

Abstract
Angiogenesis is highly sensitive to the composition of the vascular microenvironment, however, our understanding of the structural and matricellular components of the vascular microenvironment that regulate angiogenesis and the molecular mechanisms by which these molecules function remains incomplete. Our previous results described a novel pro-angiogenic activity for Microfibril-Associated Glycoprotein-2 (MAGP-2), but did not address the molecular mechanism(s) by which this is accomplished. We now demonstrate that MAGP-2 promotes angiogenic cell sprouting by antagonizing Notch signaling pathways in endothelial cells. MAGP-2 decreased basal and Jagged1 induced expression from the Notch sensitive Hes-1 promoter in ECs, and blocked Jagged1 stimulated Notch1 receptor processing in transiently transfected 293T cells. Interestingly, inhibition of Notch signaling by MAGP-2 seems to be restricted to ECs since MAGP-2 increased Hes-1 promoter activity and Notch1 receptor processing in heterologous cell types. Importantly, constitutive activation of the Notch signaling pathway blocked the ability of MAGP-2 to promote angiogenic cell sprouting, as well as morphological changes associated with angiogenesis. Collectively, these observations indicate that MAGP-2 promotes angiogenic cell spouting in vitro by antagonizing Notch signaling pathways in ECs.
Keywords: angiogenesis, MAGP-2, Notch, cancer, matricellular, ECM
Introduction
It is estimated that 70 human diseases incorporate either excessive or insufficient angiogenesis as a component of their pathology (Carmeliet 2003; Carmeliet 2005). To successfully treat these diseases, it is therefore important to develop therapeutic strategies to both promote as well as antagonize angiogenesis. Angiogenesis consists of an intricate cascade of events that culminate in the formation of new blood vessels from pre-existing blood vessels. Although significant progress has been made in understanding how growth factors, cytokines, and the intracellular signaling mechanisms stimulated by these molecules regulate angiogenesis, significantly less emphasis has been placed on identifying matricellular molecules within vascular microenvironments that regulate angiogenesis. Even less well understood are the molecular mechanisms by which these matricellular proteins manipulate angiogenesis. Given that some of the most potent regulators of angiogenesis are either matricellular proteins or proteolyzed fragment of structural ECM molecules, it is imperative to understand the molecular mechanisms by which these molecules regulate angiogenesis. Indeed, a thorough understanding of angiogenicly active extracellular matrix molecules and the signaling cascades that are manipulated by these molecules, will provide important information for the development of small molecule drugs to either promote or antagonize angiogenesis.
We recently reported the results of a microarray based screen that monitored gene expression in ECs during an in vitro angiogenesis assay conducted on tumor derived Matrigel matricies (Albig et al. 2007). In this screen, 20% of the differentially regulated transcripts identified encoded secreted molecules, several of which were subsequently shown to either promote or antagonize angiogenesis when overexpressed from ECs. In particular, Microfibril Associated Glycoprotein-2 (MAGP-2, MFAP-5, MP-25) was shown to possess significant pro-angiogenic activity both in vitro and in vivo. Consistent with this, MAGP-2 expression is increased in several different human cancers and models of human cancer (Creighton et al. 2003; Iacobuzio-Donahue et al. 2003; Bild et al. 2006) suggesting that MAGP-2 expression might be important for the acquisition of the vascular phenotype requisite for tumor growth and metastasis.
MAGP-2 and the related molecule, MAGP-1 are the only members of a family of small extracellular matrix proteins that associate with microfibrils in elastin networks. Functionally, MAGP-2 promotes collagen deposition and elastin fiber assembly in cultured fibroblasts (Lemaire et al. 2005; Lemaire et al. 2006). Consistent with this, MAGP-2 expression in increased in the tight skin mouse model of systemic sclerosis and in the skin of human systemic sclerosis patients (Lemaire et al. 2004). Mechanistically, MAGP-2 has been shown to interact with the Notch signaling pathway via direct interactions with the Notch1 receptor as well as the Jagged1, Jagged2, and Dll-1 Notch ligands. Interaction of MAGP-2 with Notch1 and Jagged1 is reported to initiate ectodomain shedding and subsequent initiation of the Notch signaling cascade (Nehring et al. 2005; Miyamoto et al. 2006).
In the classical model of Notch signaling, Notch receptors (Notch 1–4) present on the “receiving” cell are stimulated by Notch ligands (Jagged 1, 2 and Delta-like 1, 2, and 4 in mammals) that are presented on the “signaling” cell (Weinmaster 1997). Interaction between receptor and ligand stimulates cleavage of Notch receptors by ADAM and gamma-secretase proteases thus liberating the c-terminal Notch intracellular domain (NICD) that subsequently translocates to the nucleus where in participates in a transcriptional complex to drive expression of Notch responsive genes (Weinmaster 1997). Notch signaling plays an essential role in regulating normal vessel development and angiogenesis in mammals (Shawber and Kitajewski 2004; Leong and Karsan 2006). However, given that Notch activation has been shown to promote both angiogenesis as well as angiostasis (Leong and Karsan 2006; Rehman and Wang 2006), the precise role of Notch signaling during angiogenesis remains to be determined. This may be due to the fact that vasculature endothelial cells express both Notch receptors and Notch ligands (Alva and Iruela-Arispe 2004; Vorontchikhina et al. 2005; Hellstrom et al. 2007; Hofmann and Luisa Iruela-Arispe 2007) thus leading to the possibility that “cell autonomous” Notch signaling may play an important role during angiogenesis. In support of this idea, several recent reports describe an important role for cell autonomous Notch activation during angiogenesis (Sainson et al. 2005; Noguera-Troise et al. 2006; Hellstrom et al. 2007; Leslie et al. 2007). Moreover, Notch4 and Dll4 are expressed almost exclusively by endothelium (Uyttendaele et al. 1996; Shutter et al. 2000; Mailhos et al. 2001) suggesting that these proteins may serve unique functions in endothelial cells and may participate in EC specific cell autonomous Notch signaling.
Given our previous results showing that MAGP-2 promotes angiogenesis both in vitro and in vivo and that MAGP-2 has been shown to regulate Notch signaling (Miyamoto et al. 2006), we hypothesized that MAGP-2 may promote angiogenesis via its ability to modulate Notch activation in angiogenic ECs. Consistent with this hypothesis, we found that MAGP-2 decreased Notch signaling in ECs in vitro and that constitutive activation of Notch signaling reversed the hyper-angiogenic phenotype of MAGP-2 overexpressing ECs. These results indicated that MAGP-2 promotes angiogenic sprouting at least in part by virtue of its ability to inhibit anti-angiogenic Notch signaling events operant during angiogenic sprouting in vitro.
Materials and Methods
Cell Culture
Retroviral supernatants were produced by EcoPack2 retroviral packaging cells (Clontech, Mountain View, CA) and used to infect mouse brain microcapillary MB114 endothelial cells as described previously (Albig and Schiemann 2004; Albig and Schiemann 2005; Albig et al. 2006; Albig et al. 2007). Infected cells were analyzed 48 h post-infection and the highest 10% of YFP-expressing cells were collected on a MoFlo cell sorter (Cytomation, Fort Collins, CO). Afterward, isolated cells were expanded to yield stable polyclonal populations that were ≥ 95% positive for transgene expression. Human kidney 293T, MDA-MB-231 breast carcinoma, MCA102 fibrosarcoma (Wexler and Rosenberg 1979), and B16F10 melanoma cells were cultured in DMEM media supplemented with 10% fetal bovine serum (FBS), while SVEC4–10 endothelial cells (O’Connell and Edidin 1990)were cultured in DMEM media supplemented with 10% FBS that had been heat inactivated by incubation for 30 minutes at 56°C. Human umbilical vein ECs (HUVEC; passages 3–6) and human microvascular ECs (HMEC) were maintained in EGM-2 media (Cambrex Corp., East Rutherford, NJ) supplemented with EC growth factors (Bullet Kit, Cambrex).
