

Abstract
The zinc finger transcription factor Krüppel-like factor 4 (KLF4) has been implicated in both tumor suppression and progression. However, its function in pancreatic cancer has not been well characterized. Here, we show that pancreatic cancer cell lines expressed various levels of KLF4 RNA and protein. Ectopic expression of KLF4 by FG and BxPC-3 pancreatic cancer cells resulted in cell cycle arrest and marked inhibition of cell growth in vitro and attenuation of tumor growth and metastasis in an orthotopic mouse model. Overexpression of KLF4 also led to significant induction of p27Kip1 expression, at both the RNA and protein levels, in a dose- and time-dependent manner, indicating that KLF4 transcriptionally regulates the expression of p27Kip1. Chromatin immunoprecipitation assays consistently showed that KLF4 protein physically interacts with the p27Kip1 promoter. Promoter deletion and point mutation analyses indicated that a region between nucleotides −435 and −60 of the p27Kip1 promoter and intact of the three KLF4-binding sites within that region were required for the full induction of p27Kip1 promoter activity by KLF4. Our findings suggest that KLF4 transactivates p27Kip1 expression and inhibits the growth and metastasis of human pancreatic cancer.
Introduction
Pancreatic cancer is currently the fourth leading cause of cancer-related death in the United States (1). The great majority of pancreatic cancers are adenocarcinomas arising from the ductal cells of the exocrine pancreas. The etiology and pathogenesis of pancreatic adenocarcinoma remain unclear, although heterogeneous genetic alterations have been reported. The most common of these genetic alterations include point mutations of KRAS; inactivation of CDKN2A/p16/MTS1, p53, and MADH4/SMAD4/DPC4; and overexpression of several mitogenic growth factors and their tyrosine kinase receptors (2). The phosphatidylinositol 3-kinase/Akt pathway was recently found to be activated in pancreatic cancer owing to aberrant expression of Akt or phosphatase and tensin homologue (3, 4). Continued identification of signature gene alterations in pancreatic cancer will provide a conceptual framework to guide future analyses of this complex disease and the development of strategies for its early detection and effective treatment.
Krüppel-like factor 4 (KLF4; formerly known as gut-enriched Krüppel-like factor) is a zinc finger transcription factor, and its mRNA is expressed primarily in postmitotic, terminally differentiated epithelial cells in organs such as skin, lungs, and gastrointestinal tract (5, 6). In cell culture, KLF4 expression can be increased by serum deprivation, contact inhibition, and DNA damage (7, 8). Conversely, reduced expression of KLF4 has been reported in various tumors (9–12), and restoration of KLF4 expression can induce growth arrest in colon cancer cells or apoptosis in bladder or gastric cancer cells and leukemia (12–15). Accumulating clinical evidence also suggests that KLF4 functions as a tumor suppressor; genetic and epigenetic alterations of the KLF4 gene have been found in gastrointestinal cancers (12, 16–18). On the other hand, KLF4 expression is increased in primary breast ductal carcinoma (19) and in oral and dermal squamous cell carcinomas (10, 20), suggesting that KLF4 is important in tumor development and progression (21–23). Moreover, whether and how KLF4 functions in pancreatic cancer remains unknown.
In the present study, we investigated the expression and molecular function of KLF4 in human pancreatic cancer cells. Our results showed that KLF4 positively regulates p27Kip1 expression and inhibits the growth and metastasis of human pancreatic cancer.
Materials and Methods
Cell lines and culture conditions
The human pancreatic adenocarcinoma cell lines Capan-1, AsPC-1, BxPC-3, Hs766T, MiaPaCa-2, and PANC-1 and human embryonic kidney (HEK) 293 cells were purchased from the American Type Culture Collection; FG human pancreatic adenocarcinoma cell line was obtained from Michael P. Vezeridis (Brown Medical School, Providence, RI; ref. 24). All cell lines were maintained in plastic flasks as adherent monolayers in MEM supplemented with 10% fetal bovine serum (FBS), sodium pyruvate, nonessential amino acids, L-glutamine, and a vitamin solution (Flow Laboratories).
