

Abstract
Ca2+ signaling in the dyadic cleft in ventricular myocytes is fundamentally discrete and stochastic. We study the stochastic binding of single Ca2+ ions to receptors in the cleft using two different models of diffusion: a stochastic and discrete Random Walk (RW) model, and a deterministic continuous model. We investigate whether the latter model, together with a stochastic receptor model, can reproduce binding events registered in fully stochastic RW simulations. By evaluating the continuous model goodness-of-fit for a large range of parameters, we present evidence that it can. Further, we show that the large fluctuations in binding rate observed at the level of single time-steps are integrated and smoothed at the larger timescale of binding events, which explains the continuous model goodness-of-fit. With these results we demonstrate that the stochasticity and discreteness of the Ca2+ signaling in the dyadic cleft, determined by single binding events, can be described using a deterministic model of Ca2+ diffusion together with a stochastic model of the binding events, for a specific range of physiological relevant parameters. Time-consuming RW simulations can thus be avoided. We also present a new analytical model of bimolecular binding probabilities, which we use in the RW simulations and the statistical analysis.
INTRODUCTION
It is an important and contentious issue whether diffusion in signaling micro domains can be modeled deterministically and continuously, or if stochastic and discrete Random Walk (RW) methods should be employed (1–6). Signaling micro domains are used by the cell to convey information and it is important to use accurate and reliable simulation methods when these processes are studied. Traditionally, they have been studied using Fick's second law of diffusion together with macroscopic rate laws, where the latter are used to model chemical reactions. These laws provide a deterministic prediction of the changes of the average number of molecules in a process over time. The solutions are continuous functions of both space and time. Reaction diffusion processes in macroscopic environments, where fluctuations from the predicted average number of particles in a solution are small, are modeled successfully by these laws. The laws were originally empirical but they are also well founded in statistical physics (7). In recent years, as smaller and smaller subcellular domains have been studied, researchers have focused on the discreteness and stochasticity of the physiological processes. This has raised issues for the deterministic models (4,8). In subcellular micro domains, the number of involved molecules is small and the fluctuations from the predicted average number of molecules involved become dominant. Three-dimensional RW simulators have been developed to incorporate the discreteness and stochasticity of the signaling in intracellular micro domains. One well-established simulator is MCell (9,10), which has been used in some recent studies of subcellular signaling. The results of these studies illustrate clearly the fundamental discreteness and stochasticity of the studied processes (1,11,12). Another approach to modeling the discreteness and stochasticity of a subcellular process is to model the diffusion and possible buffer dynamic with a deterministic and continuous model together with a stochastic model of receptors that switch states randomly according to the concentration at the receptor site, i.e., modeling the binding of single molecules to a receptor stochastically. Different versions of this method have recently been used to study the functionality of the well-studied signaling micro domain of the dyadic cleft, in ventricular myocytes (13–15), and also in a whole cell study of the Ca2+ dynamics in the endoplasmic reticulum (16). Although this method is already in use, the fundamental problem of using a continuous and deterministic representation of a small number of diffusing molecules has not been addressed. This issue is of great concern when signaling in the dyadic cleft is studied, because the volume of this domain is in the magnitude of atto liters. This concern is illustrated by the fact that during diastole, when the myocyte is relaxing, the cytosolic [Ca2+] is as low as 0.1 μM, leaving, on average, 0.02 Ca2+ ions present in the cleft. Hereafter, we will relate to this model, i.e., the continuous and deterministic description of Ca2+ diffusion together with a stochastic and discrete description of single receptors, as “the continuous model.”
The dyadic cleft is a signaling micro domain in which the Ca2+-induced Ca2+ release mechanism is controlled tightly (17,18). A traveling action potential triggers the influx of external Ca2+ through the L-type Ca2+ channels (LCCs). From the mouth of a LCC, which are located at the membrane of a T-tubule (TT), Ca2+ diffuses into the cleft. The cleft is narrow, ∼15 nm wide (19,20), and a unitary LCC current creates a very high Ca2+ concentration in the cleft, = 10–200 μM (21), compared to the value at rest, = 0.1 μM. This Ca2+ signal triggers both the inactivation of the LCC current and further Ca2+ release from the opposing Ryanodine receptors (RyRs) (22,23), which are attached to the sarcoplasmic reticulum (SR), an intracellular Ca2+ store. What causes the reliable termination of Ca2+ release from the RyR is still a debated issue (24). However, among the proposed explanatory hypotheses, inactivation due to binding of single Ca2+ ions to receptors in the dyadic cleft, is well established (25,26). For a recent review of the Ca2+ dynamics in the cleft, see Bers and Guo (27), and the references therein.
For a long time, continuous and deterministic models have been used to study Ca2+ dynamics in the dyadic cleft (21,28–31), and its role in the release of Ca2+. Two recent studies of Ca2+ dynamics use a discrete RW model to describe the Ca2+ diffusion in the cleft (12,32). Koh et al. (12) uses MCell and argues that few Ca2+ ions in a small volume cannot properly be simulated with a continuous model of diffusion. However, they do not present any results that support this claim. Tanskanen et al. (32) present an impressive study that includes physiological details on a microscale level, such as the electrostatic force from the sarcolemmal and the geometrical structures of the large membrane proteins in the cleft, while integrating the Ca2+ release from many clefts, and thus obtaining a measure of the Ca2+ release from the whole cell. In contrast to Koh et al. (12), they explicitly address the difference between their model and an equivalent model that uses a deterministic description of Ca2+ diffusion. They do this by measuring the effect on the excitation-contraction coupling (ECC) gain when they vary the diffusion constant of Ca2+, together with the parameters that determine the influx of Ca2+ ions to the cleft. They show that the ECC gain varies with the parameters (see Fig. 12 in (32)). This result points to a “subtle but potentially significant difference in predicted macroscopic behavior arising from the underlying stochastic simulation of Ca2+ motion in the dyad” (32). The rationale for this statement is that if they had changed the same parameters in an equivalent model using a deterministic description of Ca2+ diffusion, they would not have registered any differences in ECC gain because the receptors situated in the cleft would have experienced the same level of Ca2+ concentration. In our study we examine the discrete events in the cleft that are actually modeled differently in a continuous versus a RW model of diffusion in the dyadic cleft; namely, the binding of single Ca2+ ions to single receptors. By doing this, we strip the model of Ca2+ dynamics in the dyadic cleft of many important physiological details that affect the generation and termination of a spark (12,21,32), but the comparison between the actual differences between the two diffusion models become clearer.
FIGURE 12.

(A and B) Number of registered binding events from 100 runs each, where we altered different parameters. The data were collected from a receptor 30 nm from the center and are represented by the box-plots together with a 95% confidence interval for the true means (red horizontal lines). The blue solid circles represent the expected number of binding events that are predicted by the continuous model. (A) We scaled the number of Ca2+ ions that enter the cleft, i.e., 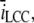 together with the diffusion constant D, with a factor represented by the x axis. The spatial resolution was constant for these simulations, σ = 5 nm. The blue asterisk denotes a statistical difference between the continuous model and the RW model for scale = 0.1. (B) We kept the scale constant at 0.1, but altered the spatial resolution (see the x axis). Here, the difference between the RW model and the continuous model increased as the mean value of the collected binding events declined with the spatial resolution. (C) We ran the simulation 100 times. We collected the mean binding rates for each run that the receptor were exposed to. The data from each set of 100 runs are presented as 95% confidence intervals for the true means. The blue horizontal lines represent the binding rates collected from runs in which we registered binding events, as in panel B. The red horizontal lines represent binding rates collected from runs in which we did not register binding events, only the rate. In these runs we could not differentiate statistically between the registered binding rates and the rates predicted from the continuous model.
together with the diffusion constant D, with a factor represented by the x axis. The spatial resolution was constant for these simulations, σ = 5 nm. The blue asterisk denotes a statistical difference between the continuous model and the RW model for scale = 0.1. (B) We kept the scale constant at 0.1, but altered the spatial resolution (see the x axis). Here, the difference between the RW model and the continuous model increased as the mean value of the collected binding events declined with the spatial resolution. (C) We ran the simulation 100 times. We collected the mean binding rates for each run that the receptor were exposed to. The data from each set of 100 runs are presented as 95% confidence intervals for the true means. The blue horizontal lines represent the binding rates collected from runs in which we registered binding events, as in panel B. The red horizontal lines represent binding rates collected from runs in which we did not register binding events, only the rate. In these runs we could not differentiate statistically between the registered binding rates and the rates predicted from the continuous model.
We also present what is, to our knowledge, a novel model of bimolecular binding probabilities between single diffusive ligands and single stationary or mobile receptors that are used in our RW simulator. The model is analytical. It depends only on the diffusion constant of the ligand, the macroscopic binding rate, the time-step of the RW algorithm, and the distance between the two molecules at the beginning of the time-step. The first three parameters are all known before a simulation starts and the binding probabilities are precomputed with respect to distance for the reactions that are included in the simulation. During a simulation, lookup tables are used. The error introduced by the model is studied thoroughly for a large set of parameters. We find that for a given time-step, the error introduced by the bimolecular interaction model is much smaller than the error introduced by the RW simulation, due to an absorbing boundary in our model. Hence, we can use larger time-steps for the time-consuming reaction process.
The results of the statistical goodness-of-fit tests reveal that the continuous model, for a specific parameter range, can reproduce the registered binding events from the RW simulations. This is somewhat unexpected, because the binding probability in the continuous model is linear with respect to the [Ca2+] at a single receptor and is also constant during steady-state simulations. This is in contrast to the binding probabilities in the RW model, which depend directly on the distance between a RyR and any nearby Ca2+ ions. We find that the large variations in binding rates at the timescale of a single time-step, equal to 1.25 × 10−4 ms, are integrated and smoothed at the timescale of binding events, equal to 0.5 ms. These results refine the statements made in a number of recent studies (2,6,10,12), which claim that when the number of participating particles in a volumes falls, a deterministic description of concentration is invalid or does not make sense, and fully stochastic methods have to be employed. Our study reveals that the extra discreteness and stochasticity that a full RW model introduces are integrated at the timescale of binding events, to the same value given by the continuous model. This also explains why the average description of the [Ca2+] in the cleft, given by the continuous model, is sufficient when the registration of single binding events is studied. The result is parameter-dependent. For small values of the diffusion constant, we find a difference between the two models similar to that which Tanskanen et al. (32) find. We further investigate the cause of this difference and the quantitative dependency of the parameters.
This article is divided into five main sections. The Introduction is followed by Theory, in which we describe the models and how we solve them. Also in that section, we derive and analyze the model of bimolecular binding probabilities. In Methods, we explain how we performed our simulations and which statistical tests we used. Our simulations and tests are presented in the Results and then revisited in the Discussion.
