

Abstract
The interpretation of ϕ-values has led to an understanding of the folding transition state ensemble of a variety of proteins. Although the main guidelines and equations for calculating ϕ are well established, there remains some controversy about the quality of the numerical values obtained. By analyzing a complete set of results from kinetic experiments with the SH3 domain of αspectrin (Spc-SH3) and applying classical error methods and error-propagation formulas, we evaluated the uncertainties involved in two-state-folding kinetic experimental parameters and the corresponding calculated ϕ-values. We show that kinetic constants in water and m values can be properly estimated from a judicious weighting of fitting errors and describe some procedures to calculate the errors in Gibbs energies and ϕ-values from a traditional two-point Leffler analysis. Furthermore, on the basis of general assumptions made with the protein engineering method, we show how to generate multipoint Leffler plots via the analysis of pH dependencies of kinetic parameters. We calculated the definitive ϕ-values for a collection of single mutations previously designed to characterize the folding transition state of the αspectrin SH3 domain. The effectiveness of the pH-scanning procedure is also discussed in the context of error analysis. Judging from the magnitudes of the error bars obtained from two-point and multipoint Leffler plots, we conclude that the precision obtained for ϕ-values should be ∼25%, a reasonable limit that takes into account the propagation of experimental errors.
INTRODUCTION
Rate-equilibrium free-energy relationships (REFERs), commonly used to characterize kinetic mechanisms in chemical processes, have been shown to provide information about transition states in protein-folding reactions. This information is provided by comparing changes in the activation and equilibrium Gibbs energies obtained upon introducing a perturbation (mutation) (1–5). The most universally known REFER is the ϕ-value, calculated from individual perturbations throughout the protein that modify energetic aspects related to them. Therefore ϕ = ΔΔG‡-U/ΔΔGF-U, where ΔΔG‡-U is the change in the Gibbs energy of activation and ΔΔGF-U the overall change in the Gibbs energy of folding on mutation. The relationship between the value obtained and the distribution of mutations along the protein sequence allows us to map the energetic organization of the transition state of folding compared to the native state. This is the so-called protein engineering method (6–10).
During the past decade, the interpretation of ϕ-values has allowed us to understand the folding transition state ensemble of a variety of proteins and to formulate some important hypotheses (11–17). Although the main guidelines for calculating ϕ are well established, there is a dearth of information concerning the quality of the numerical values deriving from them. Some controversy has recently arisen about their low reproducibility and wide margins of uncertainty (18–23). Furthermore, some other REFER strategies that take advantage of experimental limitations in the determination of the value of ϕ have emerged as alternatives, although they do not seem to solve the problem since disagreement in their values merely serves to increase the confusion (24–26).
Some globular proteins are considered to be models for carrying out folding studies. This is the case with the α-spectrin SH3 domain (Spc-SH3), which has provided us with a detailed picture of its transition-state ensemble and folding pathway (12–15,27–31). Our intention here is twofold: first, to evaluate the uncertainties involved in kinetic experimental parameters and the corresponding calculated ϕ-values by applying classical error methods to the analysis of a large set of data obtained from the Spc-SH3 domain under different pH conditions; and second, to derive some general protocols concerning routine data analysis based on the evaluation of error propagation and on the general assumptions involved in the protein engineering method (6–10).
THEORY
Estimation of errors: random errors from instrumental uncertainties
Error can be defined as the difference between an observed or calculated value and the true value. To begin to evaluate error, it is important to distinguish between the terms “accuracy”, or how close the result of the experiment is to the true value, and “precision”, or how well the result has been determined, without reference to its agreement with the true value. The accuracy of an experiment generally depends upon how well we can control or compensate for systematic errors (reproducible discrepancies). Precision is a measurement of the reproducibility of the result due to random errors. Absolute precision indicates the magnitude of the uncertainty in the result (in the same units as the result), whereas relative precision indicates uncertainty in terms of a fraction of the value of the result (normally as a percentage) (32).
The mean (μ) and the standard deviation are the general choice to characterize precision (random errors) in our experimental data set, as they are sufficient to describe our experimental data distribution. The former is considered an estimation of the true value, whereas the latter reflects the uncertainty due to fluctuations in the observations in our attempt to determine the true value.
The most probable estimation of the mean (μ) of a random set of observations can be calculated as their average, assuming a Gaussian distribution of measurements. For a set of N data points with values xi the mean can be defined as
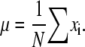 |
(1) |
In the same way, the standard deviation can be calculated using
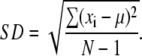 |
(2) |
Under the assumption that all data points xi are drawn from the same parent distribution and consequently were obtained with an uncertainty characterized by the same standard deviation the uncertainty in mean determination can be expressed by the standard error (σμ):
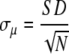 |
(3) |
from which it might be deduced that standard errors can be reduced infinitely by an increase of repetitions, whereas in practice we must make four times as many measurements to decrease uncertainty by a factor of 2.
Propagation of errors
To calculate the value of a dependent variable x, which is a function of one or more different measured variables (v, w, etc.), we must know how to propagate or carry over the uncertainties in the measured variables to determine the uncertainty in the dependent variable. The approximation for the standard error, σx, for x can be written as
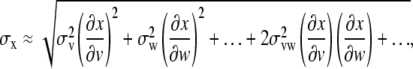 |
(4) |
where … indicates other possible measured variables. The first two terms can be considered to be an average of the squares of deviations in x, produced by uncertainties in v and w, respectively. These are the terms that usually dominate uncertainties. If the fluctuations in the measured quantities v and w… are uncorrelated, on average we should expect to find equal distributions of positive and negative values and thus the disappearance of the third term (referred to as the cross-term) at the limit of a range of random selection of observations (32).
Using the error-propagation formula (Eq. 4), we can estimate the standard error of the mean as
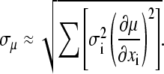 |
(5) |
This approximation neglects correlations between measurements xi as well as second- and higher-order terms in the expansion of variance 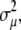 but it should be a reasonable approximation as long as none of the data points contribute a major portion to the final result. Under these considerations, this equation can be assumed, as a general rule, to be equivalent to Eq. 3 (32).
but it should be a reasonable approximation as long as none of the data points contribute a major portion to the final result. Under these considerations, this equation can be assumed, as a general rule, to be equivalent to Eq. 3 (32).
