

Abstract
Picrotoxin (PTX) is a noncompetitive antagonist of many ligand-gated ion channels, with a site of action believed to be within the ion-conducting pore. In the A-type gamma-aminobutyric acid receptor, a threonine residue in the second transmembrane domain is of particular importance for the binding of, and ultimate inhibition by, PTX. To better understand the relationship between this residue and the PTX molecule, we mutated this threonine residue to serine, valine, and tyrosine to change the structural and biochemical characteristics at this location. The known subunit stoichiometry of the A-type gamma-aminobutyric acid receptor allowed us to create receptors with anywhere from zero to five mutations. With an increasing number of mutated subunits, each amino acid substitution revealed a unique pattern of changes in PTX sensitivity, ultimately encompassing sensitivity shifts over several orders of magnitude. The electrophysiological data on PTX-mediated block, and supporting modeling and docking studies, provide evidence that an interaction between the PTX molecule and three adjacent uncharged polar amino acids at this position of the pore are crucial for PTX-mediated inhibition.
INTRODUCTION
The A-type gamma-aminobutyric acid receptor (GABAAR) belongs to the family of ligand-gated ion channels (LGIC), which is also comprised of excitatory nicotinic acetylcholine receptors (nAChR) and 5-hydroxytryptamine receptors, as well as the inhibitory glycine and glutamate receptors. A hallmark of this LGIC family is a pentameric arrangement of subunits, with each subunit comprised of a large extracellular N terminus, four transmembrane domains, a large intracellular loop, and a small extracellular C terminus (1–7). There is general agreement that the second transmembrane domain (M2) from each of the five subunits lines the pore (8), although other intracellularly located regions have been implicated in ion conduction (9,10). The native GABAAR is composed of several different subunits (α, β, γ, δ …) with the predominant receptor being α1β2γ2 at a subunit stoichiometry of 2:2:1 (11,12).
The GABAAR is a target for many classes of pharmacological compounds including benzodiazepines, barbiturates, steroids, and noncompetitive antagonists (13). These noncompetitive antagonists include β-lactam antibiotics, t-buylbicyclophosphorothionate, insecticides, and picrotoxin (PTX). PTX, which is a botanical convulsant, is an alkaloid compound capable of blocking members of the LGIC family including GABA, glycine, glutamate, 5-hydroxytryptamine, and nACh receptors (14–18).
Numerous lines of evidence have led us to believe that PTX binds in the pore of the GABAAR (17,19–21). Given that there is no defined structure for the pore-lining M2 region of the GABAAR, much of what we know about M2 structure comes from other LGIC family members. Investigations into the M2 of LGIC have suggested an α-helical structure with a slight kink near the middle (22–25). Residues that are important for the GABAAR chloride selectivity, as well as the other amino acids of M2 that face the pore, have been identified (9,25).
Using the conventional numbering system, with the cytoplasmic end of M2 being 1′ and the extracellular side being 20′ (26), residues that have been most often implicated in PTX inhibition of the GABAAR are at the 2′ and 6′ positions. Mutating these residues, particularly 6′, can dramatically decrease the sensitivity of the GABAAR for PTX (19,27–30). In addition, modeling studies that have docked PTX in the pore support a role for the 6′ ring in PTX inhibition (31,32). In this study, we investigated the nature of the interaction between PTX and the GABAAR by coexpression of wild-type and 6′ mutant subunits. Due to the known stoichiometry and arrangement of GABAAR subunits, each mutant receptor had a unique assembly of zero to five mutant subunits. As the number of mutated subunits was increased, there was a concomitant decrease in the sensitivity of the receptor for PTX. Correlation of the electrophysiological data with homology modeling and docking simulations suggests that a ring of uncharged polar amino acids at the 6′ level of the M2 transmembrane domain is a crucial component of PTX-mediated inhibition. Furthermore, we favor a model in which PTX interacts (via hydrogen bonds) with three adjacent residues, resulting in a noncompetitive inhibition of the GABAAR.
METHODS
Mutagenesis, cloning, and in vitro transcription
Rat α1, β2, and γ2 cDNA were subcloned into the pGEMHE vector (33). Mutations α1T260S, α1T260V, and α1T260Y and γ2T271S, γ2T271V, and γ2T271Y were generated using the overlap extension method (34). The β2T256S, β2T256V, and β2T256Y mutations were introduced using the QuickChange protocol (Stratagene, La Jolla, CA). All constructs were fully verified by cDNA sequencing. cDNA was linearized with Nhe I (New England BioLabs, Ipswitch, MA), and capped cRNA were transcribed from linearized cDNA using the T7 mMessage mMachine Kit (Ambion, Austin, TX). Integrity and yield of the cRNA were verified on a 1.0% agarose gel.
Xenopus oocyte expression
Female Xenopus laevis (Xenopus I, Ann Arbor, MI) were anesthetized with 0.2% 3-aminobenzoic acid ethyl ester, methanesulfonate salt. The ovarian lobes were surgically removed from the frog and placed in a calcium-free, oocyte Ringer's 2 incubation solution. The oocyte Ringer's 2 solution consisted of 92.5 mM NaCl, 2.5 mM KCl, 1 mM MgCl2, 1 mM Na2HPO4, 5 mM HEPES, 50 U/ml penicillin, and 50 μg/ml streptomycin at pH 7.5. The lobes were then cut into smaller pieces and digested with 0.3% collagenase A (Roche, Indianapolis, IN) while shaking for 1.5–2 h at room temperature. The stage VI oocytes were selected and incubated at 18°C before cRNA injection.