Generation of KLF4-expressing vectors
Adenoviral vectors were generated and used as reported previously (12). Retroviral vectors were generated using Retro-X system from Clontech. Briefly, Phoenix ecotropic packaging cells were transfected with the retroviral vector pLXSN-FLAG-KLF4 or the corresponding vector without KLF4 using Lipofectamine 2000 reagent (Invitrogen). Retroviral supernatants were collected, filtered, and used for infecting target cells according to the manufacturer’s instruction. The green fluorescent protein (GFP)-tagged vector pEGFP-KLF4 was created by inserting a HindIII-SalI fragment encoding human KLF4 into pEGFP-C1 (Clontech); sequences were confirmed by DNA sequence analysis.
Animals
Athymic BALB/c nude mice were purchased from The Jackson Laboratory and housed in laminar flow cabinets under specific pathogen-free conditions according to the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care and in accordance with the current regulations and standards of the U.S. Department of Agriculture, Department of Health and Human Services, and NIH. The xenograft experiments were done when the mice were 8 wk old.
Immunocytochemical analysis
FG cells were seeded in Falcon culture slides (Becton Dickinson) at 1 × 105 per well in DMEM supplemented with 10% FBS. Cells were transfected with pEGFP-KLF4, permeabilized in 0.1% NP40, and incubated with a specific anti-p27Kip1 serums and then Texas red–labeled secondary antibody. Nuclear staining was accomplished by incubation in a solution containing 10 μg/mL 4′,6-diamidino-2-phenylindole (Sigma-Aldrich). Fluorescence imaging was performed with a Zeiss Axiophoto 2 microscope (Carl Zeiss, Inc.), with Adobe Photoshop 8.0 used to capture the images.
Cell proliferation assay
FG and BxPC-3 cells were seeded at 4 × 105 per well in six-well culture plates and incubated for 12 h at 37°C, after which they were incubated in serum-free medium containing Ad-FLAG-KLF4 or Ad-EGFP at a multiplicity of infection (MOI) of 20 for the FG cells or 15 for the BxPC-3 cells for 2 h at 37°C. Cells were then washed with serum-free medium, replenished with complete DMEM, and incubated for 1 to 4 d at 37°C. Cells were tested for viability with trypan blue exclusion and counted daily with a hemocytometer. To measure cell doubling time, 1 × 104 cells in 1.5 mL complete medium were plated in triplicate in 24-well plates. Cells were trypsinized and then counted daily for 1 wk using a hemocytometer. Cell population doubling time (DT) in hours was determined using the following equation: DT (hours) = 0.693(t − to)/ln (Nt/No), where to = time at which exponential growth began, t = time in hours, Nt = cell number at time t, and No = initial cell number (25).
Fluorescence-activated cell sorting analysis
FG and BxPC-3 cells were prepared for fluorescence-activated cell sorting (FACS) analysis of cell cycle distribution as described previously (26). Briefly, cells were seeded at 5 ×105 per dish in 60-mm cell culture dishes and incubated at 37°C for 12 h, after which Ad-KLF4 was added at 10 or 20 MOI for the FG cells and at 7.5 or 15 for the BxPC-3 cells, and Ad-EGFP was used to adjust the total MOI to be equal in each group. At 40 h after infection, the cells were harvested and fixed in 70% ethanol. Thirty minutes before FACS analysis, the cells were stained with propidium iodide and analyzed with a FACSCalibur equipped with the CellQuest software program (Becton Dickinson).
Northern blot analysis
Northern blotting was performed using total RNA extracted from cell cultures and using p27Kip1 and KLF4 cDNA as probes. Equal loading of RNA samples was monitored by hybridizing the same membrane filter with a human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA probe (27).