Errors associated to a least-squares fitting to a straight line
Let us now assume that, instead of making a number of measurements of a single quantity x, we make a series of N measurements of a pair (xi, yi), where index i runs once more from 1 to N. Bearing in mind that the functional relationship between both variables can be approximated by a straight line of the form y = a + bx, the standard errors are estimates of uncertainties in the estimates of regression coefficients a and b, which is analogous to the standard error of the mean (defined by Eqs. 3 and 5):
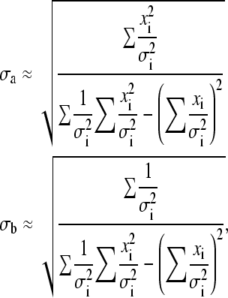 |
(6) |
where σi are the standard errors in yi values, assuming errors in xi to be negligible. The contribution of σx to errors in the yi values can be evaluated as σx × b and should be added to the value of σi.
An alternative and much simpler strategy to Eq. 6, assuming that all errors make the same contribution (σi ≈ σ), could be (32)
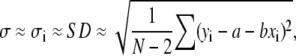 |
(7) |
where it should be noted that errors in yi can be directly estimated from data scattering and the fitting results, no knowledge of individual error bars being required. In this case, Eq. 6 can be written as
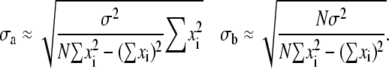 |
(8) |
Errors calculated from Eq. 8 are commonly known as “fitting errors”.
Errors associated to a least-squares fitting to an arbitrary function
In practice, the errors associated to any least-squares fitting can be easily obtained from any computer approximation capable of giving an analytical solution via a minimization procedure. The Marquardt method is the best choice among software used for the least-squares fitting of experimental kinetic traces and has therefore been included within software packages of most common stopped-flow instruments. This method has the considerable advantage of being reasonably insensitive to the starting values of the parameters and provides an estimate of standard errors based on the considerations in this section (32). The graphics program Sigma Plot 2000 (SPSS, Jandel, Chicago, IL), which was used to analyze the chevron plots and error propagation, is also based on the Marquardt method.
RESULTS AND DISCUSSION
Data set development
We considered the results of five different sets of kinetic experiments (urea unfolding and refolding) conducted with the Spc-SH3 D48G mutant in 50 mM phosphate buffer (pH 7.0, 25°C), which amounted to ∼100 independent determinations of kinetic constants, to evaluate the experimental and propagated errors for the kinetic parameters deriving from chevron plots. Our experiments were made with two different instruments (an SX.18MV-R from Applied Photophysics, Leatherhead, Surrey, UK and an SFM-3 from Bio-Logic, Claix, France) by different operators at different times over several years (Table 1), thus avoiding all possible experimental error sources, such as any possible bias deriving from wrong instrumental calibration or time-dependent tuning derivations. For comparison, we carried out a parallel analysis with chevron plots at pH 3.5 and 2.5 obtained from a single experimental set. Details about protein purification and experimental kinetic procedures done here can be obtained from previous works since some of our experiments have been published previously (12,14,30).
TABLE 1.
Details for the data set built to analyze errors in folding kinetic parameters
| Conditions | Experiment | Operator | Date (location) | Instrument |
|---|---|---|---|---|
| 50 mM phosphate pH 7.0 | 1* | JCM† | 1997 (EMBL) | SFM-3 Bio-Logic |
| 2* | JCM† | 1998 (EMBL) | SFM-3 Bio-Logic | |
| 3* | ESC‡ | 2002 (EMBL) | SFM-3 Bio-Logic | |
| 4 | ESC‡ | 2007 (UGR) | SX.18MV-R Ap Phot. | |
| 5 | AMC§ | 2007 (UGR) | SX.18MV-R Ap Phot. | |
| 50 mM glycine pH 3.5 | 1* | JCM† | 1997 (EMBL) | SFM-3 Bio-Logic |
| 50 mM glycine pH 2.5 | 1* | JCM† | 1998 (EMBL) | SFM-3 Bio-Logic |
We also built a second data set to evaluate propagated errors in ϕ-values from these kinetic parameters and to develop the pH-scanning procedure. We used a mutational analysis that we had previously carried out on Spc-SH3 (12,14,30). The fitting errors included in these previous works were the starting point for this analysis.
Errors in the first-order kinetic constants
Our intention was to obtain the standard errors estimated for every kinetic constant from a nonlinear fitting of the kinetic curves (fluorescence versus time) to the equation
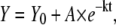 |
(9) |
where Y is the fluorescence signal, Y0 is its value at t = 0, A is the amplitude, and k is the first-order kinetic constant. An analysis of the distribution of the residuals of this fitting constitutes a criterion in discerning whether the first-order kinetic scheme is adequate. This step is fundamental since kinetic phases can be easily masked in the analysis and lead to wrong estimations of k. To improve the signal/noise ratio, a strategy involving the averaging of the different kinetic traces under the same solvent conditions is widely accepted in experimental kinetics. Although this averaging strategy reduces errors, our experience has shown us that it might be one of the main sources of a decrease in accuracy by introducing sometimes “artificial” phases.
Based on error-theory considerations, σk = ±3σi (where σi is the fitting standard error to Eq. 9) should ensure that 99% of k values for such experimental conditions would be included within the interval. In Fig. 1 some representations of the kinetic constants versus the denaturant concentration at pH 7.0 are shown, including error bars. From Fig. 1 B (σk = ±3σi), it can be observed that in practice ∼90% of the error bars overlap the fitting line, which constitutes a considerable quantity of data. In fact, this interval might constitute a realistic limit since a perusal of Fig. 1 C (σk = ± 6σi) does not reveal much better overlapping, whereas in Fig. 1 A (σk = ± 2σi) it is noticeably worse.
FIGURE 1.

Kinetic constants obtained at different urea concentrations for the Spc-SH3 D48G mutant in 50 mM phosphate buffer (pH 7.0, 25°C). Experiments were done with two different instruments by different operators at different times (see text for details). The solid line represents the least-squares fitting to an exponential function. Open and filled symbols represent data obtained from refolding and unfolding experiments, respectively. Arrows indicate data whose error bars do not overlap the fitting line. Error bars were calculated from a different weighting of the fitting errors resulting from Eq. 9. (A) Twice fitting errors (2σi); (B) three times fitting errors (3σi); (C) six times fitting errors (6σi).
