

Abstract
Antigen-specific CD8+ T cells are required for the clearance of most viral infections and several cancers. However, it is not clear in vivo whether CD8+ T cells can engage multiple targets simultaneously, engagement results in the formation of an immunologic synapse or molecules involved in CD8 function are redistributed to the synapse.We used here high-resolution microscopy to visualize interactions between virus-specific effectors and target cells in vivo. Using either in situ tetramer staining or green fluorescent protein–labeled virus-specific T cells, we have shown that a single CD8+ T cell can engage two or three targets, a synapse occurs at the site of engagement and molecules involved in attachment (lymphocyte function–associated antigen 1), signaling (Lck) and lytic activity (perforin) are differentially positioned on the T cell. In addition, we have established an in vivo approach for assessing the intricacies of antigen-specific T cell activation, migration, engagement, memory and other defining elements of adaptive immunity.
The adaptive immune response allows the host to purge itself of a diverse repertoire of invading pathogens. The effector arm of the adaptive immune response includes CD8+ T cells, which possess an arsenal of cellular weapons and are exquisitely refined in their ability to recognize target cells displaying small peptides on a protein scaffold called the major histocompatibility complex (MHC)1. Upon recognition of a foreign peptide, CD8+ T cells can use either cytopathic or noncytopathic mechanisms to rid a cell of an invading pathogen. These mechanisms include deposition of pore-forming molecules and granzyme release, engagement of the apoptosis-inducing ligands (such as Fas ligand) and the release of cytokines such as interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α)2.
The mobility of the adaptive immune response necessitates integration of numerous signals in a diversified array of cellular microenvironments. Naïve T cells that initially become activated and proliferate in secondary lymphoid tissues later acquire the capacity to migrate to organs that serve as the “breeding grounds” for invading pathogens3. Because of the overwhelming complexity of multiorgan cellular interactions and the inability to track antigen-specific cell populations, in vitro systems have been established for studying T cell responses in order to reduce the complexity. The development of strategies to visualize antigen-specific immune responses in vivo has helped to overcome this type of reductionism4–7. However, the interactions of antigen-specific cells in vivo remain largely undefined. Knowledge of these interactions will improve our understanding of immunopathogenesis, autoimmunity and vaccine strategies.
To visualize cellular immune interactions in vivo, we used a well characterized viral model that is defined by lethal immunopathology. Lymphocytic choriomeningitis virus (LCMV) is a noncytopathic natural mouse pathogen that, upon intracerebral (i.c.) inoculation, initiates peripheral expansion of virus-specific T cells that ultimately traffic to the central nervous system (CNS) and cause severe meningitis at 6–10 days8. Prodigious numbers of mononuclear cells accumulate within the ependyma, choroid plexus and meninges (the primary sites of viral replication), leaving the brain parenchyma virtually devoid of inflammation. The lethality of this disease is dependent on CD8+ T cells9 and, more specifically, the effector molecule perforin10. In addition, i.c. transfer of as few as 103 virus-specific CD8+ T cell clones is sufficient to drive this disease process11. These studies also indicate that, in the absence of an immune response, LCMV alone is incapable of inducing mortality10,11, probably as a result of its noncytopathic replication cycle. The disease process depends entirely on virus-specific CD8+ T cell effector–target cell interactions, and thus provides a powerful, physiologic in vivo model with which to visualize cellular interactions involved in adaptive immunity.
Here we used two strategies to visualize interactions between cytotoxic lymphocytes (CTLs) and LCMV-infected targets in the CNS. Six different MHC class I–restricted epitopes within the nucleoprotein (NP) and glycoprotein (GP) regions of LCMV have been mapped in C57BL/6 (B6) mice; they account for the entire virus-specific CD8+ T cell response12–14. We focused on Db-GP(33–41)-specific CD8+ T cells because a CTL clone specific for one of the dominant epitopes, Db-GP(33–41), has been used to generate a T cell receptor (TCR)–transgenic (Tg) mouse (referred hereafter to as GP33 TCR–Tg)15. We visualized Db-GP(33–41)-specific T cells in situ using MHC class I tetramers or by genetically labeling TCR-Tg cells with green fluorescent protein (GFP). Using these techniques, we have shown in a physiologic model that the cellular reorganization of CTLs is associated with target cell interactions and that a CTL has the capacity to engage multiple targets simultaneously in vivo.
Results
In situ tetramer staining of LCMV-specific T cells
Two reports describe a strategy for visualizing antigen-specific CD8+ T cell responses known as in situ MHC class I tetramer staining4,5. In both studies, the groups relied almost entirely on the use of 200 μm unfixed vibratome sections for tetramer staining, which—in our experience—results in distorted tissue anatomy and is incompatible with high-resolution visualization of cellular interactions (data not shown). Thus, we modified the technique to allow reproducible and distinct staining of antigen-specific CD8+ T cells on frozen sections (Fig. 1). When splenocytes isolated from naïve GP33 TCR–Tg mice were stained with Db-GP(33–41) tetramers and analyzed by flow cytometry, we found that 94% of the CD8+ T cells were GP(33–41)-specific (Fig. 1a). Tissue sections from the same spleen were then cut and analyzed with in situ tetramer staining (Fig. 1b–d). Using this technique, we easily observed robust tetramer staining in the splenic white pulp of naïve GP33 TCR–Tg mice, and enumeration of tetramer-labeled cells revealed that 82% (1467/1788) of the CD8+ T cells were also tetramer+. The minor difference in the percentage of tetramer+ cells obtained by the two techniques demonstrates that the in situ staining procedure is slightly less sensitive than labeling cells in suspension.
Figure 1. Tetramer staining of virus-specific CD8+ T cells in the spleens and CNS of GP33 TCR–Tg mice.

