

Abstract
While the onset and extent of epilepsy increases in the aged population, the reasons for this increased incidence remain unexplored. The present study used two inbred strains of mice (C57BL/6J and FVB/NJ) to address the genetic control of age-dependent neurodegeneration by building upon previous experiments that have identified phenotypic differences in susceptibility to hippocampal seizure-induced cell death. We determined if seizure induction and seizure-induced cell death are affected differentially in young adult, mature, and aged male C57BL/6J and FVB/NJ mice administered the excitotoxin, kainic acid. Dose response testing was performed in three-four groups of male mice from each strain. Following kainate injections, mice were scored for seizure activity and brains from mice in each age group were processed for light microscopic histopathologic evaluation seven days following kainate administration to evaluate the severity of seizure-induced brain damage. Irrespective of the dose of kainate administered or the age group examined, resistant strains of mice (C57BL/6J) continued to be resistant to seizure-induced cell death. In contrast, aged animals of the FVB/NJ strain were more vulnerable to the induction of behavioral seizures and associated neuropathology after systemic injection of kainic acid than young or middle-aged mice. Results from these studies suggest that the age-related increased susceptibility to the neurotoxic effects of seizure induction and seizure-induced injury is regulated in a strain-dependent manner, similar to previous observations in young adult mice.
Keywords: mouse strain, excitotoxicity, kainic acid, epilepsy, cell death, aging
Knowledge about the influence of aging on the susceptibility of the brain to seizure disorders is of critical importance in geriatric medicine and public health. Epilepsy is the most common neurological disorder after stroke, affecting more than 50 million persons worldwide and with a 2–3% life time risk of being given a diagnosis of epilepsy (Browne and Holmes, 2001). Currently it is well known that there is a significant incidence of epilepsy in children. However, less clearly recognized but of increasing importance is the significant incidence of epilepsy in the elderly population. Recent studies in the United States and Europe indicate that the age-specific incidence of epilepsy is higher in those over the age of 65 years than in those in the first decade of life (Tallis et al., 1991; Hauser, 1992; DeLorenzo et al., 1997; Treiman et al., 1998; Wallace et al., 1998; Epilepsy Foundation of America, 1999).
While in the elderly, this pattern can partly be explained by the age of occurrence of diseases associated with the onset of seizures (e.g. Alzheimer’s disease, cerebrovascular disease, and brain tumors), the incidence of convulsive disorders in the elderly is increased even among those with unprovoked seizures without an identified cause (reviewed in Hauser, 1992). Case studies suggest that aging alone may have an epileptogenic effect on the neuron and descriptive case studies and results from animal studies have suggested that advancing age increases the vulnerability to both seizure induction and acute neurological events for partial epilepsy and possibly for generalized-onset epilepsy, as well (reviewed in Wozniak et al., 1991; Hauser, 1992; Kerr et al., 2002). However, while it has been suggested that old age is the most common time for people to develop seizures (Hauser, 1992; Wallace et al., 1998), little is known about whether aging per se is an independent risk factor for epileptogenesis. Likewise, while epilepsy is one of the most common neurological conditions, our understanding of the molecular pathways that regulate cell death after seizure activity remain in their infancy and largely lag behind work in other areas of brain injury.
The use of animal models has been an essential component in the discovery and development of new drugs for the treatment of epilepsy. While a number of experimental animal models have been developed to help elucidate the pathophysiology of this condition (reviewed in Löscher, 1997, 2002; Coulter et al., 2002; Morimoto et al., 2004), one of the most widely used animal models of temporal lobe epilepsy (Sperk, 1994) involves the systemic administration of chemoconvulsants, such as kainic acid (KA), a potent agonist of the AMPA/kainate class of glutamate receptors. Systemic and intracerebral administration of kainic acid (KA) initially induces a characterized pattern of seizure activity (Coyle, 1983) that lasts for hours, followed by a latent seizure-free period of weeks, preceding the development of spontaneous recurrent focal seizures that begin between 3–4 weeks (Brandt et al., 2003). In addition, administration of kainic acid activates ionotropic glutamate receptors (iGluRs), and selectively induces excitotoxic cell death in the CA3 and CA1 hippocampal subfields and within the dentate hilus, while sparing neurons in the CA2 subfield and the dentate granule cell layer (Nadler et al., 1978; Schwob et al., 1980; Nadler, 1981; Ben-Ari, 1985). Furthermore, there is a direct relationship between the generation of epileptiform activity and the extent of damage in hippocampal subfields (Ben-Ari, 1985; Okazaki and Nadler, 1988). Many features of this rodent model, such as hippocampal sclerosis and mossy fiber sprouting, resemble human temporal lobe epilepsy. As a result, the kainate model replicates several phenomenological features of human temporal lobe epilepsy and can be used as an animal preparation to understand the basic mechanisms of epileptogenesis (Ben-Ari, 1985; Turski et al., 1989; Buckmaster and Dudek, 1997). While a large number of studies have addressed age-dependent effects on susceptibility to seizure-induction and seizure-induced cell damage during development (Haas et al., 2001; Baram et al., 2002; Galanopoulou et al., 2002; Lado et al., 2002), few studies have addressed an age-dependent analysis of seizure-induced cell death susceptibility. The present study was aimed at investigating the age-related induction of KA-induced seizure severity in mice and the relation to seizure-induced cell death.
EXPERIMENTAL PROCEDURES
Animals
Young (2 month old), middle-aged (12 months) and old (18 months) male C57BL/6J mice, obtained from the NIA aging colony, and FVB/NJ mice, aged in-house, were used as animal subjects. Animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Southern California. Every effort was made to minimize the number of animals used and their suffering. FVB/NJ mice were 7–8 months of age when purchased and aged in-house in the animal facilities for an additional period of 5–11 months before use.
Kainic acid administration
Kainic acid (Diagnostic Chemical, Ltd., Charlottetown, PEI) was dissolved in isotonic saline (pH 7.3) and administered subcutaneously to adult male mice. Preliminary dose response studies had defined seizure thresholds and revealed consistent seizures among all six inbred mouse strains, with a mortality of less than 25% with a dose of 25 mg/kg, s.c. (Schauwecker and Steward, 1997). Dose response testing was performed in three-four groups of male mice (n=7 mice/group) from each strain; 10-, 15-, 20-, 25- and 30-mg/kg doses of KA were injected subcutaneously (s.c.). KA solutions were prepared fresh on the day of each experiment and each animal received only one injection.
Seizure testing
Following KA injections, mice were placed in clear plastic cages and monitored every 15 min for 4 h for the onset of locomotor activity and behavioral manifestations of limbic seizure episodes. As described previously (Schauwecker, 2000; Schauwecker et al., 2000; Schauwecker, 2002a,b; Santos and Schauwecker, 2003), these episodes commenced with automatisms including staring, rigidity, and immobility, followed by jaw movements, blinking, and head bobbing, and forelimb clonus. The next stage of seizures which consisted of rearing, forelimb/head clonus, and tonic/clonic seizures, postural imbalance, and uncontrolled running and jumping defined the latency to first maximal seizure. Status epilepticus was defined as continuous behavioral seizure activity lasting at least 1 hour or a series of intermittent seizures without restoration of normal behavioral patterns between successive seizures. Mice were scored for seizure activity using a previously defined seizure scoring scale as follows (Racine, 1972): Stage 1, immobility; Stage 2, forelimb and/or tail extension, rigid posture; Stage 3, repetitive movements, head bobbing; Stage 4, rearing and falling; Stage 5, continuous rearing and falling; Stage 6, severe tonic-clonic seizures. Seizure parameters monitored included latency of convulsions, duration of seizure activity, and mortality.