Micropipettes for cRNA injection were pulled from borosilicate glass (Drummond Scientific, Broomall, PA) on a Sutter P87 horizontal puller and their tips were cut with microscissors. The cRNA was injected into oocytes with a Nanoject microinjection system (Drummond Scientific) at a total injection volume of 40–70 nl (1 μg/Λ). Injection ratios were 1:1:8 for α1:β2:γ2, respectively. This was done to ensure that all of the expressed receptors contained the γ2 subunit. Previous studies have shown that injecting a surplus of the γ2 cRNA does not alter the functional stoichiometry (11). For those experiments involving the mixing of wild-type and mutant subunits (β and βT6′V or α and αT6′V) we determined the “functionally equivalent” amount of cRNA to inject based on the maximum amplitude of the currents after the receptors (i.e., αβγ versus αβT6′Vγ) were expressed in oocytes. If the expression levels were different, the cRNA was diluted and reinjected, and the functional expression was again compared in oocytes. This was repeated until the amount of expression was equal. Oocytes were injected with a 1:1:1:8 ratio of α:αT6′V:β:γ or α:β:βT6′V:γ to create a population where 25% of the receptors would have two wild-type subunits, 25% would have two mutant subunits, and 50% would have both one wild-type and one mutant subunit. For each mixture experiment, we also injected the same amount of wild-type and mutant cRNA into oocytes to be used as a final correction factor for any differences in the relative functional expression. Knowing the fraction of current blocked by PTX for wild-type and double mutant receptors, we were then able to subtract their dose-inhibition curves from the total inhibition curve, thereby isolating the receptors with only one mutant subunit. This analysis assumed that, in terms of PTX block, the position of the mutant subunit within the pentameric receptor was inconsequential.
Voltage clamp of oocytes
We used two-electrode voltage-clamp procedures to record current two or three days after cRNA injection. The experiments were performed using an OpusXpress 6000A (Molecular Devices, Sunnyvale, CA). This system allows for automated and simultaneous impalement and voltage clamp of an octet of oocytes. The oocytes were perfused continuously with Ringer's solution containing 92.5 mM NaCl, 2.5 mM KCl, 5 mM HEPES, 1 mM CaCl2, and 1 mM MgCl2 at pH 7.5. All experiments were performed at room temperature. Recording microelectrodes were filled with 3M KCl, and their resistances ranged from 0.5 to 1.5 Mω. Oocytes were clamped at −70 mV, and agonist/antagonist–containing solutions were delivered from a 96-well plate by disposable pipette tips, with a solution exchange time 0.5–1.0 s.
Data analysis
Agonist concentration-response relationships were fit (least-squares) with a sigmoidal equation as follows:
 |
(1) |
in which I and Imax represent current at a given agonist concentration (A) and maximal agonist-induced current, respectively. EC50 is the half-maximal effective agonist concentration, and nH is the Hill coefficient. For determination of the EC50, currents were measured at their peak amplitudes.
The IC50 (concentration of PTX, which decreased the agonist-mediated current by 50%) for each receptor was determined by a least-squares fit of the relationship to the data as follows:
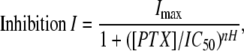 |
(2) |
where current I is a function of the inhibitor concentration, PTX. The IC50 for PTX was obtained using the experimentally determined EC50 concentration of GABA, and currents were sampled at the end of drug application. In a few cases, the currents had not quite reached steady state. At our time resolution, we were unable to distinguish if this was an incomplete block or incomplete desensitization. In select data sets, we reanalyzed data and extrapolated (via exponential fits) to a plateau value for complete block. In no case did the IC50 values differ by more than 10% from the approach of simply measuring the current at the end of the PTX application. Because we could not define the relative contribution of desensitization to the current decays, it is not clear which approach is more accurate. Regardless, this minor difference in the sensitivity of PTX block (IC50) would not alter the general conclusions of this study. All data are presented as mean ± SE with nH indicating the Hill coefficient. Standard errors reported were derived from the independent fits of individual cells as opposed to the fit for the averaged data. For some of the T6′Y mutant receptors, we were unable to inhibit 50% of the current; for this reason, the recorded IC50 was extrapolated from the available data.
Structural modeling
Internal coordinates mechanism (ICM) software (Molsoft, La Jolla, CA) was used to build a representation of the M2 region of the GABAA receptor. The homology model was built by working from the 4Å resolution structure of the nAChR transmembrane domain (35). Alignments were constructed by pairing GABA and nAChR subunits on the putative basis of their role in ligand binding. In this model, the two GABAA α1 subunits correspond to nAChR γ and δ, the two GABAA β2 subunits correspond to the two nAChR α-subunits, and the GABAA γ2 subunit corresponds to the nAChR β-subunit (45). The overall model was then energy-minimized to eliminate intrasubunit/intersubunit clashes. After introducing mutations, further energy minimizations confirmed that these perturbations did not impart any instability to the M2 region. For the mutagenesis studies, models of each individual mutant were made as described above and then combined with other wild-type– or mutant–modeled subunits. The energy would again be globally minimized and the 6′ side chain positions were locally minimized to their environment.
Docking simulations
For docking simulations, we used the homology models created in the manner described above. The resulting models were then run through the ICM protocol to find pockets that are probable drug binding sites. Of these sites, only one site was located within the pore region. Residues around this binding pocket were then analyzed for charge and hydrophobicity. Because PTX is a mixture of picrotoxin and picrotin, and picrotoxinin is the more potent compound at the GABAAR, we used a three-dimensional representation of a picrotoxinin molecule for all docking simulations. In the docking simulations for mutant receptors, the PTX molecule originated in the orientation found for the wild-type GABAAR but was not constrained during subsequent energy minimizations. ICM Monte Carlo minimization protocols were used, which ultimately provided information concerning PTX orientation, binding energy (ΔG), and probable hydrogen-bond partners. Binding energies were derived from electrophysiologically determined IC50 values by the relationship where R is the universal gas constant (8.31 J·K-1·mol−1) and T is the temperature in Kelvin as follows:
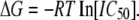 |
(3) |
RESULTS
Pore-lining amino acids and PTX block
Fig. 1 shows an alignment of the M2 region of several GABA and glycine subunits. Of particular interest is the residue at the 6 position. In PTX-sensitive subunits, there is always an uncharged polar threonine residue. Conversely, in subunits that impart PTX resistance (Fig. 1, asterisk), there is a nonpolar methionine or phenylalanine residue. This suggests that the hydrogen-bonding ability of the 6′ residue may be a feature of PTX-mediated inhibition.