Antigen-specific CD8+ T cells stained with an anti-CD8 and MHC class I tetramers were analyzed by (a) flow cytometry or (b–j) confocal microscopy. (a) By flow cytometry, 94% of CD8+T cells from the spleens of a naïve GP33 TCR–Tg mouse were tetramer+. In a nontransgenic B6 control, none of the CD8+ T cells were tetramer+. (b–d) Db-GP(33–41)-specific (green) CD8+ T cells (red) were detected in the splenic white pulp of naïve GP33 TCR–Tg mice. Overlapping fluorescence in the merged image appears in yellow. Of these T cells, 82% were tetramer+. (e–g) None of the CD8+ T cells from GP33 TCR–Tg mice were labeled with the control tetramer Db-NP(396–404). (h–j) Interactions between CD8+ (red) and Db-GP(33–41)–specific (green) effectors and LCMV-infected targets (blue) were visualized in the CNS of GP33 TCR–Tg mice on day 4 after LCMV infection. Red circles represent individual CD8+T cells; overlapping fluorescence between CD8 (red) and tetramer (green) appears in yellow. (h) Note the foci of tetramer staining (arrows) present around the cell membrane of a CTL in the area of LCMV infection. (h,i) TCR polarization to the CTL–target cell interface was also observed (arrowheads). (h, inset) More homogenous TCR distribution was observed on CTLs not engaged with virus-infected targets in the CNS. (j, arrows and inset) In rare instances (<1%), CTLs extended TCR-coated protrusions that partially wrapped around the virus-infected target.
To evaluate the specificity of the in situ tetramer technique, spleen sections from GP33 TCR–Tg mice were stained with two mismatched LCMV tetramers: Db-NP(396–404) (Fig. 1e–g) and Ld-NP(118–126) (data not shown). With both tetramers, none—Db-NP(396–404) (0/1628) and Ld-NP(118–126) (1/2392)—of the CD8+ T cells stained positive, demonstrating the high specificity of this technique.
Visualization of TCR polarization in vivo
Having established in situ tetramer staining in the TCR-Tg mice, we next asked whether this technique could be used to visualize effector–target cell interactions during LCMV infection in vivo. Brain sections were obtained from GP33 TCR–Tg mice 4 days after i.c. inoculation with 103 PFU (plaque-forming units) of LCMV Armstrong strain (referred to as LCMV). At this time point, flow cytometry studies revealed that >78% of the CD8+ T cells in the CNS stained with the tetramer. This percentage was expected, based on the high frequency of Db-GP(33–41)-specific CD8+ T cells in peripheral lymphoid tissues. No positive staining was observed on infected CNS tissue from GP33 TCR–Tg mice stained with the control tetramers Db-NP(396–404) and Ld-NP(118–126) (data not shown). In contrast to the homogenous distribution of tetramer staining on nearly all naïve splenic Db-GP(33–41)-specific T cells (Fig. 1c), foci of tetramer staining were observed on activated CD8+ T cells within the CNS (Fig. 1h). This was consistent with the aggregation of lipid rafts after TCR engagement16 and the localization of TCR within these rafts. In CNS brain sections, tetramer+ cells were also observed interacting with LCMV-infected targets within the meninges, ependyma and choroids plexus (Fig. 1h–j). In 56% (10/18) of these interactions, tetramer staining was localized primarily at the interface between the juxtaposed antigen-specific CD8+ T cell and LCMV-infected target (Fig. 1h–j; this is best illustrated in Fig. 1i).
To address whether TCR reorganization occurred solely at the sites of CTL engagement, we compared the distribution of tetramer staining on conjugates and nonconjugates in the CNS. These analyses revealed that the probability of finding polarized TCR was significantly greater (P = 0.022) for CTLs engaged with LCMV-infected targets (Fig. 1h,i) compared to those that were not engaged (Fig. 1h, inset) (2/23 and 9%, respectively). Interfacial localization of the Db-GP(33–41)-specific TCR is highly suggestive of in vivo immunologic synapse formation and is analogous to the TCR reorganization observed during immunologic synapse formation in vitro17. Finally, in rare instances we observed tetramer+ protrusions that extended partially around LCMV-infected target cells (Fig. 1j). These protrusions may potentially aid peptide-MHC (pMHC) sampling and immunologic synapse formation.
Because TCR-Tg mice were used to visualize in vivo immunologic synapse formation, it is possible that the TCR focusing was not representative of natural effector–target cell interactions. Thus, we next wanted to validate our findings in nontransgenic B6 mice. B6 mice infected intracerebrally with LCMV showed an expansion (1.8–7.9%) of Db-GP(33–41)-specific CD8+ T cells in the spleen during days 4–6 after infection (Fig. 2a). Despite the presence of virus-specific cells in the spleen on days 4 and 5, these cells were not detectable in the CNS. In fact, few mononuclear cells could be isolated from the CNS at these time points. On days 5–6 after infection, virus-specific CD8+ T cells migrated from peripheral lymphoid tissues to the CNS. A sizeable number of mononuclear cells were present in the CNS on day 6 after infection; these included Db-GP(33–41)-specific cells, which accounted for 6.5% of the total CD8+ T cell pool. The presence of virus-specific T cells in the CNS at this time point coincided with the onset of neurologic dysfunction and mortality. When in situ tetramer staining was used to visualize and enumerate Db-GP(33–41)-specific cells in the brain (Fig. 2b–d), it was calculated that 4.5% (79/1626) of the CD8+ T cells were tetramer+. Visualization of interactions between Db-GP(33–41)-specific cells and LCMV-infected targets was more labor intensive because of the lower frequency of these cells compared to the frequency in GP33 TCR–Tg mice. However, in support of our findings in the TCR-Tg mice, TCR focusing was observed at the interface between Db-GP(33–41)-specific CD8+ T cells and the LCMV-infected targets (Fig. 2e).
Figure 2. Visualization of antiviral immunity in the CNS of B6 mice during the induction of lethal meningitis.