Plasmids
The Myc-tagged mammalian expression vectors encoding murine Notch1 [pCS2+mN1FL6MT; (Mumm et al. 2000)] and Jagged-1 [pCS2+Jag1–6MT; (Mumm et al. 2000)] were kindly provided by Dr. Raphael Kopan (Washington University, St. Louis, MO). A retroviral Notch1 ICD vector was constructed by PCR amplifying the murine Notch1 ICD domain (amino acids 1744–2531 and contained in pCS2-mN1FL6MT) using a 5′ oligonucleotide that contained a unique Xho I restriction site, a Kozak consensus sequence, and a start codon (5′GGCGGCCTCGAGGCCACCATGGTGCTGCTGTCCCGC) and a 3′oligonucleotide that contained a unique Hpa I restriction site, a stop codon, and the C-terminal Myc-tag (5′GGCGGCGTTAACTCATGAATTCAAGTCCTCTTCAGA). The resulting PCR product was ligated into identical restriction sites in the bicistronic retroviral vector, pMSCV-IRES-GFP (Albig and Schiemann 2005). The pHes1-luciferase, pCMV-Hes1, and pCMV-NICD plasmids were kindly provided by Dr. Jan Jensen (University of Colorado Health Science Center, Denver, CO).
In vitro angiogenic sprouting assays
The quantitative assessment of angiogenic sprouting from quiescent monolayers of ECs was conducted with MB114 ECs as previously described (Albig and Schiemann 2004). For actin and β-catenin immunofluorescence localization studies in sprouting EC cultures, quiescent monolayers of MB114 ECs were overlayed with a dilute collagen gel (1:20 dilution of collagen concentrate into MB114 growth media (~0.1 mg/ml collagen) for 1 day whereupon the gel was gently aspirated and the remaining cells were washed and fixed overnight in PBS containing 4% formaldehyde.
Fluorescent microscopy
ECs that had been fixed overnight in PBS/4% formaldehyde were washed with PBS++ (1XPBS, 0.5mM CaCl, 0.9mM MgCl) then permeabilized with PBS++ containing 0.2% TritonX-100 for 5 minutes. To stain for β-catenin, cells were sequentially incubated with PBS++ containing 1%BSA and 200μg/ml of goat-gamma globulin (Jackson Immunoresearch Laboratories, West Grove, PA) for 1hr to block non-specific interactions, with PBS++/1%BSA containing anti-β-catenin monoclonal antibodies (1:200 dilution, BD Transduction Labs, San Diego, CA) for 2 hours, with 5μg/ml of biotinylated-goat anti-mouse antibodies (Moleculer Probes, Eugene OR), and finally with 1.2 μg/ml of Alexa Strepavadin (Moleculer Probes, Eugene OR) for 1hr. Between all incubations, the cells were washed at least three times for five minutes with PBS++. Afterward, the coverslips were mounted on glass slides with Prolong mounting media containing DAPI (Molecular Probes, Eugene, OR). For staining of the actin cytoskelton, fixed ECs were washed with PBS, then non-specific binding sites were blocked by incubation PBS containing 1.5% FBS for 1hour followed by incubated with 0.25μM Rhodamine-conjugated phalloidin (Sigma, St. Louis, MO) for an additional hour at RT. After extensive washing with PBS, cover slips were mounted on glass slides as above.
Notch Processing Assay
To monitor the effects of MAGP-2 on the processing and S3 cleavage of Notch1, human kidney 293T cells were transiently transfected in 6-well plates with LT-1 liposomes containing various combinations of 0.5 μg/well of Notch1 (pCS2+mN1FL6MT), 0.5 μg/well Jagged-1 (pCS2+Jag1–6MT), and either 1.5 μg/well of MAGP-2 (pcDNA3.1-MAGP-2/Myc-His) or empty pcDNA3.1 Myc-His. In some cases, transfections were performed in duplicate where the duplicate cells were also cotransfected with the Hes-1 luciferase and CMV-β-gal reporter vectors (50ng/vector/well). Forty-eight hr post-transfection, the cells were washed with ice-cold PBS, lysed immediately in Buffer H/1% Triton X-100 (Schiemann et al. 2002), and incubated on ice for 30 min. Afterward, insoluble material was removed by microcentrifugation and 100 μl of the resulting clarified extract was fractionated through 6% SDS-PAGE gels. The fractionated proteins were transferred electrophoretically to nitrocellulose and probed with 1:1000 dilutions of either anti-Myc 9E10 monoclonal antibodies (Covance, Princeton, NJ) to visual all Notch1 cleavage species, or with the Val1744 antibody (Cell Signaling Technologies, Boston, MA) that recognizes only cleaved Notch1 (N1ICD) receptors. For experiments monitoring Notch activation via Hes-1 luciferase activation, transfected cells were washed with 1XPBS, lysed in passive lysis buffer, and processed for luciferase activity as described below.
Luciferase assays
MB114 and HUVEC EC were seeded onto 24-well plates at a density of 25,000 cells/well and transiently transfected the following day by overnight exposure to LT1 liposomes (Mirus) containing 100 ng/well of pCMV-β-gal, together with 200 ng/well of Hes-1-luciferase. In some experiments, cells were also transfected with 10ng/well of Jagged1 cDNA and/or100ng of MAGP-2 cDNA were indicated. Afterward, the cells were fed with 0.5 ml of fresh media and the next morning, luciferase and β-gal activities contained in detergent-solubilized cell extracts where determined. To determine the effect of MAGP-2 on TGF-β induced Hes-1 or SBE promoter activity, MB114 cells were plated as described above then transiently transfected the following day with 100ng/well pCMV-β-gal together with 100ng/well of either HES-1-luciferase or SBE-luciferase and 100ng/well of MAGP-2 cDNA or empty pcDNA3.1 Myc-His6 vector. The following day, cells were washed twice with PBS and incubated overnight in SFM supplemented with or without TGF-β1 (0–5ng/ml) as indicated. The next morning, luciferase and β-gal activities contained in detergent-solubilized cell extracts were determined. Luciferase assays performed in alternative cell lines were conducted with Hes-1 luciferase constructs as described above using 25,000 cells/well for the SVEC and HMEC cell lines, and 80,000 cells/well for the MDA-MB-231, B16F10, and MCA102 cell lines.
Recombinant MAGP-2 protein preparation
Bacterial MAGP-2 expression vectors were synthesized by PCR amplifying the full-length MAGP-2 cDNA (less the signal sequences) using oligonucleotides that incorporated unique Nde I (N-terminus) or Bam HI and FLAG-tag (C-terminus) sequences. The resulting PCR fragments were ligated into identical sites in pSBET (Schenk et al. 1995). FLAG-tagged recombinant proteins were purified by passing sonicated and TBS/0.1% Triton X-100-solubilized bacterial cell extracts over a column containing immobilized FLAG-M2 monoclonal antibodies (Sigma, USA). Bound proteins were washed initially with 10 column volumes of TBS/0.1% Triton X-100, followed by an additional 20 column volumes of TBS. Afterward, recombinant proteins were eluted by addition of 2.5 column volumes of FLAG M2 peptide (100 μg/ml), which subsequently was concentrated by centrifugation against PBS (5 kDa cutoff; Sartorius, Germany). Western blot analysis of rMAGP-2 was performed with anti-mouse MAGP-2 polyclonal antibodies (kindly provided by Dr. J.M. Shipley, Washington University, St. Louis, MO) or with anti-M2 monoclonal antibodies (Sigma).
Results
MAGP-2 inhibits Notch1 signaling
To determine what effect, if any, MAGP-2 has on Notch signaling in ECs, we first utilized a Hes1-luciferase reporter gene whose expression is induced by Notch activation (Iso et al. 2003) to monitor changes in Notch signaling in the presence of exogenous MAGP-2. Recombinant FLAG-tagged MAGP-2 was purified from bacterial lysates (Fig 1A) and applied to MB114 and HUVEC ECs that had been transfected with Hes-1 luciferase. Figure 1B shows that Hes-1 driven luciferase activity was decreased in MB114 and HUVEC ECs that had been treated with either 1 or 5 μg/ml of rMAGP-2-FLAG fusion protein. We also compared Hes-1 promoter activity in both MB114 and HUVEC cells transiently transfected with either MAGP-2 cDNA or an empty vector control. Similar to our results in figure 1B, MAGP-2 decreased Hes-1 promoter activity by 20% and 30% in MB114 and HUVEC ECs respectively (data not shown). Moreover, MAGP-2 expression abrogated the ability of Jagged-1 to induce Hes1-luciferase activity in MB114 cells (Fig. 1C), suggesting that MAGP-2 functions to antagonize Jagged-1 and, consequently, Notch signaling in ECs.
Figure 1. MAGP-2 decreases Notch signaling/Hes-1 promoter activity in ECs.