Histological staining
In order to evaluate the severity of KA-induced excitotoxic brain damage in different strains of mice, brains from each strain of mice were processed for light microscopic histopathologic evaluation according to previously published methods (Schauwecker et al., 2004). Briefly, seven days after seizure induction by KA, mice were anesthetized with Avertin and transcardially perfused with 4% paraformaldehyde in 0.1M phosphate buffer (pH 7.4). Brains were removed and post-fixed in 30% sucrose for at least 12–18 hours for cryoprotection. Horizontal (35 μm) sections were cut on a sliding microtome and immersed in phosphate buffer (pH 7.4); free-floating until histological processing was started. Every sixth section (~210 μm) was processed for cresyl violet staining to assess cell loss and morphology. An alternate series of sections were stained with a modified Gallyas silver stain, which stains degenerating fibers, synaptic terminals, and cell bodies (Nadler and Evenson, 1983; as modified in Schauwecker, 2003), and examined for the appearance of degenerative debris. An additional series of sections were stained with Fluoro-Jade C, a fluorescent marker for dying neurons, according to the method outlined previously (Schmued et al., 2005). Briefly, horizontal sections were mounted from 0.1M phosphate buffer (pH 7.4) onto gelatin-coated slides and allowed to air-dry for several hours. Slides were then immersed in absolute alcohol for 3 min, followed by 70% ethanol for 2 min, and distilled water for 2 min. The slides were transferred to 0.06% potassium permanganate for 15 min. After rinsing with distilled water for 2 min, the slides were incubated for 30 min in 0.001% Fluoro-jade solution (Chemicon, Temecula, CA) made in 0.1% acetic acid. Slides were rinsed briefly in water, allowed to air dry for several minutes, cleared in xylene, and coverslipped with non-fluorescent mounting media (Kirkegaard & Perry Laboratories, Gaithersburg, MD).
Neuronal loss quantification
We counted cells in defined areas of CA1, CA3, the dentate hilus, and the dentate gyrus in a blinded manner using unbiased stereological methods on cresyl violet-stained sections as described (Schauwecker and Steward, 1997; Schauwecker et al., 2000). The number of Nissl-stained neurons in area CA3, area CA1, the dentate hilus, and the dentate gyrus were counted in both the right and left hippocampus and counting was initiated within the ventral hippocampus at the first point where hippocampal subfields could be easily identified. This level corresponded to horizontal section 54, based on the atlas of Sidman et al. (1971). Hippocampal subfields were based on Franklin and Paxinos classification (1997) and discrimination between the CA3 and dentate hilus region was based on morphological features and locations of the cells (West et al., 1991). Specifically, for dentate hilar cell counts, the hilus was operationally defined as the region bordered by the supra- and infrapyramidal granule cell layers and excluding the densely packed pyramidal neurons of area CA3.
Neuron counts were made in all subfields and the numbers for each side were averaged into single values for each animal. Surviving cells were counted only if they were contained within the pyramidal cell layer, dentate hilus, or dentate gyrus, possessed a visible nucleus and characteristic neuronal morphology, and had a cell body larger than 10 μm. Six square counting frames (200 × 200 μm) were randomly placed in the pyramidal layer of fields CA1 and CA3 or in the dentate hilus or dentate gyrus in 4–5 regularly-spaced horizontal sections from each animal. Neuronal nuclei were evaluated at three different focal planes and only those in the focal plane were counted with a 40X objective and considered as a counting unit. Stereological analysis was performed with the aid of Image-Pro Plus software (Media Cybernetics, Inc., Silver Spring, MD) in combination with a SPOT digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI), and a motorized Z-stage (Optiscan, Prior Scientific, Fairfax, VA). Final cell counts are expressed as the percentage of cells as compared to intact mice. Results were assessed statistically by one-way ANOVA using the computer program, SigmaStat (Jandel Scientific, San Rafael, CA), and intergroup differences were analyzed by the Student Newman-Keuls post hoc test. Data were considered significant at P<0.05.
RESULTS
We have previously demonstrated that certain strains of mice are strongly resistant to seizure-induced excitotoxic cell death in spite of similar seizure severity (Schauwecker and Steward, 1997; Schauwecker et al., 2000; Schauwecker, 2000; 2002a,b). Here we asked whether: 1) age can act as a risk factor to increase the susceptibility of mice to kainate-induced behavioral seizures and seizure-induced hippocampal cell loss; and 2) genetic susceptibility to seizure-induced cell death is affected by age.
Strain differences in seizure parameters among young adult mice
Young adult mice from both strains exhibited dose-dependent increases in behavioral seizure scores (see Seizure incidence in Tables 1,2) and in the duration of severe seizures (Tables 1,2). However, while dose-dependent increases in mortality were observed in both strains, a strain-dependent difference in mortality was only observed at a dose of 15 mg/kg, with FVB/NJ mice displaying a significant increase in mortality as compared to C57BL/6J mice. No other significant differences in seizure severity were observed at the other doses of kainate. Similarly, the dose of KA effective for producing limbic status epilepticus in 50% of mice in each group (ED50) was different between strains (Fig. 1). While mice of the FVB strain appeared to enter status epilepticus at a dose of 15 mg/kg of kainate, between 15–20 mg/kg of kainate elicited status in the B6 strain.
Table 1.
Effect of kainic acid administration on seizure parameters in young, middle-aged, and aged mice
| Mouse strain | Age group | Dose (mg/kg) | Seizure incidence | Latency (min.) | Duration (min.) | Mortality |
|---|---|---|---|---|---|---|
| C57BL/6J | Young | 15 | 5/7 | 55.0±7.7 | 35.0±15.5 | 0/7 |
| C57BL/6J | Young | 20 | 8/8 | 55.6±8.1 | 78.8±9.2 | 2/8 |
| C57BL/6J | Young | 25 | 8/8 | 59.3±7.3 | 90.0±14.1 | 2/8 |
| C57BL/6J | Young | 30 | 17/17 | 38.9±4.8 | 35.3±7.8 | 10/17 |
|
| ||||||
| C57BL/6J | Mature | 10 | 1/6 | 72.0±0.0 | 55.0±0.0 | 0/6 |
| C57BL/6J | Mature | 15 | 5/7 | 55.4±15.2 | 26.4±14.7 | 0/7 |
| C57BL/6J | Mature | 20 | 14/14 | 41.4±4.9 | 47.7±7.5 | 6/14 |
|
| ||||||
| C57BL/6J | Aged | 10 | 5/6 | 40.0±8.8 | 61.7±17.9 | 2/6 |
| C57BL/6J | Aged | 15 | 6/6 | 31.5±3.4 | 55.2±10.0 | 2/6 |
| C57BL/6J | Aged | 20 | 5/6 | 19.5±4.3 | 39.0±24.9 | 4/6 |
Table 2.
Effect of kainic acid administration on seizure parameters in young, middle-aged, and aged mice
| Mouse strain | Age group | Dose (mg/kg) | Seizure incidence | Latency (min.) | Duration (min.) | Mortality |
|---|---|---|---|---|---|---|
| FVB/NJ | Young | 15 | 9/9 | 48.8±5.2 | 39.7±10.2 | 4/9 |
| FVB/NJ | Young | 20 | 9/10 | 62.7±8.3 | 58.2±9.7 | 4/10 |
| FVB/NJ | Young | 25 | 10/10 | 69.4±2.2 | 70.6±8.5 | 5/10 |
| FVB/NJ | Young | 30 | 20/20 | 46.2±4.7 | 56.3±7.0 | 12/20 |
|
| ||||||
| FVB/NJ | Mature | 10 | 5/6 | 34.3±7.3 | 64.2±18.6 | 3/6 |
| FVB/NJ | Mature | 15 | 13/13 | 34.3±3.3 | 52.5±11.7 | 9/13 |
| FVB/NJ | Mature | 20 | 13/13 | 37.3±4.5 | 68.9±6.3 | 9/13 |
|
| ||||||
| FVB/NJ | Aged | 10 | 7/7 | 65.7±7.6 | 62.9±14.9 | 5/7 |
| FVB/NJ | Aged | 15 | 6/6 | 28.3±3.6 | 60.3±8.6 | 3/6 |
| FVB/NJ | Aged | 20 | 5/5 | 24.8±4.1 | 50.6±20.1 | 2/6 |
Fig. 1.

Dose-response curves of kainate-induced seizures in 3 different age groups of mice (C57BL/6J and FVB/NJ). Dose-response curves for convulsant effects of KA were expressed as the percentage of animals (n=12 mice per data point) displaying Racine Stage 5 seizures in response to systemic kainate injection (mg/kg). The dotted horizontal line depicts the ED50 value, which is a dose predicted to induce Racine Stage 5 seizures in 50% of animals.