Therefore, a suitable weighting of the standard errors deriving from our least-squares fitting seems to correctly reflect the precision of k (random errors), although some other error sources appear to contribute in the most diverging cases to the decrease in accuracy (systematic errors), mainly those related to a bias in instrumental calibration (thermostat, syringes, pipettes, refractometer, etc.) and/or those coming from a mistaken averaging of the kinetic curves. The values obtained for σk = ±3σi range from 1% to 10% in relative values (4.7% on average, Fig. 2 D). These observations agree with previously reported ones obtained by the comparison of repeated experiments, which were also made at different laboratories using different instruments but not over a lengthy period of time as ours were (23). The authors assess that fitting errors (±σi) result in a certain degree of underestimation and that more reasonable error bars might be ∼10% (approximately equivalent to ±6σi in our calculations). Nevertheless, from an analysis of our experimental data set, we conclude that, as a general rule, it might be enough to consider σk = ±3σi (≈ 5%) for common k errors.
FIGURE 2.

Chevron plot of the Spc-SH3 D48G mutant obtained in 50 mM phosphate buffer (pH 7.0, 25°C). Experiments were done in two different instruments by different operators at different times (see text for details). The solid lines in A, B, and C represent the least-squares fitting to the chevron equation (see Martinez and Serrano (14) for details). Open and solid symbols represent data obtained from refolding and unfolding experiments, respectively. Error bars were calculated from a different weighting of the fitting errors resulting from Eq. 9. (A) Twice fitting errors (2σi/k); (B) three times fitting errors (3σi/k) at three pH values: pH 7.0 (50 mM phosphate), pH 3.5, and pH 2.5 (50 mM glycine); (C) six times fitting errors (6σi/k). (D) The relative errors in k weighted in the same way as in B as a function of their respective urea concentrations. In this case the solid line represents the average value of 4.7% (0.047).
Errors in chevron plot parameters
A chevron plot represents the logarithmic dependence of kinetic rate constants upon urea concentration at a given temperature. The plot corresponding to all the experiments made with the Spc-SH3 D48G mutant is shown in Fig. 2, A–C. Error bars for the kinetic constants were calculated as set out in Fig. 1 (σk = ±2σi (panel A), σk = ±3σi (panel B), and σk = ±6σi (panel C)), σi being the standard error provided by the Marquardt algorithm after each least-squares fitting to Eq. 9. Thus, for a natural logarithm, the error will coincide with the relative error of the magnitude σlnk = σk/k.
In the chevron plots shown in Fig. 2 the least-squares fitting to the equation describing the chevron (see Martinez et al. (12) and Martinez and Serrano (14) for details) is represented as a solid line. From this analysis, we obtained the values included in Table 2 for k‡-U, k‡-F, and m‡-U. Focusing on the fittings at pH 7.0, the value of m‡-F was fixed at −0.42 M−1 (the average value for mutants studied in the ϕ-value analysis at pH 7.0; see below) due to the lack of information about the unfolding arm as a consequence of the high stability of the protein at this pH. Since experimental data have been collected from several independent sets of experiments, the errors obtained should account for “real” errors, which means ∼68% probability of including the true value. Relative errors obtained as 3σx/x (≈ 90% “real” confidence, as can be concluded from the analysis in Fig. 1) give 13% for k‡-U, 22% for k‡-F, and 4% for m‡-U estimations. The differences in the relative errors obtained for both kinetic constants (almost double) may well be related to the longer extrapolation of the unfolding arm to 0 M denaturant.
TABLE 2.
Kinetic parameters with error estimations for the Spc-SH3 D48G mutant under different buffer conditions at 25°C
| pH | Method | m‡-F (M−1) | m‡-U (M−1) | k‡-F (s−1) | k‡-U (s−1) |
|---|---|---|---|---|---|
| 50 mM phosphate pH 7.0 | Global fitting | 0.42* | −0.80 ± 0.03 (4%) | 0.00160 ± 0.00036 (22%) | 69 ± 9 (13%) |
| Individual fitting | 0.42* | −0.79 ± 0.03 (4%) | 0.00165 ± 0.00012 (8%) | 71 ± 8 (12%) | |
| Average of mutant values | 0.42 ± 0.02 (4%) | −0.90 ± 0.02 (4%) | |||
| 50 mM glycine pH 3.5 | Individual fitting | 0.50 ± 0.06 (12%) | −1.20 ± 0.09 (8%) | 0.08 ± 0.02 (25%) | 50 ± 7 (14%) |
| Average of mutant values | 0.52 ± 0.06 (12%) | −1.24 ± 0.09 (8%) | |||
| 50 mM glycine pH 2.5 | Individual fitting | 0.37 ± 0.03 (8%) | −0.92 ± 0.18 (20%) | 0.94 ± 0.18 (19%) | 11.6 ± 1.5 (13%) |
| Average of mutant values | 0.39 ± 0.06 (15%) | −0.96 ± 0.09 (9%) |
Errors are calculated as 3σx, where σx is the fitting or averaging error of x. Values in parentheses represent the relative errors calculated as (3σx/x) × 100.
Value fixed in the least-squares fitting.
An alternative approach to error analysis is possible only at pH 7.0 from the individual fitting of the different data sets and further averaging of results (Table 2). No dispersion of the data values is observed compared to the global fitting. The errors were calculated as in Eq. 3 and were approximately of the same magnitude, except for the improvement found in the error estimated for k‡-F. This improvement is a consequence of the acceptable reproducibility (low scattering) of the k‡-F data obtained from the different fitting sessions. Nevertheless, it may be unreliable since, as mentioned above, the value of m‡-F was fixed to −0.42 M−1, which noticeably reduces scattering in the k‡-F values.