(A) FLAG-tagged MAGP-2 protein was expressed in bacterial cells and isolated by anti-FLAG chromatography. MAGP-2 preparations were confirmed by comassie staining (Co), and by western blot analysis with either anti-MAGP-2 antibodies (M) or anti-FLAG M2 antibodies (F). The arrow denotes MAGP-2 protein. (B) MB114 and HUVEC cells were transiently transfected with pHes1-luciferase plus pCMV-β-gal and subsequently treated with rMAGP-2 (1 to 5 μg/ml). Afterward, luciferase and β-gal activities contained in detergent-solubilized cell extracts were measured. Data are mean (± SEM) of 2 independent experiments. (C) GFP- and MAGP-2-expressing MB114 cells were transiently transfected with pHes1-luciferase and pCMV-β-gal cDNAs, together with or without Jagged-1 cDNA as indicated. Afterward, luciferase and β-gal activities were measured as above. Data are the mean (± SEM) of four independent experiments. (*, **, ***, p < 0.05; Student’s Test).
MAGP-2 blocks Notch1 receptor activation by Jagged1
To further confirm the ability of MAGP-2 to antagonize Notch signaling, we monitored processing of the Notch1 receptor after stimulation with Jagged1 in the presence or absence of exogenous MAGP-2. Activation of Notch1 signaling involves three proteolytic processing events, termed S1, S2, and S3, that produce three distinct Notch1 fragments, termed TMIC, NEXT, and N1ICD, respectively (Mumm et al. 2000). NICD production is mediated by a gamma-secretase cleavage reaction that cuts Notch1 at a membrane proximal cytoplasmic site (Mumm et al. 2000), resulting in the release and subsequent translocation of N1ICD to the nucleus where it regulates the expression of Notch1-responsive genes, including Hes1 (Kopan 2002). Therefore, 293T cells were transiently transfected with various combinations of C-terminally myc-tagged Notch1, Jagged1, and MAGP-2 cDNAs to directly monitor the effect of MAGP-2 on N1ICD accumulation western blot analysis. Notch1 receptor processing was monitored by western blot analysis of whole cell lysates with either the Val1744 antibody that specifically detects only the NICD fragment of Notch1 (Fig 2A), or with α-myc antibodies to detect various Notch1 fragments (Fig S1). As shown in figure 2A Jagged-1 expression significantly enhanced basal Notch1 N1ICD accumulation as compared to cells solely expressing Notch1. However, the ability of Jagged-1 to induce Notch1 cleavage and N1ICD production in 293T cells was significantly reduced by co-expression of MAGP-2 (Fig. 2A). Notch1 and Jagged1 expression was not effected by MAGP-2 overexpression indicating that the ability of MAGP-2 to decrease N1ICD accumulation was not due to decreased Notch1 or Jagged1 expression. Identical results were obtained by monitoring Notch1 S1, S2, and S3 fragment accumulation with anti-Myc antibodies in the presence of overexpressed MAGP-2 protein (Figure S1).
Figure 2. MAGP-2 inhibits Notch1 receptor processing.
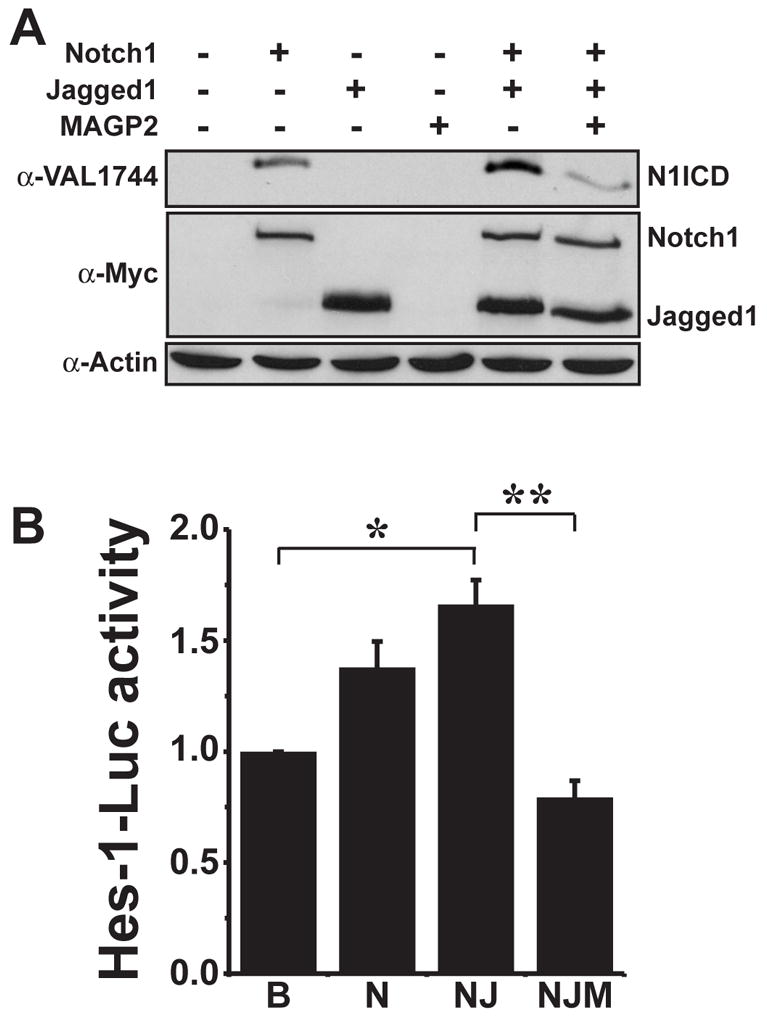
(A) 293T cells were transiently transfected with cDNAs encoding Myc-tagged versions of Notch1, Jagged-1, and MAGP-2 in various combinations as indicated. Accumulation of N1ICD was monitored by immunoblotting whole cell extracts with monoclonal antibodies (α-Val1744) that only recognize the N1ICD domain. Afterward, the blot was stripped and immunoblotted with anti-myc monoclonal antibodies to control for differences in Notch1 and Jagged1 expression or with anti-β-actin monoclonal antibodies to control differences in protein loading. Shown is a representative experiment that was performed three times in its entirety. (B) Human 293T cells were transfected with Hes-1 luciferase and CMV-β-gal reporters either alone (B=basal) or in addition to Notch1 (N), Notch1 + Jagged1 (NJ), or Notch1 + Jagged1, + MAGP-2 (NJM) as described above. Empty vector was used to normalize total DNA transfected in all wells. Afterward, luciferase and β-gal activities were measured as described (materials and methods). Data are the mean (± SEM) of three independent experiments. (*, **, p < 0.05; Student’s Test).
To indirectly monitor Notch activation in transfected cells, we performed identical transfection protocols in 293T cells as described above with the notable exception that Hes-1 luciferase and CMV-β-gal reporter constructs were supplemented to the transfection mixtures. This allowed us to indirectly monitor N1ICD accumulation in the presence or absence of exogenous MAGP-2 by measuring Hes-1 luciferase activity. Similar to the results in figure 2A, MAGP-2 decreased Jagged1 stimulated Hes-1 luciferase activity (Fig 2B). Collectively, these results support our hypothesis that MAGP-2 blocks Notch receptor activation by Jagged1.
MAGP-2 specifically blocks Notch signaling
To determine if MAGP-2 specifically blocks Notch signaling or alternatively if MAGP-2 has broader effects on cell signaling, we took advantage of recent findings showing that the ability of TGF-β to induce Hes1 promoter activity requires Smad3 to interact physically with N1ICD (Blokzijl et al. 2003), a reaction that is dispensable for canonical Smad3-mediated signaling stimulated by TGF-β (Blokzijl et al. 2003). We therefore reasoned that if MAGP-2 specifically interacts with the Notch signaling pathway, the ability of MAGP-2 to inhibit NICD production in ECs would reduce the capacity of TGF-β to induce luciferase expression driven by the Hes1 promoter, but not that driven by the synthetic Smad2/3-binding element [SBE; (Jonk et al. 1998)]. Accordingly, MAGP-2 expression in MB114 cells significantly decreased the ability of TGF-β to stimulate Hes1-luciferase activity, but had no effect on its stimulation of SBE-luciferase activity (Fig. 3). This result indicated that MAGP-2 specifically blocks Notch signaling without interfering with TGF-β signal transduction mechanisms.
Figure 3. MAGP-2 selectively antagonizes Notch signaling in MB114 ECs.