Strain differences in seizure parameters among middle-aged mice
Similar to our observations in young adult mice, irrespective of mouse strain, a dose-dependent increase in seizure incidence was observed (Tables 1,2). In contrast, latency to severe seizures decreased in a dose-dependent manner only in B6 mice, and remained unchanged irrespective of dose in FVB mice. While in general, the duration of severe seizures did not differ significantly across doses in either mouse strain, a significant strain-dependent difference in seizure duration was noted at the highest dose (20 mg/kg) of kainate in mature mice in that FVB mice exhibited seizures for a significantly longer period of time as compared to B6 mice. Similar to previous observations in young adult mice, mortality increased in both strains in a dose-dependent manner. A significant strain-dependent difference in mortality was observed at a dose of 15 mg/kg kainate with approximately 69% of FVB mice dying while no B6 mice died at this dose. Similarly to our observations in young adult mice, while the ED50 for status in mature animals was 10 mg/kg for FVB mice, it was between 15–20 mg/kg of kainate in the B6 strain (Fig. 1).
Strain differences in seizure parameters among aged mice
The most striking difference among this age group was the heightened sensitivity of aged mice, irrespective of dose and strain, to KA. Nearly all mice exhibited severe seizures at all doses of kainate. In addition, irrespective of strain, latency was decreased in a dose-dependent manner (Tables 1,2). A significant strain-dependent difference in latency was observed at the lowest dose of kainate (10 mg/kg) with FVB mice exhibiting a significantly reduced latency to onset of severe seizures. In contrast, no strain-dependent differences in either seizure duration or mortality were observed. A significant strain-dependent difference in the ED50 for status was observed in that a kainate dose of 10–15 mg/kg in B6 mice triggered status, while it was already saturated at 10 mg/kg for FVB mice (Fig. 1).
Effect of age on susceptibility to seizure-induced cell death: Young adult mice
Previously, we had discovered that commonly used mouse strains exhibit differences in susceptibility to kainic acid-induced cell death in the hippocampus. Following systemic administration of KA to young adult B6 and FVB mice, comparable seizure activity (e.g. no differences in time spent in status epilepticus or duration of seizures) is observed. However, resistant strains of mice (B6) show essentially no cell death within the hippocampus or within any other region of the brain. Our results in the young adult (2 month) group replicate these findings. In particular, irrespective of the dose of kainate administered to young adult mice, we found a significant strain-dependent difference in susceptibility to seizure-induced cell death (Figure 2; Figure 5). However, within the FVB strain, cell loss in the three hippocampal subfields (hilus, area CA3, area CA1) did not differ in a dose-dependent manner (Figure 2; Table 3).
Fig. 2.
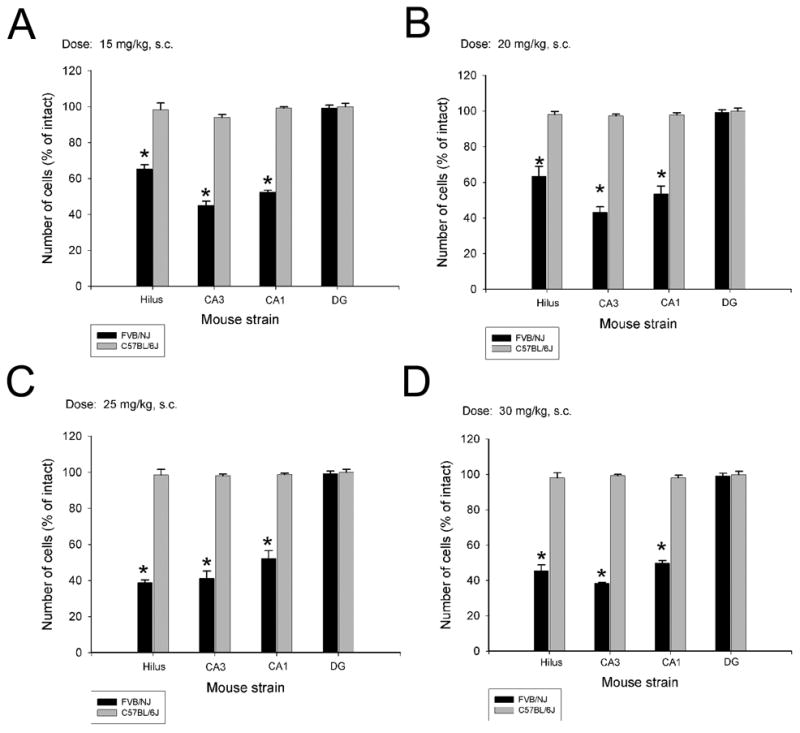
Quantification of kainate-induced neuronal damage in hippocampal subfields at 4 different doses of KA in young adult C57BL/6J and FVB/NJ mice. A strain-dependent difference in cell loss in the dentate hilus, area CA3 and area CA1 was observed at 7 days following KA administration in FVB/NJ mice irrespective of the dose of kainate administered. Data represent the mean ± SEM of at least 8 mice/strain. *P<0.05. DG, dentate gyrus; H, hilus; CA3, area CA3; CA1, area CA1.
Fig. 5.

Neuronal cell loss following kainate administration in C57BL/6J and FVB/NJ mice is strain-dependent in three different age groups of mice. Low- and high-power photomicrographs of cresyl violet-stained horizontal sections of the hippocampus showing the destruction of neurons in the CA3 and CA1 subfields and within the dentate hilus 7 days after kainate administration in all three age groups of FVB/NJ mice. In contrast, no cell loss was evident in C57BL/6J mice of any age group following kainate administration. CA3, CA3 pyramidal cell layer; CA1, CA1 pyramidal cell layer; H, hilus. Scale bars, 750 μm (low-power photomicrographs); 350 μm (high-power photomicrographs).
Table 3.
Age-response comparison of hippocampal cell loss (percentage) following kainate administration in FVB/NJ mice
| Hippocampal subregion | Dose (mg/kg) | Young Adult | Mature | Aged | P-value |
|---|---|---|---|---|---|
| Hilus | 10 | N/A | 3.3±2.2 | 50.1±13.5* | 0.001 |
| 15 | 34.8±2.5 | 64.8±14.8 | 94.4±5.6* | 0.003 | |
| 20 | 36.6±7.0 | 76.0±13.9 | 48.1±13.4 | ||
| CA3 | 10 | N/A | 0.9±1.0 | 4.5±0.1* | 0.041 |
| 15 | 55.0±2.3 | 55.8±12.1 | 79.4±6.1 | ||
| 20 | 57.0±7.3 | 78.7±12.1 | 62.2±11.2 | ||
| CA1 | 10 | N/A | 0.1±0.9 | 1.8±0.5 | |
| 15 | 47.5±1.0 | 46.6±16.9 | 72.0±6.8 | ||
| 20 | 46.5±11.5 | 64.6±16.8 | 15.6±8.8* | <0.001 |
Values represent means ± S.E.M. of at least 5 mice in each age group.
P<0.05 as compared to other age groups.
Effect of age on susceptibility to seizure-induced cell death: Middle-aged mice
Among mature (12 month old) mice, a significant strain-dependent difference in seizure-induced cell loss was still observed irrespective of the kainate dose (Figure 3; Figure 5). Among FVB mice, while cell loss was not detectable at a dose of 10 mg/kg, a significant reduction in hippocampal neurons within the dentate hilus (F=11.834; P=0.001), area CA3 (F=16.306; P<0.001) and area CA1 (F=5.895; P=0.016) was observed at both the 15- and 20-mg/kg doses of kainate (Fig. 3B,C; Figure 6). It is important to note that while we did not observe a significant increase in the extent of hippocampal cell loss between the 15- and 20-mg/kg kainate doses, cell loss was increased in a dose-dependent manner among all three hippocampal subfields (Table 3).
Fig. 3.