(a) Mononuclear cells were isolated from the CNS and spleens of B6 mice on days 4, 5 and 6 after infection and the frequencies of Db-GP(33–41)-specific CD8+ T cells were calculated. Percentage numbers of cells are indicated (n = 4). (b–d) Brain sections were cut from symptomatic B6 mice on day 6 after infection and analyzed by in situ tetramer staining (green) and confocal microscopy. Many CD8+T cells (red) were present in the meninges, ependyma and choroid plexus. (b–d) Dense meningeal infiltrate in which four CD8+ T cells were Db-GP(33–41)+ (arrows). The frequency of tetramer+ cells was calculated by sampling CNS infiltrates from three infected mice. (e) With three-color analyses,TCR focusing (yellow) was observed at the interface between a single CTL (red) and an LCMV-infected target (blue).Yellow represents overlap between the tetramer (green) and CD8 (red) stains. Note the presence of tetramer staining at the interface (arrows), but not around the remaining CD8+ T cell membrane. (f) CD8+ T cells (red) interacted with up to three LCMV-infected (green) target cells (asterisks) in the CNS. Overlapping membrane between the CTL and infected targets appears in yellow.A tetramer stain is not shown. (g) Tetramer+ cells were also found interacting with multiple targets (asterisks). Note the foci of tetramer staining (yellow) around the cell membrane of a CTL (red) that is engaged with two targets (blue). The TCR is not polarized toward either target.
Multicellular T cell engagement
Several in vitro studies have used time-lapse microcinematography to show that CTLs are “serial killers” capable of eliminating several targets within a relatively short period of time18–20. These studies have identified one potential mechanism by which a limited effector cell population can purge virus from a seemingly overwhelming number of targets. In the CNS of LCMV-infected mice, we visualized a second potential mechanism that may help a CTL in its fight against viruses. In the LCMV-infected CNS, virus-infected targets substantially outnumber effector cells. However, in this situation we commonly found single CD8+ T cells interacting with as many as three virus-infected target cells (Fig. 2f,g). These interactions suggest that a single CTL can amplify its effectiveness by engaging multiple targets in vivo.
Labeling LCMV-specific T cells
Having demonstrated that at least one aspect of immunologic synapse formation (that is, TCR polarization) occurs in vivo, we next established an in vivo model system to visualize additional molecules recruited to the synapse. In situ tetramer staining is an excellent way of identifying antigen-specific T cells in tissues of interest. However, the sensitivity of the technique likely declines after TCR internalization, which is expected to occur as virus-specific T cells come into contact with their cognate antigen in the CNS. Thus, we developed a simple strategy for tracking virus-specific CD8+ T cells that was not dependent on labeling the antigen-specific TCR.
To this end, GP33 TCR–Tg mice were crossed with B6 TgN(ACTbEGFP)10sb mice21, which express GFP under the control of the actin promoter (hereafter referred to as GFP mice). GFP+CD8+ Db-GP(33–41)-specific T cells were isolated from F1 mice, and 105 cells were adoptively transferred intravenously into naïve B6 recipients. Three days later, an i.c. inoculation with 103 PFU of LCMV was given; it resulted in a massive expansion (45-fold) of these cells in the spleen by day 5 after infection (Fig. 3a). GFP+ cells accounted for 54% of all CD8+ and 77% of all tetramer+—Db-GP(33–41)—cells in the spleen. These cells also migrated to the CNS and induced symptoms and/or mortality a day earlier than observed in B6 mice that did not receive a transfer. In the CNS, GFP+ cells represented 66% of all CD8+ and 89% of all tetramer+ cells. Additionally, immunofluorescence analysis of coronal brain sections revealed that the Db-GP(33–41)- specific GFP+ T cells trafficked primarily to areas of viral infection (the meninges, choroid plexus and ependyma) and were not distributed uniformly throughout the brain parenchyma (Fig. 3b–f). This model system is therefore suitable for visualizing the distribution of additional molecules on engaged CTLs in vivo.
Figure 3. Expansion and migration of GFP+ Db-GP(33–41)-specific T cells after an i.c. infection.

(a) On day 5 after infection, mononuclear cells were isolated from the spleens and CNS of B6 mice that were given GFP+ Db-GP(33–41)-specific T cells. Cells were stained with anti-CD8 and Db-GP(33–41) tetramers and analyzed by flow cytometry. Percentage numbers of cells are indicated (n = 4). (b–f) Reconstructions of coronal brain tissue sections were done on day 5 after infection to visualize the distribution of infiltrating GFP+ effector cells in relation to LCMV-infected targets. An overlay of nuclei (blue), LCMV (red) and GFP+ effectors (green) was generated with three-color immunofluorescence and image analysis software. The overlay (b) revealed that GFP+ effectors (d) localized primarily to the sites of LCMV infection (c). High-magnification images of b show the distribution of GFP+ effectors around the LCMV ependyma (e) and meninges (f). Each green dot represents a single antigen-specific CD8+ T cell.
Cellular reorganization of CTL in vivo
To determine the two-dimensional (2D) distribution of cell-membrane proteins involved with adhesion (lymphocyte function–associated antigen 1, or LFA-1), signaling (Lck) and effector function (perforin) in a physiologic setting, we visualized these molecules in conjugates of Db-GP(33–41)-specific GFP+ T cells and LCMV-infected CNS targets (Figs. 4 and 5). Activation of lymphocytes increases the mobility of the integrin LFA-122, which equips T cells with an antigen-independent mechanism to engage potential targets and scan for the appropriate pMHC23. We found that LFA-1 was localized to the CTL–target cell interface in 64% (23/36) of the conjugates examined (Figs. 4a–d and 5b), consistent with the integrin redistribution observed in several in vitro studies17,24. This pattern of staining was only observed on 7% (2/29) of CTLs not engaged with infected targets in the CNS (Fig. 5a), and the probability of finding LFA-1 polarization was statistically greater (P < 0.001) in conjugates compared to nonconjugates (Fig. 5f). LFA-1 polarization was observed even in effector cells that were surrounded on all sides by virus-infected targets (Fig. 5b); this demonstrated that reorganization of the plasma membrane can occur despite the complex cellular environment.