FIGURE 1.

Sequence alignments of several subunits. Alignment of the M2 domains of several ligand-gated ion channels including the rat GABA α1, β2, γ2, ρ1, ρ2 and glycine β1 subunits are shown. Subunits that impart PTX resistance are indicated by asterisks. The 6′ residue investigated in this study is highlighted by the shaded box.
To more directly address the issue of PTX-mediated inhibition, we mutated this 6′ residue in each of the GABA α1, β2, and γ2 subunits to a serine, valine, or tyrosine residue. Although serine and tyrosine residues contain a hydroxyl group, this hydroxyl is in a different orientation than in the naturally occurring threonine residue. Alternatively, the valine mutation has a very similar size to threonine but lacks the hydroxyl group. These particular mutations were used to investigate how the presence and position of a hydroxyl group at the 6′ level influences inhibition by PTX. In addition, due to the known stoichiometry and subunit arrangement of GABAAR subunits, we could examine receptors with zero to five mutations at this particular position.
Mutant agonist sensitivity
An EC50 was determined for all mutant receptors to ensure that the 6′ mutations did not have a deleterious effect on the activation of the receptor (Fig. 2). Fig. 2 A shows GABA-activated currents from oocytes expressing wild-type receptors as well as receptors where all five 6′ residues had been mutated to serine, valine, or tyrosine. In Fig. 2 B, the continuous lines are fits of the Hill equation (Eq. 1) to the dose-response relationships, and yield EC50 values of 64.6 ± 2.0 (n = 11), 81.3 ± 7.4 (n = 12), 78.3 ± 6.8 (n = 11), and 88.6 ± 4.6 (n = 11) for the wild-type, serine, valine, and tyrosine mutants, respectively. Whereas a majority of the receptors experienced no significant difference in their EC50 for GABA, only the αYβYγY receptor displayed a significant shift in GABA sensitivity (p > 0.05). EC50 values for all combinations of mutations used in this study are provided in the Table 1. In addition to ensuring normal receptor function, determination of the EC50 for each combination was necessary, as this concentration of GABA was used in the determination of the IC50 for PTX.
FIGURE 2.

GABA sensitivities of mutant receptors. (A) Representative traces of GABA-induced currents for the wild-type GABA receptor and the three different mutations examined in this study. In this case, all subunits carry the indicated mutation; serine (S), valine (V), or tyrosine (Y). (B) The GABA dose-response relationship for the wild-type receptor (open circles), as well as the receptors where all five subunits were mutated to serine (solid circles), valine (squares), or tyrosine (diamonds). The continuous lines are fits of the Hill equation (Eq. 1) to the dose-response relationships of the peak current and yielded EC50 values of 64.6 ± 2.2, 81.3 ± 7.5, 81.3 ± 6.9, and 88.7 ± 4.7 μM for wild-type, T6′S, T6′V, and T6′Y, respectively. This pore mutation did not significantly alter the sensitivity of the receptor to GABA.
TABLE 1.
Characterization of mutant GABAARs
| Receptor
|
EC50 (GABA)
|
IC50 (PTX)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| α | β | γ | n | EC50 | nH | n | IC50 | nH | n |
| Wt | Wt | Wt | 0 | 64.6 ± 2.2 | 1.8 ± 0.2 | 11 | 0.8 ± 0.1 | 1.1 ± 0.1 | 11 |
| Wt | Wt | S | 1 | 57.5 ± 9.3 | 1.7 ± 0.1 | 10 | 3.2† ± 1.9 | 0.9 ± 0.2 | 11 |
| S | Wt | Wt | 2 | 78.8 ± 8.6 | 1.5 ± 0.3 | 8 | 7.3 ± 1.7 | 0.8 ± 0.1 | 12 |
| Wt | S | Wt | 2 | 58.1 ± 3.5 | 1.6 ± 0.3 | 10 | 5.0 ± 1.4 | 1.3 ± 0.1 | 13 |
| S | Wt | S | 3 | 51.2 ± 6.6 | 1.8 ± 0.1 | 9 | 11.1 ± 2.1 | 0.7 ± 0.3 | 14 |
| Wt | S | S | 3 | 51.1 ± 5.1 | 2.1 ± 0.2 | 11 | 8.01 ± 2.1 | 0.9 ± 0.1 | 12 |
| S | S | Wt | 4 | 61.0 ± 6.2 | 1.8 ± 0.1 | 8 | 21.3 ± 1.2 | 1.0 ± 0.2 | 16 |
| S | S | S | 5 | 81.3 ± 7.4 | 1.7 ± 0.1 | 12 | 38.2 ± 2.4 | 1.1 ± 0.4 | 14 |
| Wt | Wt | V | 1 | 65.5 ± 5.4 | 2.2 ± 0.2 | 11 | 4.2† ± 2.1 | 1.2 ± 0.4 | 17 |
| V | Wt | Wt | 2 | 74.3 ± 9.6 | 1.9 ± 0.1 | 12 | 126 ± 7.5 | 1.3 ± 0.3 | 13 |
| Wt | V | Wt | 2 | 63.3 ± 8.0 | 1.7 ± 0.2 | 13 | 148 ± 12 | 1.0 ± 0.1 | 11 |
| V | Wt | V | 3 | 77.3 ± 7.1 | 2.1 ± 0.1 | 11 | 184 ± 18 | 1.3 ± 0.3 | 14 |
| Wt | V | V | 3 | 81.3 ± 4.4 | 1.6 ± 0.3 | 9 | 191 ± 11 | 1.4 ± 0.2 | 11 |
| V | V | Wt | 4 | 78.2 ± 5.6 | 1.9 ± 0.1 | 12 | 242 ± 16 | 0.8 ± 0.1 | 12 |
| V | V | V | 5 | 78.3 ± 6.8 | 1.6 ± 0.3 | 11 | 361 ± 26 | 1.2 ± 0.3 | 14 |
