

Abstract
The photoprocesses of native (phyA of oat), and of C-terminally truncated recombinant phytochromes, assembled instead of the native phytochromobilin with phycocyanobilin (PCB-65 kDa-phy) and iso-phycocyanobilin (iso-PCB-65 kDa-phy) chromophores, have been studied by femtosecond transient absorption spectroscopy in both their red absorbing phytochrome (Pr) and far-red absorbing phytochrome (Pfr) forms. Native Pr phytochrome shows an excitation wavelength dependence of the kinetics with three main picosecond components. The formation kinetics of the first ground-state intermediate I700, absorbing at ∼690 nm, is mainly described by 28 ps or 40 ps components in native and PCB phytochrome, respectively, whereas additional ∼15 and 50 ps components describe conformational dynamics and equilibria among different local minima on the excited-state hypersurface. No significant amount of I700 formation can be observed on our timescale for iso-PCB phytochrome. We suggest that iso-PCB-65 kDa-phy either interacts with the protein differently leading to a more twisted and/or less protonated configuration, or undergoes Pr to Pfr isomerization primarily via a different configurational pathway, largely circumventing I700 as an intermediate. The isomerization process is accompanied by strong coherent oscillations due to wavepacket motion on the excited-state surface for both phytochrome forms. The femto- to (sub-)nanosecond kinetics of the Pfr forms is again quite similar for the native and the PCB phytochromes. After an ultrafast excited-state relaxation within ∼150 fs, the chromophores return to the first ground-state intermediate in 400–800 fs followed by two additional ground-state intermediates which are formed with 2–3 ps and ∼400 ps lifetimes. We call the first ground-state intermediate in native phytochrome Ifr 750, due to its pronounced absorption at that wavelength. The other intermediates are termed Ifr 675 and pseudo-Pr. The absorption spectrum of the latter already closely resembles the absorption of the Pr chromophore. PCB-65 kDa-phy shows a very similar kinetics, although many of the detailed spectral features in the transients seen in native phy are blurred, presumably due to wider inhomogeneous distribution of the chromophore conformation. Iso-PCB-65 kDa-phy shows similar features to the PCB-65 kDa-phy, with some additional blue-shift of the transient spectra of ∼10 nm. The sub-200 fs component is, however, absent, and the picosecond lifetimes are somewhat longer than in 124 kDa phytochrome or in PCB-65 kDa-phy. We interpret the data within the framework of two- and three-dimensional potential energy surface diagrams for the photoisomerization processes and the ground-state intermediates involved in the two photoconversions.
INTRODUCTION
Phytochromes are the photoreceptors responsible for light-induced signal transduction in higher plants which control general functions in plant development, e.g., seed germination, hypocotyl growth, blooming behavior, and chlorophyll synthesis (1–4). Phytochromes have also been found in cyanobacteria (5) and even heterotrophic bacteria (6) where they also perform photoregulatory functions. Plant phytochromes (phys) contain an open chain tetrapyrrole, phytochromobilin (PΦB), as their chromophore and exist in two stable isomeric forms, a red-absorbing form, Pr, and a far-red absorbing form, Pfr (7,8). Upon electronic excitation of Pr with red light, PΦB undergoes a Z,Z,Z to Z,Z,E isomerization of the 15-16 double bond, probably accompanied by single-bond rotations to compensate the introduced sterical stress upon the photoisomerization (8–10).
The isomerization starts on a femtosecond timescale and initiates a sequence of conformational changes in the chromophore/protein complex which extend up to seconds (11). These thermal reactions finally lead to the physiologically active Pfr. The back reaction to Pr can either occur thermally (with a very slow rate) or can be photoinduced by far-red light. The molecular mechanisms of phytochrome photoconversions have recently been reviewed extensively (2).
The dynamics of the very early photochemical events leading to the interconversion of the two spectral forms of native Pr and Pfr have been the subject of several investigations using ultrafast spectroscopy, but most published work dealt with the kinetics of Pr only. Also, the analogous processes in recombinant phytochromes carrying modified chromophores like, e.g., phycocyanobilin (PCB), iso-phycocyanobilin (iso-PCB), or others have been studied to a much lesser extent (12).
Several studies of the excited-state dynamics of Pr by time-resolved fluorescence have been reported (13–16). In general multiexponential fluorescence kinetics with lifetimes ranging from ∼10 ps to 60 ps have been found. From studies of various parameters like, e.g., excitation wavelength dependence, temperature, and degree of photoconversion on the kinetics we concluded that there exist at least two chromophore conformations in the excited state of Pr which are in equilibrium on the picosecond timescale (13). A similar heterogeneity and complexity of the Pr → Pfr phototransformation kinetics is also observed in the following ground-state reactions and is an intrinsic property of homogeneous native phytochrome (11).
Later studies confirmed this excited-state equilibration model in the 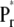 state (12,17–19). However, the insight gained from fluorescence studies alone is limited, due to the high complexity of the excited-state dynamics of the very flexible chromophore. Consequently, additional ultrafast studies have been performed on native phys using ultrafast transient absorption techniques, in particular to characterize also the excited-state dynamics of Pfr which could not be studied using fluorescence techniques due to its much faster, sub-picosecond lifetime (17–23). Phytochrome from cyanobacteria as well as recombinant phys with modified chromophores have been also been studied by femtosecond spectroscopy (12). All these studies confirmed the excited-state kinetics deduced from fluorescence data for Pr, i.e., a heterogeneous or multiexponential excited-state kinetics in the 10–60 ps time range and the formation of first ground-state intermediate produced upon excitation of Pr (I700) within 25–30 ps (13) as a general property of Pr phytochromes. In comparison, the initial decay of the excited state of Pfr was found in all studies to be in the range of several hundred femtoseconds, followed by a fairly complex sequence of events showing several lifetimes up to 1 ns.
state (12,17–19). However, the insight gained from fluorescence studies alone is limited, due to the high complexity of the excited-state dynamics of the very flexible chromophore. Consequently, additional ultrafast studies have been performed on native phys using ultrafast transient absorption techniques, in particular to characterize also the excited-state dynamics of Pfr which could not be studied using fluorescence techniques due to its much faster, sub-picosecond lifetime (17–23). Phytochrome from cyanobacteria as well as recombinant phys with modified chromophores have been also been studied by femtosecond spectroscopy (12). All these studies confirmed the excited-state kinetics deduced from fluorescence data for Pr, i.e., a heterogeneous or multiexponential excited-state kinetics in the 10–60 ps time range and the formation of first ground-state intermediate produced upon excitation of Pr (I700) within 25–30 ps (13) as a general property of Pr phytochromes. In comparison, the initial decay of the excited state of Pfr was found in all studies to be in the range of several hundred femtoseconds, followed by a fairly complex sequence of events showing several lifetimes up to 1 ns.
We present here detailed studies of native oat phyA, in both the Pr and the Pfr states, as well as of the corresponding recombinant 65-kDa phys assembled with either PCB or iso-PCB (i.e., 17-ethyl, 18-methyl PCB (24)). The recombinant proteins span the amino-acid sequence of oat phyA from position 1 until position 595. We emphasize the advanced data analysis methods, which allow for a decision between continuous distributions and more discrete multiexponential kinetics. From the comparison of the data, it is now possible to shed light on some discrepancies in the femto- to sub-nanosecond kinetics in the work reported by other authors and ourselves (13,17,23).