THEORY
Continuous model
Ca2+ diffusion in the continuous model is described by a well-known reaction-diffusion model, which consists of a set of coupled partial differential equations (33,34). Symmetry in the angular and z directions was assumed, thus reducing the full three-dimensional model to a one-dimensional model in the radial direction. If c, Bm, and Bs denote, respectively, the concentration of Ca2+, mobile buffer, and stationary buffer, the full system is given by
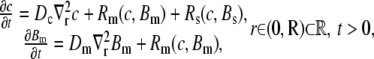 |
(1) |
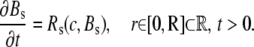 |
(2) |
The reaction terms are given by
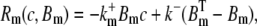 |
(3) |
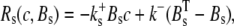 |
(4) |
where 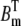 and
and 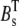 are the total concentration of the two buffer types. The values Dc and Db are the diffusion constants of Ca2+ and the mobile buffer, respectively, and
are the total concentration of the two buffer types. The values Dc and Db are the diffusion constants of Ca2+ and the mobile buffer, respectively, and 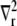 is the radial diffusion operator
is the radial diffusion operator
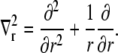 |
(5) |
The initial conditions are given by
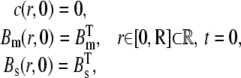 |
(6) |
and the boundary conditions are given by
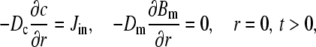 |
(7) |
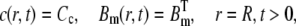 |
(8) |
where Jin is the LCC line source, Cc is the Ca2+ concentration in cytosol, and R is the radius of the cleft.
The actual values of the parameters we used in the simulations are given in Methods, below. The full system was solved using explicit finite different schemes (35).
The binding of single Ca2+ ions could not be modeled literally in the continuous model, because single Ca2+ ions do not exist in the model. However, in a Markov chain model of an RyR, the Ca2+-dependent transition from one state to another is an indirect model of the physiological event of a Ca2+ ion binding to a receptor at a channel (36,37). Given that we did not want to simulate the dynamics of the whole RyR, but only the transition between two [Ca2+]-dependent states, we reduced the channel model to only include two states: one with Ca2+ bound, cR, and one with Ca2+ unbound, R,
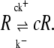 |
(9) |
The total binding rate depends on the Ca2+ concentration, c, at the position of the receptor together with the on-rate, k+. The unbinding rate depends only on the off-rate k− and is thus Ca2+-independent. The independency of [Ca2+] in the off-rate makes the transition from the bound state to the unbound state model-independent, and we could therefore exclude it from our study because we were only interested in the differences. Effectively, this meant that we removed the bound state, cR, from the receptor model, thus reducing the receptor model to a one-state model that serves as an indicator of Ca2+ binding events.
With this reduction of the channel model, we were able to represent the registration of single Ca2+ binding events at a RyR, in the continuous model, with a Poisson processes, determined only by the rate or intensity function λ(t) = c(t)k+ (38). The probability that one Ca2+ ion would bind to a RyR was modeled as 1 minus the probability of zero bindings:
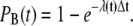 |
(10) |
We had to retain the quantity λ(t)Δt, which represents the expected number of binding events during a time-step, much smaller than 1; this was obtained by minimizing the probability of getting more than one binding event during a time-step. When the [Ca2+] was fixed at each receptor, i.e., during the steady state, we had a homogeneous Poisson process with constant rate λ = c k+. In the transient simulation, where the [Ca2+] varied at each receptor, the Poisson process was inhomogeneous with rate λ(t) = c(t)k+. The model reduction, together with the observation that the registration of binding events could be represented by a Poisson process, were used in the goodness-of-fit tests, as shown in Methods, below.
Random Walk model
Our discrete model of diffusion is based on an RW description of Brownian motion (39). The model is a simple, but powerful stochastic model of diffusion. In a simulation, the position of each diffusive ligand in the cleft is tracked. For each ligand and time-step, a random displacement, Δr = (Δx Δy, Δz), is sampled from a trivariate probability density and added to the position of the ligand. The distribution is a solution to Fick's second law of diffusion for a point source (33). With homogeneous diffusion constant, D, and a fixed time-step, Δt, the trivariate probability density is given by
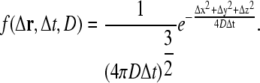 |
(11) |
The expected radial displacement of a single RW step is 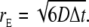 The spatial scale of the simulation is hence set by D and Δt. Three different types of diffusive ligands were simulated in the RW model: Ca2+, and a diffusive buffer with and without bound Ca2+. We used the same diffusion constant for the two buffer molecules, Db. Two different types of boundary were used: one reflective and one absorbing, ∂ΩN2 and ∂ΩD (see Fig. 6). If a particle, Ca2+ or mobile buffer, crossed one of the reflective boundaries, it was mirrored into the volume again. If a Ca2+ ion crossed the absorbing boundary, it was removed from the simulation, to allow the modeling of a Ca2+ concentration in cytosol that was assumed to be zero. During a simulation, we kept the total concentration of the mobile buffer constant in the cleft. This was achieved by not allowing a buffer molecule cross to the ∂ΩD boundary, i.e., if a buffer molecule ended outside the boundary, a new displacement was sampled until it was inside. In addition, if the buffer molecule had a Ca2+ ion attached to it when it ended outside the boundary, the Ca2+ was removed, due to the assumed zero [Ca2+] in the cytosol.
The spatial scale of the simulation is hence set by D and Δt. Three different types of diffusive ligands were simulated in the RW model: Ca2+, and a diffusive buffer with and without bound Ca2+. We used the same diffusion constant for the two buffer molecules, Db. Two different types of boundary were used: one reflective and one absorbing, ∂ΩN2 and ∂ΩD (see Fig. 6). If a particle, Ca2+ or mobile buffer, crossed one of the reflective boundaries, it was mirrored into the volume again. If a Ca2+ ion crossed the absorbing boundary, it was removed from the simulation, to allow the modeling of a Ca2+ concentration in cytosol that was assumed to be zero. During a simulation, we kept the total concentration of the mobile buffer constant in the cleft. This was achieved by not allowing a buffer molecule cross to the ∂ΩD boundary, i.e., if a buffer molecule ended outside the boundary, a new displacement was sampled until it was inside. In addition, if the buffer molecule had a Ca2+ ion attached to it when it ended outside the boundary, the Ca2+ was removed, due to the assumed zero [Ca2+] in the cytosol.
FIGURE 6.

Geometry of the disk that we used to model the dyadic cleft. The LCC ion source is included in the center of the disk as a line source.
We implemented two different possible sources of Ca2+ ions in the cleft: 1), one or more LCCs; or 2), passive influx from the cytosol. The Ca2+ ions that entered through an LCC were introduced into the center of the cleft at a random height, to mimic the line source used in the continuous model (see above). The number of Ca2+ ions entering the cleft through the current per ms is given by 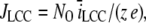 where NO is the number of open channels, e the elementary charge, and z the valence of the Ca2+ ion. The number of Ca2+ ions entering the cleft from the cytosol is given by
where NO is the number of open channels, e the elementary charge, and z the valence of the Ca2+ ion. The number of Ca2+ ions entering the cleft from the cytosol is given by 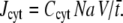 These ions were placed at a random position at the boundary ∂ΩD. Here, Ccyt is the Ca2+ concentration in cytosol, Na Avogadro's number, and V the volume of the cleft. The value
These ions were placed at a random position at the boundary ∂ΩD. Here, Ccyt is the Ca2+ concentration in cytosol, Na Avogadro's number, and V the volume of the cleft. The value 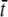 is the average time each Ca2+ ion spent in the cleft, given that it entered at the boundary ∂ΩD. When there were no buffers in the cleft, this value was found to be
is the average time each Ca2+ ion spent in the cleft, given that it entered at the boundary ∂ΩD. When there were no buffers in the cleft, this value was found to be 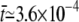 ms.
ms.
Stochastic modeling of single receptors
In addition to handling the RW of single Ca2+ ions in a continuous three-dimensional space, we wanted to let these ions bind to single receptors and study the resulting binding statistics. We did not find any software that was able to do this when we started our study, e.g., MCell 2 only supported single binding events to a density of receptors at the membrane. Therefore, we decided to develop our own model of bimolecular interactions.
Not only RyRs were treated as single receptors in the discrete RW model, but also all buffer molecules, so we had to deal with Ca2+ unbinding from receptors too. This was in contrast to the continuous case, in which only the event of Ca2+ binding to single RyR receptors was treated stochastically. The probability that a Ca2+ ion will unbind from a receptor during a time-step depends solely on the unbinding rate k− for the receptor and the size of the time-step and is given by
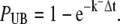 |
(12) |
The probability that a Ca2+ ion and a receptor will bind was calculated using the same macroscopic rate law that was used in the continuous case (see Eq. 9). It is counterintuitive to use a macroscopic law between single discrete molecules, because these do not have the macroscopic property of concentration. However, because the position of a diffusive ligand is given by a probability distribution between the time-steps, we used this distribution to calculate the average number density of a single diffusive particle at a certain distance and time (7). This quantity is deterministic and predicts the expected density or concentration of a particle.
Despite the fact that the concept of average number density has been used before (7), we argue that a single diffusive particle is described more appropriately in terms of its expected concentration, which is given in Molar and can thus be used in the macroscopic rate law, as intended. The word “expected” also reflects the deterministic, a priori knowledge of the contribution to the average concentration that a particle would make if the position were sampled many times.
We derived the concept of expected concentration by dividing the entire spatial domain that surrounds the diffusive ligand into N equally spaced shells. Each shell had a volume of 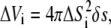 where ΔSi = i δs, δs ∝ 1/N, and i = 1…N. Fixing the time to t < Δt, we sampled the position of the diffusive ligand K times. Let Ni be the number of times the ligand occurred in the shell at ΔSi. Dividing this by K, we obtained the averaged number of times the ligand occurred in the ith shell. Then, the average number density of the particle in the same shell is given by
where ΔSi = i δs, δs ∝ 1/N, and i = 1…N. Fixing the time to t < Δt, we sampled the position of the diffusive ligand K times. Let Ni be the number of times the ligand occurred in the shell at ΔSi. Dividing this by K, we obtained the averaged number of times the ligand occurred in the ith shell. Then, the average number density of the particle in the same shell is given by
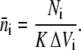 |
(13) |
Dividing this by Avogadro's number, Na, we arrived at the average concentration given in Molar. Given that we were sampling a deterministic probability distribution K times, we used this information to express the expected number of times a particle occurred in the ith shell, after time t:
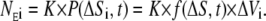 |
(14) |
Substituting Ni in Eq. 13 with this value, and letting N → ∞, we obtain the expected concentration that this ligand exerts after t ms at distance ΔS,
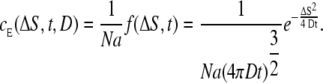 |
(15) |
Here we have divided by Avogadro's constant to obtain the concentration in Molar. We see that the cE is directly proportional to the probability distribution in Eq. 11, which makes sense. The expected concentration of a single Ca2+ ion after t = 45 ns, with D = Dc = 105 nm2 ms−1, is plotted against ΔS in Fig. 1 (left panel, solid line).
FIGURE 1.

(Left panel) Expected [Ca2+], as given by Eq. 15, experienced by a receptor situated a distance ΔS from it, at t = 45 ns. The diffusion constant of Ca2+ is Dc = 105nm2 ms−1. The solid line represents the [Ca2+] experienced by a stationary receptor and the dashed line represents the [Ca2+] experienced by a mobile receptor, with Db = Dc/2, as given by Eq. 26. The right panel shows the corresponding probabilities that a Ca2+ ion will bind to a stationary receptor (solid line) and to a mobile receptor (dashed line), as given by Eq. 24, where Δt = 45 ns and k+ = 30 μM−1 s−1. The probabilities are plotted against the distance between the Ca2+ ion and the receptor. Note the logarithmic scale used for ΔS in the right panel.