Otherwise, errors in lnk obtained at different denaturant concentrations (Fig. 2 D) are randomly distributed around the average value (4.7% in our example) because of a lack of correlation with the denaturant concentration. This is also explicable by the low range of variability in the lnk values, which definitely masks any correlation between the magnitude of these values and their corresponding errors (which should translate into a V-shape of points in Fig. 2 D), due to a predominance of other experimental uncertainties. Therefore, according to the Theory section, error bars in the kinetic constants at different urea concentrations can be considered equal on average (σi ≈ σ), and their values extrapolated at 0 M denaturant can be correctly estimated from fitting errors (σa in Eq. 8) instead of using propagation formulas (Eq. 6), all of which simplifies our error calculations. The availability of a complete set of experimental data also contributes positively to this equivalency. Thus, kinetic constants in water can also be correctly estimated from an adequate weighting of fitting errors, as previously done by Zarrine-Afsar and Davidson (33), although the 9% given therein could still be a bit of an underestimation, probably because repetitions in that case were carried out under better “reproducibility” conditions, since the experiments were conducted with the same instrument and probably by a single operator.
Comparing the results obtained at pH 7.0 to those of the analysis of single experiments at pH 3.5 and 2.5, we can conclude that the errors in the kinetic constants are similar in magnitude, whereas those associated to m values increase dramatically (Table 2). In neither case was there any restriction on parameter values, which turns these analyses into a more realistic approach to an ordinary kinetic evaluation. There is also an expected discrepancy in the magnitudes of the relative errors in k‡-U (12%–14%) and k‡-F (19%–25%). The comparatively higher error found in the unfolding kinetic constants results from the longer extrapolation to the 0 M denaturant in this case. Furthermore, the value of 25% should even be considered as the lower limit of the real experimental error since in cases of extremely high stability the corresponding kinetic constants cannot be freely determined. Thus, using the pH 7.0 data as an example, the error in the unfolding kinetic constant will be significantly larger in a fitting with a free-floating unfolding slope since fluctuations in the latter will redound on the former.
Another interesting observation is the absence of any significant influence of accuracy in the errors. Thus, if the errors obtained for kinetic parameters deriving from chevron representations at pH 7.0 (including all the data of the five different experimental sets) are compared to those estimated at pH 3.5 and 2.5 (one single experimental set) (Table 2), it can be seen that at pH 7.0, errors account for the most common and relevant effects on accuracy, together with the unavoidable fluctuations that worsen precision, whereas at pH 3.5 and 2.5 they account only for the latter. It is surprising that errors are comparable in magnitude. Thus, the possible contribution of systematic errors (accuracy) seems to be counteracted by the higher number of kinetic constants collected at pH 7.0, which might reduce the magnitude of the error (see Theory section).
Errors related to m values can also be evaluated using the data obtained for different mutants designed for ϕ-analysis, which is our only possibility in the case of m‡-F. It is to be hoped that the nondisruptive character of mutations designed for ϕ analysis (12,14,27–30) will avoid any significant effect on m values, mainly related to macroscopic exposure of hydrophobic groups (34). In fact, a comparison of the chevron plots for the different mutants does not reveal any significant differences between their slopes (see Table 1 from Martinez and Serrano (14)), which is also reflected in the low standard error of the average (Table 2). We determined the mean values of m‡-F and m‡-U according to Eqs. 3 and 5. As can be seen in Table 2, the errors edge the values toward coinciding with those obtained from the global and individual fittings.
Analysis of errors in Gibbs energies and ϕ-values
Once the values of k‡-F and k‡-U are estimated from chevron plot analysis, the values of ΔΔG and ϕ can easily be obtained using the well-known formulas
 |
(10) |
and
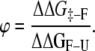 |
(11) |
ΔΔGF-U should be calculated from an independent equilibrium denaturation curve, although it can also be obtained from the kinetic data by ΔΔGF-U = ΔΔG‡-U − ΔΔG‡-F.
By means of Eqs. 4 and 10, we can obtain the following expressions for σΔΔG
 |
(12) |
where the third term is the estimated cross-term accounting for the interdependence of both errors. This, together with Eq. 11, gives rise to the following definitive formula for errors in ϕ-values
 |
(13) |
where σΔΔGF-U = σΔΔG‡-U + σΔΔG‡-F or, alternatively, the error corresponding to equilibrium unfolding experiments, which in practice should be of the same magnitude or higher. The first two terms estimate the errors both in activation and equilibrium Gibbs energies, respectively, whereas the third arises from the cross-term of Eq. 4. In all cases, we considered that σvw = σv × σw.
The error bars calculated from Eq. 13 for the different ϕ-values obtained previously for the Spc-SH3 domain (obtained from Table 1 of Martinez and Serrano (14)) under three different pH conditions (panels A, B, and C, respectively) are represented in Fig. 3. Different strategies to analyze errors have been considered (errors in bars from left to right): i), errors in black bars are drawn as the propagated errors in kinetic constants calculated by Eqs. 12 and 13 (obtained as three times the fitting standard errors from chevron plots); ii), errors in gray bars were calculated from a simplified way in which the cross-terms in Eqs. 12 and 13 were avoided; and iii), and iv), errors in dark-gray and light-gray bars are analogous to i), and ii), respectively, but take only the fitting errors into account.
FIGURE 3.

ϕ-values obtained for all mutants compared to their reference values. Fitting errors were estimated from a two-point Leffler analysis (Eq. 11) after propagation of least-squares fitting k‡-F and k‡-U standard errors (see text for details). (A) Values obtained in 50 mM phosphate buffer (pH 7.0, 25°C). (B) Values obtained in 50 mM glycine/HCl buffer (pH 3.5, 25°C). (C) Values obtained in 50 mM glycine/HCl buffer (pH 2.5, 25°C). Errors have been calculated in different ways: i), errors in black bars were drawn from propagation by Eqs. 12 and 13 of errors in kinetic constants (obtained as three times the fitting standard errors from chevron plots); ii), errors in gray bars were calculated from a simplified Eq. 13 in which the cross-term has been omitted; and iii), and iv), errors in dark-gray and light-gray bars are analogous to i), and ii), respectively, but take into account nonweighted fitting errors (σlnk = σi/k). The whole set of values was taken from Table 1 of Martinez and Serrano (14).