MATERIAL AND METHODS
Phy A was isolated from etiolated oat sprouts in its native (124 kDa) form (25). For generation of recombinant 65 kDa phytochrome, the phytochrome-A (phyA) encoding cDNA were expressed in Hansenula polymorpha and purified by a metal affinity chromatography, making use of a His6-tag attached to the C-terminus of the recombinant apoprotein (26,27). The apoproteins were assembled with PCB or iso-PCB (see Fig. S1 in Supplementary Material, Data S1): PCB-65 kDa-phy and iso-PCB-65 kDa-phy. PCB was isolated from Spirulina platensis as described by Kufer et al. (28). For the synthesis of iso-PCB, see Robben et al. (24). In case of the recombinant phytochromes, dithiothreitol and Pefa Block (Merck, Darmstadt, Germany) were added to protect the sample against proteolysis and oxidation. The samples had an absorbance of ∼0.8/mm in their Pr forms at the maximum except for iso-PCB-65 kDa-phy, which had approximately an order-of-magnitude lower absorption. They were filled in a horizontally swinging rotating cuvette with an optical path length of 1 mm, which provided a complete exchange of the sample in the volume of the laser spot between each shot. The cuvette was illuminated with far-red light using an RG9 filter (Schott, Mainz, Germany) during Pr measurements or with red light using a 667 nm (bandwidth 12 nm) interference filter during Pfr measurements to maintain a stable concentration of either form. In both cases, the light of a projector (250 W) was filtered accordingly and collimated to a beam diameter of ∼5 cm at the cuvette. All time-resolved measurements were performed at 10–11°C. The integrity of all samples was verified by measuring the absorption spectra of both Pr and Pfr before and after the measurements. Full functionality, i.e., no measurable degradation due to the measurements, was observed. The native phytochrome sample was characterized before and after the measurements also by PAGE, which did not indicate any degradation.
Overall assembly yields for PCB and iso-PCB were similar, although the reaction rate for the latter is somewhat slower. The photoisomerization quantum yields of native phytochrome and of PCB-phy are in the range 0.15–0.18 (19,29). The photoisomerization quantum yield of iso-PCB-phy has not been determined exactly. However, estimates based on the photostationary state composition (see Fig. S1 in Data S1) suggest that it may be slightly smaller than for native phy.
Ultrafast transient absorption
The femtosecond transient absorption setup has been described previously in detail (30). In brief, a regenerative Ti:Sa amplifier system generates 70–80 fs pulses at a repetition rate of 3 kHz. The main part of the pulse energy was used to drive an optical parametric amplifier (Topas, Light Conversion, Vilnius, Lithuania). The resulting pulses had a temporal width of ∼60 fs (full width at half-maximum) and a spectral width of ∼8 nm between 620–733 nm and were attenuated to a pulse energy of 1–10 nJ (3 × 1013–3 × 1014 phot/cm2/pulse), depending upon the excitation wavelength and sample. They were focused to a spot of ∼120–130 micrometer diameter on the cuvette. All measurements were performed under magic angle polarization conditions. The transient absorption spectra were recorded by a home-built high speed camera on the basis of a diode array covering a detection range of ∼125 nm at a pixel resolution of 0.5 nm. The data collection comprised three acquisition cycles for each detection wavelength interval:
An ultrashort femtosecond time range up ∼2.5 ps with a resolution of ∼13 fs per point.
A picosecond time range up to ∼20 ps with a resolution of 33 fs,
A sub-nanosecond time range up to 500 ps with a resolution of 0.8 ps.
Excitation for most samples occurred near the long-wave absorption peak, except for cases where specifically the excitation wavelength dependence was studied. For iso-PCB-phy, a shorter excitation wavelength, sufficiently away from the detection wavelength range, had to be used to overcome the problems due to sample scattering. A significant development of sample scattering over the measurement time was observed, presumably due to a higher tendency of iso-PCB-phy to aggregate. This phenomenon also leads to a slight distortion of the transient absorption signal in the very early time range, however the photoreversibility was not affected.
Data analysis
The data analysis was performed by the lifetime density approach. This allows us to distinguish discrete lifetime components on the one hand and potential lifetime distributions on the other hand as demonstrated in Croce et al. (30). The data from the three different time ranges were analyzed simultaneously. It is important to note that the resulting lifetime density maps do not contain any assumptions regarding the underlying kinetic models, i.e., no a priori assumption regarding the presence of discrete or distributed kinetics is made at that step. The model function is a quasicontinuous distribution of exponential functions, ranging from 50 fs to 10 ns, and the procedure essentially involves an inverse Laplace transform of the original three-dimensional (ΔA, λdet., time) data surface (30). Thus, the lifetime density map is a true representation of the measured kinetics, without any assumptions made regarding a specific kinetic model. Complications in the analysis may arise from the fact that the early kinetics displays large-amplitude coherent oscillations throughout the entire detection range which cannot be modeled with exponential functions (31). The oscillations are thus present in the residuals of the analysis, whereas the fitted theoretical data surfaces (representing the lifetime density maps) are devoid of these oscillations. Note that all time-dependent spectra and lifetime density maps presented in the figures are based on the fitted theoretical data surfaces, thus not showing the oscillatory contributions, which are contained in the residuals. For all further details of the lifetime density analysis, see Croce et al. (30).
RESULTS
Stationary absorption
Native 124 kDa phyA and recombinant 65-kDa phys show similar absorption characteristics in both Pr and Pfr (see Fig. S1 in Data S1). The only major differences are the absorption maxima which are shifted from 668 to 654 and 659 nm (PΦB, PCB, iso-PCB, respectively) in case of the Pr forms and from 730 to 717 and even up to 706 nm for iso-PCB in case of Pfr. All samples could be efficiently converted into the Pfr forms and back by light excitation, although the overall conversion for the phytochromes with modified chromophores is lower, i.e., ∼65% (PCB) and ∼50% (iso-PCB), instead of 70% for the native chromoprotein (24). For all phytochromes, the so-called red-light adapted forms (see Fig. S1 in Data S1, dashed lines) always consist of a mixture of the Pr and Pfr contributions in a photochemical equilibrium. Due to a higher degree of scattering of the iso-PCB-containing sample, the baseline and thus the absorption rises toward shorter wavelengths to some extent (see Fig. S1 C in Data S1).
Femtosecond transient absorption kinetics of the Pr forms
The early steps in the Pr to Pfr phototransformation are characterized by relatively simple kinetic features. An overview of the original kinetic surfaces is given in Fig. 1. Selected transient absorption spectra, corrected for chirp, are shown in Fig. 2 (see Materials and Methods). After excitation at 665 nm, native Pr shows the typical ground-state bleaching and stimulated emission decay. This signal decays in a few tens of picoseconds giving rise to a growing-in of a new absorption at ∼690–700 nm for long delay times (Fig. 1 B). The latter can be attributed to the intermediate I700 on the basis of nanosecond flash photolysis data (11). On the very early timescale, an ∼100-fs process, associated with a red shift of the bleaching due to the rise of stimulated emission above 690 nm (Figs. 1 A and 2 A), is observed. This process is associated with strong oscillations in the transient absorption signal lasting up to ∼1 ps. These oscillations will be analyzed in detail in a separate study. The kinetics of recombinant phys in general resembles those of native phy despite the significantly lower amplitude of the new absorption developing at ∼690 nm (compare, e.g., Fig. 1, C and D, with Fig. 2 B) related to I700 formation. The I700 absorption feature is nearly absent in the iso-PCB-65 kDa-phy sample (Fig. 1 D), although surprisingly there is still an approximately equal amount of chromoprotein bleached on the long timescale (Fig. 1 D and 3 C).