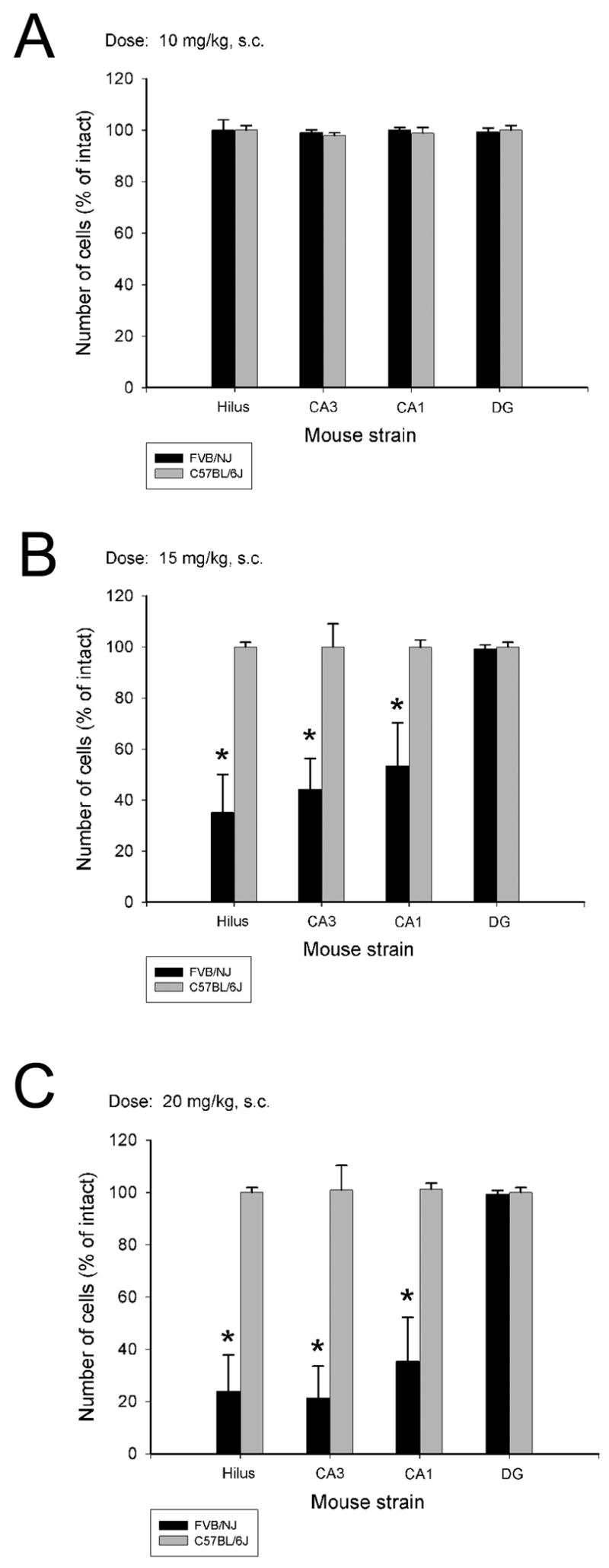
Quantitative analysis of neuronal density in hippocampal subfields following administration of 3 different doses of KA to middle-aged C57BL/6 and FVB/NJ mice. While no cell loss as observed at the lowest dose of KA (A), a significant increase in cell loss was observed in the dentate hilus, and areas CA3 and CA1 of FVB/NJ mice seven days following KA administration. Data represent the mean ± SEM of at least 8 mice/strain. *P<0.05.
Fig. 6.

Comparison of susceptibility to kainate-induced cell death in FVB/NJ mice of three different age groups. Photomicrographs of Fluoro-Jade C-stained horizontal hippocampal sections at low and high magnification showing loss of neurons within the hippocampus in three age groups of FVB/NJ mice 7 days following kainate administration. Note the degeneration of neurons in the dentate hilus, and in the CA3 and CA1 subfields of the hippocampus as demonstrated with the fluorescent marker, Fluoro-Jade C. CA1 and CA3 denote the hippocampal subfields; H, dentate hilus. Scale bars: low-power photomicrographs, 750 μm; high-power photomicrographs, 100 μm.
Effect of age on susceptibility to seizure-induced cell death: Aged mice
Aged B6 mice continued to retain their resistance to kainate-induced cell death irrespective of the dose of kainate administered (Figure 4; Figure 5). However, cell death in FVB/N mice, administered the lowest dose of kainate (10 mg/kg), was restricted solely to the dentate hilus (Fig. 4A; Table 3), with no significant signs of cell death observed in either area CA3 or CA1. The most dramatic reduction in hippocampal number across all 3 subfields was observed at a dose of 15 mg/kg (Fig. 4B; Table 3). Interestingly, cell loss was only significantly reduced in the dentate hilus and in area CA3 at a dose of 20 mg/kg kainate (Fig. 4C; Figure 6). While some hippocampal cell loss was noted in area CA1, it was not significantly reduced as compared to intact animals (Fig. 4C; Table 3; Figure 6).
Fig. 4.
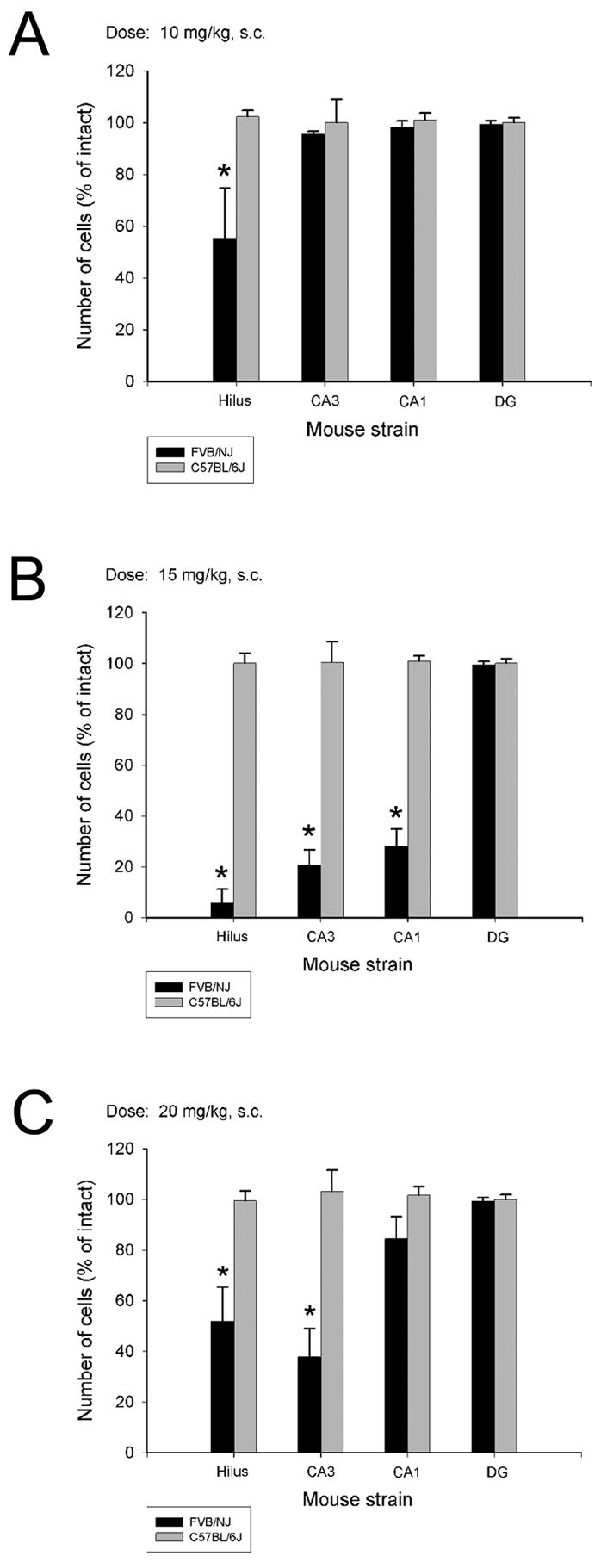
Quantitative analysis of neuronal density in hippocampal subfields following systemic administration of KA at 3 different doses to two strains of aged mice. Strain-dependent differences in cell loss in the dentate hilus were observed at all 3 doses of KA. In contrast, strain-dependent differences in cell loss in area CA3 were observed at only the two highest doses (B and C), while cell loss in area CA1 was only observed at a dose of 15 mg/kg (B). No significant differences in the extent of cell loss were observed 7 days following administration of KA, irrespective of dose, in C57BL/6J mice. Data represent the mean ± SEM of eight mice/dose for each strain. *P<0.05.
