

Abstract
Several association studies support the hypothesis that genetic variants can modify the influence of environmental factors on behavioral outcomes, i.e., G × E interaction. The case-control design used in these studies is powerful, but population stratification with respect to allele frequencies can give rise to false positive or false negative associations. Stratification with respect to the environmental factors can lead to false positives or false negatives with respect to environmental main effects and G × E interaction effects as well. Here we present a model based on Fulker et al. (1999) and Purcell (2002) for the study of G × E interaction in family-based association designs, in which the effects of stratification can be controlled. Simulations illustrate the power to detect genetic and environmental main effects, and G × E interaction effects for the sib pair design. The power to detect interaction was studied in eight different situations, both with and without the presence of population stratification, and for categorical and continuous environmental factors. Results show that the power to detect genetic and environmental main effects, and G × E interaction effects, depends on the allele frequencies and the distribution of the environmental moderator. Admixture effects of realistic effect size lead only to very small stratification effects in the G × E component, so impractically large numbers of sib pairs are required to detect such stratification.
Keywords: Gene by environment interaction, Genetic association, Sib pair-based designs
Introduction
Several studies have demonstrated that genetic variants may modify the influence of environmental factors on behavioral outcomes, or, equivalently, that environmental factors modify the effects of genes (e.g., Caspi et al. 2002, 2003; Foley et al. 2004; Huizinga et al. 2004; Yaffe et al. 2000). Recently, for example, Lasky-Su et al. (2007) reported SNP-by-socioeconomic status interaction with respect to attention hyperactivity deficit (ADHD) symptom count in and around the BDNF gene. Although some of these studies may be subject to methodological limitations (Eaves 2006), gene by environment interaction (G × E) should be considered in genetic association studies.
Most genetic association studies are based on a case-control design. While case-control designs for genetic association are powerful, they suffer from potential effects of population stratification, leading to false positives or negatives (e.g., Cardon and Bell 2001; Posthuma et al. 2004). Family-based designs, which compare genetically related subjects, are therefore preferred. Fulker et al. (1999) proposed a design for association analysis of quantitative traits in sib pair data using maximum-likelihood variance-components procedures. They showed that the design is robust against spurious association stemming from population stratification, because the association effect is decomposed into a within-family effect and a between-family effect. The within-family effect is free of the potential effects of population stratification, because sibling pairs are drawn from the same family, and thus from the same genetic stratum. This design was extended by Neale et al. (1999) to include covariates, and by Abecasis et al. (2000a) to include multiple sibs, and parental information. The Fulker model now forms the basis for widely used statistical packages such as QTDT (Abecasis et al. 2000a, b).
Just like the association between genotypes and phenotypes, the associations between the environment and a phenotype, and between the G × E interaction and a phenotype are susceptible to the effects of population stratification. If two populations with (a) different allele frequencies, (b) different environmental frequencies (categorical environmental measure) or different environmental means (continuous environmental measure), and (c) different phenotypic means, are mixed, spurious environmental effects and spurious interaction effects can result, in addition to spurious allelic effects. In the sib pair design, it is therefore expedient to decompose into orthogonal between- and within-family effects (1) the allelic association; (2) the main effect of the environment; and (3) the G × E interaction.
In the present paper, we extend the sib pair model proposed by Fulker et al. (1999) to include environmental main effects and G × E interaction effects. The measured environmental variable may be either categorical or continuous. We report simulations carried out to investigate the statistical power to detect the presence of environmental main effects and G × E interaction effects for different effect sizes, different allele frequencies, and different environmental frequencies or means. In addition, we examine the statistical power to detect spurious G × E interaction due to population stratification.
Sib pair-based association including environmental effects and G × E interaction
We assume a diallelic marker with allele A1 with frequency p, and allele A2 with frequency 1 − p = q, and genotypes A1A1, A1A2 and A2A2 with genotypic effects a, d and −a, respectively, (Fisher 1918; Falconer and Mackay 1996). For simplicity we assume throughout the paper that the marker under study is the actual quantitative trait locus (QTL), i.e., recombination fraction θ is zero. In reality, the genotypic value of a marker is unequal to zero only if the marker is the QTL, or if the marker is in linkage disequilibrium (LD) with the QTL. We assume that the observed trait value of an individual is a function of a major gene effect (QTL), an additive polygenic genetic background effect, a shared familial or ‘common environmental’ effect, and an unshared, unique environmental effect (which also includes measurement error). Furthermore we assume that the effects of the additive polygenic genetic background, the common and unique environment, and the QTL are mutually uncorrelated, and that the additive polygenic genetic background effect and the environmental effects are normally distributed with mean zero.
If data from sibling pairs are available, the additive and dominance QTL effects may be partitioned into between- and within-family effects, as specified in Fulker et al. (1999), Abecasis et al. (2000a, b), and Posthuma et al. (2004). We will now introduce parameters for the effects of the environment, and for the interaction between genotype and environment.
Categorical environment
We assume that there are two environmental levels or conditions. The probability of being in either one of the environmental conditions is assumed not to depend on one’s genotype, i.e., the correlation between genotype and environmental status (rGE) is zero. We also assume that the probability of being in either one of the environmental conditions is independent of the environmental condition of other family members.
We adopt the notation of van den Oord (1999), and model the environmental main effect (e) as the difference in the phenotypic means of environmental Conditions 1 and 2. To model the interaction effect, we assign interaction effect i to subjects with genotype A1A1 in Condition 2, interaction effect −i to subjects with genotypes A2A2 in Condition 2, and interaction effect c to the heterozygotes A1A2 in Condition 2. Modeled as such, the interaction parameter i represents the difference between genotypic value a in Condition 2, and genotypic value a in Condition 1, after the main effect of the environmental condition has been taken into account. Similarly, interaction parameter −i represents the difference between genotypic value −a in Condition 2 and genotypic value −a in Condition 1, after accounting for the environmental main effect. The interaction parameter c represents the difference between the dominance effect in Condition 2 and in Condition 1, once the main effect of the environment has been accounted for (see Mather and Jinks 1977, for a similar parameterization). For the purpose of illustration, the expected phenotypic means
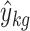 (i.e., the expected score of subjects in condition k with genotype g) are presented in Table 1 for the case of an environment with three levels.
(i.e., the expected score of subjects in condition k with genotype g) are presented in Table 1 for the case of an environment with three levels.
Table 1.
Expected phenotypic means for genotypic groups distinguished with respect to three environmental conditions
| Genotype | Mean | ||
|---|---|---|---|
| Condition 1 | Condition 2 | Condition 3 | |
| A1A1 | τi + a | τi + a + e 1 + i 1 | τi + a + e 2 + i 2 |
| A1A2 | τi + d | τi + d + e 1 + c 1 | τi + d + e 2 + c 2 |
| A2A2 | τi − a | τi − a + e 1 − i 1 | τi − a + e 2 − i 2 |
Note: τi denotes the family-specific intercept, a denotes the additive genotypic value, d denotes the dominance deviation for the heterozygotes, e 1 and e 2 denote the main effects for the environment (i.e., the in- or decrease of the phenotypic mean in Conditions 2 and 3 compared to Condition 1), i 1 and i 2 denote the G × E interaction effects for homozygotes (i.e., the difference between the genotypic values a in Conditions 2 and 3 compared to the genotypic value a in Condition 1, after the main effect for environment is accounted for), and c 1 and c 2 denote the G × E interaction for heterozygotes (i.e., the difference between the dominance effect in Conditions 2 and 3 compared to the dominance effect in Condition 1, once the main effect of the environment is accounted for)
Note that in the case of sib pair data (or data including multiple siblings and parents), various combinations of these means models are likely to be observed. When family-data are available, the effects of the QTL, the environmental measure under study, and their interaction, may be further partitioned into between- and within-family effects. To illustrate, for sib pairs and a dichotomous environment, all possible combinations are presented in Tables 2–4.
Table 3.