FIGURE 1.

Original femtosecond to (sub-)nanosecond transient absorption kinetics of three different types of phytochrome in their Pr forms in a three-dimensional surface presentation (A and B). Native phytochrome on two different timescales excited at 665 nm (note the inversed absorption scale in A). (C) PCB-65 kDa-phy and (D) iso-PCB-65 kDa-phy, both excited at 660 nm. The black lines indicate some selected transient decays.
FIGURE 2.

Transient absorption spectra after deconvolution and chirp correction at selected delay times of the Pr forms of (A) 124 kDa phytochrome (native phyA), excited at 665 nm. (B) PCB-65 kDa-phy and (C) iso-PCB-65 kDa-phy, excited at 660 and 620 nm, respectively. Note that these transient spectra have been recalculated from the lifetime density maps (Fig. 3). They do not contain the oscillatory contributions, which are rather found in the residuals of the analyses (see below).
FIGURE 3.

The lifetime density maps showing the excitation wavelength dependence of the femto- to nanosecond kinetics for native phyA in its Pr form. Excitation wavelengths are 620 nm (A), 640 nm (B), 665 nm (C), and 685 nm (D). Dark-blue color indicates negative and green/yellow-white positive amplitudes of a lifetime component while orange color reflects the zero level. The lifetime range is drawn on a logarithmic scale and extends from ∼100 fs to ∼6 ns (see Materials and Methods).
To gain more insight into the complexity of the kinetics for the I700 formation and in view of some contradicting earlier reports (19), a detailed excitation wavelength dependence was measured for native phy and subjected to the lifetime density analysis (Fig. 3, A–D). The data reveal processes with lifetimes of ∼70–100 fs, 10–80 ps in a wavelength range of 650–690 nm, 28 ps in the range of ∼680–720 nm and a nondecaying term (on our timescale up to several nanoseconds) either as a bleaching between 650 and 670 nm or as new absorption appearing in the range of ∼680–710 nm. The broad 10–80 ps feature appears to be composed of two spectrally similar components with lifetimes of ∼12–16 and 50 ps (Fig. 3, A and C). The spectrum of the third component (28-ps bleaching decay) coincides with the spectrum of the absorption of the I700 intermediate. A weak rise of a bleaching with a lifetime of ∼1–2 ps can also be found. The two decay components with ∼15- and 50-ps lifetimes centered at 650–680 nm disappear for very long excitation wavelengths (Fig. 3 D) indicating rather complex excited-state dynamics upon excitation with large vibronic excess energies and much simpler kinetics for negligible excess energies. No changes in the lifetimes are observed upon excitation with different wavelengths, but only the relative amplitudes of components change strongly.
As can be seen already from the original data surfaces (Fig. 3), the kinetics of PCB-65 kDa Pr (Fig. 4 A) resembles that of native phy (see Fig. 3, A or B), whereas that of iso-PCB-65 kDa-phy (Fig. 4 B) is characterized by the complete lack of the 28-ps component in both wavelength ranges (650–685 nm and 685–730 nm). The picosecond decay of the latter can be described by two discrete components with ∼12 and 60 ps lifetimes and an amplitude ratio of 1:1.2-to-1.8 plus a constant term. The picosecond kinetics of both native and PCB-65 kDa phy samples thus support the assignment of the 28-ps and the 40-ps components, respectively, to I700 formation. For iso-PCB-65 kDa-phy, no such component was observed, but only the two kinetic features at shorter wavelengths, which are also present in the other phys. The chromophore type could, in principle, have a critical influence not only on the yield, but also on the associated kinetics for the I700 formation. Surprisingly, this effect is born out only to a minor extent for PCB as chromophore, i.e., the lifetime is shifted in the PCB sample from ∼28 ps in native phytochrome to ∼40 ps (see Fig. 4 A at ∼695 nm).
FIGURE 4.

Femto- to nanosecond kinetics represented as lifetime density maps for (A) PCB-65 kDa-phy excited at 660 nm and (B) iso-PCB-65 kDa-phy excited at 620 nm in their Pr forms. For a description of the color coding see Fig. 3.
Femtosecond transient absorption kinetics of the Pfr forms
Although the red light-adapted states always represent a mixture of Pr and Pfr forms, the Pfr kinetics can be measured exclusively upon selective excitation of the mixtures (see Fig. S1 in Data S1), using appropriate excitation wavelengths of 733 nm for native phy (Fig. 5) and 715 nm for PCB-65 kDa-phy (not shown). Transient spectra for native and PCB-65 kDa Pfr at selected delay times are shown in Fig. 6, A and B, respectively. The Pfr kinetics of the native as well as recombinant phys have in common a strong excited-state absorption below 720 nm or 700 nm, respectively, and the stimulated emission for wavelengths longer than 730-nm decay in fs (Figs. 5 and 6). A first intermediate seems to be formed in ∼500 fs. The formation of a new weak but broad absorption centered around 675 nm, overlapping with the bleaching at ∼730 nm is observed in picoseconds (Fig. 6). A gradual shift in the peak positions and shapes of these features can be observed beyond 10 ps.
FIGURE 5.

Transient absorption kinetics of native phyA excited at 733 nm in the Pfr form in a three-dimensional surface representation.
FIGURE 6.

Transient absorption spectra at selected delay times for the Pfr forms of (A) native phyA excited at 733 nm and (B) PCB-65 kDa-phy excited at 715 nm. See note in Fig. 2 legend.
Two dominant excited-state relaxation or decay processes can be identified for native phy in the lifetime density maps, shown as bright white signals. A 150-fs component is found at ∼650–730 nm and a 400-fs component at ∼520–700 nm (positive amplitude, turning into a negative amplitude above 700 nm) (Fig. 7 A). At least two processes follow. The first one with a lifetime of ∼3 ps predominantly describes a bleaching and stimulated emission (SE) decay while the second one, with a lifetime of ∼400 ps, gives rise to a new broad absorption at 600 to 685 nm. All of them are ascribed to ground-state processes due to the lack of the stimulated emission above 730 nm and the initial excited-state absorption below 700 nm. The nondecaying component is associated with a new absorption developing below 685 nm and bleaching above 690 nm. The PCB (Fig. 7 B), and in principle also the iso-PCB-phy (Fig. 7 C) reveal quite similar kinetics in the characteristic wavelength ranges, although the 400-fs component appears to be slower (∼500 and 600 fs), and the bands are significantly broader than for native phy. Despite some small distortions (Fig. 7) due to the lower absorbance of the iso-PCB sample and some blue shift in the wavelength positions, the main features, both lifetimes and the signs of the spectral features are in agreement with the PCB and the native phy kinetics. The 400-fs and 3-ps components seem to be related to a rise and decay, respectively, of an absorption above 740 nm (Fig. 7, B and C).
FIGURE 7.

Lifetime density maps for the Pfr forms of (A) native phyA excited at 733 nm, and (B) PCB-65 kDa-phy, and (C) iso-PCB-65 kDa-phy, both excited at 715 nm. At these wavelengths the remaining Pr forms after phototransformation are not excited. For a description of the color coding see Fig. 3. (C) Lifetime range below ∼200 fs has not been included in the analysis because of problems with the coherent solvent dynamics.