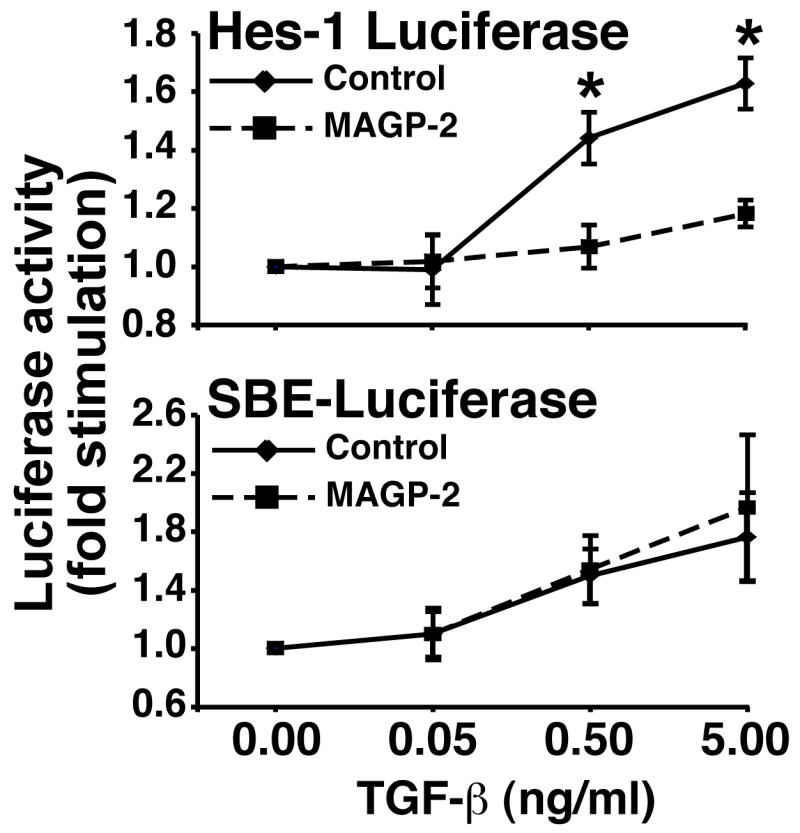
GFP- and MAGP-2-expressing MB114 cells were transiently transfected with either pHes1- or pSBE-luciferase, both together with pCMV-β-gal as indicated. Afterward, the resulting transfectants were stimulated overnight with increasing concentrations of TGF-β1 (0–5 ng/ml). Data are the mean (± SEM) of 3 independent experiments. (*, p < 0.05; Student’s T-Test).
MAGP-2 regulates Notch signaling in a cell type specific manner
In contrast to our results in ECs, Miyamoto et al, previously reported that MAGP-2 promotes Notch signaling in non-EC heterologous cell types (Miyamoto et al. 2006). Given our results presented thus far, we hypothesized that MAGP-2 might have cell type specific effects on Notch signaling activity. Therefore, we again used luciferase assays to compare the ability of MAGP-2 to regulate Hes-1 promoter activity in a variety of cell types. To accomplish this, MAGP-2 or empty vector was transfected with the Hes-1 luciferase reporter into two additional EC lines (SVEC murine ECs, and HMEC human microvascular ECs) and a variety of cancer cell types including MDA-MB-231 human breast carcinoma cells, B16F0 mouse melanoma cells, and MCA102 mouse fibrosarcoma cells. Interestingly, MAGP-2 decreased Notch signaling activity in all EC lines tested, but increased Hes-1 promoter activity in other non-EC cell types (Fig 4). Moreover, transient overexpression of MAGP-2 (Fig 4B) or addition of rMAGP-2 protein (data not shown) decreased N1ICD accumulation in SVEC ECs but increased N1ICD accumulation in B16F0 melanoma cells. We were unable to detect N1ICD fragments by western blot analysis in other cell types and were thus unable to perform similar analysis on these cell lines (data not shown). These results together with our previous reports demonstrated that MAGP-2 manipulates Notch signaling in a cell type specific manner.
Figure 4. MAGP-2 regulates Notch signaling in a cell type specific manner.
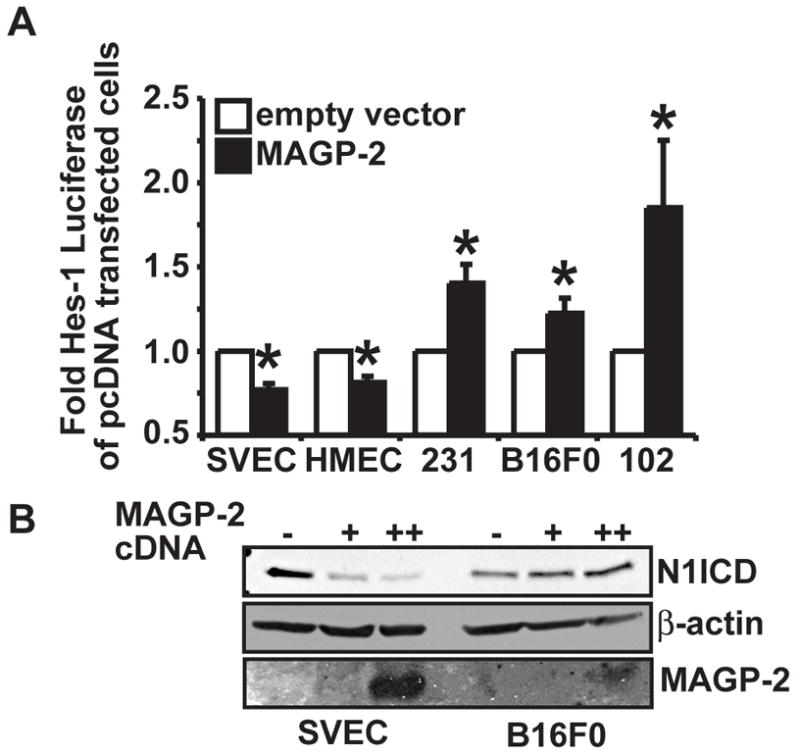
(A) Endothelial cells lines (SVEC and HMEC) and various tumor cell lines (MDA-MB-231 (231) breast carcinoma cells, B16F0 melanoma cells (B16), and MCA102 fibrosarcoma cells (102)) were transiently transfected with either empty vector (pcDNA3.1 myc-his) or MAGP-2 cDNA (pcDNA3.1-MAGP-2-myc-his) together with pHes-1 luciferase and pCMV-β-gal. Afterward, luciferase and β-gal activities were measured as above. Data are the mean (± SEM) of three independent experiments. (*, p < 0.05; Student’s Test). (B) SVEC ECs and B16F0 melanoma cells were transiently transfected with either empty vector (−) or with 1 (+) or 2.5 (++)μg of MAGP-2 cDNA and endogenous Notch1 NICD fragments were monitored by western blot analysis of whole cell lysates with anti-Val1744 antibodies. The western blot was subsequently stripped and reprobbed with anti-β-actin antibodies to monitor differences in protein loading. Conditioned media from the transfected cells was collected, precipitated with TCA, and analyzed by western blot with anti-MAGP-2 antibodies to monitor MAGP-2 expression. Shown is a representative experiment that was performed three times in its entirety with similar results.
MAGP-2 promotes angiogenic sprouting by antagonizing Notch signaling
Previous reports have established Notch signaling as an important mediator of angiostasis both in vitro and in vivo (MacKenzie et al. 2004; Noseda et al. 2004; Leong and Karsan 2006; Williams et al. 2006). We have previously demonstrated that MAGP-2 promotes angiogenic sprouting in vitro and angiogenesis in vivo (Albig et al. 2007), and our current results demonstrate that MAGP-2 antagonizes Notch signaling in ECs. Based on these findings, we hypothesized that MAGP-2 promotes angiogenesis by antagonizing Notch signaling. Given that Notch signaling has been shown to both promote and antagonize angiogenesis in a cell type specific manner, it was important to first determine whether Notch signaling is pro- or anti-angiogenic in MB114 ECs. In doing so, MB114 cells were transiently transfected with the Hes1-luciferase reporter gene, and subsequently were treated with or without the highly specific gamma-secretase inhibitor, DAPT (Sastre et al. 2001), which inhibits S3-mediated cleavage of Notch receptors and consequently, NICD-mediated induction of Hes1 expression. As anticipated, DAPT administration significantly inhibited Hes1 promoter activity in MB114 cells (Fig 5A), but more importantly, MB114 cells treated with DAPT formed significantly more angiogenic sprouts than did their untreated counterparts (Fig. 5B). These findings indicate that Notch signaling antagonizes angiogenic sprouting of MB114 cells.