The general tendency of error magnitudes for the three pH conditions is black > dark-gray ≥ gray > light-gray. Calculation of the ratio σϕ+cross/σϕ-cross (i.e., the quotient between errors black/gray and dark-gray/light-gray) for each mutant allows us to estimate the contribution of the cross-term to σϕ. Our results suggest that the cross-term increases error by a factor of 2.9 ± 0.8. Similar conclusions have been drawn from a computational study with randomly simulated chevron data (22), where the need to take into account the cross-term in ϕ-error estimations is emphasized by showing here the different error bars obtained from the analysis. Thus, avoiding covariance leads to a significant underestimation of σϕ (on average, around three times according to our estimations).
Another interesting effect appears from a comparison of the black/dark-gray and gray/light-gray quotients (Fig. 3). We obtained an average value of 3.3 ± 0.7, which affords the ratio σϕ(3σlnk)/σϕ(1σlnk). Thus, a threefold increase in the error in the kinetic constants practically translates into a parallel triplication in the ϕ-errors, as might be expected.
For a more thorough analysis of these effects, we represented the errors in the ϕ-values calculated from Eq. 13 (black bars in Fig. 3) versus the absolute errors in every ΔΔG function (Fig. 4) and also versus the global ΔΔG functions (Fig. 5). In all representations, we included the whole data set (reference and mutant proteins) under the three pH conditions assayed. Some correlation among variables can be seen (Fig. 4), as it is clear that higher errors in ϕ-values were obtained with lower ΔΔG values (Fig. 5), as expected (18). An unambiguous correlation (r = 0.87) results when the relative errors of ΔΔGF-U versus the error in the ϕ-values are represented (Fig. 6 A), which is not to be found with either of the individual activation ΔΔG values (Fig. 6, B and C). The relation found with a 99% confidence is
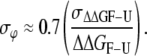 |
(14) |
FIGURE 4.

A test for correlation among errors in ϕ-values and absolute errors in the different Gibbs energy functions. (A) Absolute errors in ΔΔGF-U. (B) Absolute errors in ΔΔG‡-F. (C) Absolute errors in ΔΔG‡-U. The solid lines represent linear regression through each pH data set. (Solid circles) pH 7.0; (open circles) pH 3.5; (solid triangles) pH 2.5.
FIGURE 5.

A test for correlation between errors in ϕ-values and the different Gibbs energy functions. (A) ΔΔGF-U function. (B) ΔΔG‡-F function. (C) ΔΔG‡-U function. (Solid circles) pH 7.0; (open circles) pH 3.5; (solid triangles) pH 2.5.
FIGURE 6.

A test for correlation between errors in ϕ-values and relative errors in the different Gibbs energy functions. (A) Relative errors in ΔΔGF-U. (B) Relative errors in ΔΔG‡-F. (C) Relative errors in ΔΔG‡-U. Solid lines represent linear regression through the whole data set.
Thus, as a general rule, errors in the ϕ-values can be taken to represent 70% of the relative errors in the equilibrium Gibbs energies, which were obtained from kinetic experiments as the sum of the relative errors in the activation Gibbs energy values. This correlation clearly reveals that the lowest ΔΔGF-U values are affected by the highest relative errors (18).
Evaluation of pH dependencies via Leffler plots (pH-scanning)
As shown by Fersht et al. (20), the change in Gibbs energy on mutating a residue into a protein can be split into three main notional components: 1), the change in energy of the covalent bond that is mutated; 2), the change due to noncovalent interactions at the site of mutation plus that deriving from any additional changes due to protein reorganization; and 3), the change in solvation Gibbs energy. The authors propose that comparison of individual ϕ-values for different mutations at a particular site can be made correctly by using multipoint Leffler plots; this is also a way of detecting deviations arising from inadequate mutations for transition-state analysis. Thus, deviations from linearity are due mainly to different values of the solvation component for different mutants, as well as to some additional differences in the reorganization term.
Here we propose an alternative and fully complementary way to obtain multipoint Leffler plots using just the kinetic data for a single mutation (designed to be nondisruptive) under different pH conditions; as many points as desired are used in the Leffler representation. It is generally accepted that the differences observed in the pH stability of small proteins generally derives from purely entropic contributions to Gibbs energies and that it is possible to obtain a unique Gibbs energy function for the protein, ΔGF-U(T) or ΔGF-U(denat), using the different stability values obtained under the different pH conditions (28,35–41). From this point of view pH can be considered an almost “neutral” strategy to modify changes in the Gibbs energy of folding (both activation and equilibrium). This approach avoids any reorganization within the structure and minimizes any possible changes in solvation energies compared to the effects caused by mutational analysis, since neither the solvation nor the reorganization components of a single mutation differ significantly under the various pH conditions. The only assumption from a kinetic point of view might be the maintenance of the folding mechanism and the rate-limiting step of the folding reaction throughout the pH interval assayed, which is easily corroborated by the linearity of the Leffler plots for each mutation ((18) and references therein).
The multipoint Leffler plots for different Spc-SH3 mutants taken from Table 1 of Martinez and Serrano (14) are set out in Fig. 7. A glance at Eq. 11 reveals that the respective ϕ-values result from the slopes of every least-squares linear regression, with standard errors (Eq. 8). The regressions contained from three to four points (at three different pH conditions; Table 3) and, using the correlation coefficient to test the quality of the fittings, we always arrived at values of r ≥ 0.8, most of them higher than 0.9. Therefore, we can draw the conclusion that the folding mechanism is confined to within the pH range studied (2.5–7.0), which is confirmed by the similarity of the ϕ-values deriving from the two-point plots (Fig. 3), although the low number of data could be insufficient to definitely confirm such a linear behavior.
FIGURE 7.

Multipoint Leffler plots for all the mutations of the Spc-SH3 domain. The majority of residues have ϕ-values of between 0 and 1 and lie between the solid black lines of slopes 0 and 1. Fitting lines, calculated from linear regression to the equation y = bx, are shown in the same color code as their respective data points. The whole set of values was taken from Table 1 of Martinez and Serrano (14).
TABLE 3.