Vibrational oscillations
Strong oscillations are observed for the Pr forms during the first picosecond. They are most pronounced for native phy (see Fig. S2 in Data S1, which shows the oscillations on the short timescale as a function of the detection wavelength) and substantially weaker for the first 500 fs for PCB-65 kDa-phy (see Fig. S3 A in Data S1). They are even more reduced for iso-PCB-65 kDa-phy (see Fig. S3 B in Data S1). We do not analyze these oscillations in this work in detail, which appear to be primarily due to the movement of coherent vibrational wavepackets in the excited state. Suffice it to say that, in our view, these oscillations are largely concentrated in modes which are strongly coupled to the initial photoreactions.
DISCUSSION
Pr kinetics
The extensive analysis of the excitation-wavelength dependence turned out to be crucial for the characterization of the three close-lying picosecond processes. For native phy, only the 28-ps component describes the formation of ground-state I700, based on the following three arguments:
The rise of absorption fits spectrally the expected absorption band of I700 at ∼690 nm (Fig. 3).
The long-lived absorption and its rise kinetics are almost invariant against the excitation wavelength and remains the same for long excitation wavelengths.
The amplitude of this feature is strongly correlated with the amount of I700 formed also in recombinant phys assembled with PCB and in particular with iso-PCB, where the I700 absorption is essentially absent along with a disappearance of this kinetic feature.
At least point 1 does not seem to be fulfilled in the work of Bischoff et al. (19) where the decay-associated spectrum of the only picosecond component which could be resolved in that lifetime range, a 34-ps component, resembles in its amplitude and position more a decay of the ground-state bleaching or stimulated emission at 670 nm in its amplitude and position than to a rise of the I700 absorption. Clearly, the strong excitation wavelength dependence of the amplitudes of the other two components with 15- and 50-ps lifetimes for native and recombinant phys complicates the kinetics and, when performing only a single wavelength excitation experiment, can lead to erroneous or incomplete interpretations. This property can explain also the strong excitation wavelength dependence of the two picosecond lifetimes found by Savikhin et al. (17) (see Fig. 3 and Table 1 in that work). The longer lifetime in their description varied from 30 to 130 ps with a mean value of 50–60 ps. The faster component with a mean value of ∼16 ps fits to our fast 12–16 ps component. However, neither this fast nor the slowest picosecond component can be attributed to the formation of I700. Only the 28-ps component located between these lifetimes is associated with I700 formation. An excitation wavelength dependence for a 25–32 ps lifetime has also been reported by Rentsch et al. (23) in transient absorption measurements. Early time-resolved fluorescence and absorption studies had yielded similar lifetimes for the I700 formation based on single exponential fits, e.g., 30–40 ps and 24 ps (21).
The 15- and 50-ps components can only be assigned to excited-state equilibration processes and presumably, some competing transitions back to the ground state via different pathways. Thus, these excited-state processes reflect some complex dynamics on the excited-state potential energy surface with several local minima as is supported also by the fluorescence kinetics (13). The initial population of these states depends critically on the excitation wavelengths and also on the structural details of the chromophore. These local minima can be associated with either inactive (not leading to I700) or active channels. The system performs some complex movements on that surface as outlined in Figs. 8 A and 9, which gives rise to quite complicated structures of the excited-state potential surface, since the reaction coordinate describing the Z,Z,Z to Z,Z,E photoisomerization (32–34) is composed of many nuclear coordinates of the chromophores and of the nuclear coordinates of the protein. We have summarized these features in the cartoon of Fig. 9 which links the potential surfaces with the dynamics. Despite the complexity of this figure, the three-dimensional presentation can only give a glimpse of the actual complexity of the energy surfaces and the dynamics. The exact form of the potential surfaces shown here should not be taken literally but rather as a demonstration of the principal properties of the surfaces and the associated kinetics. Without a detailed knowledge of the ground-state structure and rather advanced calculations for the excited states, the details cannot be unraveled. However, we note that a simple picture reflecting the same principal features has been proposed for the first time by Holzwarth et al. (13). In that work, lifetimes of 4–16 and 45 ps have been found in fluorescence kinetics which is in agreement with the new data presented here.
FIGURE 8.

Two-dimensional reaction coordinate models for the phototransformations of native phyA from Pr to Pfr (left) and back from Pfr to Pr (right). The absorption maxima of ground state and intermediates are shown as vertical arrows. The lifetimes of the major reaction steps are also given and are also indicated by arrows.
FIGURE 9.

Cartoon showing a three-dimensional presentation of the excited (S1) and ground state (GS) potential energy surfaces for the Pr to Pfr phototransformation of native phyA. The S1 potential surface of Pr has several potential minima which are separated by low barriers and can be reached after photoexcitation. The model separates the reaction coordinate for chromophore isomerization from the perpendicular reaction coordinate(s) of the protein/chromophore conformations not leading to isomerization. The green arrows indicate the excitation into a state with low excess energy and direct relaxation into the active channel leading to I700. The green shaded Gaussian (reached by the green upward arrow from Pr) indicates the initially created wavepacket in the excited state. The light blue arrows shows excitation into states with higher excess energy. The dark blue arrows show movements on the excited-state potential surface—involving chromophore and protein modes—leading out of nonreactive or less reactive channels (potential minima). The red arrow indicates excitation of I700 into an excited-state surface common to I700 and Pr. The purple arrows indicate internal conversion from nonreactive potential minima back to the Pr ground state (for further discussion see text). Note that the detailed form of these potential energy surfaces should not be taken literally. The figure merely serves to illustrate the principal properties of the dynamics in the excited state and the interpretation of parallel and/or alternative processes.
The results obtained with the recombinant phytochromes are of particular interest for a better understanding of the origin of 15- and 50-ps components on the one hand, and the ∼28-ps component on the other hand. In the case of native phy, these data confirm the assignment of the 28-ps component to the formation of I700 (in the case of PCB-65 kDa-phy, an ∼40 ps-component is found). If we take the amplitude of the absorption around 690 nm as a measure of the concentration of I700, notwithstanding some possible variations in absorption coefficients for this intermediate for the different phytochromes, we can conclude that I700 formation for iso-PCB-65 kDa-phy is at best very minor and even for the PCB-65 kDa-phy the I700 yield (or the extinction coefficient) should be significantly reduced as compared to native phy. In contrast to the apparently low I700 yield, Pfr formation seems to be still quite efficient in both samples, i.e., PCB-65 kDa-phy as well as the iso-PCB-65 kDa-phy (35). The most likely reason is a reduced absorption coefficient of the chromophores in the I700 intermediates from iso-PCB-65 kDa-phy and PCB-65 kDa-phy. Such an assumption implies that the iso-PCB chromophore in the I700 intermediate would have an absorption coefficient one-order-of-magnitude lower than that of the native chromophore. A reduction of oscillator strength could be due to a distorted conformation of the chromophore in the protein binding site. The open-chain tetrapyrrole in the recently reported structure of a bacterial phytochrome (6), although carrying biliverdin as chromophore, shows a coiled conformation which is held in place by steric and electrostatic interactions and also by hydrogen bonds, already in the ground state. It can thus be assumed that steric constraints from the protein on the structurally modified photoisomerized iso-PCB chromophore may enforce an even stronger coiled conformation resulting in a reduced oscillator strength. Such an explanation would be in line with a recent study where an 17-isopropyl (ipr) substituent causes—already in the ground state—a strong hypsochromic shift of ∼100 nm, due to the strong inherent steric interaction between the large ipr-substituent and the neighboring C-ring (24). Alternatively, one may also assume that the protonation state of the chromophore might change, with a strong impact on the absorption coefficient. Although both Pr and Pfr are undoubtedly protonated in plant and bacterial phytochromes in their ground states (36), the situation for several intermediates is still under debate, as was recently outlined from a study of a bacterial phytochrome. In this chromoprotein, an intermediate in the microsecond-to-millisecond time range was found to transiently deprotonate and later reprotonate (36). One might thus assume that the hydrogen-bond network that leads to the protonation of the chromophore is disturbed in the case of iso-PCB (due to its changed substitution pattern), which would cause an easier (transient) deprotonation.