The expected concentration, cE, that a Ca2+ ion exerts upon a nearby receptor, ΔS nm away after τ ms, was used to calculate the probability of not binding during a tiny time interval Δτ ≫ Δt. For this, we used the macroscopic rate law from Eq. 9 together with the Poisson probability distribution for zero events,
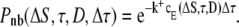 |
(16) |
The probability of not binding during the whole time-step Δt, equals the product of this quantity evaluated for τi = Δτ(i + 1/2), where i = 0, …, N, and N = Δt/Δτ. Keeping D, Δt, k+, and ΔS constant, this probability is
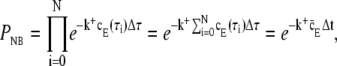 |
(17) |
where 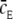 equals the average value of cE, the receptor experience during a time-step. In the limit where Δτ → 0 and N → ∞,
equals the average value of cE, the receptor experience during a time-step. In the limit where Δτ → 0 and N → ∞, 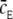 becomes
becomes
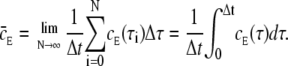 |
(18) |
Using the function for cE from Eq. 15, in this equation we get
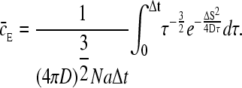 |
(19) |
With change of variables, the integral on the right-hand side can be represented by the upper incomplete γ-function (40). The lower part of such a function is defined as
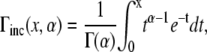 |
(20) |
and the upper part is defined from Eq. 20 by
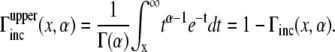 |
(21) |
After the change of variables, the integral in Eq. 19 becomes
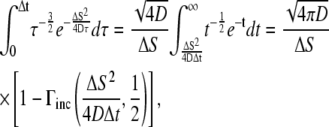 |
(22) |
where the identity of 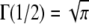 has been used. Using this in Eq. 19, we obtained an analytical expression of the average expected concentration that a receptor experiences during a time-step from a nearby ligand:
has been used. Using this in Eq. 19, we obtained an analytical expression of the average expected concentration that a receptor experiences during a time-step from a nearby ligand:
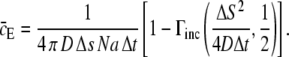 |
(23) |
Assuming that the quantity 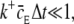 we can write the probability of registering only one binding event as
we can write the probability of registering only one binding event as
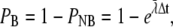 |
(24) |
where 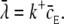 This equation is analogous to Eq. 10, applied only to a single ligand. The expected number of binding events during a time-step is
This equation is analogous to Eq. 10, applied only to a single ligand. The expected number of binding events during a time-step is 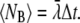 For the continuous case, we had to keep this value much smaller than one, to minimize the probability of getting two or more binding events during a time-step. See also the validity study below.
For the continuous case, we had to keep this value much smaller than one, to minimize the probability of getting two or more binding events during a time-step. See also the validity study below.
The binding probability for a Ca2+ ion near to a mobile receptor, i.e., a mobile buffer, was modeled in the same way as for the stationary receptor, with one exception. A mobile buffer moves during a time-step, which leads to a difference in the expected concentration experienced by the buffer from a nearby Ca2+ ion. Instead of evaluating cE at a single point, as for the stationary receptor, we evaluated it for all possible positions, cE(r,t), and weighted these with the probability, pm(r, t), that the buffer was present. For an arbitrary spatial point r, this quantity is
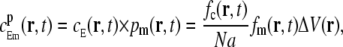 |
(25) |
where fc and fm are the values of the probability density for the Ca2+ and the mobile buffer molecule, respectively. The superscript, p, denotes the concentration at a single spatial position. Using angular symmetry, a cylindrical coordinate system was chosen to integrate 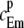 over all spatial points. The Cartesian coordinate line, z, was placed in line with the two particles (Fig. 2), and the position of the Ca2+ ion defines the origin. The distance between the two particles is ΔS. The result of the integration was the expected Ca2+ concentration experienced by a nearby mobile receptor, at time t, separated by a distance ΔS,
over all spatial points. The Cartesian coordinate line, z, was placed in line with the two particles (Fig. 2), and the position of the Ca2+ ion defines the origin. The distance between the two particles is ΔS. The result of the integration was the expected Ca2+ concentration experienced by a nearby mobile receptor, at time t, separated by a distance ΔS,
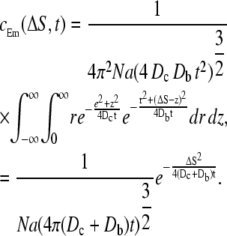 |
(26) |
Here, Dc and Db are the diffusion constants of the Ca2+ ion and the mobile buffer. Notice that this expression is identical to the expected concentration experienced by a stationary receptor, i.e., Eq. 15, with D = Dc + Db. This result made it possible to use Eq. 24 to calculate the binding probability of a Ca2+ ion to a nearby mobile receptor, merely by setting the diffusion constant, D, to the sum of the diffusion constants of the two particles. In Fig. 1 (left panel, dashed line), the expected concentration of a Ca2+ ion experienced by a nearby tentative mobile buffer is plotted. Also in Fig. 1 (right panel, dashed line), the calculated probability of a nearby Ca2+ ion to bind to the same mobile buffer, during a time-step of Δt = 45 ns, with Dc = 105 nm2 ms−1, Db = Dc/2, and k+ = 30 μM s−1, is plotted.
FIGURE 2.

Coordinate used to integrate the [Ca2+] experienced by the buffer molecules, b. The position of the Ca2+ ion defines the origin, and the distance between the two particles is ΔS.
Validity study of bimolecular binding probability
The model of the bimolecular binding probability requires that a single receptor registers only one binding event per time-step. In the continuous model, this could be controlled by keeping the expected number of binding events during a time-step, 〈NE〉 = c(t)k+Δt, much smaller than one. The corresponding probability of getting two or more binding events per time-step is then small. Using the Poisson probability distribution, this equals 1 minus the sum of the probabilities of 0 and one binding per time-step:
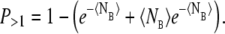 |
(27) |
Using typical large values for the physical parameters, [Ca2+] ≃ 1 mM and k+ – 100 μM−1 s−1 and a small value for the time-step Δt = 1.25 × 10−4 ms, we obtained a small expected number of bindings per time-step 〈NE〉 = 1.25 × 10−2 and a very small value for the probability of two or more binding events, P>1 ≃ 8 × 10−5.
A similar analysis for the bimolecular binding model was not straightforward. The expected number of binding events during a time-step for a single ligand, 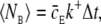 depends on the stochastic ΔS-variable, and we must ensure that the probability for more than one nearby ligand to bind to the receptor is small. The latter probability depends on the local density of ligands nearby the receptor and is also a stochastic entity.
depends on the stochastic ΔS-variable, and we must ensure that the probability for more than one nearby ligand to bind to the receptor is small. The latter probability depends on the local density of ligands nearby the receptor and is also a stochastic entity.
Depending on the parameters, 〈NE〉 can well exceed 1, which increases the probability of registering two or more binding events from a single diffusive ligand. To study this probability with arbitrary parameters, we expressed the expected number of binding events per time-step, 〈NE〉, using dimensionless units. We let the expected displacement of a single ligand in one spatial direction, 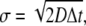 define the length scale ΔS = σΔS*. The expected number of binding events per time-step in dimensionless units is then
define the length scale ΔS = σΔS*. The expected number of binding events per time-step in dimensionless units is then
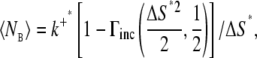 |
(28) |
where
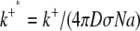 |
(29) |
and represents the dimensionless version of k+. Note that Δt is redundant because it follows D and σ. Using this in Eq. 27, we obtained the probability of getting two or more binding events from a single diffusive ligand nearby a receptor. This quantity is plotted for different values of k+* and ΔS* in the left panel of Fig. 3. The probability is sensitive to ligands that are very close to the receptor and to large values of k+*.
FIGURE 3.

Results of our study of the validity of the model of the bimolecular binding probability in Eq. 24. (Left and middle panels) Probability of registering two or more binding events at a receptor. (Left panel) Probability generated by a single ligand, with respect to the dimensionless quantities, ΔS* and k+*, whereas the center panel shows the overall probability of registering two or more binding events at a receptor in an infinite medium with constant concentration, c, for different values of k+*. (Right panel) Absolute value of the relative difference between the probability of registering a binding event from the continuous model and the RW model. (Center and right panels) Curves were computed using σ = 5 nm and D = 105 nm2 ms−1. These curves would be shifted downwards if a smaller σ were chosen.
In an infinite medium with a constant concentration, the probability that a ligand will be r dimensionless units away from a receptor is P(r) = 2πσ3 c Na r2Δr, where c is the concentration and Δr a small distance chosen to ensure that P(r) ≪ 1. The probability of not getting a binding event from a distance r equals the probability that a ligand will not be at that distance plus the probability that a ligand will be there, times the probability of not binding from that distance. Keeping k+* and c constant we get 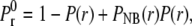 where
where 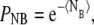 and 〈NE〉 is distance-dependent (see Eq. 28). We chose a cutoff distance of r = 5 that defines our domain and computed the probability of not registering any binding events from this volume,
and 〈NE〉 is distance-dependent (see Eq. 28). We chose a cutoff distance of r = 5 that defines our domain and computed the probability of not registering any binding events from this volume, 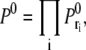 where ri = iΔr. The probability of getting one binding from a distance r is
where ri = iΔr. The probability of getting one binding from a distance r is 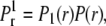 where
where 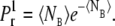 The probability of registering only one binding event from the distance r and not from any other distances equals
The probability of registering only one binding event from the distance r and not from any other distances equals 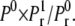 Finally, the probability of registering only one binding event from the whole domain is the sum of all these probabilities:
Finally, the probability of registering only one binding event from the whole domain is the sum of all these probabilities:
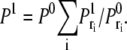 |
(30) |
The probability of registering two or more binding events from the whole domain is then P>1 = 1 – (P0 + P1). This quantity is plotted for different values of c and k+* in the center panel of Fig. 3. Because it is not straightforward to interpret a dimensionless [Ca2+], we chose to plot this variable with physical values. To do this we had to choose physical values for D and σ for the figure. These parameters were set to σ = 5 nm and D = 105 nm2 ms−1 and yielded the result shown in the plot. A smaller σ, i.e., a smaller time-step, will result in the curves shifting downwards. We see that the probability of registering more than one binding event per time-step is quite large for high concentrations; >1 mM for the largest values of k+*. In this particular case, the solid and dashed lines represent an on-rate of, respectively, 3800 and 380 μM−1 s−1, i.e., quite large values.
We were able to define the probability of registering a binding event from our test domain during a time-step as 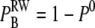 and compare this with the continuous equivalence from Eq. 10, for convenience here named
and compare this with the continuous equivalence from Eq. 10, for convenience here named 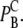 Using the same values for the parameters as above, we computed the absolute value of the relative difference between these two models,
Using the same values for the parameters as above, we computed the absolute value of the relative difference between these two models, 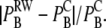 The result is shown in the right panel of Fig. 3. We see that the difference is very small and is more or less constant for different values of c. The downward bend seen for the largest values of k+* represents the difference between the two models in a parameter range in which both models produce erroneous probabilities and should, therefore, be ignored. These results indicate clearly the similarities in registered binding events between the two models for a large parameter range.