The definitive ϕ-values and error bars for the α-spectrin SH3 domain (Spc-SH3), calculated from pH-scanning analysis
| Mutation | ϕ-values (multipoint Leffler analysis)* pH-scanning | σϕ (multipoint Leffler analysis) pH-scanning | σϕ (two-point Leffler analysis) pH 7.0 | σϕ (two-point Leffler analysis) pH 3.5 | σϕ (two-point Leffler analysis) pH 2.5 | σϕ (two-point Leffler analysis) AVERAGE |
|---|---|---|---|---|---|---|
| L8S | 0.14 (4) | 0.14 | 0.21 | 0.70 | 0.44 | 0.45 |
| A11G | 0.03 (3) | 0.08 | 0.08 | 0.26 | 0.17 | |
| D14S | 0.14 (4) | 0.10 | 0.20 | 1.82 | 1.00 | |
| V23A | 0.32 (3) | 0.19 | 0.19 | 1.02 | 0.61 | |
| T24A | 0.27 (4) | 0.30 | 0.30 | 0.71 | 0.32 | 0.44 |
| D29A | 0.22 (4) | 0.05 | 0.40 | 0.73 | 0.66 | 0.60 |
| L33V | −0.28 (4) | 0.21 | ||||
| S36N | 0.26 (4) | 0.32 | 0.94 | 1.10 | 0.65 | 0.90 |
| K43A | 0.24 (4) | 0.27 | 0.32 | 1.08 | 0.42 | 0.61 |
| V44A | 0.50 (3) | 0.12 | 0.09 | 0.31 | 0.20 | |
| V46A | 1.20 (4) | 0.21 | 0.58 | 0.16 | 0.37 | |
| N47G | 0.40 (4) | 0.34 | 1.05 | 1.60 | 0.45 | 1.03 |
| D48G | 1.27 (4) | 0.44 | 2.35 | 2.35 | ||
| F52A | 0.61 (3) | 0.19 | 0.12 | 0.28 | 0.20 | |
| V53A | 0.72 (3) | 0.69 | 0.26 | 0.54 | 0.40 | |
| A55G | 0.61 (3) | 0.41 | 0.24 | 0.30 | 0.27 | |
| V58A | 0.18 (3) | 0.16 | 0.21 | 0.36 | 0.28 |
Values in parentheses indicate the number of data points in the Leffler representation (Fig. 7).
In Table 3 we considered that σϕ = ±3σi (99% confidence), in which σi is the respective fitting standard error in the slope for each mutant, together with the definitive ϕ-values obtained from multipoint Leffler analysis. Errors corresponding to the two-point analysis at the three pH values, respectively (also with 99% confidence; black bars of Fig. 3), and their averages, are also included.
Initially it should be noted that the error bars from the multipoint Leffler analyses are not the highest ones, except for mutants V53A and A55G, which were only measured under two pH conditions and show considerable dispersion (Fig. 7). In general, the errors in the ϕ-values are either clearly improved or at least stay the same (for simplicity's sake a comparison can be made with the averaged error bars). Interestingly, after a closer examination of Fig. 7 and Table 3 for all mutations, it can be seen that the improvement in the errors is better for the ϕ-values calculated from lower ΔΔGF-U values, whereas the errors with the mutations in which ΔΔGF-U > 2 kcal × mol−1 do not improve to the same extent, being in general similar to those deriving from the two-point analysis. This is the case with mutants V53A and A55G (ΔΔGF-U ≈ 2 kcal × mol−1) as well as mutants V44A, F52A, and V58A. It is also of interest that the closest positions to ϕ-values 0 and 1, corresponding to the A11G and D48G mutants, are the only two to show clear improvement.
These observations show that this approach is quite useful since errors resulting from lower ΔΔGF-U values are worse when estimated from two-point Leffler plots (as shown here and in Sanchez and Kiefhaber (18) and Fersht and Sato (20)), and through pH scanning the error in the analysis can be reduced considerably, and even more so if we obtain different ΔΔGF-U results at the different pH values assayed (as with the L8S, A11G, D14S, and V23A mutants). On the other hand, mutants with higher ΔΔGF-U values do not improve appreciably, although a better estimation can be achieved in most cases. From a statistical point of view, this effect might be explained by the higher degree of complementation of data sets in those cases where a gradation of ΔΔGF-U values exists, whereas the worst improvements result as a consequence of having similar values. Thus, in almost all cases a better improvement is achieved rather than considering some repetitions under the same experimental conditions, which could be correctly represented by the averaged values in Table 3. It is also important to mention that individual errors are frequently undefined from propagation estimations (absent errors in Table 3), mainly at pH 2.5, where the most unstable mutants (those with a higher ΔΔGF-U) have been found to have significant errors due to an inadequate amount of refolding information. In the case of mutation L33V (negative ϕ-value), the error could be determined only from Leffler analysis.
Judging by the magnitudes of the error bars shown in Table 3 and bearing in mind that the ϕ-value is a relative quantity (calculated from a quotient of two Gibbs energy changes; Eq. 11), the errors are obtained directly as relative values. The obtained values are quite variable, ranging from 5% (0.05) to 70% (0.69), being 25% on average; the errors from two-point analysis are more dispersed (17%–235%; 60% on average). In our opinion, the average value obtained of 25% might be a reasonable value for σϕ, which basically propagates both routine and typical experimental random errors. Lower errors can be obtained when a combination of optimum ΔΔGF-U values and low data dispersion give rise to symmetrical and well-defined chevron plots. Higher errors will, therefore, include contributions deriving from asymmetry and/or excessive data scattering in the chevron plots.
Therefore, precision can be significantly improved by pH-scanning analysis (as much as 35% on average), since protein stability is normally affected by changes in the pH value and thus asymmetry can be corrected. It is also evident that the more data points added to the Leffler plot the more error is reduced, probably until the limit imposed by experimental error propagation. This also happens when the data points are numerically different, which ensures a better complementation among them. The fact that in this kind of representation the fitting line must cross the origin (Fig. 7) contributes significantly to a decrease in error. Thus, in a two-point plot the fitting line is scattered only by the second value corresponding to the mutation, as it is evident that additional points positively contribute to the accuracy in line position. Of course, the farther the point is from the origin (higher ΔΔGF-U values) the lower the scattering, which translates into a lower σϕ (18).