When the 28-ps I700 rise term is absent, the bleaching decay term in the 650–680 nm range is also lacking, again demonstrating that little or no I700 is formed for iso-PCB-65 kDa-phy (see Fig. 4 B). We can also rule out that a hypothetically blue-shifted I700 absorption at 665 nm might be fully compensated by a bleaching and stimulated emission contribution of the excited Pr over the whole relevant wavelength range, since this would require the decay of the bleaching to nearly zero in that lifetime range, which is not the case. An alternative explanation would be that, in the case of iso-PCB-65 kDa-phy, only very small amounts of I700 are formed, and also in the case of PCB-65 kDa-phy the yield is substantially reduced relative to native phy. This interpretation might be supported by nanosecond to microsecond flash photolysis experiments which show only a minor I700 absorption contribution for iso-PCB-65 kDa-phy and a reduced absorption in the case of PCB-65 kDa-phy (35). This alternative interpretation would imply that the Pr-to-Pfr conversion, at least for the iso-PCB-65 kDa-phy, occurs predominantly via a different pathway and different intermediates. At present, we cannot finally exclude any of these two alternative explanations.
For the excited-state decay of Pr, we explicitly rule out a model of distributed rate constants between a single pair of excited-state minima arising either from a distribution of activation barriers in the excited-state or some ground-state inhomogeneity. Such a model has been assumed previously to explain the multiexponential kinetics of the Pr excited-state decay (12) but it does not satisfy the data presented here. In particular, it does not agree with the wavelength dependence and the kinetics of the phys with modified chromophores. Our data rather suggest an interpretation within a somewhat extended version of the simple model involving an equilibration between different excited-state potential wells that was first proposed by Holzwarth et al. based on fluorescence lifetime data and their wavelength dependence (13). The notion that an equilibration between different excited-state potential minima exists in the excited state of Pr is also supported by the finding that excitation of Pr and of I700 afford the same excited-state dynamics (17) and that I700 can be efficiently reverted to Pr photochemically (37,38).
The 100-fs and the 1–2 ps components observed in all Pr samples are related to the initiation of the isomerization and to an excited-state relaxation accompanying the formation of the stimulated emission at ∼695 nm (Fig. 2 A). The second, weaker component manifests itself as a small rise of a bleaching or stimulated emission in the absorption decays for wavelengths longer than 670 nm. The initial excited-state conformational relaxation seems to be finished after ∼2 ps. Our data show some significant deviations from results reported in the literature, as far as the short-lived relaxations are concerned. A component with a lifetime of 1.8 ps was also detected by Bischoff et al. (19) with an extremely strong negative amplitude peaking at ∼660 nm. These authors used an excitation wavelength of 610 nm, close to the one shown in Fig. 3 A. Surprisingly, the amplitude of the 1.8 ps is equally strong as the negative amplitude of the component with a lifetime of 34 ps, hence indicating a decay of a bleaching. In contrast, in our case the negative amplitude is approximately an order-of-magnitude lower and is accompanied by an equally strong positive feature at ∼690 nm, reflecting, at least in part, a rise of stimulated emission. Thus, we assume some nonlinear coherent artifacts in the data of Bischoff et al. (19), possibly due to very high excitation intensity. A similar explanation might apply for the 3.2 ps bleaching decay component found earlier by those authors (23). In a separate work of that group (39) a broad transient absorption around 725 nm was observed during the first 300 fs with an amplitude equally strong as the bleaching at 665 nm. This strong absorption was attributed to a 200-fs relaxation process in the excited state before a picosecond process. We do not observe any positive absorption of this kind in the specified wavelength range (Figs. 1 A and 2 A). However, an ultrafast excited state relaxation with an estimated lifetime of ∼100 fs, close to what we have found, was suggested by Teuchner et al. (18) based on picosecond intensity-dependent transmission measurements.
Pfr photoconversion
The Pfr forms of all studied phytochromes exhibit similar kinetics in the femtosecond as well as in the picosecond time regimes. According to the decay of the excited-state absorption (S1-Sn) below 650 nm and the simultaneous loss of the stimulated emission above 740 nm (Fig. 6), excited Pfr returns to the electronic ground state(s) with a lifetime of 400–600 fs. These ground states could either be the initial Pfr state or a first intermediate of the photoconversion, given the overall 15% yield of photoconversion. In addition, the decay of the excited-state absorption and rise of a bleaching in the range between 670 and 720 nm is indicative for a conformational relaxation process with a 150-fs lifetime before the decay of the excited state. PCB-65 kDa-phy shows kinetics very similar to native phy. However, some of the spectral features are blurred and the maxima of the spectra are blue-shifted by ∼15 nm, as is expected from the blue-shifted ground-state absorption. For iso-PCB-65 kDa-phy the data also suggest similar overall characteristics, although the sub-200 fs features could not be analyzed properly, since this time range is overlaid in our data by strong solvent dynamics due to the lower optical density of the sample. The maxima of the intermediates of iso-PCB-65 kDa-phy are blue-shifted by 10 nm versus those of PCB-65 kDa-phy.
Dynamics on the early timescale
The timescale of the transient absorption up to 2–3 ps displays the most complex kinetics for Pfr. At <300 fs a second bleaching band develops at 725 nm in addition to the initial ground-state bleaching maximum located near the 733-nm excitation wavelength. The data in Figs. 5 A and 6 A indicate that this second bleaching has a lifetime of ∼150 fs. The process occurs concomitant with the decays of both the initial strong excited-state absorption (ESA) at 670–720 nm and the bleaching above 740 nm. The latter decay is probably due to the loss of stimulated emission (SE) within ∼400 fs. This can be understood in terms of a scenario where the SE spectrally broadens and rapidly moves out into the near-IR region due to rapid conformational relaxation on the excited-state surface. This also explains the lack of any appreciable fluorescence from Pfr. The ultrafast vibrational relaxation on the excited-state surface leads to a decay of both ESA and SE, i.e., the initially excited state moves out of the Franck-Condon region for the S1→S0 and the low-lying S1→Sn transitions as a consequence of a displacement of the excited-state potential energy minima of the Franck-Condon active modes. This interpretation is also supported by the decay of the bleaching in the 700–720-nm band while the ESA below 650 nm persists. We assign this ultrafast Pfr relaxation to a partial twisting of the C15,16 double and/or C14,15 single bonds along the reactive low-frequency mode(s), which ultimately affords the final (15,16Z→E) isomerization product. Such a mechanism appears to be in line with recent resonance Raman data (10,34,40), and would agree with the transient absorption data of Bischoff et al. (19). The ultrafast decay of the Pfr excited state and the initial part of the photoisomerization also agrees well with the findings of Andel et al. (34) that the excited state and its first photoproduct state are strongly coupled.