Figure 4. Cellular reorganization of Db-GP(33–41)-specific T cells juxtaposed to LCMV-infected targets.
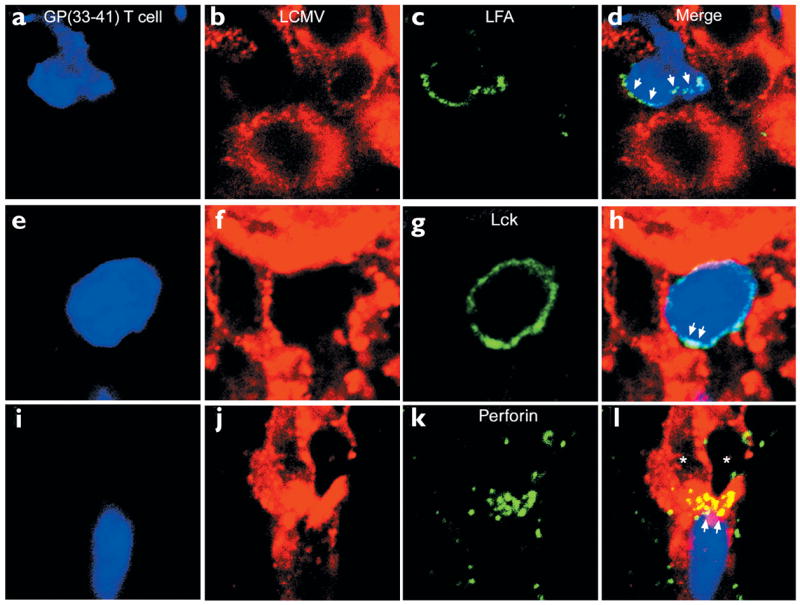
The cell membrane distribution of (a–d) LFA-1, (e–h) Lck and (i–l) perforin on GFP+ Db-GP(33–41)-specific T cells (blue) engaged with LCMV-infected targets (red) was evaluated by confocal microscopy on day 5 after infection. (a–d) Distribution of LFA-1 (green) on a single Db-GP(33–41)-specific CTL. Note the polarization of LFA-1 staining toward the LCMV-infected target cell (arrows). (e–h) The distribution of Lck in a CTL surrounded almost entirely by LCMV-infected targets. The GFP+ cell showed dense Lck staining on one side of the cell membrane (arrows). (i–l) An example of polarized perforin staining at the interface between a CTL and virus-infected target. Nearly all the perforin staining (green) is localized on two LCMV-infected targets (arrows). Asterisks indicate the LCMV-infected targets. Overlapping LCMV (red) and perforin (green) signals appear in yellow.
Figure 5. Patterns of interfacial LFA-1, Lck and perforin staining on Db-GP(33–41)-specific T cells in the CNS.

Representative staining patterns (all in green) are shown for (a,b) LFA-1, (c,d) Lck and (e) perforin. (f) The distribution of LFA-1 and Lck was quantified on conjugates and nonconjugates in the CNS.Asterisks denote a statistical difference between conjugates and nonconjugates (P < 0.001). (b) Polarization of LFA-1 (arrows) was observed on 64% (23/36) of the conjugates of Db-GP(33–41)-specific T cells (blue) and LCMV-infected targets (red). (a) In contrast, polarized LFA-1 was only observed on 7% (2/29) of the CTLs not engaged with LCMV-infected targets. (d) Aggregation of Lck was observed for 55% (21/38) of the conjugates (arrows). (c) This pattern was observed on 4% (2/45) of nonconjugates. Complete polarization of Lck was never observed. (e) CTL engagement of two LCMV-infected targets (asterisks). Note the delivery of perforin on both infected targets (arrows), which are in engaged at opposite ends of the CTL.The overlap between perforin (green) and LCMV (red) appears in yellow. Perforin staining can also be observed inside the CTL.
We next examined the distribution of the signaling kinase Lck in CTL–target cell conjugates (Figs. 4e–h and 5c,d). Lck is responsible for phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) after ligation of the TCR-CD3 complex25. In naïve CD8+ T cells, Lck is homogenously distributed in the cytosol; however, in vivo priming of naïve T cells results in redistribution of Lck to the cell membrane, where it associates with CD826,27. This redistribution may increase the efficiency of effector-memory cells by enhancing their capacity to transduce activation signals. Consistent with published observations27, we found that Lck was present primarily on the plasma membrane of virus-specific CD8+ T cells in the CNS of infected mice (Figs. 4e–h and 5c,d). When conjugates were examined, it was revealed that in contrast to LFA-1, Lck never polarized completely to the CTL–target cell interface. In most cases, Lck was present around the circumference of the effector cell membrane, although in 55% (21/38) of the conjugates a cluster of Lck was observed at an interface (Figs. 4e–h and 5d). The probability of finding this density of staining was significantly greater for CTL–target cell conjugates (P < 0.001) than for the nonconjugates (2/45 or 4%) (Fig. 5c,f). It is possible that the density of interfacial staining seen in the majority of the conjugates represents kinase aggregation at the site of an immunologic synapse. Nevertheless, the complete interfacial polarization of Lck observed on CTLs in vitro24 was never observed on CTLs engaged with virus-infected targets in vivo.