Western blot analysis
Standard Western blotting was done using whole-cell lysates and rabbit polyclonal antibodies against human KLF4, p27Kip1, cyclin D1, p21CIP1 (Santa Cruz Biotechnology), and a peroxidase-linked, species-specific, anti-rabbit IgG F(ab′)2 fragment (Amersham). Equal protein sample loading was monitored by hybridizing the same membrane filter with an anti-GAPDH antibody. Proteins were detected with Amersham’s enhanced chemiluminescence system. Kodak Digital Science Image station 440CF system was used for the acquisition of quantitative imaging data (27).
Small interfering RNA
RNA interference was done with synthetic small interfering RNA (siRNA) oligo to human KLF4 (Ambion). Briefly, BxPC-3 cells were seeded to 80% confluence in six-well plates in triplicate and transiently transfected with KLF4 siRNA or a control scrambled siRNA (10 μL/well, 20 μmol/L) using Lipofectamine 2000. Cell or protein samples were harvested 48 h after transfection and processed for FACS or Western blot analysis. In some experiments, BxPC-3 cells were infected with Mission short hairpin RNA (shRNA) lentiviral particles (Sigma-Aldrich), which carry nontargeting or specific KLF4-targeting shRNA. After puromycin selection, the antibiotic-resistant cell clones were cultured in complete DMEM without puromycin for 3 to 4 d. The cells were then seeded at 5 × 105 per dish in 60-mm cell culture dishes and incubated in DMEM containing 1.0% FBS for 24 h at 37°C before being harvested and processed for protein extraction and FACS analysis, respectively. For in vivo tumorigenicity analysis, BxPC-3 cells and BxPC-3 cells that carry nontargeting or KLF4-specific targeting shRNA were seeded at 6 × 106 per dish in 150-mm cell culture dishes and incubated in complete DMEM for 24 h at 37°C, and then single-cell suspensions were prepared for in vivo assay using the similar procedure as described below.
Tumor growth and metastasis
Tumor cells in exponential growth phase were prepared for inoculation in mice. Single-cell suspensions that were >95% viable were used. Tumor cells (1 × 106 per mouse) were then injected into the subcutis or pancreas of nude mice (10 mice per group). The animals were killed 60 d after the tumor cell injection or when they seemed moribund. Primary pancreatic tumors were then harvested and weighed. Livers were also removed and fixed in Bouin’s solution for 24 h to distinguish neoplastic lesions from organ parenchyma; metastases on the surface of the liver were counted under a dissecting microscope by an investigator blinded as to treatment condition.
Construction of p27Kip1 promoter reporter plasmids and mutagenesis
The p27Kip1 promoter constructs (both the full-length construct and a series of 5′ deletion mutants) were originally obtained from Dr. Toshiyuki Sakai (Kyoto Prefectural University of Medicine, Kyoto, Japan; ref. 28). To generate additional 5′ deletion mutants, the following primers were used for PCR amplification: 5′-gataaggtaccttgttttgttcggttttgtttttttgag-3′ (sense) and 5′-agatcgcagatctcgagcccgg-3′ (antisense). The PCR product was sub-cloned into the Asp718/BglII sites of the pGVB2 vector. To generate site-directed mutants of KLF4 elements in the proximal region of the p27Kip1 promoter, the p27-N-MB435 vector was constructed with the following primers: 5′-gataaggtaccgaactttctctatcgatcac-3′ (sense) and 5′-agatcgcagatctcgagcccgg-3′ (antisense). The PCR product was then subcloned into the Asp718/BglII sites of the pGVB2 vector, which was then used for site-directed mutation with the QuikChange Site-Directed Mutagenesis kit (Stratagene) according to the manufacturer’s instructions. Primers (with the mutated sites shown in bold) used to generate mutation in the putative KLF4-binding site 1 (designated p27-N-MB435/KLF4-m1) were 5′-gagggggttcgggccgcgtaggTTcgctttgttttgttcggttttg-3′ (sense) and 5′-caaaaccgaacaaaacaaagcgAAcctacgcggcccgaaccccctc-3′ (antisense); for mutation in the KLF4-binding site 2 (designated p27-N-MB435/KLF4-m2), 5′-gactcggacgggctttgccaTActctccgcttgcctggtcccc-3′ (sense) and 5′-ggggaccaggcaagcg-gagagTAtggcaaagcccgtccgagtc-3′ (antisense); and for mutation in the KLF4-binding site 3 (designated p27-N-MB435/KLF4-m3), 5′-cgcgggaccg-cgggcttgcaTAAgcccagactcggacgggc-3′ (sense) and 5′-gcccgtccgagtctgggcTTAtgcaagcccgcggtcccgcg-3′ (antisense). All constructs were verified by sequencing the inserts and flanking regions of the plasmids.