| Wt | Wt | Y | 1 | 52.2 ± 5.5 | 2.3 ± 0.2 | 10 | 1339 ± 39 | 1.6 ± 0.2 | 10 |
| Y | Wt | Wt | 2 | 54.2 ± 5.1 | 2.1 ± 0.4 | 9 | 1605 ± 31 | 1.3 ± 0.4 | 9 |
| Wt | Y | Wt | 2 | 51.5 ± 4.7 | 1.8 ± 0.1 | 8 | 1544 ± 46 | 1.4 ± 0.3 | 11 |
| Y | Wt | Y | 3 | 61.8 ± 9.3 | 1.9 ± 0.2 | 11 | 1594 ± 55 | 1.6 ± 0.3 | 12 |
| Wt | Y | Y | 3 | 77.6 ± 6.0 | 1.7 ± 0.2 | 11 | 1533 ± 64 | 1.7 ± 0.2 | 14 |
| Y | Y | Wt | 4 | 81.2 ± 6.9 | 1.6 ± 0.1 | 10 | 1636 ± 41 | 1.4 ± 0.3 | 14 |
| Y | Y | Y | 5 | 88.6* ± 4.6 | 1.9 ± 0.2 | 11 | 1614 ± 48 | 1.8 ± 0.2 | 11 |
The left column indicates the subunit composition used as well as the total number of mutant subunits in that receptor.
Denotes a significant difference in EC50 as tested by the Student's t-test (p < 0.05).
Indicates a lack of significant difference in the IC50 from the wild-type (p > 0.05).
6′ mutations dramatically influence PTX sensitivity
To determine the mutated receptors' sensitivities to PTX, increasing concentrations of PTX were applied in the presence of the EC50 concentration of GABA. As shown in Fig. 3 A, GABA-mediated current was reduced by PTX in a dose-dependent manner. Fig. 3 B shows a fit of the data to Eq. 2 for the wild-type receptor, as well as the case where all five of the subunits carried a serine, valine, or tyrosine mutation at the 6′ position. IC50 values for the configurations where all five subunits have been mutated were 38.2 ± 2.4 (n = 14), 361 ± 26 (n = 14), and 1614 ± 48 (n = 11) μM for the serine, valine, and tyrosine mutants, respectively (Fig. 3 B and Table 1).
FIGURE 3.

Determination of the IC50 for picrotoxin. (A) GABA-activated currents (using an EC50 concentration of GABA) in the presence of increasing concentrations of PTX are shown. Note that the mutations imparted varying amounts of picrotoxin resistance. (B) The PTX inhibition relationship for wild-type receptors (open circles), as well as the receptors where all five subunits had been mutated to serine (solid circles), valine (squares), or tyrosine (diamonds) are shown. The continuous lines are fits of Eq. 2 to the dose-response relationships. This yielded IC50 values of 0.8 ± 0.1, 38.3 ± 2.5, 361 ± 26, and 1614 ± 48 for wild-type, T6′S, T6′V, and T6′Y, respectively. For these experiments, current measurements were made at the end of agonist-antagonist application.
In the experiment discussed above, all five of the GABAAR subunits were mutated. However, the α, β, and γ-subunits, which comprised the functional GABAAR, are in a 2:2:1 ratio (11). If we coexpress mutants with wild-type subunits in various combinations, we can examine how partial, rather than comprehensive, adjustments of the biochemical environment at the 6′ level modulate inhibition by PTX.
PTX sensitivity is determined by the number of mutated subunits
In the first case, the 6′ threonine was mutated to a serine residue, maintaining the polar character of the side chain, but likely altering the orientation of the hydroxyl group. The Table 1 and Fig. 4 A, show that, as the number of T6′S mutations increases, there is a concomitant increase in the IC50 for PTX. As the number of serines increases, there is an apparent geometric increase in the IC50 ( Fig. 4 A, inset). In addition, there are two possible combinations that produce receptors with two mutant subunits (α- or β-subunits carrying the mutation) and two possible combinations that produce receptors with three mutant subunits (α- and γ- or β- and γ -subunits carrying the mutation). The fact that there is no significant difference between the IC50 values for the two or three mutant combinations indicates that PTX sensitivity is determined by the number of mutated residues rather than the identity (or position) of the subunit being mutated.
FIGURE 4.

Relationship between the IC50 for picrotoxin and the number of 6′ mutations. Graphs are shown for the (A) serine mutation, (B) valine mutation, and (C) tyrosine mutation. The solid lines in all three graphs simply connect the symbols, except for the inset in the serine graph where a linear regression was performed on a semilogarithmic plot of the data. In the case of serine, each additional mutation produced an approximate twofold increase in the IC50. Note how, in the case of valine, one mutation produced very little change in PTX sensitivity. (B) The open circles denote receptors with only one mutant α- or β-subunit.
