scholar]
- 22.Evans J, Battersby S, Ogilvie A D, Smith C A, Harmar A J, Nutt D J, Goodwin G M. Neuropharmacology. 1997;36:439–443. doi: 10.1016/s0028-3908(97)00027-0. [DOI] [PubMed] [Google Scholar]
- 23.Bellivier F, Laplanche J L, Leboyer M, Feingold J, Bottos C, Allilaire J F, Launay J M. Biol Psychiatr. 1997;41:750–752. doi: 10.1016/S0006-3223(96)00524-0. [DOI] [PubMed] [Google Scholar]
- 24.Bellivier F, Henry C, Szoke A, Schurhoff F, Nosten-Bertrand M, Feingold J, Launay J M, Leboyer M, Laplanche J L. Neurosci Lett. 1998;255:143–146. doi: 10.1016/s0304-3940(98)00677-6. [DOI] [PubMed] [Google Scholar]
- 25.Hoehe M R, Wendel B, Grunewald I, Chiaroni P, Levy N, Morris-Rosendahl D, Macher J P, Sander T, Crocq M A. Am J Med Genet. 1998;81:1–3. doi: 10.1002/(sici)1096-8628(19980207)81:1<1::aid-ajmg1>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 26.Jorm A F, Henderson A S, Jacomb P A, Christensen H, Korten A E, Rodgers B, Tan X, Easteal S. Mol Psychiatr. 1998;3:449–451. doi: 10.1038/sj.mp.4000424. [DOI] [PubMed] [Google Scholar]
- 27.Battersby S, Ogilvie A D, Smith C A, Blackwood D H, Muir W J, Quinn J P, Fink G, Goodwin G M, Harmar A J. Psychiatr Genet. 1996;6:177–181. doi: 10.1097/00041444-199624000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Battersby S, Ogilvie A D, Blackwood D H, Shen S, Muqit M M, Muir W J, Teague P, Goodwin G M, Harmar A J. J Neurochem. 1999;72:1384–1388. doi: 10.1046/j.1471-4159.1999.721384.x. [DOI] [PubMed] [Google Scholar]
- 29.Yee S P, Rigby P W. Genes Dev. 1993;7:1277–1289. doi: 10.1101/gad.7.7a.1277. [DOI] [PubMed] [Google Scholar]
- 30.Hogan B, Beddington R, Constantini F, Lacy E. Manipulating the Mouse Embryo. Plainview, NY: Cold Spring Harbor Lab. Press; 1995. [Google Scholar]
- 31.Fiskerstrand, C. E., Lovejoy, E. & Quinn, J. P. (1999) FEBS Lett., in press. [DOI] [PubMed]
- 32.Hynes M, Rosenthal A. Curr Opin Neurobiol. 1999;9:26–36. doi: 10.1016/s0959-4388(99)80004-x. [DOI] [PubMed] [Google Scholar]
- 33.Ye W, Shimamura K, Rubenstein J L, Hynes M A, Rosenthal A. Cell. 1998;93:755–766. doi: 10.1016/s0092-8674(00)81437-3. [DOI] [PubMed] [Google Scholar]
- 34.Wallace J A, Lauder J M. In: Handbook of Chemical Neuroanatomy. Bjorklund A, Hökfelt T, Tohyama M, editors. Vol. 10. Amsterdam: Elsevier; 1992. pp. 619–645. [Google Scholar]
- 35.Aitken A R, Tork I. J Comp Neurol. 1988;274:32–47. doi: 10.1002/cne.902740105. [DOI] [PubMed] [Google Scholar]
- 36.Fattori E, Lazzaro D, Musiani P, Modesti A, Alonzi T, Ciliberto G. Eur J Neurosci. 1995;7:2441–2449. doi: 10.1111/j.1460-9568.1995.tb01042.x. [DOI] [PubMed] [Google Scholar]
- 37.Yeo T T, Yang T, Massa S M, Zhang J S, Honkaniemi J, Butcher L L, Longo F M. J Neurosci Res. 1997;47:348–360. doi: 10.1002/(sici)1097-4547(19970201)47:3<348::aid-jnr13>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 38.Coome G E, Kawaja M D. Neuroscience. 1999;90:941–955. doi: 10.1016/s0306-4522(98)00499-0. [DOI] [PubMed] [Google Scholar]
- 39.Wang D M, Taylor S, Levy-Wilson B. J Lipid Res. 1996;37:2117–2124. [PubMed] [Google Scholar]
- 40.Huber M C, Kruger G, Bonifer C. Nucleic Acids Res. 1996;24:1443–1452. doi: 10.1093/nar/24.8.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walters M C, Magis W, Fiering S, Eidemiller J, Scalzo D, Groudine M, Martin D I. Genes Dev. 1996;10:185–195. doi: 10.1101/gad.10.2.185. [DOI] [PubMed] [Google Scholar]
- 42.Eissenberg J C, Elgin S C. Trends Genet. 1991;7:335–340. doi: 10.1016/0168-9525(91)90424-o. [DOI] [PubMed] [Google Scholar]
- 43.Paxinos G. The Rat Nervous System. San Diego: Academic; 1995. [Google Scholar]
- 44.Molliver M E. J Clin Psychopharmacol. 1987;7:3S–23S. [PubMed] [Google Scholar]
- 45.Byerley W, Hoff M, Holik J, Caron M G, Giros B. Hum Mol Genet. 1993;2:335. doi: 10.1093/hmg/2.3.335. [DOI] [PubMed] [Google Scholar]
- 46.Blum K, Braverman E R, Wu S, Cull J G, Chen T J, Gill J, Wood R, Eisenberg A, Sherman M, Davis K R, et al. Mol Psychiatr. 1997;2:239–246. doi: 10.1038/sj.mp.4000261. [DOI] [PubMed] [Google Scholar]
- 47.Cook E H, Jr, Stein M A, Krasowski M D, Cox N J, Olkon D M, Kieffer J E, Leventhal B L. Am J Hum Genet. 1995;56:993–998. [PMC free article] [PubMed] [Google Scholar]
- 48.Gelernter J, Kranzler H R, Satel S L, Rao P A. Neuropsychopharmacology. 1994;11:195–200. doi: 10.1038/sj.npp.1380106. [DOI] [PubMed] [Google Scholar]
- 49.Gill M, Daly G, Heron S, Hawi Z, Fitzgerald M. Mol Psychiatr. 1997;2:311–313. doi: 10.1038/sj.mp.4000290. [DOI] [PubMed] [Google Scholar]
- 50.Wallace J A. Neurotransmitters and Early Embryogenesis. Berlin: Springer; 1988. [Google Scholar]