DISCUSSION
This study used the kainate chemoconvulsant model to determine whether age is a risk factor for kainate-induced seizures and seizure-induced cell death in middle-aged and aged mice in comparison with established outcomes in young adult mice that are differentially susceptible to kainate-induced cell death. As a first step towards addressing the mechanism regulating susceptibility differences, we wanted to determine whether the variable response to kainate seizure induction and excitotoxic injury results from differences in pharmacological sensitivity to kainate. We found that, irrespective of mouse strain, aging mice showed increased behavioral seizure severity to kainate. In general, mice of both strains and all age groups exhibited the behavioral manifestations typically evoked by convulsant doses of KA. In addition, vulnerability of the hippocampus to KA increased with age in FVB mice, while no obvious damage was observed in B6 mice irrespective of their age. Results from this study highlight two important principles. First, resistance to KA continues throughout the lifespan of B6 mice, as aged mice continue to be resistant to the neurotoxic effects of KA. Second, aging increases the vulnerability to induction of KA-induced seizures and associated neuropathology in FVB mice.
Our finding that aging increased behavioral seizure sensitivity to systemic kainate administration in both mouse strains is in agreement with previous reports (Wozniak et al., 1991; Dawson and Wallace, 1992). In particular, Dawson and Wallace (1992) reported a significant reduced latency to full clonic-tonic seizures in aged Long-Evans and F-344 rats. As well, the number of seizures was also significantly increased above that of controls. Similarly, Wozniak et al. (1991) found that middle-aged and old rats were significantly more sensitive than young rats to the seizure-inducing properties of kainate. In our study, among the age groups assessed, the most significant difference was the increased sensitivity to the seizure-induction properties of kainate in the oldest age group of mice (18 mos.). Irrespective of strain, a significant upward shift in the percentage of mice achieving status was observed. However, it is interesting to note that while there was a significant age-effect among all three age groups in FVB mice, among B6 mice, a significant age-effect was only noted when comparing the aged group to the young and middle-aged groups.
The increased sensitivity of aged mice to the proconvulsant actions of KA could reflect age-related changes in limbic and extra-limbic network activity. In particular, Darbin et al. (2004) found an age-related reduction in fast frequencies in EEG recordings and behavioral characteristics elicited during kainate treatment in aged Fischer 344 rats, suggestive of a change in seizure network activity. Alterations in seizure network activity could result from age-related alterations in synaptic connectivity (Barnes and McNaughton, 1980; Rapp et al., 1999; Smith et al., 2000), electronic coupling (Barnes et al., 1987), receptor and channel properties (Pitler and Landfield, 1990; Gutiérrez et al., 1996), or the number and type of neurons (Shetty and Turner, 1998; Cadiacio et al., 2003; Stanley and Shetty, 2004).
Age-related alterations in neurotransmitter and second messenger systems in the brain could also play a role in altering the sensitivity of aged mice to the proconvulsant actions of KA. In particular, alterations in seizure sensitivity to kainate could result from the decline in glutamate receptor densities observed in aged rodents (Tamaru et al., 1991; Carpenter et al., 1992; Clark et al., 1992). Several studies have recognized age-related changes in the density and function of the different ionotropic glutamate receptors (Gonzales et al., 1991; Pittaluga et al., 1993; Nicoletti et al., 1995; Magnusson, 1998; Mitchell and Anderson, 1998; Wenk and Barnes, 2000). Although still somewhat controversial, many studies have shown that the expression of AMPA-, KA- and NMDA-sensitive receptors as well as the GABAA receptor is either constant or variably diminished in many different brain regions including the hippocampus during aging (Gonzales et al., 1991; Pittaluga et al., 1993; Le Jeune et al., 1996; Nicolle et al., 1996; Eckles-Smith et al., 2000; Kuehl-Kovarik et al., 2000; Magnusson, 2000; Sonntag et al., 2000; Wenk and Barnes, 2000; Adams et al., 2001; Clayton and Browning, 2001; Clayton et al., 2002; Lerma et al., 2001). Differences in a variety of other factors, including voltage-gated calcium channels (Vigues et al., 1999; Kelly et al., 2003), androgen levels (Mejias-Aponte et al., 2002; Ramsden et al., 2003; Ciriza et al., 2004), and GABA receptor function (MacGregor et al., 1997; Ma et al., 2001) which have been shown to modulate kainate-induced seizure activity in young animals, may modulate susceptibility in aged animals as well.
With regard to the age-dependent increase in susceptibility to KA-induced neuronal damage in FVB mice, our results are in agreement with previous studies in rats (Wozniak et al., 1991; Golden et al., 1995; Kesslak et al., 1995). Regardless of age, all FVB mice observed to be in status epilepticus, exhibited the well-documented acute neuropathologic response to convulsant doses of KA (Nadler et al., 1980a,b; Schwob et al., 1980; Nadler, 1981; Coyle, 1983; Sperk et al., 1983; Ben-Ari, 1985; Jarrard and Meldrum, 1993; Olney, 1994), while no obvious damage was observed in C57BL/6 mice. While the mechanisms whereby neuronal cells die following an excitotoxic insult are not fully understood, it has been hypothesized that a variety of cascades involving biochemical and electrophysiological events combine to produce neuronal damage (Choi, 1992; Whetsell, 1996; Michaelis, 1998; Nicholls and Budd, 1998; Sattler and Tymianski, 2000). As the neurodegeneration that occurs after kainate-induced status epilepticus has the features of delayed excitotoxic cell death (Meldrum, 1993; Aarts and Tymianski, 2003; Malva et al., 2003), current models of excitotoxicity have suggested that damage is mediated through a mechanism involving the activation of presynaptic glutamate receptors and the excessive release of glutamate (Choi, 1992, 1994). Despite findings that there is an age-related component to kainate toxicity (Auer, 1991; Kesslak et al., 1995), the molecular basis for this difference in kainate-induced seizure susceptibility is not clear.
In agreement with our reported results of greater damage in aged FVB/N mice following kainate administration, other aged animals and humans also appear to exhibit greater excitotoxic damage following experimental kainate than young controls (Auer, 1991; Wozniak et al., 1991; Kesslak et al., 1995). At present, the mechanisms responsible for age-dependent regulation of seizure–induced cell damage remain unclear. Reports using systemic administration of KA show that vulnerability of the hippocampus to KA increases with aging and while many factors may contribute, alterations in blood-brain barrier permeability may be one of these contributing factors as exemplified by the increased permeability of the blood brain barrier in aged animals (Wozniak et al., 1991; Dawson and Wallace, 1992; Kesslak et al., 1995). In particular, injection of glutamate analogs either systemically or intracerebrally can cause BBB breakdown in some regions of the brain, such as the hippocampus (Nitsch and Hubauer, 1986; Ruth, 1986; Ruth and Feinerman, 1988). Furthermore, differences in the ability of aging animals to metabolize kainate or the pharmacokinetics of drug delivery may produce alterations in CNS bioavailability of the drug.
CONCLUSION
In summary, our findings demonstrate that seizure susceptibility in both strains increases with advancing age. Among FVB mice, seizure-induced cell death becomes more pronounced with advancing age, while C57BL/6 mice continue to be resistant to KA-induced cell death. Future studies will need to reveal the risk factors and mechanisms responsible for differential vulnerability to glutamate excitotoxicity between these two mouse strains and the interaction with age. Besides differences in glutamate receptor expression or responsiveness, alterations in the activity of inhibitory pathways in the hippocampus could also underlie the differential vulnerability of the hippocampus to kainate. An improved understanding of the mechanisms responsible for aging as a risk factor is a necessary first step in the development of neuroprotective treatments that can be applied to individuals most likely at risk of developing epilepsy.
Acknowledgments
This work was supported by NIH grant AG025508 to P.E.S. and by the Rose Hills Foundation Research Fellowship to C.S.B.. The authors wish to thank Dr. Thomas H. McNeill for thoughtful comments on the manuscript.