It is important to note that the dynamics on the early timescale up to ∼2 ps seem to be wavelength-dependent and nonexponential in essence, as is indicated by the complex pattern observed in the lifetime density plots of Fig. 7. The first ground-state intermediate is formed within ∼500 fs and decays with ∼3 ps lifetime and it has an absorption peak around 630 nm, but its main long-wavelength absorption seems to be located at ∼750 nm for native Pfr. Our measurements extend only up to 755 nm for native Pfr (Fig. 7 A). At the long-wavelength end of that range, one can see the bleaching decay at ∼730 nm and some of the absorption rise around 750 nm. This is seen more clearly in the data of Bischoff et al. (19), which extend up to 850 nm. The difference spectrum of their 3.8 ps component peaks at ∼775 nm, which corresponds to a maximum of the formed intermediate of ∼750 nm. We thus call this first ground-state intermediate Ifr-750. Presumably, the chromophore conformation in this species is already the (15,16Z) isomer, with some torsional strain remaining. In the data of iso-PCB-65 kDa-phy the long-wavelength absorption of the intermediate corresponding to Ifr-750 in native phy (note the blue-shifted spectra of iso-PCB) is clearly seen in our data, thus supporting the notion of very similar Pfr decay kinetics in all the studied phys.
The decay of the Ifr-750 intermediates with 3-ps lifetime produces a new intermediate with an absorption band around 675 nm. We call this intermediate Ifr-675 since it has distinctly different characteristics from Ifr-750, but it does not yet resemble the Pr ground-state absorption. Interestingly, there are two bleaching maxima in the transient spectra of native Pfr at 725 and 740 nm in our data which decay with similar but not exactly the same kinetics. That might indicate two slightly different conformational states. The postulate of two Ifr-750 subpopulations would fall in line with the independent observation (11) that there are two spectrally distinguishable, noncorrelated and nonequilibrating Pfr pools (Pfr-722 and Pfr-730) that differ with regard to their capability to revert thermally to Pr on a timescale of hours.
Dynamics on longer timescales
The lifetime density plot shows a pronounced lifetime component of ∼400 ps which extends over the entire detection wavelength range. It has a negative amplitude below 670 nm, and a positive amplitude above 700 nm. The negative part gives rise to the development of a new absorption band with a maximum at 667 nm (Fig. 6 A). This feature is indicative of the formation of a new intermediate. Its absorption maximum and spectral shape are quite similar to those of the Pr ground state as judged from the difference spectrum (Fig. 6 A). It is certainly not yet the fully relaxed form of Pr, since relaxation is known to continue at least into the millisecond range (41). Nevertheless, the spectrum of this early 500-ps intermediate indicates that the configurational and conformational changes of the chromophore are essentially completed on that timescale and that slower protein relaxation follows the rapid change in chromophore structure. We thus call this intermediate, the last one observable on the timescale of our measurement, a pseudo-Pr state. The protein relaxations have some influence on spectral details including transition strength of the chromophore. They are consequently observed as a modest spectral development of the chromophore absorption over a long period (41).
After 600 ps, the 740-nm ground-state bleaching band has not yet reached the shape of the Pfr ground-state absorption. Since there is no clear evidence for a longer-lived (several hundred picosecond lifetime) intermediate absorbing in the same wavelength region, we conclude that conformation equilibration and hole-filling in the Pfr ground state may be rather slow, probably limited by the protein dynamics. We note again that the overall photoconversion yield is only 15%. This raises the interesting question where this substantial quantum yield loss occurs directly from the Pfr excited state or from one or more of the later intermediates. Inspection of the data shows that ∼20% of the quantum yield loss seems to occur directly from the Pfr excited state. The major part of the quantum yield loss seems to occur from the thermal reversion of the pseudo-Pr state(s) to Pfr. This sheds interesting light on the potential energy surface of that ground state.
Potential energy diagram
The data permit us to draw a fairly detailed potential energy diagram for the initial steps of the Pfr photoreaction (Fig. 8 B). This diagram is based on the following findings: The SE decays rapidly in the Franck-Condon region and probably moves out into the NIR region within 200–300 fs. The potential surfaces of ground and first excited states should be strongly displaced with respect to each other. The excited state then decays from the potential minimum corresponding to a C15 twisted conformation of Pfr (Pfr twisted), with a main kinetics of ∼500 fs to a ground-state product, Ifr-750, which should have a pronounced absorption band at shorter wavelengths (630 nm). A potential barrier separates this state from the next intermediate, Pfr675, which appears with a kinetics of ∼3 ps and decays in 400 ps into a further intermediate, pseudo-Pr, absorbing at 667 nm. The potential barriers in the ground state could be caused either by the protein potential or by an internal potential barrier within the chromophore. The pseudo-Pr state is already reminiscent of the ground state Pr spectrum. Most likely, it is identical to an intermediate called lumi-F observed as the first intermediate after irradiation of Pfr stabilized at −90°C, with an absorption at 673 nm (42). Low-temperature resonance Raman data independently have confirmed that the chromophores of lumi-F and the subsequent intermediate meta-F have already undergone E → Z isomerization, but have not yet reached room-temperature equilibrium geometry (43).
Comparison with other systems
Some features of the phytochrome kinetics, in particular the Pfr kinetics, show similarities to other biological systems, e.g., bacteriorhodopsin and rhodopsin. Rapid vibrational relaxation with loss of SE has also been observed for these two systems (44–47). The major difference in phytochrome might be that a larger part of the isomerization process in Pfr seems to occur on the excited-state surface as compared to the other systems. However, all of these systems undergo ultrafast photoisomerization initiated by partial isomerization (rotation around double- and/or single-bond) on the excited-state surface, followed by crossing to the ground state, and completion of the first reaction step to a primary ground-state product. This first product is succeeded by further intermediates, the kinetics and conformation of which are most probably determined by protein dynamics and potential (48).
CONCLUSIONS
A 28-ps component describes the formation of the first intermediate I700 in the Pr state of native phy, which is characterized by absorption around 693 nm. This process is accompanied by strong oscillations presumably reflecting the excited-state dynamics necessary to prepare the complex for a favorable protein/chromophore conformation during the first picoseconds. In particular, the tight interactions between chromophore and protein and the mutual influence on the conformational changes are supported by the recently presented three-dimensional structure (6). It thus appears reasonable that the protein conformational changes induced by the photoisomerization process of the tetrapyrrole chromophore(s) are strongly coupled to a few vibrational chromophore modes which appear in the oscillation patterns on the excited-state surface. The formation time of I700 in PCB-phy is quite similar.
Both the less efficient I700 formation in recombinant phytochromes and the higher complexity of the kinetics for native phy at short excitation wavelengths indicate that other relaxation pathways exist in the excited state associated with lifetimes of 15 and 50 ps. These processes lead to the reversible population of several local potential minima on the excited-state surface that do not directly give rise to photoisomerization via the I700 intermediate. Direct relaxation out of these potential wells might lead back to the ground state directly, or alternatively, might eventually contribute to Pfr formation via a first intermediate different than I700. The various local potential minima on the excited-state surface seem to be more or less in equilibrium with each other; at least, they interconvert partially. The cartoon shown in Fig. 9 presents schematically the different pathways on the excited- and ground-state hypersurfaces. It appears that the return to the ground state may proceed from different protein conformations and that I700 might be formed on various parallel pathways, possibly also ending up in different I700 states characterized by different protein/chromophore conformations.