Both sibs in environmental Condition 2
| Genotype | Genotypic effect | Environmental effect | Additive and interaction | Dominance and interaction | Environment | Partitioned effects | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sib1 | Sib2 | Sib1 | Sib2 | Sib1 | Sib2 | Between mean | Within diff/2 | Between mean | Within diff/2 | Between mean | Within diff/2 | Sib1 (B + W) | Sib2 (B − W) |
| A1A1 | A1A1 | a + i | a + i | e | e | ab + ib | 0 | 0 | 0 | eb | 0 | [ab + ib] + eb | [ab + ib] + eb |
| A1A1 | A1A2 | a + i | d + c | e | e | ½(ab + ib) | ½(aw + iw) | ½(db + cb) | −½(dw + cw) | eb | 0 | ½[ab + aw + ib + iw] + ½[db − dw + cb − cw)] + eb | ½[ab − aw + ib − iw] + ½[db + dw + cb + cw] + eb |
| A1A1 | A2A2 | a + i | −a − i | e | e | 0 | aw + iw | 0 | 0 | eb | 0 | [aw + iw] + eb | −[aw + iw] + eb |
| A1A2 | A1A1 | d + c | a + i | e | e | ½(ab + ib) | −½(aw + iw) | ½(db + cb) | ½(dw + cw) | eb | 0 | ½[ab − aw + ib − iw] + ½[db + dw + cb + cw] + eb | ½[ab + aw + ib + iw] + ½[db − dw + cb − cw] + eb |
| A1A2 | A1A2 | d + c | d + c | e | e | 0 | 0 | db + cb | 0 | eb | 0 | [db + cb] + eb | [db + cb] + eb |
| A1A2 | A2A2 | d + c | −a − i | e | e | −½(ab + ib) | ½(aw + iw) | ½(db + cb) | ½(dw + cw) | eb | 0 | −½[ab − aw + ib − iw] + ½[db + dw + cb + cw] + eb | −½[ab + aw + ib + iw] + ½[db − dw + cb − cw)] + eb |
| A2A2 | A1A1 | −a − i | a + i | e | e | 0 | −(aw + iw) | 0 | 0 | eb | 0 | −[aw + iw] + eb | [aw + iw] + eb |
| A2A2 | A1A2 | −a − i | d + c | e | e | −½(ab + ib) | −½(aw + iw) | ½(db + cb) | −½(dw + cw) | eb | 0 | −½[ab + aw + ib + iw] + ½[db − dw + cb − cw] + eb | −½[ab − aw + ib − iw] + ½[db + dw + cb + cw)] + eb |
| A2A2 | A2A2 | −a − i | −a − i | e | e | −(ab + ib) | 0 | 0 | 0 | eb | 0 | −[ab + ib] + eb | −[ab + ib] + eb |
The main effect for the environmental and the G × E interaction are non-zero in both sibs
Table 2.
Both sibs in environmental Condition 1
| Genotype | Genotypic effect | Environmental effect | Additive and interaction | Dominance and interaction | Environment | Partitioned effects | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sib1 | Sib2 | Sib1 | Sib2 | Sib1 | Sib2 | Between mean | Within diff/2 | Between mean | Within diff/2 | Between mean | Within diff/2 | Sib1 (B + W) | Sib2 (B − W) |
| A1A1 | A1A1 | a | a | 0 | 0 | ab | 0 | 0 | 0 | 0 | 0 | ab | ab |
| A1A1 | A1A2 | a | d | 0 | 0 | ½ ab | ½ aw | ½ db | −½ dw | 0 | 0 | ½[ab + aw] + ½[db − dw] | ½[ab − aw] + ½[db + dw] |
| A1A1 | A2A2 | a | −a | 0 | 0 | 0 | aw | 0 | 0 | 0 | 0 | aw | −aw |
| A1A2 | A1A1 | d | a | 0 | 0 | ½ ab | −½ aw | ½ db | ½ dw | 0 | 0 | ½[ab − aw] + ½[db + dw] | ½[ab + aw] + ½[db − dw] |
| A1A2 | A1A2 | d | d | 0 | 0 | 0 | 0 | db | 0 | 0 | 0 | db | db |
| A1A2 | A2A2 | d | −a | 0 | 0 | −½ ab | ½ aw | ½ db | ½ dw | 0 | 0 | ½[−ab + aw] + ½[db + dw] | −½[ab + aw] + ½[db − dw] |
| A2A2 | A1A1 | −a | a | 0 | 0 | 0 | −aw | 0 | 0 | 0 | 0 | −aw | aw |
| A2A2 | A1A2 | −a | d | 0 | 0 | −½ ab | −½ aw | ½ db | −½ dw | 0 | 0 | −½[ab + aw] + ½[db − dw] | ½[−ab + aw] + ½[db + dw] |
| A2A2 | A2A2 | −a | −a | 0 | 0 | −ab | 0 | 0 | 0 | 0 | 0 | −ab | −ab |
The main effect for the environmental and the G × E interaction are zero in both sibs
Table 4.
Sibs in different environmental conditions: sib1 in Condition 1 and sib2 in Condition 2
| Genotype | Genotypic effect | Environmental effect | Additive and interaction | Dominance and interaction | Environment | Partitioned effects | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sib1 | Sib2 | Sib1 | Sib2 | Sib1 | Sib2 | Between mean | Within diff/2 | Between mean | Within diff/2 | Between mean | Within diff/2 | Sib1 (B + W) | Sib2 (B − W) |
| A1A1 | A1A1 | a | a + i | 0 | e | ab + ½ ib | −½ iw | 0 | 0 | ½ eb | −½ ew | [ab + ½ ib − ½ iw] + ½[eb − ew] | [ab + ½ ib + ½ iw] + ½[eb + ew] |
| A1A1 | A1A2 | a | d + c | 0 | e | ½ ab | ½ aw | ½ db + ½ cb | −½ dw − ½ cw | ½ eb | −½ ew | ½[ab + aw] + ½[db − dw + cb − cw] + ½[eb − ew] | ½[ab − aw] + ½[db + dw + cb + cw] + ½[eb + ew] |
| A1A1 | A2A2 | a | −a − i | 0 | e | −½ ib | aw + ½ iw | 0 | 0 | ½ eb | −½ ew | [aw − ½ ib + ½iw] + ½[eb − ew] | [−aw − ½ ib − ½ iw] + ½[eb + ew] |
| A1A2 | A1A1 | d | a + i | 0 | e | ½ ab + ½ ib | −½ aw − ½ iw | ½ db | ½ dw | ½ eb | −½ ew | ½[ab − aw + ib − iw] + ½[db + dw] + ½[eb − ew] | ½[ab + aw + ib + iw] + ½[db − dw] + ½[eb + ew] |
| A1A2 | A1A2 | d | d + c | 0 | e | 0 | 0 | db + ½ cb | −½ cw | ½ eb | −½ ew | [db + ½ cb − ½ cw] + ½[eb − ew] | [db + ½ cb + ½ cw] + ½[eb + ew] |
| A1A2 | A2A2 | d | −a − i | 0 | e | −½ ab − ½ ib | ½ aw + ½ iw | ½ db | ½ dw | ½ eb | −½ ew | −½[ab − aw + ib − iw] + ½[db + dw] + ½[eb − ew] | −½[ab + aw + ib + iw] + ½[db − dw] + ½[eb + ew] |
| A2A2 | A1A1 | −a | a + i | 0 | e | ½ ib | −aw − ½ iw | 0 | 0 | ½ eb | −½ ew | −[aw − ½ ib + ½ iw] + ½[eb − ew] | [aw + ½ ib + ½ iw] + ½[eb + ew] |
| A2A2 | A1A2 | −a | d + c | 0 | e | −½ ab | −½ aw | ½ db + ½ cb | −½ dw − ½ cw | ½ eb | −½ ew | −½[ab + aw] + ½[db − dw + cb − cw] + ½[eb − ew] | −½[ab − aw] + ½[db + dw + cb + cw] + ½[eb + ew] |
| A2A2 | A2A2 | −a | −a − i | 0 | e | −ab − ½ ib | ½ iw | 0 | 0 | ½ eb | −½ ew | −[ab + ½ ib − ½ iw] + ½[eb − ew] | [−ab − ½ ib − ½ iw] + ½[eb + ew] |
The main effect for the environmental and the G × E interaction are zero in sib1 and non-zero in sib2
In the case of the sib pair association design, the phenotypic score y ijkg (i.e., the observed score y of subject j from family i in condition k with genotype g) is modeled as:
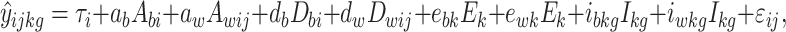 |
1 |
where τi is the family-specific intercept, and ɛ ij the residual term, i.e., the part of the phenotypic score y ijkg that is not explained by the measured QTL, the environmental measure, or the interaction between these two, and which may be due to background genetic, or background environmental effects, unmodeled interactions, or measurement error. The parameters a b and a w are the estimated between- and within-family additive genetic effects of the marker, which are weighted by the derived coefficients A bi and A wij, respectively. These coefficients are either −1, −½, 0, ½ or 1, as calculated in the 7th and 8th column of Tables 2–4 (see Fulker et al. 1999). Parameters d b and d w are the estimated between- and within-family dominance genetic effects of the marker, which are weighted by the derived coefficients D bi and D wij, respectively. These coefficients are either 0, ½ or 1, as calculated in the 9th and 10th column of Tables 2–4 (see Posthuma et al. 2004). Similarly, the parameters e bk and e wk represent the between- and within-family effects of environmental condition k, which are weighted by the derived coefficient E k. This coefficient is either −½, 0, ½ or 1, as calculated in the 11th and 12th column of Tables 2–4. The parameters i bkg and i wkg represent the between- and within-family effects of the interaction of genotype g and environmental Condition k, which are weighted by the derived coefficient I kg. This coefficient is either −½, 0, ½ or 1, as calculated in the 7th to 10th column of Tables 2–4.