The result is shown in the right panel of Fig. 3. We see that the difference is very small and is more or less constant for different values of c. The downward bend seen for the largest values of k+* represents the difference between the two models in a parameter range in which both models produce erroneous probabilities and should, therefore, be ignored. These results indicate clearly the similarities in registered binding events between the two models for a large parameter range.
Reflecting boundaries
The reflecting property of a membrane increases the expected concentration of a nearby Ca2+ ion. A receptor at or close to the membrane will therefore experience a higher concentration from a single Ca2+ ion and hence a larger probability of binding. The increase was included by mirroring the location of a receptor close to a membrane, to the opposite side, as illustrated in Fig. 4. The probability of binding was then calculated for this mirrored position and added to the initial probability,
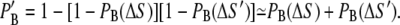 |
(31) |
Here, ΔS is the distance between the Ca2+ ion and the actual position of the receptor and ΔS′ is the distance between the ion and the mirrored receptor. The approximation in Eq. 31 holds for probabilities ≪1. If the receptor is situated at the membrane, we have ΔS = ΔS′. For simplicity, we mirrored all buffers in the upper part of the cleft to the opposite side of the SR membrane and all buffers in the lower part of the cleft to the opposite side of the TT membrane.
FIGURE 4.

How the reflection of a receptor near a membrane is modeled. The value ∂ΩN2 is the reflecting boundary of the membrane; r and r′ are the position of the receptor at its actual position and at its mirrored position. The values ΔS and ΔS′ represent the distance between the Ca2+ ion and the actual position of the receptor and the position of the mirrored one.
Monte Carlo simulation of binding
To speed up the Monte Carlo simulations of the reaction, we precomputed the probability of unbinding and binding of a single Ca2+ ion for each type of receptor included in the simulation. The unbinding probability for each buffer type was very small, which allowed us to assume that only one Ca2+ ion could unbind during a full time-step. With this assumption, we only had to sample one uniform random number per time-step for the unbinding reactions. This number was compared to a lumped unbinding probability that is given by
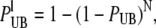 |
(32) |
where PUB is given by Eq. 12 and N is the number of buffer molecules that have a Ca2+ ion bound to it.
The probabilities of binding were precomputed with respect to ΔS and a lookup table was used during the simulation. To speed up this process even more, only Ca2+ ions within a certain maximal distance to the receptor were considered. This distance was chosen so that the probability of binding at this distance equaled 10−6. The actual Monte Carlo sampling was performed as follows: 1), traversing the empty receptors in a random order each time-step; 2), for each empty receptor, calculating the probability of binding for all Ca2+ ions within the maximal distance; 3), distributing these probabilities in a cumulative distribution, 0 < Cp1 < Cp2… < CpN < 1, where N is the number of Ca2+ ions within the maximal distance and Cpi is the cumulative binding probability of the ith Ca2+ ion; and finally, 4), drawing a uniformly distributed random number between 0 and 1. No Ca2+ ion was bound if the random number was larger than CpN. If the random number was in-between Cp(i–1) and Cpi, the ith Ca2+ ion was bound to the receptor. By choosing a small enough time-step, Δt, we ensured that both the single binding probability and the sum of all binding probabilities always was much smaller than 1. This minimized the error made in assuming that only one Ca2+ ion could bind to one receptor during a time-step.
Random Walk algorithm
A full step in our RW algorithm is presented schematically in Fig. 5. First, any Ca2+ that is scheduled to enter the cleft at this time-step is added to the variable that keeps track of all Ca2+ ions. After that, we check whether any Ca2+ ions were bound to mobile or stationary buffers or to the included RyRs, using the precomputed binding probabilities from Eq. 24. Then, we update the mobile buffers and the Ca2+ ions with new positions, using the Monte Carlo method presented above. The first procedure (the reaction loop) operated on a larger timescale than the second (the diffusion loop). A single step in the reaction loop took much longer and the accuracy was not so sensitive to the time-step, which allowed us to simulate this procedure at a larger timescale. The sampling of new displacement in the diffusion loop was cheap, but the escape rate of the Ca2+ ions leaving the cleft by the absorbing boundary ∂ΩD, was underestimated (41). This error was time-step dependent and was therefore minimized by using smaller time-steps in this loop.
FIGURE 5.

Time-step in the RW algorithm. The upper part, above the dashed line, shows the reaction loop and the lower part shows the diffusion loop. The reaction loop is simulated with a coarser time-step, Dt = 125 ns, than the diffusion loop, dt = 5 ns.
METHODS
All simulations, plots, and statistical tests were done using MATLAB (The MathWorks, Natick, MA) on a GNU/Linux laptop, with 1 GB Ram and a 2.1 GHz Pentium M processor.
Morphology and boundaries
Following Soeller and Cannell (21), we modeled the dyadic cleft as a disk (Fig. 6), with h = 15 nm and R = 100 nm. The diffusion constant of Ca2+ was set to Dc = 105 nm2 ms−1 (28). The single LCC current amplitude was chosen to be 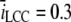 pA (43), and was released in the center of the disk along the dashed line in Fig. 6. In one of the simulations, we included both mobile and stationary endogenous buffers, using rates and concentration parameters from a previous study (44) (see Table 1). The diffusion constant of the mobile buffer, calmodulin, was set to Dm = 0.1 × Dc (21). Several open LCCs were modeled by multiplying the source amplitude by the number of open channels. The binding rate for the RyRs was set to 5 μM−1 s−1, which corresponds to binding rates previously used in models for both RyR and LCC (36,45). The TT and SR membranes were modeled as reflective, no-flux, boundaries, ∂ΩN2 in Fig. 6. The cytosol was included in the model either as a zero concentration boundary, when a LCC Ca2+ source was used, or as a constant level corresponding to diastolic [Ca2+] of 0.1 μM (see ∂ΩD in Fig. 6).
pA (43), and was released in the center of the disk along the dashed line in Fig. 6. In one of the simulations, we included both mobile and stationary endogenous buffers, using rates and concentration parameters from a previous study (44) (see Table 1). The diffusion constant of the mobile buffer, calmodulin, was set to Dm = 0.1 × Dc (21). Several open LCCs were modeled by multiplying the source amplitude by the number of open channels. The binding rate for the RyRs was set to 5 μM−1 s−1, which corresponds to binding rates previously used in models for both RyR and LCC (36,45). The TT and SR membranes were modeled as reflective, no-flux, boundaries, ∂ΩN2 in Fig. 6. The cytosol was included in the model either as a zero concentration boundary, when a LCC Ca2+ source was used, or as a constant level corresponding to diastolic [Ca2+] of 0.1 μM (see ∂ΩD in Fig. 6).
TABLE 1.
Ca2+ buffer parameters
| k+ | k− | BT | |
|---|---|---|---|
| Ca2+ buffer | [μM−1 s−1] | [s−1] | [μM] |
| Calmodulin | 100 | 38 | 24 |
| SL membrane | 115 | 1000 | 1124 |
Simulation setups and binding event registrations
As mentioned in the Introduction, we considered the event of a single Ca2+ ion binding to a receptor to be the stochastic event that determines the functional properties of the dyadic cleft. We tested how well the continuous model fits the equivalent binding events registered from the RW model. We used four tentative RyRs, positioned from the center of the cleft to the rim, to test whether the radial position of single receptors had any effect on the event registrations. We performed three different set of simulations, in which binding events were registered under different physiological conditions. These conditions were as follows: 1), steady-state [Ca2+] response due to one open LCC; 2), uniform [Ca2+] due to passive diffusion from cytosol, using very low diastolic [Ca2+] = 0.1 μM; and 3), transient [Ca2+] response from three different LCCs, which alternated between closed and open during the simulations. The statistical results from these three sets of simulations are presented in Figs. 8–10.
FIGURE 8.

Statistical data for binding events registered from Random Walk simulations with one open LCC, which acted as the Ca2+ source, situated in the center of the cleft. The binding events are registered at four different RyRs, positioned at 10, 30, 50, and 70 nm from the center of the cleft. Binding events are collected from 100 simulation runs. The registration started when the [Ca2+] had reached the steady state. The total time simulated was 30 ms. (A) Box-plot of the number of binding events from the runs at each receptor, together with a 95% confidence interval for the true means (red horizontal lines). The blue solid circles represent the expected number of binding events predicted by the continuous model. These values were computed on the basis of the fixed [Ca2+] at each receptor (see inset). In the box-plot, the green line represents the median of the data and the blue horizontal lines the limit of the upper and lower quartiles. The whiskers, i.e., the black lines extending from the blue boxes, represent the rest of the data up to a maximum length of 1.5 times the size of the two center quartiles. The green plus-signs are outliers. (B1–B4) Inter-event intervals (IEIs) from all runs presented in scaled histogram plots, corresponding to the receptor at positions 10, 30, 50, and 70 nm from the center of the cleft. The heights of the bars are scaled so the total area of a whole histogram = 1. The red lines show the probability distribution of the IEI from an homogeneous Poisson distribution with a rate based on the steady-state value of the [Ca2+] at the receptor. The blue asterisk indicates a significant difference, at 5% level, between the collected IEIs and the corresponding exponential function using a Kolmogorov-Smirnov test.
FIGURE 9.

Statistical data for binding events that are registered from Random Walk simulations. The Ca2+ source was passive diffusion from the cytosol during diastole, i.e., the resulting [Ca2+] was, on average, 0.1 μM. The binding events are registered at four different RyRs, positioned at 10, 30, 50, and 70 nm from the center of the cleft. The binding events were collected from 100 simulation runs. The total time simulated was 20 s. (A) Box-plot of the number of binding events from the runs at each receptor, together with a 95% confidence interval for the true means (red horizontal lines). The blue solid circles represent the expected number of binding events predicted by the continuous model. These values were computed on the basis of the fixed [Ca2+] at each receptor. (For an explanation of the box-plot, see the legend of Fig. 8.) (B1–B4) Inter-event intervals from all runs presented in scaled histogram plots, corresponding to the receptor at positions 10, 30, 50, and 70 nm from the center of the cleft. (For an explanation of the histogram, see the legend of Fig. 8.)
FIGURE 10.

Statistical data for binding events registered from Random Walk simulations. The Ca2+ source is zero to three open LCCs, situated in the center of the cleft (A, inset). The binding events are registered at four different RyRs, positioned at 10, 30, 50, and 70 nm from the center of the cleft. The binding events were collected from 100 simulation runs. The total time simulated was 16 ms. (A) Box-plot of the number of binding events from the runs at each receptor, together with a 95% confidence interval for the true means (red horizontal lines). The blue solid circles represent the expected number of binding events predicted by the varying [Ca2+] from the continuous model at each receptor. (See inset for the [Ca2+] at the RyR at 70 nm.) No significant differences, at 5% level, were detected. (For an explanation of the box-plot, see the legend of Fig. 8.) (B1–B4) Transformed inter-event intervals from all runs presented in scaled histogram plots, corresponding to the receptor at positions 10, 30, 50, and 70 nm from the center of the cleft. (For an explanation of the histogram, see the legend of Fig. 8.)
Each set of simulations had different deterministic Ca2+ influxes, corresponding to each physiological situation, and was run 100 times. Stochastic binding events from four different RyRs were registered. The RyRs were located along the same axis at radial distances of 10, 30, 50, and 70 nm.