The T6′V mutation (Fig. 4 B) removes a hydroxyl group and results in a GABAAR that is much more resistant to inhibition by PTX compared to wild-type or serine mutants. Mutating only one subunit (γ) in the pentamer to valine has little effect on sensitivity (p < 0.05), with an IC50 of only 4.2 ± 2.1 μM (n = 17). This suggests that PTX does not require all five 6′ residues for inhibition. However, if two or more residues are mutated to valine, we observe a dramatic increase in the IC50 for PTX. As with the serine mutations, PTX sensitivity was determined by the number of subunits mutated. For example, the α mutation on its own exhibited an IC50 of 126 ± 7.5 μM (n = 13), and the β mutation on its own exhibited an IC50 of 148 ± 12 μM (n = 11).
Mutation of the 6′ residue to a large aromatic residue that still has a hydroxyl group, was accomplished with a tyrosine substitution. In a previous study, we established that the insertion of a large nonpolar 6′ residue (phenylalanine) completely abolished PTX sensitivity (19), and that the insertion of only one of these residues was sufficient to make the GABAAR resistant to PTX (28). The Table 1 and Fig. 4 C shows that the tyrosine mutation has the same dominant negative effect, despite the fact that it is a potential hydrogen-bonding partner. That is, even a single substitution at the 6′ position imparts near PTX resistance.
Mutation of only one subunit (γ) to valine resulted in a minor increase in IC50, whereas mutation of two subunits (α or β) increased the IC50 by nearly two orders of magnitude. Our working hypothesis, which will be considered in the Discussion, predicts that any single subunit carrying the valine mutation will demonstrate a modest decrease in PTX sensitivity comparable to that observed for the γ-subunit. Owing to the 2:2:1 stoichiometry of the α1β2γ2 GABA receptor, the only straightforward method for expressing a GABA receptor with a single mutation is to mutate the γ-subunit. To circumvent this limitation, we devised the following strategy for determining PTX IC50 values for GABA receptors containing a single mutant α-subunit or a single mutant β-subunit. Briefly, this was achieved by mixing equivalent amounts of wild-type and mutant cRNA (α and αT6′V, for example) with wild-type β and γ. Ignoring subunit order, this results in a heterogeneous population of receptors of which 25% are all wild-type, 25% are all mutant, and the remaining 50% contain a single wild-type and a single mutant α-subunit. Since the PTX sensitivity of the all wild-type and all mutant receptors is known, we can subtract their scaled (see Methods) dose-inhibition relationships from that of the total curve. This leaves the dose-inhibition relationship of the remaining sub-population, that containing a single wild-type and single mutant α-subunit. The same can be accomplished for the β-subunit.
Fig. 5 shows the dose-inhibition relationship for the wild-type (open circles), the extracted dose-inhibition relationships determined for the single α and the single β-mutant subunits (open and solid squares) along with that of the γ-mutant (gray squares). As was true for the γ-mutant, the PTX sensitivity of the singly mutant α- and β-subunit was only modestly increased with IC50 values of 9.2 ± 2.8 μM (n = 7) and 12.2 ± 3.4 μM (n = 5), respectively. These IC50 values are plotted as open circles in Fig. 4 B. These data support the observation derived from the γ-mutant studies—only a modest shift in IC50 is observed when a single subunit carried the valine mutation.
FIGURE 5.

Dose-inhibition relationship for single subunit mutations for PTX. The open circles plot the picrotoxin-mediated antagonism of the wild-type receptor. Due to the 2:2:1 stoichiometry of the α1β2γ2 GABA receptor, mutation of γ and coexpression with its wild-type counterparts produce a receptor with a single 6′ mutation. To examine the sensitivity of PTX block of receptors with a single mutant α or single mutant β-subunit, mixing studies were performed in which the wild-type and mutant α-subunit (for example) was mixed in equal ratios and expressed with wild-type β and γ. Because the sensitivity of block for the wild-type and double mutant combinations is known, their inhibition curves can be subtracted from the total heterogenous population of receptors (see Methods) leaving the dose-inhibition curve for the singly mutant combination. Note the similarity of the PTX sensitivity regardless of the particular subunit carrying the mutation. The IC50 values for PTX antagonism were 9.2 ± 2.8 μM (n = 7), 12.2 ± 3.4 μM (n = 5), and 4.2 ± 2.1 (n = 17) for α, β, and γ, respectively.
DISCUSSION
Our goal in this study was to examine the PTX-mediated inhibition of the GABAAR and, in particular, how the nature of the amino acid at the 6′ level influences PTX inhibition. Because the GABAAR is a heteromeric pentamer, with a fixed stochiometry, we could examine subtle changes in PTX sensitivity as the number of mutated residues was increased from zero to five. We interpret these changes in sensitivity as being due to the repositioning, or elimination, of hydroxyl groups attached to the 6′ residue in the pore.
Docking of PTX within the GABAA receptor pore
In an attempt to rationalize the shifts in IC50 with the underlying structural changes, we constructed a homology model of the wild-type GABAAR into which a PTX molecule could be docked. This docking simulation was restricted to the cytoplasmic region of the GABAAR pore (residues 2′–9′). As shown in Fig. 6, the PTX molecule is preferentially docked in the vicinity of the 6′ residue. In its ideal docked location, the PTX molecule's oxygen-containing groups form at least three hydrogen bonds with three adjacent 6′ threonines. The isopropyl group is situated toward the cytoplasmic side of the pore, where it may interact with amino acids at the 2′ level (31,32).
FIGURE 6.

PTX molecule docked in the GABAAR pore. (A) Extracellular view of docking simulation carried out on a homology model of the GABAAR M2 domain composed of α1 (red), β2 (blue), and γ2 (green) subunits. Predicted hydrogen bonds are displayed as green dashes. (B) Side view of a PTX molecule docked at the 6′ level.
Correlation of electrophysiological and modeling data
The mutations changed the amino acid side chain at the 6′ residue, which the homology model and docking simulations suggest to be a key structural component for PTX inhibition. To further investigate how these mutations affected the interaction of PTX with pore-lining residues, homology models were constructed for each mutant receptor, and docking simulations were carried out in each case.