Figure 5. Constitutive Notch signaling antagonizes angiogenic sprouting mediated by MAGP-2 in MB114 cells.
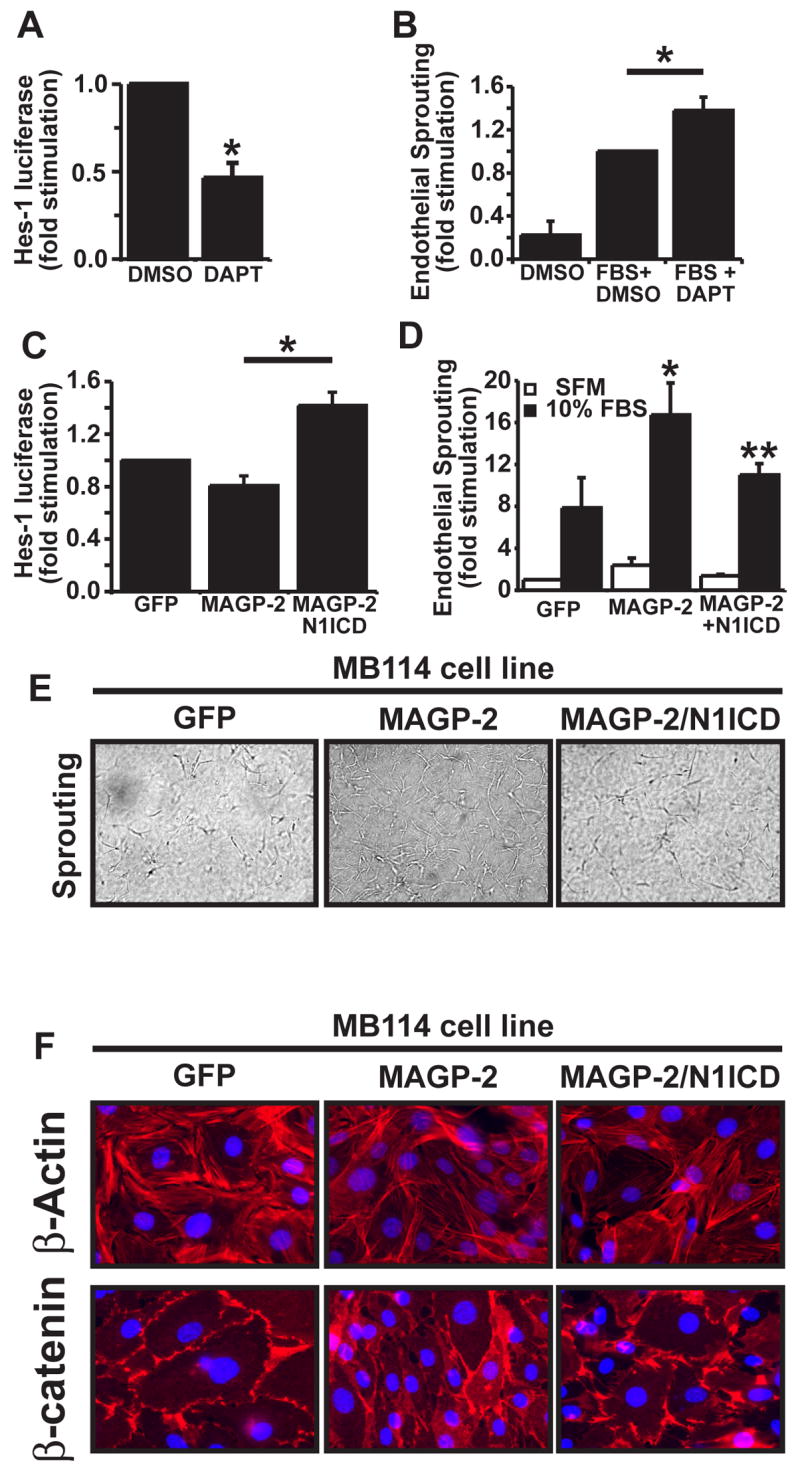
(A) MB114 cells were transiently transfected with pHes1-luciferase and pCMV-β-gal cDNAs, and subsequently were incubated overnight in the absence or presence of DAPT (10 μM). Afterward, luciferase and β-gal activities were determined. Data are the mean (± SEM) of two independent experiments. *, p<.05; student’s T-Test). (B) Quiescent MB114 cell monolayers were overlaid with rat tail collagen matrices, and were induced to form angiogenic sprouts by addition of 10% FBS supplemented with or without DAPT (10 μM). Five days later the number of invading angiogenic spouts were quantified by manual counting on a light microscope. Data are the mean (± SEM) of four independent experiment. (*, p < 0.05; Student’s T-Test). (C) GFP-, MAGP-2-, and MAGP-2/N1ICD-expressing MB114 cells were transiently transfected with pHes1-luciferase and pCMV-β-gal cDNAs. Luciferase and β-gal activities were determined 48 h post-transfection. Data are the mean (± SEM) of two independent experiments. (D) Quiescent monolayers of GFP-, MAGP-2-, and MAGP-2/N1ICD-expressing MB114 cells were overlaid with rat tail collagen matrices containing 10% FBS for 5 days. Afterward, the number of invading angiogenic sprouts were determined by manual counting under a light microscope. Data are the mean (± SEM) of four independent experiments. (*, **, p < 0.05; Student’s T-Test). (E) Quiescent monolayers of GFP-, MAGP-2-, and MAGP-2/N1ICD-expressing MB114 cells were either left untreated (quiescent) or overlayed with rat tail collagen matricies containing 10% FBS (sprouting) to induce angiogenic cell sprouting. After 5 days, angiogenic sprouts (100X magnification) were counted under a light microscope. (F) Quiescent monolayers of GFP-, MAGP-2-, and MAGP-2/N1ICD-expressing MB114 cells were prepared for immunofluorescent microscopy by incubating with rhodamine-conjugated phalloidin (upper panel) to visualize actin fibers (red) and DAPI to visualize nuclei (blue), or with β-catenin monoclonal antibodies (lower panel) to visualize cell-cell junctions (red) and DAPI (blue). Representative images are shown at 630X magnification from a single experiment that was conducted three times in its entirety.
Having shown that Notch signaling mediates angiostasis in MB114 cells, we next asked whether MAGP-2 promotes angiogenesis in MB114 cells via its ability to antagonize Notch signaling. To do so, we engineered MAGP-2-expressing MB114 cells to constitutively express active Notch1 NICD (N1ICD) fragment in an attempt to overcome the block of Notch processing mediated by MAGP-2. As we observed previously, MAGP-2 expression reduced Hes1-luciferase activity in MB114 cells, a result that was bypassed by co-expression of N1ICD in these cells (Fig. 5C) indicating that N1ICD overexpression bypassed the MAGP-2 mediated block of Notch signaling activity in these cells. More importantly however, the ability of MAGP-2 to promote angiogenic sprouting was significantly reduced by constitutive N1ICD expression in MB114 cells (Figs. 5D, 5E).
A hallmark of quiescent EC monolayers is the peripheral localization of β-catenin to adherens junctions where it serves as an adaptor molecule linking VE-cadherin to the cortical actin cytoskelton (Bazzoni and Dejana 2004). Activation of quiescent monolayers results in a loosening of adherens junctions and the concomitant loss of β-catenin from the cell periphery (Bazzoni and Dejana 2004). Simultaneously, actin fibers of the cortical actin cytoskelton in quiescent EC monolayers are de-polymerized and reformed as the long stress fibers that are requisite for cellular mobility in activated ECs. Therefore, redistribution of β-catenin and actin from cellular peripheries is an indication of angiogenesis activation. To further explore the link between MAGP-2, Notch signaling, and angiogenesis, we used fluorescent microscopy to compare actin and β-catenin staining patterns in control, MAGP-2, or MAGP-2-N1ICD overexpressing MB114 ECs. As shown in figure 5F, quiescent MB114-GFP cells exhibited a strong pattern of cortical actin staining and β-catenin localization that was primarily restricted to regions of cell-cell junctions. In comparison and consistent with the pro-angiogenic activity of MAGP-2, quiescent monolayers of MB114-MAGP-2 ECs demonstrated reduced cortical actin staining, increased actin stress fiber polymerization, and reduced peripheral β-catenin localization as compared to cultures of quiescent MB114-GFP cells. Moreover, these cells packed more closely together suggesting a reduced ability to form proper cell-cell junctions and quiescent monolayers. Constitutive activation of Notch signaling by co-expression of N1ICD in MB114-MAGP-2 ECs resulted in the striking reinstatement of β-catenin to sites of cellular contact, and a reestablishment of quiescent monolayers similar to those observed in MB114-GFP cultures. Interestingly, N1ICD overexpression failed to restore cortical actin staining in quiescent monolayers. Collectively, these results demonstrate that Notch1 activation antagonizes angiogenesis in MB114 cells, and most importantly, these results further support our hypothesis that MAGP-2 promotes angiogenesis at least in part via its ability to antagonize Notch1 processing and signaling in ECs.