Continuous environment
If the environmental measure is continuous in nature, rather than categorical, the model as presented in Eq. 1, is altered as follows. The between and within-family environmental parameters e b and e w are simply weighted by the subject’s score on the continuous environmental measure, E j, just as the genotype-dependent between and within-family interaction parameters i bg and i wg. The continuous environmental measure is now modeled as a continuous moderator, in the manner proposed by Purcell (2002). In the case of a continuous environmental measure, the phenotypic score y ijg is modeled as:
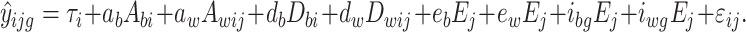 |
2 |
Given these additional effects of the environment and the G × E interaction, the variance-covariance matrix for siblings j and m of the ith family, Σ i, is given as:
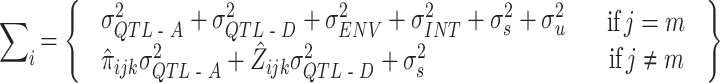 |
3 |
where σ2QTL-A is the variance due to the additive genetic effect of the marker, σ2QTL-D is the variance due to the dominance effects of the marker, σ2ENV is the variance due to the measured environmental indicator, and σ2INT is the variance due to the interaction between the marker and the environmental measure. After all these measured effects are accounted for, σ2s denotes the residual sibling resemblance, which is due to shared alleles other than the QTL alleles under study, shared environmental effects other than the measured environmental variable under study, or covariance between these two sources. Finally, σ2u denotes all variance that is not shared by siblings from the same family, and which is due to unshared alleles, and unshared environmental effects. The covariance between the phenotypic scores of siblings equals the additive and dominance QTL variance, weighted by 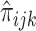 (the estimated proportion of alleles that siblings j and m from family i share IBD, i.e. p(IBD = 2) + ½ p(IBD = 1)) and
(the estimated proportion of alleles that siblings j and m from family i share IBD, i.e. p(IBD = 2) + ½ p(IBD = 1)) and 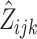 (the probability of complete IBD sharing between siblings j and m, i.e., p(IBD = 2)), respectively. Because we assumed the environmental effect under study to be unrelated to genotype (i.e. rGE = 0) or to family membership, the environmental effect and the interaction effect only contribute to the variance through σ2ENV and σ2INT, but not to the covariance between siblings j and m. Note that in practice, σ2ENV, σ2INT, and σ2u cannot be estimated individually (i.e., only the sum of them can be estimated). Note also that when the marker under study is indeed the actual QTL, as is assumed throughout this paper, and the environmental measure is an accurate reflection of the true environmental moderator, the expected variance–covariance matrix Σ
i reduces to
(the probability of complete IBD sharing between siblings j and m, i.e., p(IBD = 2)), respectively. Because we assumed the environmental effect under study to be unrelated to genotype (i.e. rGE = 0) or to family membership, the environmental effect and the interaction effect only contribute to the variance through σ2ENV and σ2INT, but not to the covariance between siblings j and m. Note that in practice, σ2ENV, σ2INT, and σ2u cannot be estimated individually (i.e., only the sum of them can be estimated). Note also that when the marker under study is indeed the actual QTL, as is assumed throughout this paper, and the environmental measure is an accurate reflection of the true environmental moderator, the expected variance–covariance matrix Σ
i reduces to
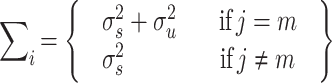 |
4 |
(Fulker et al. 1999), because the family variance–covariance matrices Σ i are formed conditionally on the marker genotypes of the siblings, and conditionally on their environmental status. Conditionally on the marker genotype and the environmental status of the siblings, there no longer is any variation in marker genotype or environmental status, so these variables no longer explain any variance. The effects of the marker and the environment are then modeled via the mean structure only, per Eqs. 1 and 2.
In the variance-components approach, the means and variances of related individuals are modeled simultaneously, as a function of the set of parameters θ which equals θ = {τi, a b, a w, d b, d w, e b, e w, i bg, i wg, σ2s, σ2u}, if the marker under study is the QTL. Maximum likelihood estimation can be used to obtain parameter estimates, and likelihood ratio tests can be used to test specific constraints on the parameters (Azzelini 1996). For example, one can test whether the regression weight for the between-family additive genetic marker effect, a b, is equal to the within-family additive genetic marker effect, a w, the idea being that a b only differs from a w when population stratification significantly influences the results of the test for genetic association.
Sib pair association models including a measured environment and G × E interaction effects can readily be implemented in the Mx software package1 (Neale et al. 2003). Appendices I and II include example Mx-scripts for the case of sib pair data and a dichotomous environment or a continuous environment, respectively. Adaptation of these scripts for the modeling of more than two siblings, or categorical environments with multiple levels is quite straightforward. Extension of these scripts to include data from nuclear families (parents and offspring; Abecasis et al. 2000a) requires some modifications which are spelled out in the Mx-script provided by Posthuma et al. (2004) in their Appendix II. An example script for the modeling of data from monozygotic and dizygotic twin pairs is available online.2 Note that whereas sib pair data only allow distinction between σ2s and σ2u, twin data allow a more detailed decomposition of the background variance into variance due to additive genetic effects (σ2A), common environmental effects (σ2C) or dominance genetic effects (σ2D), and unshared environmental effects (σ2E).
Power calculations for the G × E model
We performed a series of simulation studies to investigate the power of the extended sib pair model to detect the G × E interaction effects. We considered both a dichotomous environmental measure and a continuous environmental measure. All simulations were based on simulated sibling pairs only, and the simulated marker was assumed to be the actual QTL. The power analyses are thus limited to the detection of effects on the means (association), not the variances (linkage).