Lastly, we examined localization of the effector molecule perforin, a pore-forming protein present in the lytic granules of CTLs2 (Figs. 4i–l and 5e–f). High-resolution microcinematography has shown that CTL granules are polarized toward a target cell only after engagement28; also, lytic granules occupy a distinct cellular subdomain during immunologic synapse formation24. We found that in the CNS of LCMV-infected mice, perforin was present on both effector and target cells, consistent with the deposition of perforin onto LCMV-infected targets (Fig. 4i–l and 5e). Because of the heterogeneous staining pattern, we did not quantify perforin localization in effector–target cell conjugates. However, there were examples of CTLs that had focused their lytic granules to the effector–target cell interface (Fig. 4i–l). CTLs also appeared capable of depositing perforin on multiple targets that were engaged simultaneously in vivo (Figs. 4l and 5e). This is best illustrated in Fig. 5e, where a CTL has deposited perforin on two LCMV-infected targets that are engaged on opposite sides of the CTL. One in vitro study has shown that the microtubule-organizing center, which is required for unidirectional CTL killing, can oscillate between two simultaneously engaged targets29. This finding supports our in vivo data and establishes the functional relevance of multicellular CTL engagement. CTLs are not only capable of engaging multiple targets simultaneously, but can also deliver an effector molecule to each of the targets, thus improving the efficiency of CTLs in infected tissues.
3D analysis of CTL interactions in vivo
In vitro studies have used three-dimensional (3D) microscopy to reveal the exquisite spatial organization of various molecules in the immunologic synapse. Based on these studies, it could be argued that our 2D dataset does not adequately represent the total cellular reorganization that occurs after CTL engagement in vivo. To address this concern we used high-resolution 3D microscopy to examine in greater detail the interfacial distribution of LFA-1 and Lck on CTL–target cell conjugates. The results of the 3D analyses confirmed our previous observations by demonstrating that plasma membrane reorganization is only associated with CTL engagement (Fig. 6 and Web Movies 1–5 online). Examination of LFA-1 staining revealed polarization at the interface between a CTL and at least three LCMV-infected targets (Fig. 6a–c and Web Movies 1 and 2 online). The distribution of LFA-1 at the interface resembled a “ring-like” structure (Fig. 6b, inset). CTLs were also capable of extending an LFA-1–coated process that may assist in target cell engagement and permit efficient pMHC scanning.
Figure 6. LFA-1 and Lck 3D localization on Db-GP(33–41)-specific T cells in the CNS.
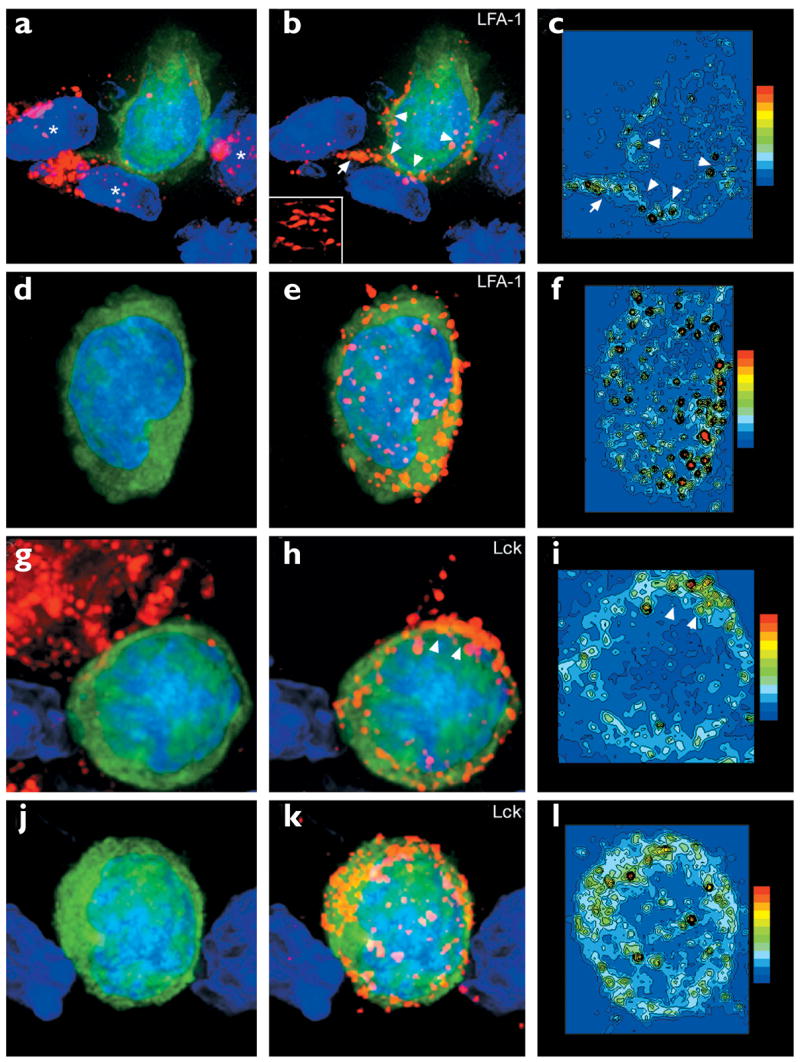
Maximal projections of 3D datasets show the total cellular distribution of (a–f) LFA-1 and (g–l) Lck, which was examined on both CNS (a–c, g–i) conjugates and (d–f, j–l) nonconjugates. Pseudocolored contour plots show the distribution of (c,f) LFA-1 and (i,l) Lck on the GFP+ Db-GP(33–41)-specific T cells.All maximal projections showed Db-GP(33–41)-specific T cells (green) and nuclei (blue). The left-hand panels show the virus in red; the middle panels show LFA-1 or Lck in red. (a) A Db-GP(33–41)-specific T cell in juxtaposition with at least three LCMV-infected targets (asterisks). (b,c) Polarization of LFA-1 was observed at the interface with each of these virus-infected targets (arrowheads) and on a process that extended from the CTL (arrow). (b) Inset shows the distribution of LFA-1 at the interface with one of the virus-infected targets. (d–f) A nonconjugate in the CNS; (e,f) note the presence of evenly distributed LFA-1 clusters around the cell membrane. (g) Another CTL–target cell conjugate. (h,i) Lck aggregation was observed at the interface between the CTL and the virus-infected target (arrowheads). (j–l) A more homogenous distribution of Lck was observed on nonconjugates.