Discussion
Angiogenesis is a highly regulated process during which new blood vessels sprout from pre-existing vessels. Angiogenesis is regulated by the contents of the vascular microenvironment which collectively activate and repress a multitude of intercellular signaling pathways. Previously, we demonstrated that MAGP-2 promotes and enhances angiogenesis both in vitro and in vivo (Albig et al. 2007) but did not address the molecular mechanism by which this is accomplished. We now propose that MAGP-2 accomplishes this by antagonizing Notch signaling in ECs. In support of this conclusion, we demonstrate that MAGP-2 antagonizes EC Notch signaling and that the pro-angiogenic activity of MAGP-2 is diminished by constitutive activation of Notch signaling. Collectively, these findings further support our overall hypothesis, that MAGP-2 is a novel activator of angiogenesis, doing so in part via its ability to inhibit the Notch1 signaling pathway.
Recently, it has become clear that Notch signal transduction mechanisms play an important, although poorly understood regulatory role during angiogenesis. Indeed, Notch activation has been shown to mediate angiostasis (Zimrin et al. 1996; Leong et al. 2002; Noseda et al. 2004; Liu et al. 2006; Williams et al. 2006) as well as angiogenesis (Shawber and Kitajewski 2004; Leong and Karsan 2006). However, the mechanisms whereby Notch mediates such disparate activities in ECs remains unclear, but may reflect a complex integration of cellular and environmental cues. Indeed, Notch signaling is subject to regulation by several factors such as the relative expression levels of various Notch receptors (Duarte et al. 2004; Delaney et al. 2005), the extent and form of Notch receptor glycosylation (Haines and Irvine 2003), the availability of various Notch ligands within vascular microenvironments, and the presence of various cytoplasmic or membrane localized Notch activators and inhibitors such as, NRARP, MINT, Numb, and Notch glycosyltransferase proteins or the Fringe family (Kadesch 2004). Moreover, Notch receptors are not equivalently activated by all Notch ligands leading to the possibility that various Notch receptor-ligand combinations are pro-angiogenic while other combinations are anti-angiogenic (Weinmaster 1997). In addition, there seems to be a new paradigm of Notch regulation emerging in which activation of Notch receptors is regulated by components of the local microenvironment. Indeed NOV/ccn3, and proteolyzed soluble Notch ligands have been shown to be regulators of Notch activation (Sakamoto et al. 2002; Kadesch 2004), and interestingly, also regulators of angiogenesis (Brigstock 2002), suggesting that regulation of Notch signal transduction by stromal proteins may also be an important mechanism by which angiogenesis is regulated in vivo. The data presented herein, further develops this idea by demonstrating that the stromal protein MAGP-2 promotes angiogenic sprouting by antagonizing Notch signaling in ECs. Future challenges for understanding how Notch signaling regulates angiogenesis will be to define which combinations of Notch receptors and ligands promote angiogenesis, which combinations of Notch receptors and ligands antagonize angiogenesis, and finally to determine which of these interactions are disrupted (or perhaps enhanced) by MAGP-2.
We currently do not understand the precise mechanism whereby MAGP-2 antagonizes Notch signaling. However, MAGP-2 does not seem to effect the expression of Notch receptors (Notch1) or Notch ligands (Jagged1) (Miyamoto et al. 2006), thus ruling out the possibility that MAGP-2 regulates Notch signaling by controlling expression of these signaling molecules. However, we cannot rule out the possibility that MAGP-2 might regulate the expression of other Notch receptors and/or ligands. More interestingly, recent studies using heterologous cell expression systems have shown MAGP-2 to interact physically with Notch1 and its ligand, Jagged-1, resulting in their shedding from the cell surface (Nehring et al. 2005; Miyamoto et al. 2006). Although we made no attempt to measure Notch1 and/or Jagged-1 extracellular domain shedding in response to MAGP-2, the production of such soluble Notch1 and Jagged-1 extracellular domains readily inhibits Notch signaling (Rebay et al. 1993; Small et al. 2001). Thus, it is tempting to speculate that MAGP-2 promotes angiogenesis in part by inducing Notch1 and/or Jagged-1 ectodomain shedding in ECs. However, in contrast to our findings in ECs, Miyamoto et al (Miyamoto et al. 2006) recently found that MAGP-2 not only induces Notch1 ectodomain shedding in heterologus Cos-7 and NIH-3T3 cells, but also Notch1 processing and NICD production, leading to transcriptional activation of the Hes5 and CSL promoters. The reasons underlying this discrepancy are currently unknown, but given that we also documented the ability of MAGP-2 to increase Notch signaling in a variety of non-EC heterologus cell types (Fig. 4), it is clear that MAGP-2 regulates Notch signaling in a cell type specific manner. We envision two possible explanations that could account for this paradox. First, differential expression patterns of Notch receptors and ligands in various cell types, coupled with the possibility that MAGP-2 may have variable effects of specific Notch receptor:ligand interactions, could lead to variable effects of MAGP-2 on Notch signaling in a cell type specific manner. This is especially important given that the Notch-4 (N4) receptor and Delta-like 4 (Dll4) Notch ligand are predominantly expressed in vascular tissue where their signaling activities promote angiostasis (Noguera-Troise et al. 2006; Ridgway et al. 2006; Williams et al. 2006; Hellstrom et al. 2007). Thus, in the context of our results, MAGP-2 mediated inhibition of Dll4 or N4 signaling might promote angiogenesis. To address this possibility, it will be important in future studies to examine the effect of MAGP-2 on a range of Notch receptor-ligand interactions in addition to the Notch1-Jagged1 interaction described herein and elsewhere (Nehring et al. 2005; Miyamoto et al. 2006). Secondly, differences in microenvironmental factors secreted by various cell types may influence the interactions between MAGP-2 and Notch receptors and/or ligands thus leading to cell type specific effects of MAGP-2 on Notch signaling. For instance, differential expression of Notch glycosyltransferases between various cell lines could potentially change the molecular interactions between MAGP-2 and Notch receptors thus leading to either enhanced or diminished Notch activation. Although testing this hypothesis will be challenging, the identification of other microenvironmental proteins that can influence Notch signaling will provide important information to further our understanding of how Notch signaling is controlled by microenvironmental cues.
As demonstrated in figure 5, constitutive activation of Notch signaling only partially blocked the pro-angiogenic phenotype of MAPG-2 expressing MB114 cells. Thus, it is likely that MAGP-2 promotes angiogenesis by multiple mechanisms. The most attractive of these alternative mechanisms is based on the fact that MAGP-2 contains a RGD integrin binding domain that has been shown to interact with the pro-angiogenic αvβ3 integrin (Gibson et al. 1999). Ligation of αvβ3 integrins promote a variety of pro-angiogenic activities such as cytoskeletal rearrangement and enhancement of VEGF signaling downstream from the KDR/VEGFR2 receptor (Masson-Gadais et al. 2003; Le Boeuf et al. 2004). This hypothesis is consistent with our previous observation that MAGP-2 enhances VEGF signal transduction mechanisms in a variety of EC lines (Albig et al. 2007). However, we have been unable to detect any differences in KDR phosphorylation in HUVEC cells stimulated with either VEGF alone, or with VEGF and MAGP-2 (data not shown). Nonetheless, future experiments will address this hypothesis by determining if MAGP-2 employs a dual mechanism involving both Notch inhibition and by integrin ligation to promote angiogenesis.