Procedures
Simulations involved a diallelic marker locus with frequency p of the increaser allele A1 being .5 or .2. Except where noted, QTL dominance effects were absent. In the case of a dichotomous environment, the frequency b 1 of Condition 1 was either .5 or .2. The continuous environmental measure was standard normally distributed, i.e., Environment ∼ N(0,1). Simulated environmental values were uncorrelated to the simulated genotypes (e.g., rGE = 0). The continuous phenotype was standard normally distributed when all measured allelic and environmental effects were zero. When these effects are not zero, the phenotypic mean and variance deviate from 0 and 1, respectively. The degree of deviation depends on their effect size.
The QTL effect, the environmental effect, and the interaction were manipulated so that in isolation, these factors each explained 1%, 2.5% or 5% of the total phenotypic variance in the total sample. In the simulations with a dichotomous environment, these effect sizes were determined for the case that both environmental conditions and alleles were evenly distributed (i.e., b 1 = b 2 = .5 and p = q = .5). Note, however, that the percentage of explained variance depends on the allele frequencies and the distribution of the environmental variable. For instance, if the parameters representing the genotypic effect of the QTL locus are chosen such that the locus explains 5% of the variance in the case that p = q = .5, this same locus (i.e., same genotypic values) only explains 3.3% of the variance in the case that p = .2. Likewise, an environmental effect that explains 5% of the variance if b 1 = b 2 = .5, explains only 3.3% of the variance if b 1 = .2. For the simulations including a continuous environment, effect sizes were determined for the case that p =q = .5.
Where noted, population stratification was generated by mixing two samples (A and B) of equal proportions, with different phenotypic means (μA and μB), and different marker allele frequencies (p A = .7, p B = .3). In the case of a dichotomous environment, environmental frequencies differed between samples A and B (b A1 = .3, b B1 = .7). In the case of a continuous environment, the environmental means differed between samples A and B (μenvA = 0, μenvB = 2). The phenotypic means of samples A and B were selected such that admixture accounted for 20% of the total phenotypic variance in the combined population, i.e., (μA − μB)2/4σ2TOT = .20 (see Abecasis et al. 2000a).3 The mixture of these two samples with different phenotypic means, different allele frequencies, and different environmental frequencies or means, results in spurious allelic, spurious environmental, and spurious interaction effects. The emphasis in these simulations is thus on the detection of false positives, but false negatives are theoretically possible (e.g., Posthuma et al. 2004; Neale et al. 1999).
For all simulations, background variance was modeled such that, after accounting for the QTL-effect, the environmental main effect, and the interaction, 30% of the remaining variance was attributable to additive polygenic genetic effects (A), and 70% was due to non-shared environmental effects (E). Covariance between the sibs due to shared environmental components (C) was fixed to zero, so all resemblance between the sibs was due to genetic factors only (i.e., the QTL and other unidentified genes). Because A and C cannot be distinguished unless the sample includes monozygotic twins, in addition to regular siblings or dizygotic twins, the term σ2s will include all the siblings’ resemblance due to shared genes other than the QTL, and common environmental influences. The term σ2u then includes all variance due to unidentified non-shared genes and non-shared environmental effects. Note that, in general, the power to detect the effects of interest increases as the residual sibling resemblance σ2s increases, even if the exact nature of resemblance (genes or environment) cannot be distinguished. This is because, as a result of increasing σ2s, the non-shared component σ2u decreases, and less unshared variance implies less “noise” (i.e., unexplained variance), which increases statistical power. The choice to fix shared environmental effects to zero in all simulations, thus results in conservative estimations of the power to detect the effects of interest.
All data simulations were performed in the R program,4 and exact data simulation was used for all analyses (van der Sluis et al. 2008; Bollen and Stine 1993; Dolan et al. 2005). Exact data simulation can be used when sufficient summary statistics are available in theory, i.e., when all information present in the raw data can be summarized sufficiently in the variance covariance matrix Σ, and the means vector μ. Exact data simulation implies the simulation of raw data that are transformed to fit the true model exactly. Consequently, when the true model is fitted to these data, all parameter estimates used to simulate the data are recovered exactly. Subsequently, the constrained, nested (wrong) model is fitted to the data, in which parameters of interest are fixed to zero, or constrained to be equal. Minus twice the difference in the log-likelihoods of the true model and the nested model asymptotically equals the non-centrality parameter λ of the non-central χ2-distribution, with df equal to the difference in the number of parameters estimated. This non-centrality parameter can subsequently be used to calculate the sample size N required for a chosen power level, given a chosen critical value α (Saris and Satorra 1993).5
The results of power analyses based on exact data simulation equal exactly the results obtained through the analysis of (population or expected) summary statistics Σ and μ. Also, as in power calculations based on summary statistics, these results are asymptotically similar to results obtained through Monte Carlo simulation (depending on the number of runs, and the sample size N used in the Monte Carlo procedure). In contrast to Monte Carlo simulation, however, exact data simulation obviates the requirement to replicate the analyses in different runs because the quasi-randomly generated data are transformed to fit the true model exactly. Exact data simulation is therefore not only easy to perform but also computationally light compared to Monte Carlo simulation, which is why we chose to use exact data simulation here. We refer to Van der Sluis et al. (2008) for an extensive discussion on exact data simulation.
Given non-centrality parameter λ, the Mx program computes the total sample size that would be required, given the reported proportion of subjects in each distinguishable group, to reject the tested hypothesis at various power levels, ranging from .25 to .99. Here, we focus on the conditions required for a power of 80%. For all statistical tests, α was chosen to equal .05.
Patterns of G × E interaction
The power to detect G × E interaction was studied given eight different patterns of interactions (see also Van den Oord 1999; Khoury et al. 1988, 1993). These eight designs are illustrated in Fig. 1 for a dichotomous environment. Design (i) concerns the situation that all effects are zero except the interaction effect for the homozygotes. As a result, the phenotypic means are equal across genotypes in Condition 1, but they are increased or decreased in the homozygotes in the second environmental condition. Design (i = c) represents a variation on Design(i); here the interaction effect in the heterozygotes is also assumed to be non-zero. More specifically, the interaction effect in the heterozygotes is set to equal the effect in the A1 homozygotes (i.e., ‘complete interaction dominance’). The phenotypic mean of the heterozygotes (A1A2) therefore equals the phenotypic mean of the group with genotype A1A1 in both the first and the second environmental condition. Design (i,e) applies when the environmental main effect and the interaction effect in the homozygotes are greater than zero. Design (i,a) is a function of a non-zero allelic effect (A1 being the increaser allele), and a non-zero interaction effect. As a result, the phenotypic means of the three genotypic groups differ in Condition 1, and fan out even more in Condition 2. Design (i,a,d) is a variation on Design(i,a), with the difference that complete genetic dominance is present under environmental Condition 1, while the interaction effect in the heterozygotes remains zero. As a consequence, the phenotypic means in the groups with genotype A1A1 and A1A2 are equal in Condition 1, but differ in Condition 2 due to different interaction effects. In Design (i,a,e), allelic effects, environmental main effects and interaction effects are non-zero, and dominance is absent for all effects. Design (−i,a) is a variation on Design(i,a), where both allelic effects and interaction effects are non-zero. For Design(−i,a), however, the signs of the interaction effects are reversed, resulting in crossing lines. As a consequence, the group with the highest phenotypic mean in environmental Condition 1, has the lowest phenotypic mean in environmental Condition 2, and vice versa. Design (−i,a,e) resembles Design(−i,a), except that in addition, environmental main effects are non-zero as well.
Fig. 1.

Different patterns of genotype-environment interaction. Design (i): interaction effect for homozygotes, no main effects; Design (i = c): interaction effect for homozygotes and heterozygotes, with interaction effect heterozygotes equal to effect A1 homozygotes, no main effects; Design (i,e): interaction effects homozygotes, and main effect environment; Design (i,a): interaction effect homozygotes, and QTL effect; Design (i,a,d): interaction effect homozygotes, and main effect QTL including dominance; Design (i,a,e): interaction effects homozygotes, and main effects environment and QTL; Design (−i,a): reversed interaction effects homozygotes, and main effect QTL; Design (−i,a,e): reversed interaction effects homozygotes, and main effects environment and QTL
Results
All tables with results of power analyses (Tables 5–7) show the number of sib pairs required for a power of 80% given α = .05; non-centrality parameters are not reported here but are available online.6
Table 5.