In summary, a proper error analysis such as the one presented here constitutes an easy way to evaluate the precision of each ϕ-value, whereas an analysis of pH dependencies of kinetic parameters allows a further improvement wherever necessary. Proceeding in this way, the three-class gradation of individual ϕ-determinations (weak, medium, and strong), as suggested by Fersht (42) and also derived from our two-point Leffler analysis, can be increased to four or even five classes. Moreover, it could be a useful tool to discard wrong ϕ-value estimations. Certainly, it involves an increase in experimental work, but a multiple mutational analysis additionally includes cloning and purification procedures, as well as possible uncertainties deriving from the differential nondisruptivity of the mutations. For example, in the Ala→Gly scanning, Gly can induce additional conformational freedom within the protein backbone compared to Ala (43). In any case, Ala→Gly and pH scanning emerge as fully compatible methods, and their combination will allow multipoint Leffler plots when necessary.
Strategies for experimental work and error analysis
On the basis of the arguments outlined above, a correct error analysis (≥90% confidence in practice) can be easily made starting from σlnk = ±3σi/k, where σi represents the fitting standard errors in k‡-U and k‡-F, which are obtained from chevron plots. Eqs. 12 and 13 can then provide the estimations of errors in the ΔΔG functions and ϕ-values, respectively. A more straightforward approach could be the use of Eq. 14 instead of Eq. 13 for ϕ-errors. In principle, no limitations for this application are envisioned from the analysis here. It is important to bear in mind that an independent determination of equilibrium Gibbs energies through equilibrium denaturation experiments not only reduces errors in ϕ but also provides an alert for systematic instrumental deviations (33). Anyhow, in cases where higher precision might be necessary, a pH-scanning analysis through Leffler representations could be done, paying special attention to their linear behavior, which verifies the maintenance of the folding mechanism (25). As we have shown here, errors in this case can be easily obtained as three times the fitting standard errors to a straight line. Proceeding in this way, ϕ-errors should not be higher than 25%.
Acknowledgments
We thank Drs. Luis Serrano, Javier Ruiz-Sanz, and Claire T. Friel for critical reading of the manuscript. We also acknowledge Dr. Jon Trout for revising the English text.
This work was financed by grants BIO2003-04274 and BIO2006-15517-C02-01 from the Spanish Ministry of Science and Education, INTAS 03-51-5569 from the European Union, and FQM-123 from the Andalusian Regional Government, Spain. E.S.C. is the recipient of a “Juan de la Cierva” research contract from the Spanish Ministry of Science and Education and A.M.C. is a predoctoral fellow with the Spanish Ministry of Science and Education.
Editor: José Onuchic.
References
- 1.Leffler, J. E. 1953. Parameters for the description of transition states. Science. 117:340–341. [DOI] [PubMed] [Google Scholar]
- 2.Jencks, D. A., and W. P. Jencks. 1977. On the characterization of transition states by structure-reactivity coefficients. J. Am. Chem. Soc. 99:7948–7960. [Google Scholar]
- 3.Sanchez, I. E., and T. Kiefhaber. 2003. Non-linear rate-equilibrium free energy relationships and Hammond behavior in protein folding. Biophys. Chem. 100:397–407. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez, I. E., and T. Kiefhaber. 2003. Hammond behavior versus ground state effects in protein folding: evidence for narrow free energy barriers and residual structure in unfolded states. J. Mol. Biol. 327:867–884. [DOI] [PubMed] [Google Scholar]
- 5.Kiefhaber, T., I. E. Sanchez, and A. Bachmann. 2005. Characterization of protein folding barriers with rate-equilibrium free-energy relationships. In Protein Folding Handbook. J. Buchner and T. Kiefhaber, editors. Wiley, Weinheim, Germany. 411–453.
- 6.Matouschek, A., J. T. Kellis Jr., L. Serrano, and A. R. Fersht. 1989. Mapping the transition state and pathway of protein folding by protein engineering. Nature. 340:122–126. [DOI] [PubMed] [Google Scholar]
- 7.Fersht, A. R., A. Matouschek, and L. Serrano. 1992. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J. Mol. Biol. 224:771–782. [DOI] [PubMed] [Google Scholar]
- 8.Serrano, L., J. T. Kellis Jr., P. Cann, A. Matouschek, and A. R. Fersht. 1992. The folding of an enzyme. II. Substructure of barnase and the contribution of different interactions to protein stability. J. Mol. Biol. 224:783–804. [DOI] [PubMed] [Google Scholar]
- 9.Serrano, L., A. Matouschek, and A. R. Fersht. 1992. The folding of an enzyme. III. Structure of the transition state for unfolding of barnase analysed by a protein engineering procedure. J. Mol. Biol. 224:805–818. [DOI] [PubMed] [Google Scholar]
- 10.Matouschek, A., L. Serrano, and A. R. Fersht. 1992. The folding of an enzyme. IV. Structure of an intermediate in the refolding of barnase analysed by a protein engineering procedure. J. Mol. Biol. 224:819–835. [DOI] [PubMed] [Google Scholar]
- 11.Fersht, A. R. 1997. Nucleation mechanisms in protein folding. Curr. Opin. Struct. Biol. 7:3–9. [DOI] [PubMed] [Google Scholar]
- 12.Martinez, J. C., M. T. Pisabarro, and L. Serrano. 1998. Obligatory steps in protein folding and the conformational diversity of the transition state. Nat. Struct. Biol. 5:721–729. [DOI] [PubMed] [Google Scholar]
- 13.Grantcharova, V. P., D. S. Riddle, J. V. Santiago, and D. Baker. 1998. Important role of hydrogen bonds in the structurally polarized transition state for folding of the src SH3 domain. Nat. Struct. Biol. 5:714–720. [DOI] [PubMed] [Google Scholar]
- 14.Martinez, J. C., and L. Serrano. 1999. The folding transition state between SH3 domains is conformationally restricted and evolutionarily conserved. Nat. Struct. Biol. 6:1010–1016. [DOI] [PubMed] [Google Scholar]