One open LCC, steady-state [Ca2+]
In the first set of RW simulations, we registered the binding events from the steady-state response of a single open LCC in the cleft. The Ca2+ influx in these simulations consisted of one open LCC situated at the center of the cleft. Initially, the cleft had zero Ca2+ ions, so registration was started after 0.2 ms, after the steady state was achieved, and the runs were stopped after 30 ms. In these runs, we were only interested in the binding events during the steady-state [Ca2+] in the cleft, so we excluded both stationary and mobile buffers from the simulations, thereby achieving a significant gain in speed. The mobile buffer actually lowers the steady-state [Ca2+]. Therefore, it could be argued that it should have been included in these simulations (21). However, neither its inclusion nor exclusion would influence the results of the comparison study, which was the main focus. The steady-state solution of [Ca2+] from the continuous model, which was used in the comparison study (see below), is presented in the inset of Fig. 8 A.
Diastolic steady-state [Ca2+]
In the second set of RW simulations, we tested the effect on the binding events when [Ca2+] was extremely low. Instead of Ca2+ influx through a channel, we had passive Ca2+ influx from the cytosol. The value of the [Ca2+] that we used corresponded to a diastolic concentration of 0.1 μM. In these simulations, we did not include any buffers, because the Ca2+ response was stationary. Due to the small number of Ca2+ ions in the cleft, ∼0.02 on average, each run had to be long (30 s) to produce reliable statistics for the tests.
Transient [Ca2+]
In the third and last set of RW simulations, we studied binding events that were registered during a transient response in the cleft. Both stationary and mobile buffers were included in these simulations. The Ca2+ influx came through three LCCs that alternated between open and closed; see inset of Fig. 10 A for the resulting LCC current. Each run lasted for 22 ms.
Comparison methods
Two different hypotheses about the statistical outcome of the binding events were formed for each RyR and for each set of simulations: 1), the mean number of events during a simulation run are the same for both models; and 2), the inter-event intervals (IEIs) of the registered binding events are the same for both models. Each of these hypotheses was tested for each RyR and for each set of simulations.
To perform the tests, we needed the solution of the continuous concentrations at each RyR. In the first set of simulations, Ca2+ entered the cleft from one single LCC and no buffers were present. Setting 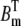 and
and 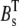 to zero in Eqs. 1–4, the steady-state solution could be solved analytically with respect to r (see Fig. 8 A, inset). In the second case, in which Ca2+ entered the cleft passively through the cytosol, we fixed the concentration at the same level as for the cytosol, 0.1 μM for all RyRs. In the third case, we needed the [Ca2+] at every time-step, c, for each RyR. We simulated the full system in Eqs. 1–4 with the same input current as was used in the RW simulations. The [Ca2+] for the ith RyR and nth time-step,
to zero in Eqs. 1–4, the steady-state solution could be solved analytically with respect to r (see Fig. 8 A, inset). In the second case, in which Ca2+ entered the cleft passively through the cytosol, we fixed the concentration at the same level as for the cytosol, 0.1 μM for all RyRs. In the third case, we needed the [Ca2+] at every time-step, c, for each RyR. We simulated the full system in Eqs. 1–4 with the same input current as was used in the RW simulations. The [Ca2+] for the ith RyR and nth time-step, 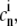 was registered.
was registered.
Test of mean number of events
Using the central limit theorem, we compared the mean number of binding events from each RyR against the expected number of binding events from the continuous model, with a one-sample Student's t-test. The continuous solution of [Ca2+] was used to compute the expected number of binding events, μ, of a whole run for each RyR. We calculated a 95% confidence interval for the expected mean from the data collected from the RW simulations, together with the corresponding p-values for the Student's t-test. The expected number of binding events during a run of length T simulated with a homogeneous Poisson with rate λi is given by
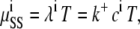 |
(33) |
where c is the [Ca2+] at the ith receptor and k+ is the macroscopic binding rate (38). In the last simulation setup, where the [Ca2+] varied, we had to integrate the rate function to get the expected number of binding events, which is given by
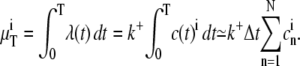 |
(34) |
Here, 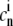 is the value of the [Ca2+] in the nth time-step and Δt the length of each step.
is the value of the [Ca2+] in the nth time-step and Δt the length of each step.
Test of same inter-event intervals
IEIs were calculated from the binding event data from each RyR in the RW simulations. All IEIs from one RyR collected during one set of simulations were combined to form one distribution. The equivalent expected distributions from the continuous model were computed for each RyR, for all three simulation setups. The goodness-of-fit of the expected distributions was tested against the registered IEI distributions collected from the RW simulations, using a Kolmogorov-Smirnov (KS) test (46).
In the first two simulation setups, the [Ca2+] at each RyR was fixed and the resulting binding rates for each RyR were constant, forming homogeneous Poisson processes. The IEIs from an homogeneous Poisson process are distributed exponentially with the same rate as the Poisson process itself (38). The expected IEI distribution for the ith RyR is given by
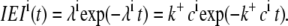 |
(35) |
These were used to compute the p-values of the KS tests (see Table 2).
TABLE 2.
Binding event statistics
| Student's t-test
|
KS-test | |||
|---|---|---|---|---|
| RyR positions | μ | 95% CI | p-values | p-values |
| One open LCC, steady-state [Ca2+] | ||||
| 10 nm | 56.8 | (54.3, 57.3) | 0.22 | 0.17 |
| 30 nm | 29.8 | (28.4, 30.8) | 0.74 | 0.042* |
| 50 nm | 17.1 | (15.6, 17.2) | 0.059 | 0.56 |
| 70 nm | 8.79 | (8.29, 9.41) | 0.83 | 0.060 |
| Diastolic steady-state [Ca2+] | ||||
| 10 nm | 15.0 | (14.0, 15.5) | 0.51 | 0.10 |
| 30 nm | 15.0 | (14.1, 15.4) | 0.49 | 0.54 |
| 50 nm | 15.0 | (14.0, 15.5) | 0.48 | 0.055 |
| 70 nm | 15.0 | (13.8, 15.3) | 0.25 | 0.56 |
| Transient [Ca2+] | ||||
| 10 nm | 68.3 | (66.5, 70.1) | 0.96 | 0.31 |
| 30 nm | 35.6 | (33.7, 36.1) | 0.27 | 0.39 |
| 50 nm | 20.4 | (19.2, 21.0) | 0.52 | 0.64 |
| 70 nm | 10.4 | (9.75, 10.8) | 0.52 | 0.087 |
Significant difference.
In the third simulation setup, the [Ca2+] at each RyR was not fixed, yielding inhomogeneous Poisson processes. The resulting IEI distribution from such a process does not follow an exponential distribution. A useful method for evaluating models of point processes in neural spike train data analysis, the time-rescaling theorem, was introduced by Brown et al. (47). They used this theorem to transform registered event times from an inhomogeneous Poisson process to represent realizations of a homogeneous Poisson processes with unit rate. Given a series of time events 0 < t1 < t2 <, …, < tn < T that realizes an inhomogeneous Poisson process with rate λ(t) > 0 for all t ∈ (0, T], the transformed realization of a homogeneous Poisson process with unit rate is
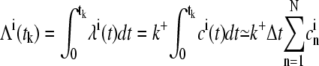 |
(36) |
for k = 1, …, n. The IEIs of this process are τk = Λ(tk) – Λ(tk–1) and they are exponentially distributed with unit rate. We used the rate from the continuous model to transform the IEIs registered from the RW model. These were then used in a goodness-of-fit test of an exponential function with unit rate.
Bonferroni procedure
We performed three different sets of RW simulations, collected binding events from four different RyRs, and performed two different statistical tests for each receptor. This left us with a total of 24 statistical hypotheses. For every test, the H0 hypothesis was that the continuous model either predicted the mean number of binding events or fitted the IEI distributions with an appropriate exponential function. The overall hypothesis of how well the continuous model fitted the sampled binding event data from the full RW model had to be determined on the basis of these tests. The number of binding events during a run was not independent of the IEI distributions. If an IEI distribution is known to follow an exponential distribution, the expected number of events follows directly from the rate of this distribution, thus reducing the number of independent tests to 12. Given that we were doing 12 independent tests, each at α = 5%, there was a probability P = 1–0.9512 = 0.46, of getting at least one false rejection. The α level for each subtest was therefore adjusted such that our main hypothesis was tested at the 5% level by using the conservative but simple Bonferroni procedure (46). The new α-level for each subtest was acquired by dividing the total α-level by the number of subtests. This gave us an α-level of 0.42% for each subtest.
RESULTS
Random Walk versus continuous solutions
To confirm that the solution from the continuous model coincided with the mean concentrations from the RW model, we did one run with the continuous model and 40 runs of the RW model, using the same parameters (Fig. 7 for result). The black lines are the concentration in the cleft given by the continuous model of, respectively, Ca2+ (solid line), mobile buffer (dashed line), and stationary buffer (dash-dotted line). The colored lines, partly covered by the black lines, are 1), the concentration results from a single RW simulation (green lines); and 2), the average results from 40 RW runs (red lines). Note that the scale for the stationary buffer traces is given in the right y axis. One LCC was opened at t = 0, to act as a Ca2+ source in the cleft. After ∼1 ms, the steady state, in which most of the stationary buffers were bound to Ca2+, was achieved. After 2 ms, a second LCC was opened. This time, the steady state occurred more quickly, due to the fact that less stationary buffer was available. We see that the [Ca2+] in the single RW run fluctuates a great deal in the steady-state period, but the mean concentration does not. After 4 ms, both LCCs were turned off and the Ca2+ left the cleft quickly. Some Ca2+ remained, due to the unbinding of Ca2+ from the stationary buffer.
FIGURE 7.

Simulation results from the continuous model (black lines) and from the Random Walk (RW) model (colored lines). The results represent the average concentrations from the whole cleft. The same simulation setup was used for the two models, including buffers from Table 1. One LCC is open from the start. After 2 ms, one more opens. Then, after 4 ms, both close. The black lines are the results from one simulation of the continuous model. Each line, solid, dashed, and dash-dotted, represents the concentration of Ca2+, mobile buffer, and stationary buffer, respectively. The right y axis shows the scale for the stationary buffer. The colored lines are the mean concentrations from 40 runs of the RW model (red lines), and the concentrations from a single RW simulation (green lines).
The result confirms what others have pointed out, that the continuous solution coincides with the mean result from several RW simulations (1–3). We did see a difference between the mean concentration of the stationary buffer registered from the RW runs, and the corresponding concentration from the continuous solution. This error was introduced in the RW model, because we did not account for the absorbing boundary when calculating the probabilities that Ca2+ ions and the stationary buffer molecules near the rim would bind. By placing the RyRs well inside the cleft, the outermost being 30 nm from the rim, we avoided this error when binding probabilities for the receptors were calculated.
It is interesting to note that the large Ca2+ flux to the buffers, primarily to the stationary buffer, fluctuates significantly less than the outflux of Ca2+ ions from the cleft. This is an effect of the low binding rate compared to the exit rate. The latter is approximately equal to the influx from the LCC during the steady state, not including the small outflux through the mobile buffer. The influx when two channels were open, between 2 and 4 ms, was ∼JLCC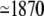 ions per ms, and the binding rate to the stationary buffer during the same steady state was JSB = CSS × k+ ≃ 20 ions per ms. This means that the outflux is 100-times larger than the flux to the buffers.
ions per ms, and the binding rate to the stationary buffer during the same steady state was JSB = CSS × k+ ≃ 20 ions per ms. This means that the outflux is 100-times larger than the flux to the buffers.