To correlate our experimental and modeling data, the IC50 values were converted into free energy of binding (ΔG) by Eq. 3 (Table 1). Fig. 7 displays the lowest energy of binding for different numbers of serine and valine mutations, as determined both experimentally (symbols) and through docking simulations (lines). Fig. 7 shows that mutating the 6′ ring from threonine to serine produced relatively modest effects, causing only a small reduction in the free energy of binding (Fig. 7, circles). For this T6′S mutation, the docking simulations predicted a similar modest progression in ΔG (Fig. 7, solid line). In the case of the T6′V mutations, we observed a steeper decrease in binding energy (Fig. 7, squares) as each threonine was replaced in the 6′ ring. This decrease in ΔG corresponds well with the energies determined by the docking simulations (Fig. 7, dashed line). Lastly, it should be noted that, in both the experimental data and predictions from the docking, the valine mutation in only one subunit has little effect on inhibition by PTX. Due to the presumed steric hindrance, energies of binding were not calculated for the T6′Y mutants.
FIGURE 7.

PTX binding energy at the 6′ level. The symbols are the experimentally determined free energy of binding of PTX versus the number of 6′ residues that carry the particular mutation. These free energies were calculated from the IC50 values using Eq. 3. The T6′S mutation (circles) had a less disruptive effect on PTX binding than that of the T6′V mutation (squares). The predicted value of the binding energy from the docking studies is illustrated by the solid (serine) and dashed (valine) lines. Although the experimental and predicted values are shifted somewhat, the pattern of the relationships are similar. Also note that the modeling and electrophysiological studies demonstrate a near lack of an effect of a single valine substitution.
Mechanism of PTX-mediated inhibition
For both the serine and valine mutants, there was only a very modest change in the IC50 for PTX when only one residue was mutated. This implies that the removal of only one 6′ threonine has little to no effect on PTX sensitivity. In addition, docking simulations of PTX into GABAARs with only one mutant subunit (Fig. 7) suggest only a modest reduction in binding energy compared to the wild-type receptor. Due to the subunit arrangement of the GABAAR, the mutation of the α- or β-subunit creates a receptor that no longer has three adjacent wild-type residues. Only the wild-type or γ-mutant receptor has three adjacent wild-type residues. We therefore devised an approach that would allow us to infer the PTX sensitivity of receptors containing only one mutant α-subunit or one mutant β-subunit based on wild-type and mutant coexpression. In these two cases, similar to the γ-subunit mutation, there was only a modest decrease in PTX sensitivity. In the wild-type receptor, there are five possible adjacent triplets that can interact with PTX. With a single mutant subunit, the potential number of adjacent triplets decreases to two. This reduction in the number of potential binding orientations might account for the significant, albeit modest, decrease in PTX sensitivity with a single mutant subunit. Nevertheless, these functional data are consistent with the docking studies that propose the PTX molecule preferentially interacts with three adjacent subunits (Fig. 6).
As the number of hydroxyl groups was reduced, the formation of GABAAR-PTX interactions became less energetically favorable. This is evident when examining both the docking and IC50 data, and suggests an alteration in receptor kinetics. Although there were apparent differences in the decay toward steady state with PTX inhibition, the question of changes in PTX association versus dissociation rates is heavily confounded by the concomitant GABAAR desensitization. An experimental system that allows for rapid solution exchange (e.g., macropatches) would be required for a rigorous kinetic analysis of PTX inhibition.
The T6′V GABAARs maintained some sensitivity to PTX, even when all five of the subunits were mutated. We attribute this to the fact that PTX was still able to reach, and interact, with secondary binding sites, most likely the 2′ residues, where hydrophobic interactions have been shown to play a role in stabilizing the PTX molecule in the pore (20,31,32,36,37). However, for the T6′Y mutations, it seems very unlikely that a molecule the size of PTX would be able to penetrate through the ring of tyrosines to the 2′ level. Therefore, the inhibition seen at higher PTX concentrations might be due a low affinity component of PTX inhibition at some other site, as has been described (38).
Previously, our lab has demonstrated that the presence of a single phenylalanine residue at the 6′ position could eliminate PTX sensitivity (19,29). It was unclear, however, if this resistance to PTX inhibition was due to a steric hindrance or the lack of a hydroxyl group for hydrogen bonding. These results clarify this issue in that, despite having a hydroxyl group, a large aromatic residue (tyrosine) at the 6′ position inhibits the binding of PTX. The tyrosine residues, similar to the phenylalanine, most likely prevent PTX inhibition by forming a physical barrier, not allowing the molecule to penetrate the pore and reach its binding site.
Although residues discussed in this study are found deep within the pore of the GABAAR, our results do not necessarily confirm the idea that PTX is acting as a pore-blocker (16,17). PTX has also been labeled a mixed (competitive and noncompetitive) antagonist (39–42) and an allosteric modulator that acts to stabilize the closed or desensitized state (42–45). Further support of an allosteric mechanism is beyond the scope of this study. However, the fact that the 6′ residue is cytoplasmic to the proposed 9′ gate of the GABA receptor (46) provides an explanation for the observed use-dependence in the PTX block of the GABAAR. In addition, it is difficult to imagine a scenario in which the presence of an ellipsoid PTX molecule with a major diameter of ∼5.4 Å, within a region of the pore that narrows to 7.2 Å, could allow for the permeation of any ion species. It is for this reason that we propose a model in which the PTX molecule is stabilized by three adjacent 6′ residues, inhibiting ion flow through a noncompetitive, pore-blocking mechanism.
Editor: Richard W. Aldrich.