Abbreviations
- ANOVA
analysis of variance
- B6
C57BL/6J
- FVB
FVB/NJ
- iGluRs
ionotropic glutamate receptors
- KA
kainic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aarts MM, Tymianski M. Novel treatment of excitotoxicity: targeted disruption of intracellular signaling from glutamate receptors. Biochem Pharmacol. 2003;66:877–886. doi: 10.1016/s0006-2952(03)00297-1. [DOI] [PubMed] [Google Scholar]
- Adams MM, Smith TD, Moga D, Gallagher M, Wang Y, Wolfe BB, Rapp PR, Morrison JH. Hippocampal dependent learning ability correlates with N-methyl-D-aspartate (NMDA) receptor levels in CA3 neurons of young and aged rats. J Comp Neurol. 2001;432:240–243. doi: 10.1002/cne.1099. [DOI] [PubMed] [Google Scholar]
- Auer RN. Excitotoxic mechanisms, and age-related susceptibility to brain damage in ischemia, hypoglycemia and toxic mussel poisoning. Neurotoxicol. 1991;12:541–546. [PubMed] [Google Scholar]
- Baram TZ, Eghbal-Ahmadi M, Bender RA. Is neuronal death required for seizure-induced epileptogenesis in the immature brain? Prog Brain Res. 2002;135:365–375. doi: 10.1016/S0079-6123(02)35033-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CA, Rao G, McNaughton BL. Increased electrotonic coupling in aged rat hippocampus: a possible mechanism for cellular excitability changes. J Comp Neurol. 1987;259:549–558. doi: 10.1002/cne.902590405. [DOI] [PubMed] [Google Scholar]
- Barnes CA, McNaughton BL. Physiological compensation for loss of afferent synapses in rat hippocampal granule cells during senescence. J Physiol. 1980;309:473–485. doi: 10.1113/jphysiol.1980.sp013521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- Brandt C, Potschka H, Löscher W, Ebert U. N-methyl-D-aspartate receptor blockade after status epilepticus protects against limbic brain damage but not against epilepsy in the kainate model of temporal lobe epilepsy. Neuroscience. 2003;118:727–740. doi: 10.1016/s0306-4522(03)00027-7. [DOI] [PubMed] [Google Scholar]
- Browne TR, Holmes GL. Epilepsy. New Engl J Med. 2001;344:1145–1151. doi: 10.1056/NEJM200104123441507. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE. Neuron loss, granule cell axon reorganization, and functional changes in the dentate gyrus of epileptic kainate-treated rats. J Comp Neurol. 1997;383:385–404. [PubMed] [Google Scholar]
- Cadiacio CL, Milner TA, Gallagher M, Pierce JP. Hilar neuropeptides Y interneuron loss in the aged rat hippocampal formation. Exp Neurol. 2003;183:147–158. doi: 10.1016/s0014-4886(03)00126-2. [DOI] [PubMed] [Google Scholar]
- Carpenter MK, Parker I, Miledi R. Messenger RNAs coding for receptors and channels in the cerebral cortex of adult and aged rats. Mol Brain Res. 1992;13:1–5. doi: 10.1016/0169-328x(92)90038-d. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res. 1994;100:47–51. doi: 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- Choi DW. Bench to bedside: The glutamate connection. Science. 1992;258:241–243. doi: 10.1126/science.1357748. [DOI] [PubMed] [Google Scholar]
- Ciriza I, Carrero P, Azcoitia I, Lundeen SG, Garcia-Segura LM. Selective estrogen receptor modulators protect hippocampal neurons from kainic acid excitotoxicity: differences with the effect of estradiol. J Neurobiol. 2004;61:209–221. doi: 10.1002/neu.20043. [DOI] [PubMed] [Google Scholar]
- Clark AS, Magnusson KR, Cotman CW. In vitro autoradiography of hippocampal excitatory amino acid binding in aged Fischer 344 rats: relationship to performance on the Morris water maze. Behav Neurosci. 1992;106:324–335. doi: 10.1037//0735-7044.106.2.324. [DOI] [PubMed] [Google Scholar]
- Clayton DA, Grosshans DR, Browning MD. Aging and surface expression of hippocampal NMDA receptors. J Biol Chem. 2002;277:14367–14369. doi: 10.1074/jbc.C200074200. [DOI] [PubMed] [Google Scholar]
- Clayton DA, Browning MD. Deficits in the expression of the NR2B subunit in the hippocampus of aged Fisher 344 rats. Neurobiol Aging. 2001;22:165–168. doi: 10.1016/s0197-4580(00)00196-2. [DOI] [PubMed] [Google Scholar]
- Coulter DA, McIntyre DC, Löscher W. Animal models of limbic epilepsies: what can they tell us? Brain Pathol. 2002;12:240–256. doi: 10.1111/j.1750-3639.2002.tb00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT. Neurotoxic action of kainic acid. J Neurochem. 1983;41:1–11. doi: 10.1111/j.1471-4159.1983.tb11808.x. [DOI] [PubMed] [Google Scholar]
- Darbin O, Naritoku D, Patrylo PR. Aging alters electroencephalographic and clinical manifestations of kainate-induced status epilepticus. Epilepsia. 2004;45:1219–1227. doi: 10.1111/j.0013-9580.2004.66103.x. [DOI] [PubMed] [Google Scholar]
- Dawson R, Jr, Wallace DR. Kainic acid-induced seizures in aged rats: neurochemical correlates. Brain Res Bull. 1992;29:459–468. doi: 10.1016/0361-9230(92)90083-a. [DOI] [PubMed] [Google Scholar]
- DeLorenzo RJ, Hauser WA, Towne AR, Boggs JG, Pellock JM, Penberthy L, Garnett L, Fortner CA, Ko D. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology. 1997;46:1029–1035. doi: 10.1212/wnl.46.4.1029. [DOI] [PubMed] [Google Scholar]
- Eckles-Smith K, Clayton D, Bickford P, Browning MD. Caloric restriction prevents age-related deficits in LTP and in NMDA receptor expression. Mol Brain Res. 2000;78:154–162. doi: 10.1016/s0169-328x(00)00088-7. [DOI] [PubMed] [Google Scholar]
- Epilepsy Foundation of America. Epilepsy, a report to the nation. Landover, MD: 1999. [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. New York, NY: Academic Press; 1997. [Google Scholar]
- Galanopoulou AS, Vidaurre J, Moshe SL. Under what circumstances can seizures produce hippocampal injury: evidence for age-specific effects. Dev Neurosci. 2002;24:355–363. doi: 10.1159/000069047. [DOI] [PubMed] [Google Scholar]
- Golden GT, Smith GG, Ferraro TN, Reyes PF. Rat strain and age differences in kainic acid induced seizures. Epilepsy Res. 1995;20:151–159. doi: 10.1016/0920-1211(94)00079-c. [DOI] [PubMed] [Google Scholar]
- Gonzales RA, Brown LM, Jones TW, Trent RD, Westbrook SL, Leslie S. N-methyl-D-aspartate mediated responses decrease with age in F-344 rat brain. Neurobiol Aging. 1991;12:219–225. doi: 10.1016/0197-4580(91)90100-x. [DOI] [PubMed] [Google Scholar]
- Gutiérrez A, Khan ZU, Ruano D, Miralles CP, Vitorica J, De Blas AL. Aging-related subunit expression changes of the GABAA receptor in the rat hippocampus. Neuroscience. 1996;74:341–348. doi: 10.1016/0306-4522(96)00137-6. [DOI] [PubMed] [Google Scholar]
- Haas KZ, Sperber EF, Opanashuk LA, Stanton PK, Moshe SL. Resistance of immature hippocampus to morphologic and physiologic alterations following status epilepticus or kindling. Hippocampus. 2001;11:615–625. doi: 10.1002/hipo.1076. [DOI] [PubMed] [Google Scholar]
- Hauser WA. Seizure disorders: the changes with age. Epilepsia. 1992;33:S6–S14. doi: 10.1111/j.1528-1157.1992.tb06222.x. [DOI] [PubMed] [Google Scholar]
- Jarrard LE, Meldrum BS. Selective excitotoxic pathology in the rat hippocampus. Neuropathol Appl Neurobiol. 1993;19:381–389. doi: 10.1111/j.1365-2990.1993.tb00458.x. [DOI] [PubMed] [Google Scholar]