Statistics of single binding events
As seen in Table 2, where the results of the Student's t-tests and the KS-tests are presented, the predicted distributions of binding events from the continuous model fit the corresponding distributions of registered binding events from the RW model. The p-values and 95% CI are included for the t-tests and the p-values are included for the KS-tests. We found only one significant difference at the 5% level and none at our Bonferroni adjusted 0.4% level. Statistics of the binding event data are also presented graphically in Fig. 8–10. These figures also visually support the results from the statistical tests presented in Table 2.
All three figures present the data in the same manner. In Figs. 8–10, panels A, the number of binding events is presented in one box-plot for each RyR, together with a 95% CI of the true mean (red horizontal lines) and the expected number of binding events predicted by the continuous model (blue solid circles). The distributions of IEI, for each RyR, are presented in scaled histograms in Figs. 8–10, panels B1–B4, in each of the three figures. The heights of the bars are scaled so that the total area of the histograms equals 1. This scaling enabled us to compare the distributions of IEIs with the expected distributions from the continuous model (red lines). Fig. 8 shows the results from the first set of simulations, where the Ca2+ source was one LCC that was open constantly. The inset in Fig. 8 A, shows the steady-state [Ca2+] in the cleft from the continuous model, where the concentration at each RyR is marked by solid blue circles. Fig. 9 presents the results from the second set of simulations, where the Ca2+ source was a passive influx from cytosol, which resulted in a [Ca2+] in the cleft that corresponds to a diastolic value of 0.1 μM. The last figure, Fig. 10, presents the results from the last set of simulations. Here, the Ca2+ source was three LCCs, which alternated between open and closed in the same manner during all simulations. The inset of Fig. 10 A shows the varying [Ca2+] from the continuous model, at the RyR positioned at 70 nm from the center (red line), together with the shifting LCC current.
We observe that the number of binding events and the expected IEI distributions depend on the radial positions of the RyRs, for the first and third set of simulations, both of which are driven by a LCC current. This is not surprising, because the [Ca2+] are higher the closer they are to the channel. Perhaps more interesting, the RyRs in the cleft actually discriminate the [Ca2+] from a single Ca2+ source. This is important for accounting for when the cleft is treated as a single compartment with the same lumped average [Ca2+] (15). There is also no over- or underregistration of binding events on a certain RyR within each set of simulations. This means that the continuous model reproduces the binding events from the RW model independently of the radial position of the RyRs.
Mean binding rate registered at a single receptor
Our goodness-of-fit tests revealed that there are no significant differences among the registration of stochastic binding events in the two models. This is not an average result, but a result that holds on the level of single runs and at the level of IEI. To acquire a better understanding of how this could be true, we examined what we called a lumped binding rate, 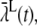 registered by a single RyR positioned 10 nm from the center during a run with one constantly open LCC.
registered by a single RyR positioned 10 nm from the center during a run with one constantly open LCC.
Each Ca2+ ion within a maximum distance of the receptor contributes, to a small extent, to the probability that a binding event will occur. This allows us to formulate the overall probability that a binding event will occur as a sum of small probabilities, where each is of the form 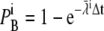 ; see Eq. 24. Again, given small binding probabilities, this formulation can be approximated with
; see Eq. 24. Again, given small binding probabilities, this formulation can be approximated with 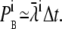 The resulting lumped binding probability is then
The resulting lumped binding probability is then 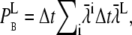 where
where 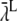 represents the lumped binding rate.
represents the lumped binding rate.
During one simulation, we registered 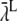 at each time-step. These values are plotted against time in Fig. 11 A. The right y axis gives the corresponding binding probabilities. The stochastic and discrete nature of the rates may be seen clearly in these chaotic data. The rate varies from time-step to time-step, as shown in the enlargement of the figure for t = [0, 0.01] ms, shown in Fig. 11 B. The mean rate registered for the whole run was
at each time-step. These values are plotted against time in Fig. 11 A. The right y axis gives the corresponding binding probabilities. The stochastic and discrete nature of the rates may be seen clearly in these chaotic data. The rate varies from time-step to time-step, as shown in the enlargement of the figure for t = [0, 0.01] ms, shown in Fig. 11 B. The mean rate registered for the whole run was 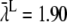 ms−1. In 80% of the time-steps, the rate was smaller than this value, and in 11% of the time-steps, the rate equaled zero. In only 4.1% of the time-steps was the rate >10 ms−1 and the maximal registered rate for this run was 414 ms−1. These rates seem large but the resulting binding probabilities,
ms−1. In 80% of the time-steps, the rate was smaller than this value, and in 11% of the time-steps, the rate equaled zero. In only 4.1% of the time-steps was the rate >10 ms−1 and the maximal registered rate for this run was 414 ms−1. These rates seem large but the resulting binding probabilities, 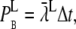 were, as seen in the right y axis, all ≪1. We used the same size of time-step as earlier, Δt = 1.25 × 10−4 ms. The binding probability that corresponded with the mean rate for the whole run was 2.4 × 10−4. To be able to take the average of the binding rates over several time-steps, it has to make sense to take the sum of several binding rates. This measure is justified by the small binding probabilities that each receptor experiences every time-step (Fig. 11, A and B). The crucial issue was how the average binding rate fluctuates on a larger timescale, i.e., do the large variations in binding rates in each time-step average-out at a larger timescale and if so, how small can this timescale be?
were, as seen in the right y axis, all ≪1. We used the same size of time-step as earlier, Δt = 1.25 × 10−4 ms. The binding probability that corresponded with the mean rate for the whole run was 2.4 × 10−4. To be able to take the average of the binding rates over several time-steps, it has to make sense to take the sum of several binding rates. This measure is justified by the small binding probabilities that each receptor experiences every time-step (Fig. 11, A and B). The crucial issue was how the average binding rate fluctuates on a larger timescale, i.e., do the large variations in binding rates in each time-step average-out at a larger timescale and if so, how small can this timescale be?
FIGURE 11.

(A and B) Lumped binding rates for each time-step, registered from one RyR during a single Random Walk simulation. In the simulation, one constantly open LCC channel was used and the RyR was positioned 10 nm from the center of the cleft. (B) Enlargement of panel A for t = [0, 0.01] ms. The mean binding rate fluctuates a lot for each time-step. (C) Filtered version of the binding rate. A Gaussian kernel with σ = 0.26 ms, corresponding to the scale of the registered IEIs, was chosen for the filtering. (A–C) Corresponding binding probabilities are given by the right y axis. For the ith time-step, this quantity is computed by 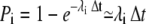 .
.
The averaged binding rate did not vary much from run to run. The mean averaged rate from 100 runs was [1.904 ± 0.019] ms−1. This value did not differ significantly from the constant rate from the continuous model, λc = 1.91 ms−1, p-value = 0.74. This result corresponds to the failure of detecting a significant difference between the average number of binding events that was registered in the RW simulations and the number given by the continuous model (see the results of the Student's t-tests in Table 2). The variations in the binding rate at the timescale of a whole simulation run thus averaged-out and were statistically indistinguishable from the continuous constant rate.
On a smaller timescale, we would expect the averaged rate to fluctuate more. For example, the mean rate for the interval shown in Fig. 11 B, i.e., t = [0, 0.01], was 2.70 ms−1. This is greater than the average rate for the whole run, which was 1.90 ms−1. However, the fluctuation at this timescale does not tell us much, because the expected number of binding events with this rate at this timescale is 0.027. To investigate the effect on the actual binding events, we have to take the average on a larger timescale. A proper scale would be the mean IEI registered in the simulation runs. This was found to be 0.52 ms (Fig. 8 B1). We filtered the registered rate with a Gaussian filter, which acts as a weighted mean over a certain time window defined by the width of the filter, σ (48). This width was set to half the size of the mean IEI, 0.26 ms. The result is presented in Fig. 11 C, together with the constant rate from the continuous model. The filtered rate is a continuous function of time and does not vary nearly as much as the unfiltered rate in Fig. 11 A. The maximal value of the filtered signal was 2.1 ms−1, the minimal was 1.7 ms−1, and the standard deviation from the mean, which of course was the same as the unfiltered rate, was 0.1 ms−1. This small variation explains why the IEIs of the registered binding events from the RW model were statistically indistinguishable from those of the continuous model.
Parameter sensitivity
To check the dependency of some of the parameters we have used, we made five runs in which we altered the diffusion constant, Dc, together with the maximal input current from one open LCC, 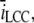 in the same manner as Tanskanen et al. (32) did. We scaled the Dc and
in the same manner as Tanskanen et al. (32) did. We scaled the Dc and 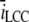 by factors of [5, 2, 1, 0.5, 0.1] and ran 100 runs of the steady-state condition, in which one LCC was open. The spatial resolution for the registration of binding events was set to σ = 5 nm for every run. We compared the number of registered binding events with the expected number from the continuous model. The number for the latter was constant in all runs, because the concentrations at the receptors were the same under the scaling. The result is shown in Fig. 12 A. The figure shows a box-plot of the number of binding events registered at the receptor 30 nm from the center of the cleft versus the scale on the x axis. There are no significant differences for scale = [5, 2, 1, 0.5], but for scale = 0.1, there is. To investigate the dependency of the parameters further, we also altered σ. We used [5, 2, 1, 0.5] for σ and also did 100 runs for each different value. The result is presented in a similar box-plot in Fig. 12 B. Note that the leftmost datapoints in this figure are identical to the rightmost datapoints from the previous figure. From the figure, we see that the number of registered binding events falls steadily. This illustrates that the binding event registration depends, not only on physical parameters, but also on the spatial resolution of the RW method. This observation coincides with the parameters used in the dimensionless on-rate Eq. 29.
by factors of [5, 2, 1, 0.5, 0.1] and ran 100 runs of the steady-state condition, in which one LCC was open. The spatial resolution for the registration of binding events was set to σ = 5 nm for every run. We compared the number of registered binding events with the expected number from the continuous model. The number for the latter was constant in all runs, because the concentrations at the receptors were the same under the scaling. The result is shown in Fig. 12 A. The figure shows a box-plot of the number of binding events registered at the receptor 30 nm from the center of the cleft versus the scale on the x axis. There are no significant differences for scale = [5, 2, 1, 0.5], but for scale = 0.1, there is. To investigate the dependency of the parameters further, we also altered σ. We used [5, 2, 1, 0.5] for σ and also did 100 runs for each different value. The result is presented in a similar box-plot in Fig. 12 B. Note that the leftmost datapoints in this figure are identical to the rightmost datapoints from the previous figure. From the figure, we see that the number of registered binding events falls steadily. This illustrates that the binding event registration depends, not only on physical parameters, but also on the spatial resolution of the RW method. This observation coincides with the parameters used in the dimensionless on-rate Eq. 29.
One large difference between the continuous model and the RW model is that in the RW model, a binding event actually leads to a removal of an ion from the cleft, in contrast to the continuous model where nothing happens. To test whether this difference is crucial for the registered difference between the two models as seen in Fig. 12 B, we performed the same simulations, but without registering any binding events. Instead we registered the mean binding rate from each run and compared this with the rate predicted from the continuous model. In Fig. 12 C, the red lines represent 95% confidence intervals of the true mean binding rate from the 100 runs. The rate predicted from the continuous model is represented by the blue solid circles. We cannot differentiate statistically between the collected mean binding rates and those of the continuous model. We also collected the mean binding rates from the simulations we did in Fig. 12 B, in which ions were removed from the solution after they were bound. The 95% confidence interval of the true mean for these binding rates is represented by the blue horizontal lines. Here, we see that the binding rates follow the number of registered binding events from Fig. 12 B, and not the predicted rate from the continuous model. These results illustrate why the RW model starts to differ from the continuous model for low values of the diffusion constant together with small values of the spatial resolution.