This idea is supported by the observation that TCRs are also present on these processes (Fig. 1j). In contrast to the pattern observed on CTL–target cell conjugates, a more homogenous distribution of LFA-1 was observed on nonconjugates (Fig. 6d–f and Web Movie 3 online). Similar findings were observed with the tyrosine kinase Lck. As mentioned above, Lck never polarized completely towards an LCMV-infected target. Rather, kinase aggregation was observed at the CTL–target cell interface (Fig. 6g–i and Web Movie 4). The contour plot illustrates this pattern of staining (Fig. 6i). In contrast to the interfacial kinase aggregation observed on CNS conjugates, a more even cellular distribution of Lck was observed on nonconjugates (Fig. 6j–l and Web Movie 5). Thus, 3D analyses support the fact that CTL reorganization occurs in vivo and is associated with target-cell engagement.
Discussion
In recent years the immunologic synapse field has advanced rapidly and generated important insights regarding the interactions between T cells and antigen-presenting cells. For example, high-resolution 3D microscopy studies in vitro have shown that formation of the immunologic synapse, as a CD4+ T cell engages an antigen-presenting cell17 or planar bilayer30, results in reorganization of the cell membrane and the assembly of interfacial concentric protein arrays. Signaling kinases (for example, Lck and protein kinase C-θ) as well as the TCR-CD3 complex localize in the central supramolecular activation cluster (cSMAC), and the entire structure appears to be stabilized by a surrounding peripheral ring (pSMAC) that consists of adhesion molecules (such as LFA-1). The flat and extensive surface area maintained during immunologic synapse formation may provide a favorable molecular environment for sustained signaling (>1 h) and activation of naïve T cells31. Conversely, activated CTLs do not require sustained signaling for effector functions. Despite this, one in vitro study showed that an immunologic synapse similar to the one described for CD4+ T cells formed within 5 min of CTL–target cell contact24. In CTL–target cell conjugates, lytic granules occupied a distinct domain within the cSMAC.
Despite contemporary analyses of immunologic synapse formation, it is not known whether the remarkable organization of the in vitro immunologic synapse is representative of physiological interactions that occur in vivo. We have established in vivo that cellular reorganization of a CTL is associated with engagement of LCMV-infected targets. It is possible that the patterns of various molecules observed on CTLs juxtaposed with LCMV-infected targets simply reflect an altered membrane organization that results from the heightened activation state of the CTL. However, this explanation is not likely, as nearby CTLs (which should have a similar activation profile) surrounded by uninfected cells did not show evidence of cellular reorganization. Thus, the fact that cellular reorganization was observed primarily on CTLs adjacent to LCMV-infected cells suggests that a target cell interaction is required. However, our study relies solely on analyses of static interactions or “snap-shots” in time, which makes it difficult to establish the precise sequence of events in vivo. With recent advances in real-time in vivo tracking methodologies as a foundation32–34, it may soon become possible to examine dynamic interactions at high resolution in complex cellular microenvironments.
In conclusion, the integration of signals from a multitude of cellular sources should occur as antigen-specific T cells become activated and traffic to affected organs. Because in vitro systems often oversimplify cellular microenvironments, we need to develop strategies that allow the evaluation of antigen-specific T cell responses in a physiologic setting. To date, much of what we know about the immunologic synapse stems from analysis of cell-cell interactions in vitro23,31. These analyses do not account for the complex 3D tissue architecture through which an antigen-specific T cell must migrate to specifically locate the appropriate targets. In addition, in vitro studies often visualize molecules recruited to the interface between an antigen-specific T cell and a single target. However, we have demonstrated in vivo instances in which CTLs interact with up to three targets or are completely surrounded by virus-infected cells. Multicellular engagement may confer on an outnumbered effector pool the ability to clear a pathogen from multiple cells simultaneously, yet this may also alter the formation of the highly ordered immunologic synapse described in vitro. Thus, analysis of antigen-specific T cells in vivo is required for the validation of immunologic concepts that emerge from simplified culture systems.
To this end, we have described a new approach for visualizing a fluorescently labeled population of antigen-specific T cells that expand massively after viral infection and home specifically to areas of viral replication. Through the use of high-resolution microscopy, the presence and distribution of various molecules within the plasma membrane of these cells can be determined in anatomically intact tissue sections. This approach should prove useful in future in vivo studies designed to address a number of key questions in cellular immunology.
Methods
Mice
B6 mice were from The Scripps Research Institute (La Jolla, CA); GP33 TCR–Tg mice were from the Jackson Laboratories (Bar Harbor, ME) and were backcrossed over ten generations on the B6 background; B6 TgN(ACTbEGFP)10sb mice were also from the Jackson Laboratories. Mouse handling conformed to the requirements of the National Institutes of Health and The Scripps Research Institute Animal Research Committee.
Virus
The Armstrong 53b strain of LCMV was used for all studies; 103 PFU was injected intracerebrally into mice at 8 weeks of age.
Adoptive transfer of GFP+Db-GP(33–41) T cells
CD8+ T cells were purified from (GP33 TCR–Tg × GFP)F1 mouse splenocytes by negative selection (Stem Cell Technologies, Vancouver, Canada). After enrichment, we determined that 98% of the cells were CD8+ and 77% of the transferred cells were double-positive for both the Db-GP(33–41) tetramer and GFP. We transferred 105 cells intravenously into naïve B6 recipients. Three days later the mice received an i.c. inoculation with 103 PFU of LCMV.
Flow cytometry
Splenocytes and brain-infiltrating lymphocytes were stained with phycoerthyrin (PE)–anti-CD8 (BD PharMingen, San Diego, CA) and allophycocyanin–Db-GP(33–41) tetramers. Tetramers were produced as described35. Cells were acquired with the FACSCalibur flow cytometer (Becton Dickson, San Jose, CA) and analyzed with FlowJo software (Treestar, San Carlos, CA).