Alternatively, our observation that β-catenin localization to cellular peripheries is decreased by MAGP-2 but rescued by overexpression of the N1ICD domain (Fig. 5) suggests that Notch signaling may be able to influence β-catenin localization to cellular peripheries and subsequent downstream β-catenin signaling activities. In support of this, it has been reported that Notch signaling both negatively and positively effects β-catenin/WNT signaling in a variety of cell lines (Ross and Kadesch 2001; Hayward et al. 2005; Masckauchan et al. 2005; Deregowski et al. 2006). Interestingly, the WNT-β-catenin-LEF signaling pathway has also been shown to participate in the regulation of angiogenesis (Cheng et al. 2003; Masckauchan et al. 2006; Goodwin et al. 2007). Thus, our observation that MAGP-2 overexpressing cells exhibit less β-catenin localization to cellular peripheries, may indicate that MAGP-2 can indirectly interact with β-catenin/WNT signaling pathways, perhaps via manipulation of Notch signaling pathways. In support of this, our preliminary results suggest that transient overexpression of MAGP-2 in ECs increases TOPFLASH-luciferase reporter activity (i.e. LEF-1 promoter activity) (data not shown).
In summary, we have determined that the stromal protein MAGP-2 promotes angiogenic cell sprouting and perhaps angiogenesis at least in part via its ability to block Notch signaling in EC lines. Future studies will focus on defining the range of Notch receptors and ligands that are acted upon by MAGP-2.
Supplementary Material
Acknowledgments
Members of the Schiemann and Albig Laboratories are thanked for critical reading of the manuscript. This research was supported in part by grants from the National Institutes of Health (CA095519) and the Cancer League of Colorado to W.P.S., and by a fellowship from the National Institutes of Health (CA99321) to A.R.A.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- Albig AR, Neil JR, et al. Fibulins 3 and 5 antagonize tumor angiogenesis in vivo. Cancer Res. 2006;66(5):2621–9. doi: 10.1158/0008-5472.CAN-04-4096. [DOI] [PubMed] [Google Scholar]
- Albig AR, Roy TG, et al. Transcriptome analysis of endothelial cell gene expression induced by growth on matrigel matrices: identification and characterization of MAGP-2 and lumican as novel regulators of angiogenesis. Angiogenesis. 2007 doi: 10.1007/s10456-007-9075-z. [DOI] [PubMed] [Google Scholar]
- Albig AR, Schiemann WP. Fibulin-5 antagonizes vascular endothelial growth factor (VEGF) signaling and angiogenic sprouting by endothelial cells. DNA Cell Biol. 2004;23(6):367–79. doi: 10.1089/104454904323145254. [DOI] [PubMed] [Google Scholar]
- Albig AR, Schiemann WP. Identification and characterization of regulator of G protein signaling 4 (RGS4) as a novel inhibitor of tubulogenesis: RGS4 inhibits mitogen-activated protein kinases and vascular endothelial growth factor signaling. Mol Biol Cell. 2005;16(2):609–25. doi: 10.1091/mbc.E04-06-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alva JA, Iruela-Arispe ML. Notch signaling in vascular morphogenesis. Curr Opin Hematol. 2004;11(4):278–83. doi: 10.1097/01.moh.0000130309.44976.ad. [DOI] [PubMed] [Google Scholar]
- Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84(3):869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- Bild AH, Yao G, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439(7074):353–7. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- Blokzijl A, Dahlqvist C, et al. Cross-talk between the Notch and TGF-beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J Cell Biol. 2003;163(4):723–8. doi: 10.1083/jcb.200305112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigstock DR. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61) Angiogenesis. 2002;5(3):153–65. doi: 10.1023/a:1023823803510. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9(6):653–60. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438(7070):932–6. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- Cheng CW, Smith SK, et al. Wnt-1 signaling inhibits human umbilical vein endothelial cell proliferation and alters cell morphology. Exp Cell Res. 2003;291(2):415–25. doi: 10.1016/j.yexcr.2003.07.006. [DOI] [PubMed] [Google Scholar]
- Creighton C, Kuick R, et al. Profiling of pathway-specific changes in gene expression following growth of human cancer cell lines transplanted into mice. Genome Biol. 2003;4(7):R46. doi: 10.1186/gb-2003-4-7-r46. Epub 2003 Jun 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney C, Varnum-Finney B, et al. Dose-dependent effects of the Notch ligand Delta1 on ex vivo differentiation and in vivo marrow repopulating ability of cord blood cells. Blood. 2005;106(8):2693–9. doi: 10.1182/blood-2005-03-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deregowski V, Gazzerro E, et al. Notch 1 overexpression inhibits osteoblastogenesis by suppressing Wnt/beta-catenin but not bone morphogenetic protein signaling. J Biol Chem. 2006;281(10):6203–10. doi: 10.1074/jbc.M508370200. [DOI] [PubMed] [Google Scholar]
- Duarte A, Hirashima M, et al. Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 2004;18(20):2474–8. doi: 10.1101/gad.1239004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson MA, Leavesley DI, et al. Microfibril-associated glycoprotein-2 specifically interacts with a range of bovine and human cell types via alphaVbeta3 integrin. J Biol Chem. 1999;274(19):13060–5. doi: 10.1074/jbc.274.19.13060. [DOI] [PubMed] [Google Scholar]
- Goodwin AM, Kitajewski J, et al. Wnt1 and Wnt5a affect endothelial proliferation and capillary length; Wnt2 does not. Growth Factors. 2007;25(1):25–32. doi: 10.1080/08977190701272933. [DOI] [PubMed] [Google Scholar]
- Haines N, Irvine KD. Glycosylation regulates Notch signalling. Nat Rev Mol Cell Biol. 2003;4(10):78697. doi: 10.1038/nrm1228. [DOI] [PubMed] [Google Scholar]
- Hayward P, Brennan K, et al. Notch modulates Wnt signalling by associating with Armadillo/beta-catenin and regulating its transcriptional activity. Development. 2005;132(8):1819–30. doi: 10.1242/dev.01724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstrom M, Phng LK, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445(7129):776–80. doi: 10.1038/nature05571. [DOI] [PubMed] [Google Scholar]
- Hofmann JJ, Luisa Iruela-Arispe M. Notch expression patterns in the retina: An eye on receptor-ligand distribution during angiogenesis. Gene Expr Patterns. 2007;7(4):461–70. doi: 10.1016/j.modgep.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobuzio-Donahue CA, Ashfaq R, et al. Highly expressed genes in pancreatic ductal adenocarcinomas: a comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Res. 2003;63(24):8614–22. [PubMed] [Google Scholar]
- Iso T, Kedes L, et al. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003;194(3):237–55. doi: 10.1002/jcp.10208. [DOI] [PubMed] [Google Scholar]
- Jonk LJ, Itoh S, et al. Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-beta, activin, and bone morphogenetic protein-inducible enhancer. J Biol Chem. 1998;273(33):21145–52. doi: 10.1074/jbc.273.33.21145. [DOI] [PubMed] [Google Scholar]
- Kadesch T. Notch signaling: the demise of elegant simplicity. Curr Opin Genet Dev. 2004;14(5):506–12. doi: 10.1016/j.gde.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Kopan R. Notch: a membrane-bound transcription factor. J Cell Sci. 2002;115(Pt 6):1095–7. doi: 10.1242/jcs.115.6.1095. [DOI] [PubMed] [Google Scholar]
- Le Boeuf F, Houle F, et al. Regulation of vascular endothelial growth factor receptor 2-mediated phosphorylation of focal adhesion kinase by heat shock protein 90 and Src kinase activities. J Biol Chem. 2004;279(37):39175–85. doi: 10.1074/jbc.M405493200. [DOI] [PubMed] [Google Scholar]
- Lemaire R, Bayle J, et al. Microfibril-associated MAGP-2 stimulates elastic fiber assembly. J Biol Chem. 2006 doi: 10.1074/jbc.M609692200. [DOI] [PubMed] [Google Scholar]