The initial kinetics of the Pfr to Pr isomerization occurs on a much faster timescale than the forward Pr to Pfr reaction, and on the nanosecond timescale, several conversion processes in the electronic ground states are resolved. The first ground-state intermediate appears with an ultrafast 500-fs lifetime and is likely accomplished via a conical intersection (49–51). It seems that this first ground-state intermediate already contains the almost completely photoisomerized chromophore, which should be similar to the chromophore conformation of the Pr form. To partly compensate this rapid geometric change, it might be possible that the initial step occurs via a so-called Hula-twist mechanism, which involves also (partial) single-bond rotations (52). The later ground-state intermediates differ primarily with respect to altered surrounding protein conformations which have some influence on the absorption properties.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
The skilled work of Helene Steffen, Wilhelm Schlamann, Tanja Berndsen, and Gül Koc in the preparation of the recombinant phytochrome types and the chromophores (PCB) is gratefully acknowledged. We also thank Prof. Silvia Braslavsky for discussions and suggestions.
Editor: Barry R. Lentz.
References
- 1.Batschauer, A. 1998. Photoreceptors of higher plants. Planta. 206:479–492. [DOI] [PubMed] [Google Scholar]
- 2.Gärtner, W., and S. E. Braslavsky. 2004. The phytochromes: spectroscopy and function. In Photoreceptors and Light Signaling. A. Batschauer, editor. Royal Society of Chemistry, Cambridge, UK.
- 3.Moller, S. G., P. J. Ingles, and G. C. Whitelam. 2002. The cell biology of phytochrome signaling. New Phytol. 154:553–590. [DOI] [PubMed] [Google Scholar]
- 4.Kevei, E., and F. Nagy. 2003. Phytochrome controlled signaling cascades in higher plants. Physiol. Plant. 117:305–313. [DOI] [PubMed] [Google Scholar]
- 5.Hughes, J., T. Lamparter, F. Mittmann, E. Hartmann, W. Gärtner, A. Wilde, and T. Börner. 1997. A prokaryotic phytochrome. Nature. 386:663. [DOI] [PubMed] [Google Scholar]
- 6.Wagner, J. R., J. S. Brunzelle, K. T. Forrest, and R. D. Vierstra. 2005. A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature. 438:325–331. [DOI] [PubMed] [Google Scholar]
- 7.Schaffner, K., S. E. Braslavsky, and A. R. Holzwarth. 1991. Protein environment and photophysics and photochemistry of the prosthetic chromophores of biliproteins. In Frontiers in Supramolecular Organic Chemistry and Photochemistry. H.-J. Schneider and H. Dürr, editors. VCH Verlagsgesellschaft, Weinheim, Germany.
- 8.Braslavsky, S. E., W. Gärtner, and K. Schaffner. 1997. Phytochrome photoconversion. Plant Cell Environ. 20:700–706. [Google Scholar]
- 9.Song, P.-S. 1994. Phytochrome as a light switch for gene expression in plants. Spectrum. 7:1–7. [Google Scholar]
- 10.Mroginski, M. A., D. H. Murgida, D. von Stetten, C. Kneip, F. Mark, and P. Hildebrandt. 2004. Determination of the chromophore structures in the photoinduced reaction cycle of phytochrome. J. Am. Chem. Soc. 126:16734–16735. [DOI] [PubMed] [Google Scholar]
- 11.Schmidt, P., T. Gensch, A. Remberg, W. Gärtner, S. E. Braslavsky, and K. Schaffner. 1998. The complexity of the Pr to Pfr phototransformation kinetics is an intrinsic property of native phytochrome. Photochem. Photobiol. 68:754–761. [Google Scholar]
- 12.Heyne, K., J. Herbst, D. Stehlik, B. Esteban, T. Lamparter, J. Hughes, and R. Diller. 2002. Ultrafast dynamics of phytochrome from the cyanobacterium Synechocystis, reconstituted with phycocyanobilin and phycoerythrobilin. Biophys. J. 82:1004–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holzwarth, A. R., E. Venuti, S. E. Braslavsky, and K. Schaffner. 1992. The phototransformation process in phytochrome. I. Ultrafast fluorescence component and kinetic models for the initial Pr→Pfr transformation steps in native phytochrome. Biochim. Biophys. Acta. 1140:59–68. [Google Scholar]
- 14.Colombano, C. G., S. E. Braslavsky, A. R. Holzwarth, and K. Schaffner. 1990. Fluorescence quantum yields of 124-kDa phytochrome from oat upon excitation within different absorption bands. Photochem. Photobiol. 52:19–22. [Google Scholar]
- 15.Song, P.-S., N. Tamai, and I. Yamazaki. 1986. Viscosity dependence of primary photoprocesses of 124 KDalton phytochrome. Biophys. J. 49:645–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruzsicska, B. P., A. R. Holzwarth, J. Wendler, S. E. Braslavsky, and K. Schaffner. 1985. Photophysics and photochemistry of degraded and native phytochrome. In Primary Photo-Processes in Biology and Medicine. R. V. Bensasson, G. Jori, E. J. Land, and T. G. Truscott, editors. Plenum, New York.
- 17.Savikhin, S., T. Wells, P.-S. Song, and W. S. Struve. 1993. Ultrafast pump-probe spectroscopy of native etiolated oat phytochrome. Biochemistry. 32:7512–7518. [DOI] [PubMed] [Google Scholar]
- 18.Teuchner, K., M. Schulz, H. Stiel, M. Maisch, and W. Rüdiger. 1995. Excited state behavior of Phytochrome Pr. Photochem. Photobiol. 62:1076–1080. [Google Scholar]
- 19.Bischoff, M., G. Hermann, S. Rentsch, and D. Strehlow. 2001. First steps in the phytochrome phototransformation: a comparative femtosecond study on the forward (Pr→Pfr) and back reaction (Pfr→Pr). Biochemistry. 40:181–186. [DOI] [PubMed] [Google Scholar]
- 20.Lippitsch, M. E., G. Hermann, H. Brunner, E. Müller, and F. R. Aussenegg. 1993. Picosecond events in the phototransformation of phytochrome—a time-resolved absorption study. J. Photochem. Photobiol. B. 18:17–25. [Google Scholar]
- 21.Kandori, H., K. Yoshihara, and S. Tokutomi. 1992. Primary process of phytochrome: initial step of photomorphogenesis in green plants. J. Am. Chem. Soc. 114:10958–10959. [Google Scholar]
- 22.Andel, F., V. C. Hasson, F. Gai, P. A. Anfinrud, and R. A. Mathies. 1997. Femtosecond time-resolved spectroscopy of the primary photochemistry of phytochrome. Biospectroscopy. 3:421–433. [Google Scholar]
- 23.Rentsch, S., G. Hermann, M. Bischoff, D. Strehlow, and M. Rentsch. 1997. Femtosecond spectroscopic studies on the red light-absorbing form of oat phytochrome and 2,3-dihydro biliverdin. Photochem. Photobiol. 66:585–590. [Google Scholar]
- 24.Robben, U., I. Lindner, W. Gärtner, and K. Schaffner. 2001. Analysis of the topology of the chromophore binding pocket of phytochromes by variation of the chromophore substitution pattern. Angew. Chem. Int. Ed. 40:1048–1050. [DOI] [PubMed] [Google Scholar]
- 25.Brock, H., B. P. Ruzsicska, T. Arai, W. Schlamann, A. R. Holzwarth, S. E. Braslavsky, and K. Schaffner. 1987. Fluorescence lifetimes and relative quantum yields of 124-kiloDalton oat phytochrome in H2O and D2O solutions. Biochemistry. 26:1412–1417. [Google Scholar]
- 26.Kneip, C., P. Hildebrandt, D. Mozley, W. Schlamann, W. Gärtner, T. Lamparter, J. Hughes, S. E. Braslavsky, and K. Schaffner. 1997. Resonance Raman spectroscopic study of recombinant phytochromes. In Spectroscopy of Biological Molecules: Modern Trends. P. Carmona, editor. Kluwer Academic Publishers, Dordrecht, The Netherlands. 129–130.