Number of sib pairs required to detect main effects of QTL and environment, and G × E interaction effects of different effect sizes, in the context of different allele frequencies, and different types of environments (categorical versus continuous) for power of .80 with α = .05 when all other effects are 0
| Effect size | Zero effects freely estimated | Zero effects fixed to zero | |||||
|---|---|---|---|---|---|---|---|
| Environment | Environment | ||||||
| b 1 = .2 | b 1 = .5 | Continuous | b 1 = .2 | b 1 = .5 | Continuous | ||
| Main effect QTLa | |||||||
| 1% | p = .2 | 3,607 | 1,441 | 737 | 600 | 600 | 600 |
| 2.5% | 1,400 | 561 | 288 | 235 | 235 | 235 | |
| 5% | 664 | 266 | 142 | 116 | 116 | 116 | |
| 1% | p = .5 | 2,290 | 921 | 473 | 385 | 385 | 385 |
| 2.5% | 890 | 359 | 185 | 151 | 151 | 151 | |
| 5% | 422 | 171 | 92 | 75 | 75 | 75 | |
| Main effect environmentb | |||||||
| 1% | p = .2 | 3,864 | 2,468 | 2,581 | 600 | 384 | 386 |
| 2.5% | 1,509 | 964 | 1,010 | 228 | 146 | 152 | |
| 5% | 716 | 458 | 490 | 110 | 71 | 75 | |
| 1% | p = .5 | 1,157 | 743 | 770 | 600 | 384 | 386 |
| 2.5% | 452 | 291 | 302 | 228 | 146 | 152 | |
| 5% | 216 | 139 | 147 | 110 | 71 | 75 | |
| Interactiona | |||||||
| 1% | p = .2 | 1,111 | 710 | 725 | 149 | 186 | 278 |
| 2.5% | 415 | 266 | 289 | 57 | 70 | 111 | |
| 5% | 186 | 120 | 142 | 27 | 33 | 55 | |
| 1% | p = .5 | 706 | 454 | 463 | 117 | 296 | 378 |
| 2.5% | 265 | 171 | 186 | 44 | 70 | 151 | |
| 5% | 119 | 77 | 92 | 21 | 33 | 75 | |
aThese power calculations assume 2 degrees of freedom
bThese power calculations assume 1 degree of freedom
Table 7.
Number of sib pairs required to detect spurious (H1:B = W vs. H0: B≠W) and genuine (H1:B≠W = 0 vs. H0: B≠W≠0) G × E interaction effects in eight different conditions for power of .80 with α = .05a
| Dichotomous environment | Continuous environment | |||
|---|---|---|---|---|
| B = W | B≠W = 0 | B = W | B≠W = 0 | |
| Design(i) | 4,745 | 274 | 2,543 | 184 |
| Design(i = c) | 4,129 | 192 | 3,277 | 126 |
| Design(i,e) | 3,832 | 274 | 4,000 | 184 |
| Design(i,a) | 3,470 | 274 | 2,071 | 184 |
| Design(i,a,d) | 3,658 | 274 | 2,146 | 184 |
| Design(i,a,e) | 2,914 | 274 | 3,045 | 184 |
| Design(−i,a) | 6,922 | 274 | 3,235 | 184 |
| Design(−i,a,e) | 5,318 | 274 | 5,563 | 184 |
B = W, test for presence of population stratification; B≠W = 0, test for significance of within-family interaction effect only
aAll tests assume two degrees of freedom
To start with, we studied the power to detect specific effects in the situation where all other effects are zero. The simulated data included either a main effect for the QTL, or a main effect of the environment, or a G × E interaction effect (i.e., interaction in the absence of main effects). Within this context, we studied the effects on the power of allele frequencies, the scale of the environmental measures (dichotomous or continuous), and in the case of a dichotomous environment, the frequencies of the environmental conditions. Knowledge of the power to detect isolated effects of given effect sizes, provides a useful guide to subsequent analyses, where interaction effects are tested in the presence of other effects. Data were simulated such that the specific effects explained 1%, 2.5% or 5% of the variance when p = .5 and, if applicable, b 1 = .5. Note that these simulations included no population stratification. All between and within parameters could thus be constrained to be equal without loss of fit (given the exact data simulation, this implies χ2 = 0 for all tests concerning admixture effects). Recall that the background variance (i.e., the variance not due to the marker under study, the environmental measure under study, or their interaction) was simulated to consist of 30% additive polygenic genetic effects (σ2s) and 70% environmental effects not-shared by the siblings (σ2u). In addition, note that in determining the power to detect the effects of interest, we first fitted the full model, i.e., the model including all effects, both zero and non-zero effects. Subsequently we fitted the model in which only the parameters of interest were constrained to zero.
The results are presented in the first three columns of Table 5. With respect to the main effects of the QTL, all tests have 2 degrees of freedom (df), as parameters for both additive and dominance allelic effects are constrained to zero. The power is greatest when p = q = .5, and when the environment is a continuous measure. A more uneven distribution of alleles is detrimental to the power to detect allelic effects, as is an uneven distribution of environmental conditions in the case of a dichotomous environmental measure. Interestingly, the distribution of the environmental variable influences the power to detect the QTL main effect, even though association between the environment and the phenotype is absent. These results are consistent with those in Table 6 of Neale et al. (1999).
All tests for environmental main effects have one degree of freedom. As can be seen from the first three columns of Table 5, the power to detect main effects of the environment is somewhat lower when the environmental effects are continuous, compared to a dichotomous environment with equally distributed conditions. The power to detect environmental main effects is lowest when both the alleles and the environmental conditions are unevenly distributed. Evidently, the allele frequencies influence the power to detect the environmental main effect when the genotypic effects are estimated freely, even though association between the QTL and the phenotype is absent.
All tests for interaction effects are 2 df-tests as both the interaction effects in the homozygotes and the heterozygotes are constrained to zero. The first three columns of Table 5 show that the power to detect interaction effects is greatest when both the allele frequencies and the environmental frequencies are evenly distributed. The power to detect interaction in the context of a continuous environment is only slightly lower.
In conclusion, if alleles are approximately evenly distributed, representative samples of about 200–400 sibling pairs are sufficient to detect main effects for the QTL or the environment, or interaction effects with effect sizes as small as 2.5–5% of the variance.
For illustrative purposes, the last three columns of Table 5 show the sample sizes required to detect the isolated effects with a power of 80% when all zero-effects are actually fixed to zero. As knowledge about which effects are actually zero is usually absent in practice, this is not a realistic situation. It does however illustrate two interesting points. First, the power to detect the effects of interest is much better in the context of a more constrained model. Practically, this implies that the order in which constraints are imposed on the model, may determine the probability to detect effects. This is something to bear in mind when deciding on model fitting procedures. Second, we previously noted that the power to detect effects (e.g., a QTL main effect, an environmental main effect) depends on the distribution of other variables in the model (e.g., the environmental variable, allele frequencies), even when these other variables are not actually associated with the phenotype under study. Naturally, this effect disappears when these zero-effects are excluded from the model.