As discussed above, the difference between the two models is parameter-sensitive. This sensitivity can be expressed by the dimensionless on-rate, k+*, from Eq. 29. This value depends on the diffusion constant, D, the macroscopic on-rate, k+, and the spatial resolution, σ. For the simulations for which we registered a difference between the RW model and the continuous model, i.e., for scale = 0.1 in Fig. 12 A, k+* = 0.013. This value indicates an upper limit for when the two models start to diverge. The spatial resolution is, in a sense, a free parameter. One could, in theory, make it as small as one likes, thus forcing a difference between the two models. Alternatively, one could make it large to smooth out a potential difference. In practice, the value of this parameter is determined by the level of spatial detail that is required in the simulation.
DISCUSSION
We have compared a RW model and a continuous model of Ca2+ diffusion in the dyadic cleft, using the distributions of stochastic events of single Ca2+ ions binding to single receptors as the measurement. We showed that for a large range of physiologically relevant parameters, there are no significant differences between the continuous model and the RW model with respect to these binding events. This is a somewhat unexpected result, considering the small number of ions included in the discrete model of [Ca2+] and the inherent variation in their position. In one set of simulations, the average number of Ca2+ ions in the cleft is 0.02, corresponding to a diastolic [Ca2+] of 0.1 μM. Thus, most of the time, there are no Ca2+ ions in the cleft. Despite this, the corresponding constant binding rate from the continuous model can reproduce the binding events registered in the RW model.
New method for computing bimolecular binding probabilities
We present a method for bimolecular binding probabilities that is, to the best of our knowledge, novel. The proposed model is based on a macroscopic rate law that we use in our RW simulations. The model is analytical and gives the binding rate between two molecules exactly. To obtain this result, it is necessary only to ensure that the probability that more than one binding event per time-step will be registered is small. The method is used to calculate the probability that a diffusive ligand will bind to a receptor, which can be stationary or mobile. The binding rate depends only on the on-rate, k+, the diffusion constant(s), Dc (Dm), the size of the time-step, Δt, and the distance between the two molecules, ΔS. The only parameter not known before a simulation is ΔS, and our knowledge of the other parameters allows us to precompute the binding probabilities with respect to ΔS. The method also lets us use larger time-steps for the computationally expensive reaction process. We also investigated thoroughly the physical parameters for which the model is applicable.
Comparison method from hazard analysis and neuroscience
RW methods and continuous methods are models of diffusion at two different levels. They have been compared before, but here we used a quantitative goodness-of-fit measurement in the comparison study. The statistical method that we used was originally developed for the evaluation of point process models, e.g., errors in industrial processes, so-called hazard analysis (49). The method has also recently been used in neuroscience for evaluating models in the analysis of data for neural spike trains (47). The method is straightforward to use and could be employed in similar studies where discrete and stochastic models are compared.
Stochastic Random Walk versus deterministic continuous modeling of [Ca2+]
A number of recent publications claim that when the number of participating particles in a volume falls, a deterministic description of concentration is invalid or does not make sense, and fully stochastic methods have to be employed (2,6,10,12). Our conclusion refines these statements.
The continuous model predicts the average number of particles involved in a process. For processes that involve a small number of particles, RW simulations show that the variation in the number of particles can be of the same magnitude or larger than this average, in a single simulation (1–3,7). This is a strong argument against using a continuous description of [Ca2+] in small volumes such as the dyadic cleft, but only if the precise position of a certain diffusive ligand is important for the physiological process. We show that this is not the case for ligands with sufficiently large diffusion constants. The important receptors in the cleft that register the Ca2+ signal do not switch states according to whether there are Ca2+ ions close to them or not, but rather according to whether there are any Ca2+ ions bound to them or not. These events set the right timescale for the discreteness and stochasticity of the signaling in the cleft. In our study, we showed that these events can be simulated perfectly well by a continuous model of [Ca2+], for a given range of model parameters. The binding events occur on a larger timescale, hiding the huge variations in the single binding rates connected to each diffusing Ca2+ ion in the RW simulations (illustrated in Fig. 11, A–C). Fig. 11, A and B, show the strongly fluctuating binding rates. Fig. 11 C shows the same rate but filtered through a Gaussian filter, with σ equaling half the mean IEI, the timescale for the binding events. Here we see that the fluctuations on the scale of IEIs are small and follow the constant rate of the continuous model. In this way, the receptor acts as an integrator of the fluctuations in the binding rates. We also show that the radial positions of the receptors are important for determining the rate of binding events at each receptor. This is important to bear in mind when, as in some models, the dyadic cleft is treated as one compartment with the same lumped [Ca2+] (15). The concentration may reach a steady-state level quickly, but not all receptors sense the same [Ca2+] inside the cleft.
Tanskanen et al. (32) present results where their RW model shows a different result for the ECC gain, when the diffusion constant of Ca2+ and the influx of Ca2+ ions in the cleft are varied with the same amount. This difference is most probably caused by a different number of Ca2+ ions binding to the RyRs in the different runs. If the same had been done in a simulation in which the Ca2+ diffusion was modeled deterministically, a significant difference would not have been noticed, because the [Ca2+] at the RyR would have been the same, or more precisely, would have varied with the same mean, in each run. The authors claim that this is a “subtle but potentially significant difference in predicted macroscopic behavior arising from the underlying stochastic simulation of Ca2+ motion in the dyad” (32). We scaled the parameters in the same way as they did and we also recognized a difference, but only for the smallest value of the scaling, i.e., scale = 0.1 (Fig. 12 A). In addition, we changed the spatial resolution of the RW simulation and found that the number of binding events also depends on this parameter (Fig. 12 B). Finally, we showed that the difference between the models depends on the fact that an ion is removed from the solution after it is bound (Fig. 12 C). The difference becomes significant for large values of the dimensionless k+* parameter (see Eq. 29), i.e., small values of D and σ, and large values of k+. When k+* is too large, a single ion's contribution to the total binding rate becomes significant and the removal of the ion after a binding event will thus alter the total rate. We found that when k+* > 0.013, the two models registered different numbers of binding events. This is probably a conservative measure, because in our simulations we did not close a receptor for registration after an ion was bound. This made the effect of removing an ion from the vicinity of an unbound receptor larger than it would have been if the receptor had been in a bound state. For example, when k+* = 0.026 for the binding of Ca2+ ions to the stationary buffer in the transient simulation, we did not register any difference between the two models. It is important to bear in mind that the on-rate k+ for the RyRs is difficult to measure, and thus is often a free parameter. This makes the actual difference between the two models more fuzzy in real modeling, because one probably could fit the two models' macroscopic behavior to the same data, just by using slightly different parameters.
There are limitations in the continuous model that relate to the representation of more details. Such details could, for example, be the electrostatic interaction between single molecules (50), diffusion limitations due to excluded volumes (51), or diffusion in environments with large tortuosity and with possible molecule traps (52). However, the introduction of these extra details must be accompanied by an argument for the necessity of their inclusion. The study by Nicholson et al. (52) actually incorporates the micro-level effects into an effective diffusion constant. The authors thereby sanction the use of a macroscopic model of a micro-level phenomenon.
Limitations in our model of the dyadic cleft and Ca2+ dynamics
Our study of the dyadic cleft is limited, because it only examined the distribution of binding events and not the result of this event, i.e., the whole physiological signaling pathway of the Ca2+-induced Ca2+ release. However, these extra dynamics have nothing to do with diffusion. Thus, their inclusion would only introduce redundant information into our comparison study. We did include the dramatic event of a channel opening and closing during the simulation (Fig. 10).
Our physiological model of the cleft does not incorporate all present knowledge about the cleft, e.g., the electrostatic effect on diffusion due to the charged phospholipids in the membrane (53,54), or the obstructing effect that the large feet of the RyRs obviously have in the cleft (55). However, the aim of the study was not to present a state-of-the-art model of the dyadic cleft, but rather to use the cleft as a well-studied model system for our comparison study between the RW model and the continuous model. The effects of these extra details can, however, be included in both models, again only introducing redundant information. The electrostatic effect of the membrane is probably the easiest to include in the continuous model, as Soeller and Cannell (21) have done in their study of Ca2+ diffusion in the cleft. Our cleft model is also one-dimensional. Others have simulated the Ca2+ dynamics in the cleft using both two and three dimensions (12,21,29). We could have expanded our study to both two and three dimensions and added the geometric effects of the large feet of the RyRs, but our intention was not to present the most accurate model of the cleft. The dimension we included in our study was in the radial direction, because it is in this direction that the gradient in [Ca2+] is largest when a channel is open.
Neither did we include the effect of crowding (56,57) in the small and fuzzy cleft space (58). However, a Ca2+ ion is much smaller than the other diffusing macromolecules that are supposed to be in the cleft (59). A single Ca2+ ion can thus probably utilize most of the volume, making the excluded-volume argument regarding crowded environments (51) less forceful for Ca2+.
The binding of single Ca2+ ions to the RyRs are not reflected in the solution of the continuous model. Each RyR should introduce a small Ca2+ sink to the nearby environment when an external Ca2+ source is turned on, and introduce a small source when the external Ca2+ source is turned off. Due to the large diffusion constant to Ca2+, and the low affinity of the RyR, this sink is very small compared to the outflux of Ca2+ ions from the cleft. We performed RW simulations in which a Ca2+ ion was removed from the solution when it was registered as bound to a RyR, and the same simulation where the Ca2+ ion was not removed. We could not distinguish between the results. This sink is also only present during a transient face of a [Ca2+]. During the steady state, the bind flux is balanced by the unbind flux from the receptors.
CONCLUSION
The discrete and stochastic Ca2+ signaling in the physiological important dyadic cleft can be modeled accurately using a deterministic model of [Ca2+] together with a discrete and stochastic receptor model, for a certain range of parameters. Our study is the first to use the discrete binding event of single Ca2+ ions as a direct quantitative measure in a comparison study between an RW model and a continuous model of [Ca2+] in a small signaling micro domain. We also contribute a model of bimolecular binding probabilities that can be used in RW simulations. This model is, to the best of our knowledge, novel. The model is analytical; hence, the results do not depend on the size of the time-step. The study as a whole contributes both to the development of intracellular reaction-diffusion simulators (6) and the fundamental understanding of what the models actually represent (2).
Editor: Herbert Levine.