- Kelly KM, Ikonomovic MD, Abrahamson EE, Kharlamov EA, Hentosz TM, Armstrong DM. Alterations in hippocampal voltage-gated calcium channel α1 subunit expression patterns after kainate-induced status epilepticus in aging rats. Epilepsy Res. 2003;57:15–32. doi: 10.1016/j.eplepsyres.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Kerr DS, Razak A, Crawford N. Age-related changes in tolerance to the marine algal excitotoxin domoic acid. Neuropharm. 2002;43:357–366. doi: 10.1016/s0028-3908(02)00088-6. [DOI] [PubMed] [Google Scholar]
- Kesslak JP, Yuan D, Neeper S, Cotman CW. Vulnerability of the hippocampus to kainate excitotoxicity in the aged, mature and young adult rat. Neuroscience Lett. 1995;188:117–120. doi: 10.1016/0304-3940(95)11415-s. [DOI] [PubMed] [Google Scholar]
- Kuehl-Kovaric MC, Magnusson KR, Premkumar LS, Partin KM. Electrophysiological analysis of NMDA receptor subunit change in the aging mouse cortex. Mech Ageing Dev. 2000;115:39–59. doi: 10.1016/s0047-6374(00)00104-4. [DOI] [PubMed] [Google Scholar]
- Lado FA, Laureta EC, Moshe SL. Seizure-induced hippocampal damage in the mature and immature brain. Epileptic Disord. 2002;4:83–97. [PubMed] [Google Scholar]
- Le Jeune H, Cecyre D, Rowe W, Meaney MJ, Quirion R. Ionotropic glutamate receptor subtypes in the aged memory-impaired and unimpaired Long-Evans rat. Neuroscience. 1996;74:349–363. doi: 10.1016/0306-4522(96)00213-8. [DOI] [PubMed] [Google Scholar]
- Lerma J, Paternain AV, Rodriguez-Moreno A, Lopez-Garcia JC. Molecular physiology of kainate receptors. Physiol Rev. 2001;81:971–998. doi: 10.1152/physrev.2001.81.3.971. [DOI] [PubMed] [Google Scholar]
- Löscher W. Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Res. 2002;50:105–123. doi: 10.1016/s0920-1211(02)00073-6. [DOI] [PubMed] [Google Scholar]
- Löscher W. Animal models of intractable epilepsy. Prog Neurobiol. 1997;53:239–258. doi: 10.1016/s0301-0082(97)00035-x. [DOI] [PubMed] [Google Scholar]
- Ma Y, Hu JH, Zhao WJ, Fei J, Yu Y, Zhou XG, Mei ZT, Guo LH. Overexpression of gamma-aminobutyric acid transporter subtype I leads to susceptibility to kainic acid-induced seizure in transgenic mice. Cell Res. 2001;11:61–67. doi: 10.1038/sj.cr.7290067. [DOI] [PubMed] [Google Scholar]
- MacGregor DG, Graham DI, Stone TW. The attenuation of kainate-induced neurotoxicity by chromethiazole and its enhancement by dizocilpine, muscimol, and adenosine receptor agonists. Exp Neurol. 1997;148:110–123. doi: 10.1006/exnr.1997.6625. [DOI] [PubMed] [Google Scholar]
- Magnusson KR. Declines in mRNA expression of different subunits may account for differential effects of aging on agonist and antagonist binding to the NMDA receptor. J Neurosci. 2000;20:1666–1674. doi: 10.1523/JNEUROSCI.20-05-01666.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson KR. The aging of the NMDA receptor complex. Front Biosci. 1998;3:e70–e80. doi: 10.2741/a368. [DOI] [PubMed] [Google Scholar]
- Malva JO, Silva AP, Cunha RA. Presynaptic modulation controlling neuronal excitability and epileptogenesis: role of kainate, adenosine and neuropeptides Y receptors. Neurochem Res. 2003;28:1501–1515. doi: 10.1023/a:1025618324593. [DOI] [PubMed] [Google Scholar]
- Mejias-Aponte CA, Jimenez-Rivera CA, Segarra AC. Sex differences in models of temporal lobe epilepsy: role of testosterone. Brain Res. 2002;944:210–218. doi: 10.1016/s0006-8993(02)02691-4. [DOI] [PubMed] [Google Scholar]
- Meldrum BS. Excitotoxicity and selective neuronal loss in epilepsy. Brain Pathol. 1993;3:405–412. doi: 10.1111/j.1750-3639.1993.tb00768.x. [DOI] [PubMed] [Google Scholar]
- Michaelis EK. Molecular biology of glutamate receptors in the central nervous system and their role in excitotoxicity, oxidative stress and aging. Prog Neurobiol. 1998;54:369–415. doi: 10.1016/s0301-0082(97)00055-5. [DOI] [PubMed] [Google Scholar]
- Mitchell JJ, Anderson KJ. Age-related changes in [3H]MK-801 binding in the Fischer 344 rat brain. Neurobiol Aging. 1998;19:259–265. doi: 10.1016/s0197-4580(98)00058-x. [DOI] [PubMed] [Google Scholar]
- Morimoto K, Fahnestock M, Racine RJ. Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol. 2004;73:1–60. doi: 10.1016/j.pneurobio.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Nadler JV. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci. 1981;29:2031–2042. doi: 10.1016/0024-3205(81)90659-7. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Evenson DA. Use of excitatory amino acids to make axon-sparing lesions of the hypothalamus. Methods Enzymol. 1983;103:393–400. doi: 10.1016/s0076-6879(83)03027-x. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Perry BW, Gentry C, Cotman CW. Loss and reacquisition of hippocampal synapses after selective destruction of CA3-CA4 afferents with kainic acid. Brain Res. 1980a;191:387–403. doi: 10.1016/0006-8993(80)91289-5. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Perry BW, Gentry C, Cotman CW. Degeneration of hippocampal CA3 pyramidal cells induced by intraventricular kainic acid. J Comp Neurol. 1980b;192:333–359. doi: 10.1002/cne.901920209. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Perry BW, Cotman CW. Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature. 1978;271:676–677. doi: 10.1038/271676a0. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal glutamate excitotoxicity. Biochim Biophys Acta. 1998;1366:97–112. doi: 10.1016/s0005-2728(98)00123-6. [DOI] [PubMed] [Google Scholar]
- Nicoletti VG, Tendi EA, Lalicata C, Reale S, Costa A, Villa RF, Ragusa N, Giuffrida Stella AM. Changes of mitochondrial cytochrome c oxidase and FoF1 ATP synthase subunits in rat cerebral cortex during aging. Neurochem Res. 1995;20:1465–1470. doi: 10.1007/BF00970595. [DOI] [PubMed] [Google Scholar]
- Nicolle MM, Bizon JL, Gallagher M. In vitro autoradiography of ionotropic glutamate receptors in hippocampus and striatum of aged Long-Evans rats: relationship to spatial learning. Neuroscience. 1996;74:741–756. doi: 10.1016/0306-4522(96)00147-9. [DOI] [PubMed] [Google Scholar]
- Nitsch C, Hubauer H. Distant blood-brain barrier opening in subfields of the rat hippocampus after intrastriatal injections of kainic acid but not ibotenic acid. Neurosci Lett. 1986;64:53–58. doi: 10.1016/0304-3940(86)90662-2. [DOI] [PubMed] [Google Scholar]
- Okazaki MM, Nadler JV. Protective effects of mossy fiber lesions against kainic acid-induced seizures and neuronal degeneration. Neuroscience. 1988;26:763–781. doi: 10.1016/0306-4522(88)90097-8. [DOI] [PubMed] [Google Scholar]
- Olney JW. Excitatory transmitter neurotoxicity. Neurobiol Aging. 1994;15:259–260. doi: 10.1016/0197-4580(94)90127-9. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Landfield PW. Aging-related prolongation of calcium spike duration in rat hippocampal slice neurons. Brain Res. 1990;508:1–6. doi: 10.1016/0006-8993(90)91109-t. [DOI] [PubMed] [Google Scholar]