References
- 1.Cully, D. F., D. K. Vassilatis, K. K. Liu, P. S. Paress, L. H. Van der Ploeg, J. M. Schaeffer, and J. P. Arena. 1994. Cloning of an avermectin-sensitive glutamate-gated chloride channel from Caenorhabditis elegans. Nature. 371:707–711. [DOI] [PubMed] [Google Scholar]
- 2.Grenningloh, G., A. Rienitz, B. Schmitt, C. Methfessel, M. Zensen, K. Beyreuther, E. D. Gundelfinger, and H. Betz. 1987. The strychnine-binding subunit of the glycine receptor shows homology with nicotinic acetylcholine receptors. Nature. 328:215–220. [DOI] [PubMed] [Google Scholar]
- 3.Maricq, A. V., A. S. Peterson, A. J. Brake, R. M. Myers, and D. Julius. 1991. Primary structure and functional expression of the 5HT3 receptor, a serotonin gated ion channel. Science. 254:432–436. [DOI] [PubMed] [Google Scholar]
- 4.Noda, M., H. Takahashi, T. Tanabe, M. Toyosato, Y. Furutani, and T. Hirose. 1982. Primary structure of α subunit precursor of Torpedo californica acetylcholine receptor deduced from cDNA sequence. Nature. 299:793–797. [DOI] [PubMed] [Google Scholar]
- 5.Qian, H., and J. Dowling. 1994. Pharmacology of novel GABA receptors found on rod horizontal cells of the white perch retina. J. Neurosci. 14:4299–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schofield, P. R., M. G. Darlison, N. Fujita, D. R. Burt, F. A. Stephenson, H. Rodriguez, L. M. Rhee, J. Ramachandran, V. Reale, T. A. Glencorse, P. H. Seeburg, and E. A. Barnard. 1987. Sequence and functional expression of the GABAA receptor shows a ligand-gated receptor superfamily. Nature. 328:221–227. [DOI] [PubMed] [Google Scholar]
- 7.Davies, P. A., W. Wang, T. G. Hales, and E. W. Kirkness. 2003. A novel class of ligand gated ion channel is activated by Zn2+. J. Biol. Chem. 278:712–717. [DOI] [PubMed] [Google Scholar]
- 8.Leonard, R. J., C. G. Labarca, P. Charnet, N. Davidson, and H. A. Lester. 1988. Evidence that the M2 membrane-spanning region lines the ion channel pore of the nicotinic receptor. Science. 242:1578–1581. [DOI] [PubMed] [Google Scholar]
- 9.Filippova, N., V. E. Wotring, and D. S. Weiss. 2004. Evidence that the TM1–TM2 loop contributes to the ρ1 GABA receptor pore. J. Biol. Chem. 279:20906–20914. [DOI] [PubMed] [Google Scholar]
- 10.Kelley, S. P., J. I. Dunlop, E. F. Kirkness, J. J. Lambert, and J. A. Peters. 2003. A cytoplasmic region determines single-channel conductance in 5-HT3 receptors. Nature. 424:321–324. [DOI] [PubMed] [Google Scholar]
- 11.Chang, Y. C., R. Wang, S. Barot, and D. S. Weiss. 1996. Stoichiometry of a recombinant GABAA receptor. J. Neurosci. 16:5415–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKernan, R. M., and P. J. Whiting. 1996. Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci. 19:139–143. [DOI] [PubMed] [Google Scholar]
- 13.Cascio, M. 2006. Modulating inhibitory ligand-gated ion channels. AAPS J. 8:353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong, C. J., and F. S. Werblin. 1996. Use-dependent and use-independent blocking actions of picrotoxin and zinc at the GABAC receptor in retinal horizontal cells. Vision Res. 36:3997–4005. [DOI] [PubMed] [Google Scholar]
- 15.Das, P., C. L. Bell-Horner, T. K. Machu, and G. H. Dillon. 2003. The GABAA receptor antagonist picrotoxin inhibits 5-hydroxytryptamine type 3A receptors. Neuropharmacology. 44:431–438. [DOI] [PubMed] [Google Scholar]
- 16.Etter, A., D. F. Cully, K. K. Liu, B. Reiss, D. K. Vassilatis, J. M. Schaeffer, and J. P. Arena. 1999. Picrotoxin blockade of invertebrate glutamate-gated chloride channels: subunit dependence and evidence for binding within the pore. J. Neurochem. 72:318–326. [PubMed] [Google Scholar]
- 17.Inoue, M., and N. Akaike. 1988. Blockade of γ-aminobutyric acid-gated chloride current in frog sensory neurons by picrotoxin. Neurosci. Res. 5:380–394. [DOI] [PubMed] [Google Scholar]
- 18.Erkkila, B. E., D. S. Weiss, and V. E. Wotring. 2004. Picrotoxin-mediated inhibition of the nicotinic α3β4 and α7 receptors. Neuroreport. 129:47–53. [DOI] [PubMed] [Google Scholar]
- 19.Gurley, D., J. Amin, P. C. Ross, D. S. Weiss, and G. White. 1995. Point mutations in the M2 region of the α, β or γ subunit of the GABAA channel that abolish block by picrotoxin. Receptors Channels. 3:13–20. [PubMed] [Google Scholar]
- 20.Ffrench-Constant, R. H., T. A. Rocheleau, J. C. Steichen, and A. E. Chalmers. 1993. A point mutation in a Drosophila GABA receptor confers insecticide resistance. Nature. 363:449–451. [DOI] [PubMed] [Google Scholar]
- 21.Enz, R., and J. Bormann. 1995. A single point mutation decreases picrotoxinin sensitivity of the human GABA receptor ρ1 subunit. Neuroreport. 6:1569–1572. [DOI] [PubMed] [Google Scholar]
- 22.Akabas, M. H., C. Kauffmann, P. Archdeacon, and A. Karlin. 1994. Identification of acetylcholine receptor channel lining residues in the entire M2 segment of the α subunit. Neuron. 13:919–927. [DOI] [PubMed] [Google Scholar]
- 23.Reeves, D. C., E. N. Goren, M. H. Akabas, and S. C. Lummis. 2001. Structural and electrostatic properties of the 5-HT3 receptor pore revealed by substituted cysteine accessibility mutagenesis. J. Biol. Chem. 276:42035–42042. [DOI] [PubMed] [Google Scholar]