Analysis of p27Kip1 promoter activity
The activity of the full-length and mutant p27Kip1 promoter constructs was analyzed as described previously (27). Briefly, plasmids containing firefly luciferase reporters were cotransfected into tumor cells in triplicate, with pMiniTK-RL as an internal control, by using Lipofectamine 2000. The pMiniTK-RL contained a full-length Renilla luciferase gene under the control of a minimal thymidine kinase (TK) promoter (636–757 bp from pTK-RL; Promega). In some experiments, an Sp1 promoter reporter (FOR2; ref. 29) and p27Kip1 reporters were cotransfected with pcDNA3.1-KLF4 or pcDNA3.1. The activity of both the firefly and Renilla luciferase reporters was determined 48 h later using a Dual Luciferase Assay kit (Promega). Specific promoter activity was expressed as the relative ratio of firefly luciferase activity to Renilla luciferase activity. The specific promoter activity was expressed as the fold changes of the experimental group versus the control group.
Chromatin immunoprecipitation assay
For these experiments, BxPC-3 and FG cells were subjected to chromatin immunoprecipitation (ChIP) with the ChIP Assay kit (Upstate Cell Signaling Solutions). Briefly, formaldehyde was used to cross-link proteins with DNA, and cells were lysed in SDS lysis buffer. The cell lysate was sonicated to shear the DNA to 400- to 600-bp lengths. Chromatin samples were then precleared with a salmon sperm DNA/protein A agarose-50% slurry for 30 min at 4°C and immunoprecipitated overnight with no antibody, an anti-KLF4 antibody (H-180), or an anti-FLAG antibody (M2). The region between −426 and −20 nucleotides of the p27Kip1 promoter was amplified with a pair of primers (designated as P1): 5′-aagggggctcgtcttttcg-3′ (sense) and 5′-gagtgcgagagaggcggtc-3′ (antisense). The 3′-untranslated region (UTR) region of p27Kip1 gene was amplified as control for ChIP assay using the following primer set (designated as P2): 5′-aagcgttggatgtagcattatgc-3′ (sense) and 5′-agatcaccagatctcccaaatgag-3′ (antisense). The PCR products were separated on a 1.5% agarose gel, stained with ethidium bromide, and visualized under UV light.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assays (EMSA) were done with Promega’s Gel Shift Assay System. Briefly, nuclear protein was extracted from BxPC-3 cells after 40 h of transduction with Ad-KLF4 at a MOI of 5 as described previously (27). Double-stranded oligonucleotides corresponding to KLF4-binding sites in the p27Kip1 promoter region were used as probes. For competition assays, a 50-fold molar excess of unlabeled oligonucleotides (cold probe) was added to the binding reaction. For the supershift assay, specific Sp1 (PEP2), Sp3 (D-29), KLF4 (H-180), and FLAG (M2; Sigma-Aldrich) antibodies were added to a final concentration of 0.1 μg/μL. Samples were then loaded onto a 4.5% polyacrylamide gel and subjected to electrophoresis. Gels were then dried and exposed to Kodak film (Eastman Kodak).