Subsequently, we examined the power to detect genuine G × E interaction effects in the eight different designs distinguished by van den Oord (1999, see Fig. 1). For these simulations, parameter values for all non-zero effects were chosen such that in isolation, these effects would explain 2.5%. However, in the case of a dichotomous environment, the presence of other effects influences the percentage of variance explained by the G × E interaction. Using the same parameter values, the actual percentage of variance explained by the G × E interaction varied from 2.1% for Design(i,a,e), to 3.5% for Design(i = c). Also, as is well known in the context of ANOVA analysis, interaction effects can show up as main effects. In this case, the interaction effects show up as allelic main effects when the environment is dichotomous. Consequently, for Design(i) through Design(i,a,e), the main effects of the QTL deviated from zero, with effect sizes ranging from 2.5% (Design(i)) to 8.6% (Design(i,a)). For Design(−i,a) and Design(−i,a,e) on the other hand, the QTL main effect explained 0% of the variance as the actual effect of the QTL was nullified entirely by the reversed interaction effect. The G × E interaction only turned up as a main environmental effect in Design(i = c). In all cases that the main effect of the environment was specifically modeled to be larger than zero (Design(i,e), Design(i,a,e) and Design(−i,a,e)), the effect was slightly lower than 2.5% (2.3, 2.0 and 2.4, respectively) due to the presence of the G × E interaction effect. Again, the background variance was simulated to consist of 30% additive polygenic genetic effects (σ2s), and 70% environmental effects not-shared by the siblings (σ2u) in all conditions. These simulations included no population stratification, so all between and within parameters could be constrained to be equal without loss of fit.
The results of these simulations are in Table 6. All tests are 2 df-tests, as both interaction effects for the homozygotes and the heterozygotes are constrained to zero. Note that, irrespective of the allele frequencies, and the measurement scale of the environment, the power to detect interaction effects is higher for Design(i = c) than for Design(i). This makes sense, because the heterozygous group only contributes to the power to detect G × E interaction if the heterozygous interaction effect deviates from zero (Design(i = c) and not Design(i)). Note also that the power to detect the interaction in the context of complete interaction dominance (Design(i = c)) is higher given p = .2 than given p = .5. This is because the distribution of the informative genotypic groups is more even in the case of p = .2 (i.e., A1A1 + A1A2 vs. A2A2: .51:.49) than in the case of p = .5 (i.e., A1A1 + A1A2 vs. A2A2: .75:.25), which increases the power to detect the effects of interest.
Table 6.
Number of sib pairs required to detect G × E interaction effects in eight different conditions (see Fig. 1) for power of .80 with α = .05a
| Environment | ||||||
|---|---|---|---|---|---|---|
| b 1 = .2 | b 1 = .5 | Continuous | ||||
| p = .2 | p = .5 | p = .2 | p = .5 | p = .2 | p = .5 | |
| Design(i) | 410 | 261 | 263 | 169 | 289 | 185 |
| Design(i = c) | 144 | 175 | 93 | 113 | 102 | 124 |
| Design(i,e); Design(i,a); Design(i,a,d); Design(i,a,e); Design(−i,a); Design(−i,a,e) | 410 | 261 | 263 | 169 | 289 | 185 |
aAll tests assume two degrees of freedom
The power to detect the interaction effect is not influenced by the presence or absence of an environmental main effect (Design(i,e) versus Design(i), and Design(i,a,e) and Design(−i,a,e) versus Design(i,a), Design(i,a,d) and Design(−i,a)). This is understandable, given that the environmental main effect only influences the phenotypic means of the genotypic groups, but not the differences in phenotypic means between the genotypic groups. The environmental main effect may thus be viewed as a constant, which does not influence the power to detect interaction.
The presence or absence of a main effect of the QTL also has no influence on the power to detect G × E interaction. (To assure that this finding was not due to the size of the allelic effect, additional analyses including a larger allelic effect, explaining 10% and 20% of the variance rather than 2.5%, were run, which showed similar results.)
Finally, we studied the power to detect population stratification with respect to the interaction component of the model. As described above, we mixed two subsamples of equal proportions, which differed with respect to allele frequencies (p A = .7, p B = .3), and environmental distribution (in case of a dichotomous environmental measure, b A1 = .3, b B1 = .7; in case of a continuous environmental measure, μenvA = 0, μenvB = 2), choosing phenotypic means of the subsamples such that the admixture accounted for 20% of the total phenotypic variance in the combined sample. When these admixture proportions were used to simulate data in which the actual effects (allelic, environmental and interaction) were zero, spurious allelic, environmental, and interaction effects were observed in the combined sample due to the admixture. For the dichotomous environment, the between family effects deviated from the within family effects, with the stratification effect being largest for the allelic effects (N = 184 for 80% power), intermediate for the environmental effects (N = 1,465 for 80% power), and smallest for the interaction effects (N = 7,028 for 80% power). For the continuous environment, the between family effects also deviated from the within family effects, with the stratification effect being largest for the environmental effects (N = 278 for 80% power), medium for the allelic effects (N = 576 for) and smallest for the interaction effects (N = 3,233 for 80% power). It is clear that very large numbers of sib pairs are required to detect stratification effects in the interaction component. It is also noteworthy that the allele frequencies in the subsamples determine how the spurious G × E interaction is expressed. With the present admixture settings (p A = .7, p B = .3, i.e., contrasting allele frequencies), spurious G × E is only apparent with respect to the interaction parameter for the heterozygous group, while the interaction parameter for the homozygous group obtained in the combined sample does not deviate from its actual value in the subsamples. However, if the allele frequencies in the subsamples are not contrasting (e.g., p A = .3, p B = .5), both interaction parameters for the heterozygous and homozygous groups are informative about spurious interaction.
Given population stratification, we again considered the eight different interaction designs (Fig. 1) to study (a) the power to detect stratification effects with respect to the interaction component of the model (tests with 2 df as both homozygote and heterozygote interaction effects are constrained to be equal within and across families) and (b) the power to detect genuine interaction effects (tests with 2 df as both homozygote and heterozygote interaction effects are constrained to be zero within families, while the between-family effects are freely estimated). For all conditions, the background variance was again simulated to consist of 30% additive polygenic genetic effects (σ2s), and 70% environmental effects not-shared by the siblings (σ2u).
The results are presented in Table 7. Besides confirming the observation that prohibitively large samples of sib pairs are required to detect spurious interaction (B = W), it is shown that the power to detect the spurious interaction due to population admixture varies across the eight differentiated subtypes. Overall, the power to detect spurious interaction is somewhat higher when the environment is continuous in nature, but the sample sizes required to detect stratification with respect to the interaction effect are prohibitively large in all simulated scenarios.
An indication of the power to detect the genuine interaction effect is obtained by freely estimating the between-family effect, while the within-family effect is constrained to be zero (B≠W = 0). The results in Table 7 show that the power to detect G × E effects on the means is about as large as one would expect given the previous results presented in Table 6, and the distribution of the genotypes in the mixed population (i.e., freq(A1A1) = (.72 + .32)/2 = .29; freq(A1A2) = ((2 * .3 * .7) + (2 * .3 * .7))/2 = .42; freq(A2A2) = (.32 + .72)/2 = .29).
Discussion
In this paper, the family-based association design was extended to include G × E interaction effects and environmental main effects. Power calculation showed that allele frequencies, and characteristics of environment (e.g., measurement level, and in the case of a categorical environment, the frequencies of the environmental conditions) affect the power to detect G × E interaction. Relatively small interaction effects, explaining 2.5–5% of the phenotypic variance in the total sample, can be detected with reasonably small sample sizes (200–400 sib pairs, respectively), if alleles are evenly distributed. The power to detect main effects and interaction effects generally is reasonable, particularly when all zero-effects are removed from the model first.
Throughout the paper, we assumed that the marker locus under study is the actual QTL. In practice, this will often not be the case and markers will usually be more or less strongly in LD with the QTL. Also, a criterion level α of .05 was employed in the simulation studies. Often, however, one will not test for association in one, but several marker loci, and α will be adjusted downwards to control for Type I errors. The power results presented here thus concern the most favorable conditions, and in practice, larger sample sizes may be required to obtain a power of 80%.
Modeling measured environmental effects in association studies is standard (e.g., Caspi et al. 2002, 2003; Foley et al. 2004; Huizinga et al. 2006; Lasky-Su et al. 2007; Yaffe et al. 2000). The use of the extended sib pair model in such studies has the advantage of controlling for population stratification, and excluding spurious main effects of the QTL and the environment, and, given sufficiently large sample size, spurious interaction effects. This extension can be implemented readily in packages such as Mx (Appendices I and II), or, in case of a categorical environmental factor, in SPSS (Beem and Boomsma 2006).