- 27.Mozley, D., A. Remberg, and W. Gärtner. 1997. Large-scale generation of affinity-purified recombinant phytochrome chromopeptide. Photochem. Photobiol. 66:710–715. [DOI] [PubMed] [Google Scholar]
- 28.Kufer, W., and H. Scheer. 1979. Chemical modification of biliprotein chromophores. Z. Naturforsch. 34:776–781. [DOI] [PubMed] [Google Scholar]
- 29.Gensch, T., M. S. Churio, S. E. Braslavsky, and K. Schaffner. 1996. Primary quantum yield and volume change of phytochrome—a phototransformation determined by laser-induced optoacoustic spectroscopy. Photochem. Photobiol. 63:719–725. [Google Scholar]
- 30.Croce, R., M. G. Müller, R. Bassi, and A. R. Holzwarth. 2001. Carotenoid-to-chlorophyll energy transfer in recombinant major light-harvesting complex (LHC II) of higher plants. I. Femtosecond transient absorption measurements. Biophys. J. 80:901–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pollard, W. T., and R. A. Mathies. 1992. Analysis of femtosecond dynamic absorption spectra of nonstationary states. Annu. Rev. Phys. Chem. 43:497–523. [DOI] [PubMed] [Google Scholar]
- 32.Kneip, C., W. Schlamann, S. E. Braslavsky, P. Hildebrandt, and K. Schaffner. 2000. Resonance Raman spectroscopic study of the tryptic 39-kDa fragment of phytochrome. FEBS Lett. 482:252–256. [DOI] [PubMed] [Google Scholar]
- 33.Kneip, C., P. Hildebrandt, W. Schlamann, S. E. Braslavsky, F. Mark, and K. Schaffner. 1999. Protonation state and structural changes of the tetrapyrrole chromophore during the Pr→Pfr phototransformation of phytochrome: a resonance Raman spectroscopic study. Biochemistry. 38:15185–15192. [DOI] [PubMed] [Google Scholar]
- 34.Andel, F., J. T. Murphy, J. A. Haas, M. T. McDowell, I. van der Hoef, J. Lugtenburg, J. C. Lagarias, and R. A. Mathies. 2000. Probing the photoreaction mechanism of phytochrome through analysis of resonance Raman vibrational spectra of recombinant analogues. Biochemistry. 39:2667–2676. [DOI] [PubMed] [Google Scholar]
- 35.Lindner, I. 2000. Total synthesis of novel chromophores of the plant photoreceptor phytochrome and characterization of the biochemical and spectral properties of the chromoproteins. In Publication series Max-Planck-Institut für Strahlenchemie Nr.133 ISSN 0932–5131. 1–236.
- 36.Borucki, B., D. von Stetten, S. Seibeck, T. Lamparter, N. Michael, M. A. Mroginski, H. Otto, D. H. Murgida, M. P. Heyn, and P. Hildebrandt. 2005. Light-induced proton release of phytochrome is coupled to the transient deprotonation of the tetrapyrrole chromophore. J. Biol. Chem. 280:34358–34364. [DOI] [PubMed] [Google Scholar]
- 37.Scurlock, R. D., S. E. Braslavsky, and K. Schaffner. 1993. A phytochrome study using two-laser/two-color flash photolysis: I700 is a mandatory intermediate in the Pr→Pfr phototransformation. Photochem. Photobiol. 57:690–695. [Google Scholar]
-
38.Scurlock, R. D., C. H. Evans, S. E. Braslavsky, and K. Schaffner. 1993. A phytochrome phototransformation study using two-laser/two color flash photolysis: analysis of the decay mechanism of
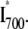 Photochem. Photobiol. 58:106–115. [Google Scholar]
Photochem. Photobiol. 58:106–115. [Google Scholar] - 39.Büchler, R., G. Hermann, D. V. Lap, and S. Rentsch. 1995. Excited state relaxations of phytochrome studied by femtosecond spectroscopy. Chem. Phys. Lett. 233:514–518. [Google Scholar]
- 40.Fodor, S. P. A., J. C. Lagarias, and R. A. Mathies. 1990. Resonance Raman analysis of the Pr and Pfr forms of phytochrome. Biochemistry. 29:11141–11146. [DOI] [PubMed] [Google Scholar]
- 41.Chen, E., V. N. Lapko, J. W. Lewis, P.-S. Song, and D. S. Kliger. 1996. Mechanism of native oat phytochrome photoreversion: a time-resolved absorption investigation. Biochemistry. 35:843–850. [DOI] [PubMed] [Google Scholar]
- 42.Eilfeld, P., and W. Rüdiger. 1985. Absorption spectra of phytochrome intermediates. Z. Naturforsch. 40C:109–114. [Google Scholar]
- 43.Matysik, J., P. Hildebrandt, W. Schlamann, S. E. Braslavsky, and K. Schaffner. 1995. Fourier-transform resonance Raman spectroscopy of intermediates of the phytochrome photocycle. Biochemistry. 34:10497–10507. [DOI] [PubMed] [Google Scholar]
- 44.Peteanu, L. A., R. W. Schoenlein, Q. Wang, R. A. Mathies, and C. V. Shank. 1993. The first step in vision occurs in femtoseconds—complete blue and red spectral studies. Proc. Natl. Acad. Sci. USA. 90:11762–11766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rothschild, K. J., H. Marrero, M. S. Braiman, and R. Mathies. 1984. Primary photochemistry of bacteriorhodopsin: comparison of Fourier transform infrared difference spectra with resonance Raman spectra. Photochem. Photobiol. 40:675–679. [DOI] [PubMed] [Google Scholar]
- 46.Polland, H.-J., M. A. Franz, W. Zinth, W. Kaiser, E. Kölling, and D. Oesterhelt. 1986. Early picosecond events in the photocycle of bacteriorhodopsin. Biophys. J. 49:651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kukura, P., D. W. McCamant, S. Yoon, D. B. Wandschneider, and R. A. Mathies. 2005. Structural observation of the primary isomerization in vision with femtosecond-stimulated Raman. Science. 310:1006–1009. [DOI] [PubMed] [Google Scholar]
- 48.Mathies, R. A., S. W. Lin, J. B. Ames, and W. T. Pollard. 1991. From femtoseconds to biology: mechanism of bacteriorhodopsin's light-driven proton pump. Annu. Rev. Biophys. Chem. 20:491–518. [DOI] [PubMed] [Google Scholar]
- 49.Domcke, W., and G. Stock. 1997. Theory of ultrafast nonadiabatic excited-state processes and their spectroscopic detection in real time. Adv. Chem. Phys. 100:1–169. [Google Scholar]
- 50.Bernardi, F., M. Olivucci, J. Michl, and M. A. Robb. 1996. Conical intersections in the theory of organic singlet photochemistry. Spectrum. 9:1–6. [Google Scholar]
- 51.Meyer, H.-D. 1983. A classical model of vibronic coupling: The ultrafast non-radiative decay via a conical intersection. Chem. Phys. 82:199–205. [Google Scholar]
- 52.Liu, R. S. H. 2002. Photoisomerization by hula-twist. Photoactive biopigments. Pure Appl. Chem. 74:1391–1396. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.