Statistical analysis
Each experiment was performed independently at least twice with similar results; findings from one representative experiment are presented. Significant differences between in vitro treatment conditions were assessed with Student’s t test (two tailed), and differences between in vivo treatment groups were assessed with two-tailed Mann-Whitney U tests. To examine the relationship between two quantitative variables, the Pearson’s linear regression analysis was performed using Statistical Package for the Social Sciences statistical software (version 11.05; SPSS, Inc.). P ≤ 0.05 was deemed significant.
Results
KLF4 suppresses pancreatic cancer cell growth in vitro
First, we measured the expression of KLF4 in various pancreatic cancer cell lines at mRNA and protein levels (Fig. 1A). KLF4 was expressed at different levels by these cell lines. Then, we examined the correlation between endogenous KLF4 levels and in vitro growth behavior of AsPC-1, BxPC-3, MiaPaCa-2, and FG cells and found that a higher expression level of KLF4 protein correlated with a longer cell doubling time (Supplementary Table S1 and Supplementary Fig. S1), indicating a possible negative effect of KLF4 on cell growth. Next, FG cells, which expressed relatively little endogenous KLF4, and BxPC-3, which expressed relatively high amount of endogenous KLF4, were transduced with adenoviral KLF4. Overexpression of KLF4 was consistent with growth suppression of both FG cells (Fig. 1B1) and BxPC-3 cells (Fig. 1B2) in vitro. These findings show that ectopic expression of KLF4 led to suppression of tumor cell growth.
Figure 1.
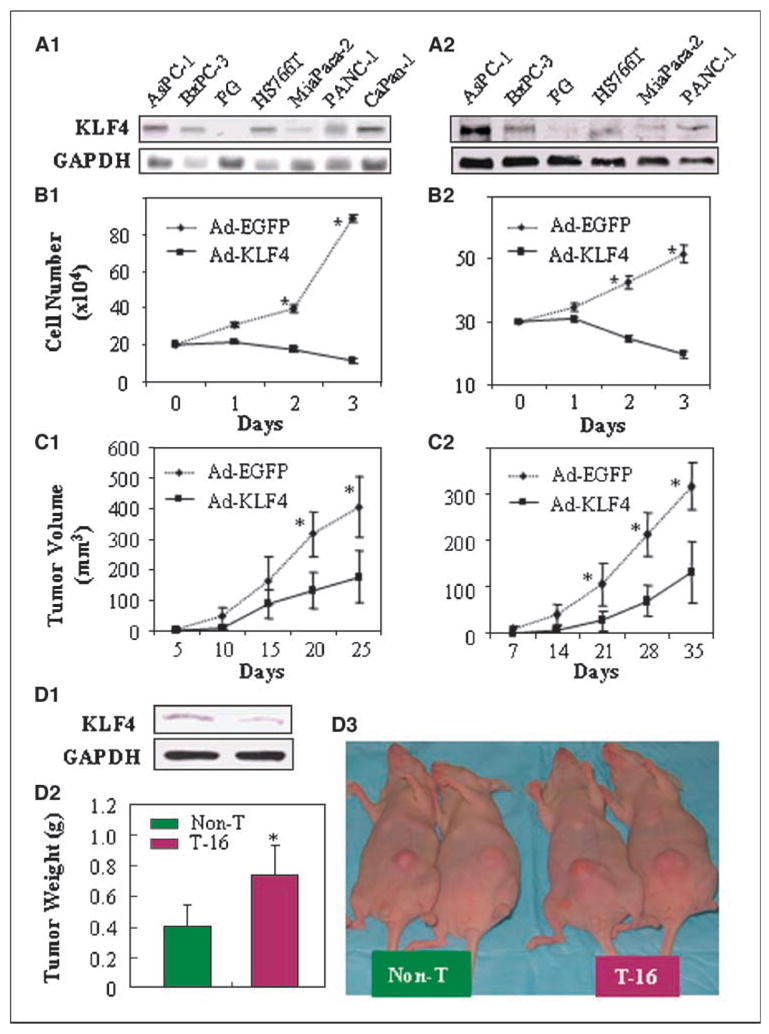
KLF4 suppresses human pancreatic cancer cell growth in vitro and in vivo. A, KLF4 expression in human pancreatic cancer cell lines at the mRNA level (by Northern blot analysis; A1) and at the protein level (by Western blot analysis; A2). B, FG and BxPC-3 cells were transduced with Ad-KLF4 or Ad-EGFP at MOIs of 20 (FG cells; B1) or 15 (BxPC-3 cells; B2) and assessed for proliferation by cell counting at 1 to 3 d after transduction. C, growth kinetics of KLF4-transduced FG cells (C1) and BxPC-3 cells (C2) injected into the subcutis of nude mice. D, BxPC-3 cells with reduced KLF4 protein expression (by Western blot analysis; D1) grew larger tumors in nude mice (D2, representative tumor sizes; D3, averages of tumor weight). Bars, SE. *, P < 0.05 versus Ad-EGFP or nontarget control (Non-T).
KLF4 inhibits pancreatic cancer growth and metastasis in vivo