Some caveats are in order. First, it has often been shown that non-normality can result in spurious interaction effects (e.g., Boomsma and Martin 2002; Martin 1999; Purcell 2002; van den Berg et al. 2007; van der Sluis et al. 2006). However, the actual presence of G × E can also render the distribution non-normal (e.g., Purcell 2002; van der Sluis et al. 2006), resulting in the problem that non-normality of the data can either indicate the presence of G × E (i.e., G × E being the source of the non-normality) or mimic the presence of G × E (i.e., non-normality due to e.g. censoring or poor scaling of the phenotypic measure). The model presented here is equally susceptible to this phenomenon.
Although there is no ready solution to this problem, researchers should at least investigate alternative reasons for the non-normality of their data than the presence of G × E (e.g., poor measurement scale, selective sampling, etc.). As has been argued before (e.g., Martin 1999; van der Sluis et al. 2006), transformation of the data is no solution as it will remove both spurious and genuine G × E from the data.
Here we presented a model with measured genotypes and a measured environment. If these measured variables are indeed the ones involved in the G × E interaction, and thus the ones causing the heteroscedasticity, then accounting for these measures (i.e., modeling their effects) should render the remaining variance (as summarized in Eq. 4) homoscedastic. In a previous paper (van der Sluis et al. 2006), marginal maximum likelihood showed to be useful in the detection of heteroscedasticity. If heteroscedasticity is present before modeling the genotypic and environmental effects, but absent when these effects are controlled for, then this can be taken to indicate that the heteroscedasticity was due to the interaction between the locus and environment under study. Yet, if the heteroscedasticity is still present, this can mean (a) that the heteroscedasticity is caused by scaling problems in the instrument used to measure the phenotype, or (b) that G × E interaction is present but the genes and environment controlled for are not the ones involved in this interaction, or only ‘rough approximations’ of the actual gene/environment involved (e.g., a poorly designed environmental measure, or a marker that is only slightly in LD with the actual QTL). Important in this context is an issue discussed by Eaves (1984) in the light of plant studies, that the genes that control average performance (i.e., main effects) may not be the genes that control the sensitivity to the environment (i.e., the genes involved in the interaction effect, giving rise to the heteroscedasticity, see Berg et al. 1989, for a similar distinction between ‘level’ and ‘variability’ genes). Within a design as discussed here, where both genes and environment are measured entities, level and variability genes can be distinguished. This distinction may be important in understanding the biological basis of the G × E interaction.
Second, throughout this paper, we assumed that the environmental measure is independent of genotype and family membership. Using so-called family-level environmental measures, i.e., environmental measures that are by definition equal for all siblings within a family, is problematic in the sib pair design discussed here, because the decomposition in between and within family environmental effects (e b vs. e w) depends on siblings that are discordant with respect to the environmental measure (see Tables 2–4). The use of family-level environmental measures thus excludes the possibility to test for stratification effects in family-level environmental components, such as socioeconomic status, divorce status of the parents, domestic violence, and loss of a parent. However, stratification with respect to the allelic effects and the interaction effect can still be controlled for, and one can still test the significance of the interaction effect, and allelic and environmental main effects. In this context it is important to note that there is ample debate about whether genuine family-level environmental measures actually exist. For example, the fact that divorce status of the parents is necessarily equal for siblings from the same family does not necessarily imply that this event has similar effects on the siblings, or is experienced in exactly the same manner by all siblings. We refer to Turkheimer et al. (2005) for an extensive discussion of this subject.
Third, the model presented so far does not account for the presence of gene-environment correlation (rGE). rGE represents the genetic liability to experience different environmental events, or the genetic control of exposure to different environments (e.g., Kendler and Eaves 1986; Plomin et al. 1977). Genetic factors have been found to substantially influence individual differences in, for example, the likelihood of experiencing stressful life events, lack of social support, participation in leisure activities, martial status, and age of first sexual intercourse (see Rutter and Silberg 2002 for a review). The finding that so many diverse ‘environmental’ measures are under genetic control, suggests that the present sib pair model may prove to be of limited use. Extension of this model to include the possibility to test and account for rGE is therefore desirable. For now, we advise researchers to test for the presence of rGE before they proceed. For instance, one can test whether the genotypic groups differ with respect to their environmental mean (ANOVA), or, if the environmental measure is categorical, with respect to the distribution of subjects across environmental conditions (χ2 test for equal frequencies). If differences with respect to the environmental measure are absent, one can proceed with the extended sib pair model as presented here.
Gene by environment interaction studies are relatively new and such studies are often characterized by difficulties concerning measurement and modeling (e.g., Eaves 2006). In general however, researchers seem to agree that studies aimed at revealing the sources of individual differences in specific qualities need to take G × E interaction into account, in order to arrive at a full account of individual differences (e.g., Caspi et al. 2006, Moffitt et al. 2005, 2006). Tests for G × E interaction are thus likely to become standard in future (association) studies.
Acknowledgements
Preparation of this manuscript was financially supported by NWO/MbGW VIDI-016-065-318, MH-65322, and DA-18673.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Appendix 1



Appendix 2



Footnotes
The Mx program is freely available at http://www.vcu.edu/mx/.
www.psy.vu.nl/u/s.van.der.sluis.
Effect size is defined as the variance explained by the effect, divided by the total variance, i.e., [½(μA − μG)2 + ½(μB − μG)2]/σ2TOT, where μA and μB are the means of the different subpopulations, and μG is the mean of the combined population, which is defined as (μA + μB)/2 = ½μA + ½μB. Substitution of μG with ½μA + ½μB gives (μA − μB)2/4σ2TOT.
R is a freely accessible software environment for statistical computing and graphics, see http://www.r-project.org/.
A small R-program is available online (www.psy.vu.nl/u/s.van.der.sluis), which can be used to calculate sample size N required to obtain a chosen level of power, given non-centrality parameter λ, and critical value α.
http://www.psy.vu.nl/u/s.van.der.sluis.
Edited by Stacey Cherny.