- 15.Riddle, D. S., V. P. Grantcharova, J. V. Santiago, E. Alm, I. Ruczinski, and D. Baker. 1999. Experiment and theory highlight role of native state topology in SH3 folding. Nat. Struct. Biol. 6:1016–1024. [DOI] [PubMed] [Google Scholar]
- 16.Chiti, F., N. Taddei, P. M. White, M. Bucciantini, F. Magherini, M. Stefani, and C. M. Dobson. 1999. Mutational analysis of acylphosphatase suggests the importance of topology and contact order in protein folding. Nat. Struct. Biol. 6:1005–1009. [DOI] [PubMed] [Google Scholar]
- 17.Fersht, A. R., and V. Daggett. 2002. Protein folding and unfolding at atomic resolution. Cell. 108:573–582. [DOI] [PubMed] [Google Scholar]
- 18.Sanchez, I. E., and T. Kiefhaber. 2003. Origin of unusual φ-values in protein folding: evidence against specific nucleation sites. J. Mol. Biol. 334:1077–1085. [DOI] [PubMed] [Google Scholar]
- 19.Garcia-Mira, M. M., D. Boehringer, and F. X. Schmid. 2004. The folding transition state of the cold shock protein is strongly polarized. J. Mol. Biol. 339:555–569. [DOI] [PubMed] [Google Scholar]
- 20.Fersht, A. R., and S. Sato. 2004. φ-value analysis and the nature of protein-folding transition states. Proc. Natl. Acad. Sci. USA. 101:7976–7981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Settanni, G., F. Rao, and A. Caflisch. 2005. φ-value analysis by molecular dynamics simulations of reversible folding. Proc. Natl. Acad. Sci. USA. 102:628–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruczinski, I., T. R. Sosnick, and K. W. Plaxco. 2006. Methods for the accurate estimation of confidence intervals on protein folding φ-values. Protein Sci. 15:2257–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de los Rios, M. A., B. K. Muralidhara, D. Wildes, T. R. Sosnick, S. Marqusee, P. Wittung-Stafshede, K. W. Plaxco, and I. Ruczinski. 2006. On the precision of experimentally determined protein folding rates and φ-values. Protein Sci. 15:553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fersht, A. R. 2004. φ value versus ψ analysis. Proc. Natl. Acad. Sci. USA. 101:17327–17328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bodenreider, C., and T. Kiefhaber. 2005. Interpretation of protein folding ψ values. J. Mol. Biol. 351:393–401. [DOI] [PubMed] [Google Scholar]
- 26.Sosnick, T. R., B. A. Krantz, R. S. Dothager, and M. Baxa. 2006. Characterizing the protein folding transition state using ψ analysis. Chem. Rev. 106:1862–1876. [DOI] [PubMed] [Google Scholar]
- 27.Viguera, A. R., L. Serrano, and M. Wilmanns. 1996. Different folding transition states may result in the same native structure. Nat. Struct. Biol. 3:874–880. [DOI] [PubMed] [Google Scholar]
- 28.Martinez, J. C., A. R. Viguera, R. Berisio, M. Wilmanns, P. L. Mateo, V. V. Filimonov, and L. Serrano. 1999. Thermodynamic analysis of α-spectrin SH3 and two of its circular permutants with different loop lengths: discerning the reasons for rapid folding in proteins. Biochemistry. 38:549–559. [DOI] [PubMed] [Google Scholar]
- 29.Vega, M. C., J. C. Martinez, and L. Serrano. 2000. Thermodynamic and structural characterization of Asn and Ala residues in the disallowed II′ region of the Ramachandran plot. Protein Sci. 9:2322–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cobos, E. S., V. V. Filimonov, M. C. Vega, P. L. Mateo, L. Serrano, and J. C. Martinez. 2003. A thermodynamic and kinetic analysis of the folding pathway of an SH3 domain entropically stabilised by a redesigned hydrophobic core. J. Mol. Biol. 328:221–233. [DOI] [PubMed] [Google Scholar]
- 31.Fernandez-Escamilla, A. M., M. S. Cheung, M. C. Vega, M. Wilmanns, J. N. Onuchic, and L. Serrano. 2004. Solvation in protein folding analysis: combination of theoretical and experimental approaches. Proc. Natl. Acad. Sci. USA. 101:2834–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bevington, P. R., and D. K. Robinson. 2003. Data Reduction and Error Analysis for the Physical Sciences. McGraw-Hill, New York.
- 33.Zarrine-Afsar, A., and A. R. Davidson. 2004. The analysis of protein folding kinetic data produced in protein engineering experiments. Methods. 34:41–50. [DOI] [PubMed] [Google Scholar]
- 34.Myers, J. K., C. N. Pace, and J. M. Scholtz. 1995. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4:2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Privalov, P. L., and N. N. Khechinashvili. 1974. A thermodynamic approach to the problem of stabilization of globular protein structure: a calorimetric study. J. Mol. Biol. 86:665–684. [DOI] [PubMed] [Google Scholar]
- 36.Pace, C. N. 1975. The stability of globular proteins. CRC Crit. Rev. Biochem. 3:1–43. [DOI] [PubMed] [Google Scholar]
- 37.Privalov, P. L. 1979. Stability of proteins: small globular proteins. Adv. Protein Chem. 33:167–241. [DOI] [PubMed] [Google Scholar]
- 38.Privalov, P. L., and S. J. Gill. 1988. Stability of protein structure and hydrophobic interaction. Adv. Protein Chem. 39:191–234. [DOI] [PubMed] [Google Scholar]
- 39.Privalov, P. L. 1989. Thermodynamic problems of protein structure. Annu. Rev. Biophys. Biophys. Chem. 18:47–69. [DOI] [PubMed] [Google Scholar]
- 40.Pace, C. N. 1990. Conformational stability of globular proteins. Trends Biochem. Sci. 15:14–17. [DOI] [PubMed] [Google Scholar]
- 41.Pace, C. N., B. A. Shirley, M. McNutt, and K. Gajiwala. 1996. Forces contributing to the conformational stability of proteins. FASEB J. 10:75–83. [DOI] [PubMed] [Google Scholar]
- 42.Fersht, A. R. 1995. Characterizing transition states in protein folding: an essential step in the puzzle. Curr. Opin. Struct. Biol. 5:79–84. [DOI] [PubMed] [Google Scholar]
- 43.Scott, K. A., D. O. Alonso, S. Sato, A. R. Fersht, and V. Daggett. 2007. Conformational entropy of alanine versus glycine in protein denatured states. Proc. Natl. Acad. Sci. USA. 104:2661–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]