- 24.Xu, M., and M. H. Akabas. 1993. Amino acids lining the channel of the γ-aminobutyric acid type A receptor identified by cysteine substitution. J. Biol. Chem. 268:21505–21508. [PubMed] [Google Scholar]
- 25.Xu, M., and M. H. Akabas. 1996. Identification of channel-lining residues in the M2 membrane-spanning segment of the GABA(A) receptor α1 subunit. J. Gen. Physiol. 107:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lester, H. A. 1992. The permeation pathway of neurotransmitter-gated ion channels. Annu. Rev. Biophys. Biomol. Struct. 21:267–292. [DOI] [PubMed] [Google Scholar]
- 27.Pribilla, I., T. Takagi, D. Langosch, J. Bormann, and H. Betz. 1992. The atypical M2 segment of the β subunit confers picrotoxinin resistance to inhibitory glycine receptor channels. EMBO J. 11:4305–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das, P., and G. H. Dillon. 2005. Molecular determinants of picrotoxin inhibition of 5-hydroxytryptamine type 3 receptors. J. Pharmacol. Exp. Ther. 314:320–328. [DOI] [PubMed] [Google Scholar]
- 29.Sedelnikova, A., B. E. Erkkila, H. Harris, S. O. Zakharkin, and D. S. Weiss. 2006. Stoichiometry of a pore mutation that abolishes picrotoxin-mediated antagonism of the GABAA receptor. J. Physiol. 577:569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang, D., Z. H. Pan, A. D. Brideau, and S. A. Lipton. 1995. Cloning of a γ-aminobutyric acid type C receptor subunit in rat retina with a methionine residue critical for picrotoxinin channel block. Proc. Natl. Acad. Sci. USA. 92:11756–11760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen, L., K. A. Durkin, and J. E. Casida. 2006. Structural model for γ-aminobutyric acid receptor noncompetitive antagonist binding: widely diverse structures fit the same site. Proc. Natl. Acad. Sci. USA. 103:5185–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhorov, B. S., and P. D. Bregestovski. 2000. Chloride channels of glycine and GABA receptors with blockers: Monte Carlo minimization and structure-activity relationships. Biophys. J. 78:1786–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liman, E. R., J. Tytgat, and P. Hess. 1992. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 9:861–871. [DOI] [PubMed] [Google Scholar]
- 34.Kammann, M., J. Laufs, J. Schell, and B. Gronenborn. 1989. Rapid insertional mutagenesis of DNA by polymerase chain reaction (PCR). Nucleic Acids Res. 17:5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyazawa, A., F. Yoshinori, and N. Unwin. 2003. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 423:949–955. [DOI] [PubMed] [Google Scholar]
- 36.O'Mara, M., B. Cromer, M. Parker, and S. Chung. 2005. Homology model of the GABAA receptor examined using Brownian dynamics. Biophys. J. 88:3286–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu, M., D. F. Covey, and M. H. Akabas. 1995. Interaction of picrotoxin with GABAA receptor channel-lining residues probed in cysteine mutants. Biophys. J. 69:1858–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smart, T. G., and A. Constanti. 1986. Studies on the mechanism of action of picrotoxinin and other convulsants at the crustacean muscle GABA receptor. Proc. R. Soc. Lond. B. Biol. Sci. 227:191–216. [DOI] [PubMed] [Google Scholar]
- 39.Constanti, A. 1978. The ‘mixed’ effect of picrotoxin on the GABA dose/conductance relation recorded from lobster muscle. Neuropharmacology. 17:159–167. [DOI] [PubMed] [Google Scholar]
- 40.Yoon, K., D. Covey, and S. Rothman. 1993. Multiple mechanisms of picrotoxin block of GABA-induced currents in rat hippocampal neurons. J. Physiol. 464:423–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dillon, G. H., W. B. Im, D. B. Carter, and D. D. McKinley. 1995. Enhancement by GABA of the association rate of picrotoxin and tert-butylbicyclophosphorothionate to the rat cloned α1β2γ2 GABAA receptor subtype. Br. J. Pharmacol. 115:539–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krishek, B. J., S. J. Moss, and T. G. Smart. 1996. A functional comparison of the antagonists bicuculline and picrotoxin at recombinant GABAA receptors. Neuropharmacology. 35:1289–1298. [DOI] [PubMed] [Google Scholar]
- 43.Ikeda, T., K. Nagata, T. Shono, and T. Narahashi. 1998. Dieldrin and picrotoxinin modulation of GABAA receptor single channels. Neuroreport. 9:3189–3195. [PubMed] [Google Scholar]
- 44.Shan, Q., J. L. Haddrill, and J. W. Lynch. 2001. A single β subunit M2 domain controls the picrotoxin sensitivity of αβ heteromeric glycine receptor chloride channels. J. Neurochem. 76:1109–1120. [DOI] [PubMed] [Google Scholar]
- 45.Porter, N. M., T. P. Angelotti, R. E. Twyman, and R. L. Macdonald. 1992. Kinetic properties of α1β1 γ-aminobutyric acid A receptor channels expressed in Chinese hamster ovary cells: regulation by pentobarbital and picrotoxin. Mol. Pharm. 42:872–881. [PubMed] [Google Scholar]
- 46.Chang, Y., and D. S. Weiss. 1998. Substitutions of the highly conserved M2 leucine create spontaneously opening ρ1 γ-aminobutyric acid receptors. Mol. Pharm. 53:511–523. [DOI] [PubMed] [Google Scholar]