- Lemaire R, Farina G, et al. Mutant fibrillin 1 from tight skin mice increases extracellular matrix incorporation of microfibril-associated glycoprotein 2 and type I collagen. Arthritis Rheum. 2004;50(3):915–26. doi: 10.1002/art.20053. [DOI] [PubMed] [Google Scholar]
- Lemaire R, Korn JH, et al. Increased expression of type I collagen induced by microfibril-associated glycoprotein 2: novel mechanistic insights into the molecular basis of dermal fibrosis in scleroderma. Arthritis Rheum. 2005;52(6):1812–23. doi: 10.1002/art.21059. [DOI] [PubMed] [Google Scholar]
- Leong KG, Hu X, et al. Activated Notch4 inhibits angiogenesis: role of beta 1-integrin activation. Mol Cell Biol. 2002;22(8):2830–41. doi: 10.1128/MCB.22.8.2830-2841.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong KG, Karsan A. Recent insights into the role of Notch signaling in tumorigenesis. Blood. 2006;107(6):2223–33. doi: 10.1182/blood-2005-08-3329. [DOI] [PubMed] [Google Scholar]
- Leslie JD, Ariza-McNaughton L, et al. Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Development. 2007;134(5):839–44. doi: 10.1242/dev.003244. [DOI] [PubMed] [Google Scholar]
- Liu ZJ, Xiao M, et al. Inhibition of endothelial cell proliferation by Notch1 signaling is mediated by repressing MAPK and PI3K/Akt pathways and requires MAML1. Faseb J. 2006;20(7):1009–11. doi: 10.1096/fj.05-4880fje. [DOI] [PubMed] [Google Scholar]
- MacKenzie F, Duriez P, et al. Notch4-induced inhibition of endothelial sprouting requires the ankyrin repeats and involves signaling through RBP-Jkappa. Blood. 2004;104(6):1760–8. doi: 10.1182/blood-2003-12-4244. [DOI] [PubMed] [Google Scholar]
- Mailhos C, Modlich U, et al. Delta4, an endothelial specific notch ligand expressed at sites of physiological and tumor angiogenesis. Differentiation. 2001;69(2–3):135–44. doi: 10.1046/j.1432-0436.2001.690207.x. [DOI] [PubMed] [Google Scholar]
- Masckauchan TN, Agalliu D, et al. Wnt5a signaling induces proliferation and survival of endothelial cells in vitro and expression of MMP-1 and Tie-2. Mol Biol Cell. 2006;17(12):5163–72. doi: 10.1091/mbc.E06-04-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masckauchan TN, Shawber CJ, et al. Wnt/beta-catenin signaling induces proliferation, survival and interleukin-8 in human endothelial cells. Angiogenesis. 2005;8(1):43–51. doi: 10.1007/s10456-005-5612-9. [DOI] [PubMed] [Google Scholar]
- Masson-Gadais B, Houle F, et al. Integrin alphavbeta3, requirement for VEGFR2-mediated activation of SAPK2/p38 and for Hsp90-dependent phosphorylation of focal adhesion kinase in endothelial cells activated by VEGF. Cell Stress Chaperones. 2003;8(1):37–52. doi: 10.1379/1466-1268(2003)8<37:ivrfva>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto A, Lau R, et al. Microfibrillar proteins MAGP-1 and MAGP-2 induce Notch1 extracellular domain dissociation and receptor activation. J Biol Chem. 2006;281(15):10089–97. doi: 10.1074/jbc.M600298200. [DOI] [PubMed] [Google Scholar]
- Mumm JS, Schroeter EH, et al. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell. 2000;5(2):197–206. doi: 10.1016/s1097-2765(00)80416-5. [DOI] [PubMed] [Google Scholar]
- Nehring LC, Miyamoto A, et al. The extracellular matrix protein MAGP-2 interacts with Jagged1 and induces its shedding from the cell surface. J Biol Chem. 2005;280(21):20349–55. doi: 10.1074/jbc.M500273200. [DOI] [PubMed] [Google Scholar]
- Noguera-Troise I, Daly C, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444(7122):1032–7. doi: 10.1038/nature05355. [DOI] [PubMed] [Google Scholar]
- Noseda M, Chang L, et al. Notch activation induces endothelial cell cycle arrest and participates in contact inhibition: role of p21Cip1 repression. Mol Cell Biol. 2004;24(20):8813–22. doi: 10.1128/MCB.24.20.8813-8822.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell KA, Edidin M. A mouse lymphoid endothelial cell line immortalized by simian virus 40 binds lymphocytes and retains functional characteristics of normal endothelial cells. J Immunol. 1990;144(2):521–5. [PubMed] [Google Scholar]
- Rebay I, Fehon RG, et al. Specific truncations of Drosophila Notch define dominant activated and dominant negative forms of the receptor. Cell. 1993;74(2):319–29. doi: 10.1016/0092-8674(93)90423-n. [DOI] [PubMed] [Google Scholar]
- Rehman AO, Wang CY. Notch signaling in the regulation of tumor angiogenesis. Trends Cell Biol. 2006 doi: 10.1016/j.tcb.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Ridgway J, Zhang G, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444(7122):1083–7. doi: 10.1038/nature05313. [DOI] [PubMed] [Google Scholar]
- Ross DA, Kadesch T. The notch intracellular domain can function as a coactivator for LEF-1. Mol Cell Biol. 2001;21(22):7537–44. doi: 10.1128/MCB.21.22.7537-7544.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainson RC, Aoto J, et al. Cell-autonomous notch signaling regulates endothelial cell branching and proliferation during vascular tubulogenesis. Faseb J. 2005;19(8):1027–9. doi: 10.1096/fj.04-3172fje. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Yamaguchi S, et al. The nephroblastoma overexpressed gene (NOV/ccn3) protein associates with Notch1 extracellular domain and inhibits myoblast differentiation via Notch signaling pathway. J Biol Chem. 2002;277(33):29399–405. doi: 10.1074/jbc.M203727200. [DOI] [PubMed] [Google Scholar]
- Sastre M, Steiner H, et al. Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2001;2(9):835–41. doi: 10.1093/embo-reports/kve180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk PM, Baumann S, et al. Improved high-level expression system for eukaryotic genes in Escherichia coli using T7 RNA polymerase and rare ArgtRNAs. Biotechniques. 1995;19(2):196–8. 200. [PubMed] [Google Scholar]
- Schiemann WP, Blobe GC, et al. Context-specific effects of fibulin-5 (DANCE/EVEC) on cell proliferation, motility, and invasion. Fibulin-5 is induced by transforming growth factor-beta and affects protein kinase cascades. J Biol Chem. 2002;277(30):27367–77. doi: 10.1074/jbc.M200148200. [DOI] [PubMed] [Google Scholar]
- Shawber CJ, Kitajewski J. Notch function in the vasculature: insights from zebrafish, mouse and man. Bioessays. 2004;26(3):225–34. doi: 10.1002/bies.20004. [DOI] [PubMed] [Google Scholar]
- Shutter JR, Scully S, et al. Dll4, a novel Notch ligand expressed in arterial endothelium. Genes Dev. 2000;14(11):1313–8. [PMC free article] [PubMed] [Google Scholar]
- Small D, Kovalenko D, et al. Soluble Jagged 1 represses the function of its transmembrane form to induce the formation of the Src-dependent chord-like phenotype. J Biol Chem. 2001;276(34):32022–30. doi: 10.1074/jbc.M100933200. [DOI] [PubMed] [Google Scholar]
- Uyttendaele H, Marazzi G, et al. Notch4/int-3, a mammary proto-oncogene, is an endothelial cell-specific mammalian Notch gene. Development. 1996;122(7):2251–9. doi: 10.1242/dev.122.7.2251. [DOI] [PubMed] [Google Scholar]
- Vorontchikhina MA, Zimmermann RC, et al. Unique patterns of Notch1, Notch4 and Jagged1 expression in ovarian vessels during folliculogenesis and corpus luteum formation. Gene Expr Patterns. 2005;5(5):701–9. doi: 10.1016/j.modgep.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Weinmaster G. The ins and outs of notch signaling. Mol Cell Neurosci. 1997;9(2):91–102. doi: 10.1006/mcne.1997.0612. [DOI] [PubMed] [Google Scholar]
- Wexler H, Rosenberg SA. Pulmonary metastases from autochthonous 3-methylcholanthrene-induced murine tumors. J Natl Cancer Inst. 1979;63(6):1393–5. [PubMed] [Google Scholar]
- Williams CK, Li JL, et al. Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood. 2006;107(3):931–9. doi: 10.1182/blood-2005-03-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimrin AB, Pepper MS, et al. An antisense oligonucleotide to the notch ligand jagged enhances fibroblast growth factor-induced angiogenesis in vitro. J Biol Chem. 1996;271(51):32499–502. doi: 10.1074/jbc.271.51.32499. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.