- Pittaluga A, Fedele E, Risiglione C, Raiteri M. Age-related decrease of the NMDA receptor-mediated noradrenaline release in rat hippocampus and partial restoration by D-cycloserine. Eur J Pharmacol. 1993;231:129–134. doi: 10.1016/0014-2999(93)90693-c. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor Seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Ramsden M, Shin TM, Pike CJ. Androgens modulate neuronal vulnerability to kainate lesion. Neuroscience. 2003;122:573–578. doi: 10.1016/j.neuroscience.2003.08.048. [DOI] [PubMed] [Google Scholar]
- Rapp PR, Stack EC, Gallagher M. Morphometric studies of the aged hippocampus: I. Volumetric analysis in behaviorally characterized rats. J Comp Neurol. 1999;403:459–470. [PubMed] [Google Scholar]
- Ruth RE. Extravasated protein as a cause of limbic seizure-induced brain damage: an evaluation using kainic acid. Adv Exp Med Biol. 1986;203:211–221. doi: 10.1007/978-1-4684-7971-3_16. [DOI] [PubMed] [Google Scholar]
- Ruth RE, Feinerman GS. Foreign and endogenous serum protein extravasation during harmaline tremors or kainic acid seizures in the rat: a comparison. Acta Neuropathol. 1988;76:380–387. doi: 10.1007/BF00686975. [DOI] [PubMed] [Google Scholar]
- Santos JB, Schauwecker PE. Protection provided by cyclosporin A against excitotoxic neuronal death is genotype dependent. Epilepsia. 2003;44:993–1000. doi: 10.1046/j.1528-1157.2003.66302.x. [DOI] [PubMed] [Google Scholar]
- Sattler R, Tymianski M. Molecular mechanisms of calcium-dependent excitotoxicity. J Mol Med. 2000;78:3–13. doi: 10.1007/s001090000077. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE. Differences in ionotropic glutamate receptor subunit expression are not responsible for strain-dependent susceptibility to excitotoxin-induced injury. Mol Brain Res. 2003;112:70–81. doi: 10.1016/s0169-328x(03)00048-2. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE. Complications associated with genetic background effects in models of experimental epilepsy. Prog Brain Res. 2002a;135:139–148. doi: 10.1016/s0079-6123(02)35014-3. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE. Modulation of cell death by mouse genotype: Differential vulnerability to excitatory amino acid-induced lesions. Exp Neurol. 2002b;178:219–235. doi: 10.1006/exnr.2002.8038. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE. Seizure-induced neuronal death is associated with induction of c-Jun N-terminal kinase and is dependent on genetic background. Brain Res. 2000;884:116–128. doi: 10.1016/s0006-8993(00)02888-2. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE, Williams RW, Santos JB. Genetic control of sensitivity to hippocampal cell death induced by kainic acid: a quantitative trait loci analysis. J Comp Neurol. 2004;477:96–107. doi: 10.1002/cne.20245. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE, Ramirez JJ, Steward O. Genetic dissection of the signals that induce synaptic reorganization. Exp Neurol. 2000;161:139–152. doi: 10.1006/exnr.1999.7251. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: Implications for gene targeting approaches. Proc Natl Acad Sci USA. 1997;94:4103–4108. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Stowers CC, Scallet AC, Xu L. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 2005;1035:24–31. doi: 10.1016/j.brainres.2004.11.054. [DOI] [PubMed] [Google Scholar]
- Schwob JE, Fuller T, Price JL, Olney JW. Widespread patterns of neuronal damage following systemic or intracerebral injections of kainic acid: a histological study. Neuroscience. 1980;9:991–1014. doi: 10.1016/0306-4522(80)90181-5. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Hippocampal interneurons expressing glutamic acid decarboxylase and calcium-binding proteins decrease with aging in Fischer 344 rats. J Comp Neurol. 1998;394:252–269. [PubMed] [Google Scholar]
- Sidman RL, Angevine JB, Taber Pierce E. Atlas of the mouse brain and spinal cord. Cambridge, MA: Harvard University Press; 1971. [Google Scholar]
- Smith AC, Gerrard JL, Barnes CA, McNaughton BL. Effect of age on burst firing characteristics of rat hippocampal pyramidal cells. Neuroreport. 2000;11:3865–3871. doi: 10.1097/00001756-200011270-00052. [DOI] [PubMed] [Google Scholar]
- Sonntag WE, Bennett SA, Khan AS, Thornton PL, Xu X, Ingram RL, Brunso-Bechtold JK. Age and insulin-like growth factor-1 modulate N-methyl-D-aspartate receptor subtype expression in rats. Brain Res Bull. 2000;51:331–338. doi: 10.1016/s0361-9230(99)00259-2. [DOI] [PubMed] [Google Scholar]
- Sperk G. Kainic acid seizures in the rat. Prog Neurobiol. 1994;42:1–32. doi: 10.1016/0301-0082(94)90019-1. [DOI] [PubMed] [Google Scholar]
- Sperk G, Lassmann H, Baran H, Kish SJ, Seitelberger F, Hornykiewicz O. Neurochemical and histopathological changes. Neuroscience. 1983;10:1301–1315. doi: 10.1016/0306-4522(83)90113-6. [DOI] [PubMed] [Google Scholar]
- Stanley DP, Shetty AK. Aging in the rat hippocampus is associated with widespread reductions in the number of glutamate decarboxylase-67 positive interneurons but not interneuron degeneration. J Neurochem. 2004;89:204–216. doi: 10.1111/j.1471-4159.2004.02318.x. [DOI] [PubMed] [Google Scholar]
- Tallis RC, Hall G, Craig I. How common are epileptic seizures in old age? Age Ageing. 1991;20:442–448. doi: 10.1093/ageing/20.6.442. [DOI] [PubMed] [Google Scholar]
- Tamaru M, Yoneda Y, Ogita K, Shimizu J, Nagata Y. Age-related decreases of the N-methyl-D-aspartate receptor complex in the rat cerebral cortex and hippocampus. Brain Res. 1991;542:83–90. doi: 10.1016/0006-8993(91)91001-h. [DOI] [PubMed] [Google Scholar]
- Treiman DM, Meyers PD, Walton NY, Collins JF, Colling C, Rowan AJ, Handforth A, Faught E, Calabrese VP, Uthman BM, Ramsay RE, Mamdani MB. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med. 1998;339:792–798. doi: 10.1056/NEJM199809173391202. [DOI] [PubMed] [Google Scholar]
- Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro EA. Cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse. 1989;3:154–171. doi: 10.1002/syn.890030207. [DOI] [PubMed] [Google Scholar]
- Vigues S, Gastaldi M, Chabret C, Massacrier A, Cau P, Valmier J. Regulation of calcium channel alpha (1A) subunit splice variant mRNAs in kainate-induced temporal lobe epilepsy. Neurobiol Disease. 1999;6:288–301. doi: 10.1006/nbdi.1999.0248. [DOI] [PubMed] [Google Scholar]
- Wallace H, Shorvon S, Tallis R. Age-specific incidence and prevalence rates of treated epilepsy in an unselected population of 2,052,922 and age-specific fertility rates of women with epilepsy. Lancet. 1998;352:1970–1973. doi: 10.1016/S0140-6736(98)04512-7. [DOI] [PubMed] [Google Scholar]
- Wenk GL, Barnes CA. Regional changes in the hippocampal density of AMPA and NMDA receptors across the lifespan of the rat. Brain Res. 2000;885:1–5. doi: 10.1016/s0006-8993(00)02792-x. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Whetsell WO., Jr Current concepts of excitotoxicity. J Neuropathol Exp Neurol. 1996;55:1–13. doi: 10.1097/00005072-199601000-00001. [DOI] [PubMed] [Google Scholar]
- Wozniak DF, Steward GR, Miller JP, Olney JW. Age-related sensitivity to kainate neurotoxicity. Exp Neurol. 1991;114:250–253. doi: 10.1016/0014-4886(91)90042-b. [DOI] [PubMed] [Google Scholar]