References
- Abecasis GR, Cardon LR, Cookson WOC. A general test of association for quantitative traits in nuclear families. Am J Human Genet. 2000;66:279–292. doi: 10.1086/302698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abecasis GR, Cookson WOC, Cardon LR. Pedigree tests of transmission disequilibrium. Eur J Human Genet. 2000;8:545–551. doi: 10.1038/sj.ejhg.5200494. [DOI] [PubMed] [Google Scholar]
- Azzelini A. Statistical inference based on the likelihood. London: Chapman and Hall; 1996. [Google Scholar]
- Beem AL, Boomsma DI. Implementation of a combined association-linkage model for quantitative traits in Linear Mixed Model procedures of statistical packages. Twin Res Human Genet. 2006;9(3):325–333. doi: 10.1375/twin.9.3.325. [DOI] [PubMed] [Google Scholar]
- Berg K, Kondo I, Drayna D, Lawn R. ‘Variability gene’ effect of cholesteryl ester transfer protein (CEPT) genes. Clin Genet. 1989;35:437–445. doi: 10.1111/j.1399-0004.1989.tb02969.x. [DOI] [PubMed] [Google Scholar]
- Bollen KA, Stine R. Bootstrapping goodness of fit measures in structural equation models. In: Bollen KA, Long JS, editors. Testing structural equation models. Newbury Park, CA: Sage; 1993. pp. 111–135. [Google Scholar]
- Boomsma DI, Martin NG. Gene-environment interactions. In: D’haenen H, den Boer JA, Willner P, editors. Biological psychiatry. London: John Wiley & Sons; 2002. pp. 181–187. [Google Scholar]
- Cardon LR, Bell JI. Association study designs for complex diseases. Nat Genet. 2001;2:91–99. doi: 10.1038/35052543. [DOI] [PubMed] [Google Scholar]
- Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, Taylor A, Poulton R. Role of genotype[e in the cycle of violence in maltreated children. Science. 2002;297(5582):851–854. doi: 10.1126/science.1072290. [DOI] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: Moderation by a polymorphism in the 5-htt gene. Science. 2003;301(5631):386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Caspi A, Moffitt TE. Gene-environment interactions in psychiatry: joining forces with neuroscience. Nat Neurosci. 2006;7:583–590. doi: 10.1038/nrn1925. [DOI] [PubMed] [Google Scholar]
- Dolan CV, van der Sluis S, Grasman R. A note on normal theory power calculation in SEM with data missing completely at random. Struct Equat Model. 2005;12(2):245–262. doi: 10.1207/s15328007sem1202_4. [DOI] [Google Scholar]
- Eaves LJ. The resolution of genotype-environment interaction in segregation analysis of nuclear families. Genet Epidemiol. 1984;1:215–228. doi: 10.1002/gepi.1370010302. [DOI] [PubMed] [Google Scholar]
- Eaves LJ. Genotype × environment interaction in psychopathology: fact or artifact? Twin Res Human Genet. 2006;9(1):1–8. doi: 10.1375/183242706776403073. [DOI] [PubMed] [Google Scholar]
- Falconer DS, Mackay TFC. Introduction to quantitative genetics. 4th . Essex, England: Pearson Education Ltd; 1996. [Google Scholar]
- Fisher RA. The correlation between relatives on the supposition of Mendelian inheritance. Trans R Soc Edinb. 1918;52:399–433. [Google Scholar]
- Foley DL, Eaves LJ, Wormley B, Silberg JL, Maes HH, Kuhn J, Riley B. Childhood adversity, monoamine oxidase A genotype, and risk for conduct disorder. Arch Genet Psychiatry. 2004;61:738–744. doi: 10.1001/archpsyc.61.7.738. [DOI] [PubMed] [Google Scholar]
- Fulker DW, Cherny SS, Sham PC, Hewitt JK. Combined linkage and association sib-pair analysis for quantitative traits. Am J Human Genet. 1999;64:259–267. doi: 10.1086/302193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizinga D, Haberstick BC, Smolen A, Menard S, Young SE, Corley RP, Stallings MC, Grotpeter J, Hewitt JK. Childhood maltreatment, subsequent antisocial behavior, and the role of monoamine oxidase A genotype. Biol Psychiatry. 2006;60:677–683. doi: 10.1016/j.biopsych.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Kendler K, Eaves L. Models for the joint effect of genotype and environment on liability to psychiatric illness. Am J Psychiatry. 1986;143:279–289. doi: 10.1176/ajp.143.3.279. [DOI] [PubMed] [Google Scholar]
- Khoury MJ, Adams MJ, Flanders WD. AN epidemiologic approach to ecogenetics. Am J Human Genet. 1988;42:89–95. [PMC free article] [PubMed] [Google Scholar]
- Khoury MJ, James LM. Population and family relative risks of disease associated with environmental factors in the presence of gene-environment interaction. Am J Epidemiol. 1993;137:1241–1250. doi: 10.1093/oxfordjournals.aje.a116626. [DOI] [PubMed] [Google Scholar]
- Lasky-Su J, Faraone SV, Lange C, Tsuang MT, Doyle AE, Smoller JW, Laird NM, Biedermand J. A study of how socioeconomic status moderates the relationship between SNPs encompassing BDNF and ADHD symptom count in ADHD families. Behav Genet. 2007;37:487–497. doi: 10.1007/s10519-006-9136-x. [DOI] [PubMed] [Google Scholar]
- Mather K, Jinks JL. Introduction to biometrical genetics. London: Chapman and Hall; 1977. [Google Scholar]
- Martin N. Gene-environment interaction and twin studies. In: Spector TD, Snieder H, MacGregor AJ, editors. Advances in twin and sib-pairanalysis. London: Greenwich Medical Media; 1999. [Google Scholar]
- Moffitt TE, Caspi A, Rutter M. Strategy for investigating interactions between measured genes and measured environments. Arch Genet Psychiatry. 2005;62:473–481. doi: 10.1001/archpsyc.62.5.473. [DOI] [PubMed] [Google Scholar]
- Moffitt TE, Caspi A, Rutter M. Measured gene-environment interactions in psychopathology: concepts, research strategies, and implications for research, intervention, and public understanding of genetics. Perspect Psychol Sci. 2006;1(1):5–27. doi: 10.1111/j.1745-6916.2006.00002.x. [DOI] [PubMed] [Google Scholar]
- Neale MC, Cherny SS, Sham PC, Whitfield JB, Heath AC, Birley AJ, Martin NG. Distinguishing population stratification from genuine allelic effects with Mx: association of ADH2 with alcohol consumption. Behav Genet. 1999;29(4):233–243. doi: 10.1023/A:1021638122693. [DOI] [Google Scholar]
- Neale MC, Boker SM, Xie G, Maes HH. Mx: statistical modeling. 6. Richmond, VA: Department of Psychiatry; 2003. [Google Scholar]
- Plomin R, DeFries JC, Loehlin JC. Genotype-environment interaction and correlation in the analysis of human behavior. Psychol Bull. 1977;84:309–322. doi: 10.1037/0033-2909.84.2.309. [DOI] [PubMed] [Google Scholar]
- Posthuma D, de Geus EJC, Boomsma DI, Neale MC. Combined linkage and association tests in Mx. Behav Genet. 2004;34(2):179–196. doi: 10.1023/B:BEGE.0000013732.19486.74. [DOI] [PubMed] [Google Scholar]
- Purcell S. Variance components models for gene-environment interaction in twin analysis. Twin Res. 2002;5:554–571. doi: 10.1375/136905202762342026. [DOI] [PubMed] [Google Scholar]
- Rutter M, Silberg JL. Gene-environment interplay in relation to emotional and behavioral disturbance. Ann Rev Psychol. 2002;53:463–490. doi: 10.1146/annurev.psych.53.100901.135223. [DOI] [PubMed] [Google Scholar]
- Saris WE, Satora A. Power evaluations in structural equation models. In: Bollen KA, Long JS, editors. Testing structural equation models. Newbury Park, CA: Sage; 1993. pp. 181–204. [Google Scholar]
- Turkheimer E, D’Onofrio BM, Meas HH, Eaves LJ. Analysis and interpretation of twin studies including measures of the shared environment. Child Dev. 2005;76(6):1217–1233. doi: 10.1111/j.1467-8624.2005.00846.x. [DOI] [PubMed] [Google Scholar]
- Van den Berg SM, Glas CAW, Boomsma DI. Variance decomposition using an IRT measurement model. Behav Genet. 2007;37:604–616. doi: 10.1007/s10519-007-9156-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Oord EJCG. Method to detect genotype-environment interactions for quantitative trait loci in association studies. Am J Epidemiol. 1999;150(11):1179–1187. doi: 10.1093/oxfordjournals.aje.a009944. [DOI] [PubMed] [Google Scholar]
- Van der Sluis S, Dolan CV, Neale MC, Posthuma D. Detecting genotype-environment interaction in monozygotic twin data: comparing the Jinks and Fulker test and a new test based on marginal maximum likelihood estimation. Twin Res Human Genet. 2006;9(3):377–392. doi: 10.1375/183242706777591218. [DOI] [PubMed] [Google Scholar]
- Van der Sluis S, Dolan CV, Neale MC, Posthuma D. Power calculations using exact data simulation: a useful tool for genetic study designs. Behav Genet. 2006;38:202–211. doi: 10.1007/s10519-007-9184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe K, Haan M, Byers A, Tangen C, Kuller L. Estrogen, apoe, and cognitive decline: evidence for gene-environment interaction. Neurology. 2000;54:1949–1954. doi: 10.1212/wnl.54.10.1949. [DOI] [PubMed] [Google Scholar]