In situ tetramer staining
Organs of interest were frozen in OCT (Tissue-Tek, Torrance, CA) on dry ice. Sections (6 μm) were then cut and incubated overnight at 4 °C in a 2% fetal bovine serum (FBS) solution containing pMHC class I tetramers conjugated to allophycocyanin (0.2 μg/ml) and rat anti-CD8 (0.2 μg/ml, Pharmingen). Tissues were then fixed with 2% formaldehyde, washed, incubated at 4 °C for 3 h with a allophycocyanin–rabbit antibody (1:1000, Biomeda, Hayward, CA), then washed and incubated at 4 °C for 3 h with Cy5–rabbit antibody (1:500) and rhodamine Red-X–rat antibody (1:500). For three-color analyses, a guinea pig anti-LCMV (1:1000) was added to the primary antibodies and a fluorescein isothiocyanate (FITC)–guinea pig antibody (1:500) was added to the secondary antibodies. Stains were visualized with an MRC1024 confocal microscope (BioRad, Richmond, CA) fitted with a krypton-argon mixed gas laser (excitation at 488, 568 and 647 nm) and a ×40 or ×63 oil objective. All 2D confocal images show a single Z section captured at a position approximating the CTL midline. To compare TCR polarization on CNS conjugates versus nonconjugates, random fields were captured from infected CNS tissue. CTLs juxtaposed with LCMV-infected targets were considered conjugates, whereas CTLs in the same CNS tissue sections—which were not juxtaposed with LCMV-infected targets—were considered nonconjugates. Statistical analysis of TCR polarization in conjugates versus nonconjugates was done with a Fisher Exact Text (P < 0.05).
Brain reconstructions
Three-color reconstructions of coronal brain sections were done with an Axiovert S100 immunofluorescence microscope (Zeiss, Thornwood, NY) fitted with an automated xy stage, a Axiocam color digital camera and a ×5 objective. To obtain tissues, mice received an intracardiac perfusion with 4% paraformaldehyde. Organs were removed and incubated for 24 h in 4% paraformaldehyde and an additional 24 h in 30% sucrose. After freezing tissues in OCT, 6-μm frozen sections were cut and stained overnight at 4 °C with a guinea pig anti-LCMV (1:1000). Tissues were then washed, incubated at 4 °C for 3 h with a rhodamine Red-X–guinea pig antibody (1:500) and washed and incubated with DAPI (1 μg/ml). Three registered images (DAPI, GFP and rhodamine Red-X) were captured for each field on the coronal brain section, and reconstructions were done with the MosaiX function in the KS300 image analysis software.
Immunocytochemical analysis of CTL engagement
For analysis of LFA-1, Lck and perforin on GFP+ CTLs, tissues were processed and cut as described above. Rat anti–LFA-1 (3 μg/ml, Pharmingen), rabbit anti-Lck (1:100, Cell Signaling, Beverly, MA) or goat anti-perforin (2 μg/ml, Santa Cruz Biotechnology, Santa Cruz, CA) antibody was added to the primary mixture containing the anti-LCMV (1:1000). LFA-1, Lck and perforin primary antibodies were labeled with the appropriate secondary antibody conjugated to Cy5. Three-color (GFP, rhodamine Red-X and Cy5) confocal analyses were done to visualize cell-cell interactions as described above. Statistical analyses of LFA-1 and Lck staining patterns in conjugates versus nonconjugates were done with a Fisher Exact Text (P < 0.05). Four-color (DAPI, GFP, rhodamine Red-X and Cy5) 3D datasets were collected with a DeltaVision system (Applied Precision, Issaquah, WA); this consisted of an Olympus IX-70 fluorescence microscope, a motorized high-precision xyz stage, a 100-W mercury lamp and KAF1400 chip-based cooled charge-coupled device camera. Exposure times were 0.1–0.8 s (2-binning), and images were obtained with a ×100 oil objective. 3D reconstructions were generated by capturing 150-nm serial sections along the z-axis. Images were deconvolved (based on the Agard-Sadat inverse matrix algorithm) and analyzed with softWorX Version 2.5.
Acknowledgments
Supported by NIH grant AI09484, training grant AG00080 (to D. B. M.) and Juvenile Diabetes Research Foundation Award 3-2000-510 (to U. C.)
Footnotes
Note: Supplementary information is available on the Nature Immunology website.
Competing interests statement
The authors declare that they have no competing financial interests.