References
- 1.Franks, K. M., and T. J. Sejnowski. 2002. Complexity of calcium signaling in synaptic spines. Bioessays. 24:1130–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Slepchenko, B. M., J. C. Schaff, J. H. Carson, and L. M. Loew. 2002. Computational cell biology: spatiotemporal simulation of cellular events. Annu. Rev. Biophys. Biomol. Struct. 31:423–441. [DOI] [PubMed] [Google Scholar]
- 3.Turner, T. E., S. Schnell, and K. Burrage. 2004. Stochastic approaches for modeling in vivo reactions. Comput. Biol. Chem. 28:165–178. [DOI] [PubMed] [Google Scholar]
- 4.Bhalla, U. S. 2004. Signaling in small subcellular volumes. I. Stochastic and diffusion effects on individual pathways. Biophys. J. 87:733–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takahashi, K., S. N. V. Arjunan, and M. Tomita. 2005. Space in systems biology of signaling pathways—towards intracellular molecular crowding in silico. FEBS Lett. 579:1783–1788. [DOI] [PubMed] [Google Scholar]
- 6.Lemerle, C., B. D. Ventura, and L. Serrano. 2005. Space as the final frontier in stochastic simulations of biological systems. FEBS Lett. 579:1789–1794. [DOI] [PubMed] [Google Scholar]
- 7.Nelson, P. 2003. Biological Physics: Energy, Information, Life. W. H. Freeman, New York.
- 8.Rao, C. V., D. M. Wolf, and A. P. Arkin. 2002. Control, exploitation and tolerance of intracellular noise. Nature. 420:231–237. [DOI] [PubMed] [Google Scholar]
- 9.Stiles, J. R., D. V. Helden, T. M. Bartol, E. E. Salpeter, and M. M. Salpeter. 1996. Miniature endplate current rise times less than 100 microseconds from improved dual recordings can be modeled with passive acetylcholine diffusion from a synaptic vesicle. Proc. Natl. Acad. Sci. USA. 93:5747–5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stiles, J. R., and T. M. Bartol. 2001. Monte Carlo methods for simulating realistic synaptic microphysiology using MCell. In Computational Neuroscience: Realistic Modeling for Experimentalists. E. D. Schutter, editor. CRC Press, Boca Raton, FL.
- 11.Coggan, J. S., T. M. Bartol, E. Esquenazi, J. R. Stiles, S. Lamont, M. E. Martone, D. K. Berg, M. H. Ellisman, and T. J. Sejnowski. 2005. Evidence for ectopic neurotransmission at a neuronal synapse. Science. 309:446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koh, X., B. Srinivasan, H. S. Ching, and A. Levchenko. 2006. A 3D Monte Carlo analysis of the role of dyadic space geometry in spark generation. Biophys. J. 90:1999–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stern, M. D., L. S. Song, H. Cheng, J. S. Sham, H. T. Yang, K. R. Boheler, and E. Ríos. 1999. Local control models of cardiac excitation-contraction coupling. a possible role for allosteric interactions between ryanodine receptors. J. Gen. Physiol. 113:469–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greenstein, J. L., and R. L. Winslow. 2002. An integrative model of the cardiac ventricular myocyte incorporating local control of Ca2+ release. Biophys. J. 83:2918–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinch, R., J. L. Greenstein, A. J. Tanskanen, L. Xu, and R. L. Winslow. 2004. A simplified local control model of calcium-induced calcium release in cardiac ventricular myocytes. Biophys. J. 87:3723–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Means, S., A. J. Smith, J. Shepherd, J. Shadid, J. Fowler, R. J. H. Wojcikiewicz, T. Mazel, G. D. Smith, and B. S. Wilson. 2006. Reaction diffusion modeling of calcium dynamics with realistic ER geometry. Biophys. J. 91:537–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fabiato, A. 1983. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 245:C1–C14. [DOI] [PubMed] [Google Scholar]
- 18.Stern, M. D. 1992. Theory of excitation-contraction coupling in cardiac muscle. Biophys. J. 63:497–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fawcett, D. W., and N. S. McNutt. 1969. The ultrastructure of the cat myocardium. I. Ventricular papillary muscle. J. Cell Biol. 42:1–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Franzini-Armstrong, C., F. Protasi, and V. Ramesh. 1999. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys. J. 77:1528–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soeller, C., and M. B. Cannell. 1997. Numerical simulation of local calcium movements during L-type calcium channel gating in the cardiac diad. Biophys. J. 73:97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balke, C. W., and W. G. Wier. 1991. Ryanodine does not affect calcium current in guinea pig ventricular myocytes in which Ca2+ is buffered. Circ. Res. 68:897–902. [DOI] [PubMed] [Google Scholar]
- 23.Beuckelmann, D. J., and W. G. Wier. 1988. Mechanism of release of calcium from sarcoplasmic reticulum of guinea-pig cardiac cells. J. Physiol. 405:233–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stern, M. D., and H. Cheng. 2004. Putting out the fire: what terminates calcium-induced calcium release in cardiac muscle? Cell Calcium. 35:591–601. [DOI] [PubMed] [Google Scholar]
- 25.Lukyanenko, V., T. F. Wiesner, and S. Gyorke. 1998. Termination of Ca2+ release during Ca2+ sparks in rat ventricular myocytes. J. Physiol. 507:667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sham, J. S., L. S. Song, Y. Chen, L. H. Deng, M. D. Stern, E. G. Lakatta, and H. Cheng. 1998. Termination of Ca2+ release by a local inactivation of ryanodine receptors in cardiac myocytes. Proc. Natl. Acad. Sci. USA. 95:15096–15101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bers, D. M., and T. Guo. 2005. Calcium signaling in cardiac ventricular myocytes. Ann. N. Y. Acad. Sci. 1047:86–98. [DOI] [PubMed] [Google Scholar]
- 28.Langer, G. A., and A. Peskoff. 1996. Calcium concentration and movement in the diadic cleft space of the cardiac ventricular cell. Biophys. J. 70:1169–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lines, G. T., J. B. Sande, W. E. Louch, H. K. Mørk, P. Grøttum, and O. M. Sejersted. 2006. Contribution of the Na+/Ca2+ exchanger to rapid Ca2+ release in cardiomyocytes. Biophys. J. 91:779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peskoff, A., J. A. Post, and G. A. Langer. 1992. Sarcolemmal calcium binding sites in heart: II. Mathematical model for diffusion of calcium released from the sarcoplasmic reticulum into the diadic region. J. Membr. Biol. 129:59–69. [DOI] [PubMed] [Google Scholar]
- 31.Hinch, R., J. L. Greenstein, and R. L. Winslow. 2005. Multi-scale models of local control of calcium induced calcium release. Prog. Biophys. Mol. Biol. 90:136–150. [DOI] [PubMed] [Google Scholar]
- 32.Tanskanen, A. J., J. L. Greenstein, A. Chen, S. X. Sun, and R. L. Winslow. 2007. Protein geometry and placement in the cardiac dyad influence macroscopic properties of calcium-induced calcium release. Biophys. J. 92:3379–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crank, J. 1979. Mathematics of Diffusion, 2nd Ed. Oxford University Press, Oxford, UK.
- 34.Smith, G. D., L. Dai, R. M. Miura, and A. Sherman. 2001. Asymptotic analysis of buffered calcium diffusion near a point source. SIAM J. Appl. Math. 61:1816–1838. [Google Scholar]
- 35.Langtangen, H. P. 2003. Computational Partial Differential Equations—Numerical Methods and Diffpack Programming, 2nd Ed. Springer-Verlag, Berlin, Germany.
- 36.Saftenku, E., A. J. Williams, and R. Sitsapesan. 2001. Markovian models of low and high activity levels of cardiac ryanodine receptors. Biophys. J. 80:2727–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zahradník, I., S. Györke, and A. Zahradníková. 2005. Calcium activation of ryanodine receptor channels—reconciling RyR gating models with tetrameric channel structure. J. Gen. Physiol. 126:515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor, H. E., and S. Karlin. 1998. An Introduction to Stochastic Modeling, 3rd Ed. Academic Press, New York.
- 39.Frey, E., and K. Kroy. 2005. Brownian motion: a paradigm of soft matter and biological physics. Annalen der Physik. 14:20–50. [Google Scholar]
- 40.Miller, I., and M. Miller. 1999. John E. Freund's Mathematical Statistics, 6th Ed. Prentice Hall, Englewood Cliffs, NJ.
- 41.Redner, S. 2001. A Guide to First-Passage Processes. Cambridge University Press, Cambridge, UK.
- 42.Reference deleted in proof.
- 43.Wang, S. Q., L. S. Song, E. G. Lakatta, and H. Cheng. 2001. Ca2+ signaling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature. 410:592–596. [DOI] [PubMed] [Google Scholar]
- 44.Smith, G. D., J. E. Keizer, M. D. Stern, W. J. Lederer, and H. Cheng. 1998. A simple numerical model of calcium spark formation and detection in cardiac myocytes. Biophys. J. 75:15–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson, J. D., C. Snyder, M. Walsh, and M. Flynn. 1996. Effects of myosin light chain kinase and peptides on Ca2+ exchange with the N- and C-terminal Ca2+ binding sites of calmodulin. J. Biol. Chem. 271:761–767. [DOI] [PubMed] [Google Scholar]
- 46.Lehmann, E., and J. P. Romano. 2005. Testing Statistical Hypotheses, 3rd Ed. Springer-Verlag, New York.
- 47.Brown, E. N., R. Barbieri, V. Ventura, R. E. Kass, and L. M. Frank. 2002. The time-rescaling theorem and its application to neural spike train data analysis. Neural Comput. 14:325–346. [DOI] [PubMed] [Google Scholar]
- 48.Dayan, P., and L. F. Abbott. 2001. Theoretical Neuroscience: Computational and Mathematical Modeling of Neural Systems. The MIT Press, Cambridge, MA.
- 49.Karr, A. F. 1991. Point Processes and Their Statistical Inference Probability, Pure and Applied. Marcel Dekker, New York.
- 50.Gabdoulline, R. R., and R. C. Wade. 2002. Biomolecular diffusional association. Curr. Opin. Struct. Biol. 12:204–213. [DOI] [PubMed] [Google Scholar]
- 51.Minton, A. P. 2001. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J. Biol. Chem. 276:10577–10580. [DOI] [PubMed] [Google Scholar]
- 52.Nicholson, C., K. C. Chen, S. Hrabetová, and L. Tao. 2000. Diffusion of molecules in brain extracellular space: theory and experiment. Prog. Brain Res. 125:129–154. [DOI] [PubMed] [Google Scholar]
- 53.McLaughlin, S. G., G. Szabo, and G. Eisenman. 1971. Divalent ions and the surface potential of charged phospholipid membranes. J. Gen. Physiol. 58:667–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langner, M., D. Cafiso, S. Marcelja, and S. McLaughlin. 1990. Electrostatics of phosphoinositide bilayer membranes. theoretical and experimental results. Biophys. J. 57:335–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wagenknecht, T., and M. Radermacher. 1997. Ryanodine receptors: structure and macromolecular interactions. Curr. Opin. Struct. Biol. 7:258–265. [DOI] [PubMed] [Google Scholar]
- 56.Ellis, R. J. 2001. Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 26:597–604. [DOI] [PubMed] [Google Scholar]
- 57.Schnell, S., and T. E. Turner. 2004. Reaction kinetics in intracellular environments with macromolecular crowding: simulations and rate laws. Prog. Biophys. Mol. Biol. 85:235–260. [DOI] [PubMed] [Google Scholar]
- 58.Lederer, W. J., E. Niggli, and R. W. Hadley. 1990. Sodium-calcium exchange in excitable cells: fuzzy space. Science. 248:283. [DOI] [PubMed] [Google Scholar]
- 59.Wriggers, W., E. Mehler, F. Pitici, H. Weinstein, and K. Schulten. 1998. Structure and dynamics of calmodulin in solution. Biophys. J. 74:1622–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]