References
- 1.Zinkernagel RM, Doherty PC. Restriction of in vitro T cell–mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature. 1974;248:701–702. doi: 10.1038/248701a0. [DOI] [PubMed] [Google Scholar]
- 2.Kagi D, Ledermann B, Burki K, Zinkernagel RM, Hengartner H. Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Annu Rev Immunol. 1996;14:207–232. doi: 10.1146/annurev.immunol.14.1.207. [DOI] [PubMed] [Google Scholar]
- 3.von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med. 2000;343:1020–1034. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- 4.Skinner PJ, Daniels MA, Schmidt CS, Jameson SC, Haase AT. Cutting edge: In situ tetramer staining of antigen-specific T cells in tissues. J Immunol. 2000;165:613–617. doi: 10.4049/jimmunol.165.2.613. [DOI] [PubMed] [Google Scholar]
- 5.Haanen JB, et al. In situ detection of virus- and tumor-specific T-cell immunity. Nature Med. 2000;6:1056–1060. doi: 10.1038/79573. [DOI] [PubMed] [Google Scholar]
- 6.Flugel A, et al. Migratory activity and functional changes of green fluorescent effector cells before and during experimental autoimmune encephalomyelitis. Immunity. 2001;14:547–560. doi: 10.1016/s1074-7613(01)00143-1. [DOI] [PubMed] [Google Scholar]
- 7.Reinhardt RL, Khoruts A, Merica R, Zell T, Jenkins MK. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 2001;410:101–105. doi: 10.1038/35065111. [DOI] [PubMed] [Google Scholar]
- 8.Allan JE, Dixon JE, Doherty PC. Nature of the inflammatory process in the central nervous system of mice infected with lymphocytic choriomeningitis virus. Curr Top Microbiol Immunol. 1987;134:131–143. doi: 10.1007/978-3-642-71726-0_6. [DOI] [PubMed] [Google Scholar]
- 9.Fung-Leung WP, Kundig TM, Zinkernagel RM, Mak TW. Immune response against lymphocytic choriomeningitis virus infection in mice without CD8 expression. J Exp Med. 1991;174:1425–1429. doi: 10.1084/jem.174.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kagi D, et al. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin–deficient mice. Nature. 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- 11.Joly E, Mucke L, Oldstone MB. Viral persistence in neurons explained by lack of major histocompatibility class I expression. Science. 1991;253:1283–1285. doi: 10.1126/science.1891717. [DOI] [PubMed] [Google Scholar]
- 12.Gallimore A, et al. A protective cytotoxic T cell response to a subdominant epitope is influenced by the stability of the MHC class I/peptide complex and the overall spectrum of viral peptides generated within infected cells. Eur J Immunol. 1998;28:3301–3311. doi: 10.1002/(SICI)1521-4141(199810)28:10<3301::AID-IMMU3301>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 13.van der Most RG, et al. Identification of Db- and Kb-restricted subdominant cytotoxic T-cell responses in lymphocytic choriomeningitis virus-infected mice. Virology. 1998;240:158–167. doi: 10.1006/viro.1997.8934. [DOI] [PubMed] [Google Scholar]
- 14.Gairin JE, Mazarguil H, Hudrisier D, Oldstone MB. Optimal lymphocytic choriomeningitis virus sequences restricted by H-2Db major histocompatibility complex class I molecules and presented to cytotoxic T lymphocytes. J Virol. 1995;69:2297–2305. doi: 10.1128/jvi.69.4.2297-2305.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 16.Janes PW, Ley SC, Magee AI. Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J Cell Biol. 1999;147:447–461. doi: 10.1083/jcb.147.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 18.Sanderson CJ. The mechanism of T cell mediated cytotoxicity. II Morphological studies of cell death by time-lapse microcinematography. Proc R Soc Lond B. 1976;192:241–255. doi: 10.1098/rspb.1976.0011. [DOI] [PubMed] [Google Scholar]
- 19.Rothstein TL, Mage M, Jones G, McHugh LL. Cytotoxic T lymphocyte sequential killing of immobilized allogeneic tumor target cells measured by time-lapse microcinematography. J Immunol. 1978;121:1652–1656. [PubMed] [Google Scholar]
- 20.Hahn K, et al. Antigen presentation and cytotoxic T lymphocyte killing studied in individual, living cells. Virology. 1994;201:330–340. doi: 10.1006/viro.1994.1298. [DOI] [PubMed] [Google Scholar]
- 21.Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ’Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- 22.Kucik DF, Dustin ML, Miller JM, Brown EJ. Adhesion-activating phorbol ester increases the mobility of leukocyte integrin LFA-1 in cultured lymphocytes. J Clin Invest. 1996;97:2139–2144. doi: 10.1172/JCI118651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krummel MF, Davis MM. Dynamics of the immunological synapse: finding, establishing and solidifying a connection. Curr Opin Immunol. 2002;14:66–74. doi: 10.1016/s0952-7915(01)00299-0. [DOI] [PubMed] [Google Scholar]
- 24.Stinchcombe JC, Bossi G, Booth S, Griffiths GM. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity. 2001;15:751–761. doi: 10.1016/s1074-7613(01)00234-5. [DOI] [PubMed] [Google Scholar]
- 25.Kane LP, Lin J, Weiss A. Signal transduction by the TCR for antigen. Curr Opin Immunol. 2000;12:242–249. doi: 10.1016/s0952-7915(00)00083-2. [DOI] [PubMed] [Google Scholar]
- 26.Veillette A, Bookman MA, Horak EM, Bolen JB. The CD4 and CD8 T cell surface antigens are associated with the internal membrane tyrosine-protein kinase p56lck. Cell. 1988;55:301–308. doi: 10.1016/0092-8674(88)90053-0. [DOI] [PubMed] [Google Scholar]
- 27.Bachmann MF, et al. Developmental regulation of Lck targeting to the CD8 coreceptor controls signaling in naive and memory T cells. J Exp Med. 1999;189:1521–1530. doi: 10.1084/jem.189.10.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yannelli JR, Sullivan JA, Mandell GL, Engelhard VH. Reorientation and fusion of cytotoxic T lymphocyte granules after interaction with target cells as determined by high resolution cinemicrography. J Immunol. 1986;136:377–382. [PubMed] [Google Scholar]
- 29.Kuhn JR, Poenie M. Dynamic polarization of the microtubule cytoskeleton during CTL-mediated killing. Immunity. 2002;16:111–121. doi: 10.1016/s1074-7613(02)00262-5. [DOI] [PubMed] [Google Scholar]
- 30.Grakoui A, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 31.Bromley SK, et al. The immunological synapse. Annu Rev Immunol. 1902;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 32.Miller MJ, Wei SH, Parker I, Cahalan MD. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science. 2002;296:1869–1873. doi: 10.1126/science.1070051. [DOI] [PubMed] [Google Scholar]
- 33.Stoll S, Delon J, Brotz TM, Germain RN. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science. 2002;296:1873–1876. doi: 10.1126/science.1071065. [DOI] [PubMed] [Google Scholar]
- 34.Bousso P, Bhakta NR, Lewis RS, Robey E. Dynamics of thymocyte-stromal cell interactions visualized by two-photon microscopy. Science. 2002;296:1876–1880. doi: 10.1126/science.1070945. [DOI] [PubMed] [Google Scholar]
- 35.Busch DH, Pilip IM, Vijh S, Pamer EG. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity. 1998;8:353–362. doi: 10.1016/s1074-7613(00)80540-3. [DOI] [PubMed] [Google Scholar]