Next, FG or BxPC-3 cells were injected s.c. into nude mice. FG and BxPC-3 cells transduced with control Ad-EGFP showed progressive growth, whereas FG and BxPC-3 cells transduced with KLF4 grew more slowly (Fig. 1C1 and C2). In an orthotopic model, FG and BxPC-3 cells were injected into the pancreas of mice (10 per group). FG and BxPC-3 cells transduced with Ad-EGFP produced large tumors and hepatic metastases, whereas FG and BxPC-3 cells transduced with KLF4 produced only localized small tumors (data not shown). In addition, we also determined the effect of knocking down KLF4 expression on tumor growth. BxPC-3 cells transduced with specific KLF4 shRNA vector (T-16) had reduced KLF4 expression and grew larger tumors compared with that of BxPC-3 cells that transduced with nontargeting vector (Non-T; Fig. 1D). There was no significant difference in tumor growth when compared with the parental BxPC-3 with BxPC-3-Non-T cells (data not shown). In addition, we also examined the effect of physiologic up-regulation of KLF4 expression on tumor growth using retroviral gene transfer and found that stable KLF4-transduced FG cells grew much slower in nude mice than the parental FG cells or FG cells stably transduced with Neo gene (Supplementary Fig. S2). All these findings suggest that KLF4 in pancreatic cancer cells suppressed the growth and metastasis of those cells in these ectopic and orthotopic mouse models.
KLF4 suppresses cell cycle progression of pancreatic cancer cells
To investigate the mechanism by which KLF4 inhibited pancreatic cancer cell growth, we tested the effects of KLF4 expression on cell cycle distribution. Overexpression of KLF4 induced G1 arrest in both FG cells (Fig. 2A, left) and BxPC-3 cells (Fig. 2B) and reduced the proportion of FG cells in S phase (Fig. 2A, right) and the percentage of BxPC-3 cells in G2-M (Fig. 2B). Additionally, lentivirus-mediated shRNA transient transfection specifically knocked down KLF4 expression in BxPC-3 cells (data not shown). After serum deprivation, knocking down of KLF4 expression in those cells significantly reduced the percentage of cells in G1 and G2 phases but increased the percentage of cells in S phase when compared with those in parental BxPC-3 cells or cells that carry nontargeting shRNA (Non-T; Fig. 2C). Alternatively, a direct transduction of KLF4 siRNA into BxPC-3 cells resulted in a similar effect on cell cycle progression in a dose-dependent manner (Supplementary Fig. S3). These results are consistent with those from our in vitro proliferation experiments, in which KLF4 expression suppressed tumor cell proliferation.
Figure 2.
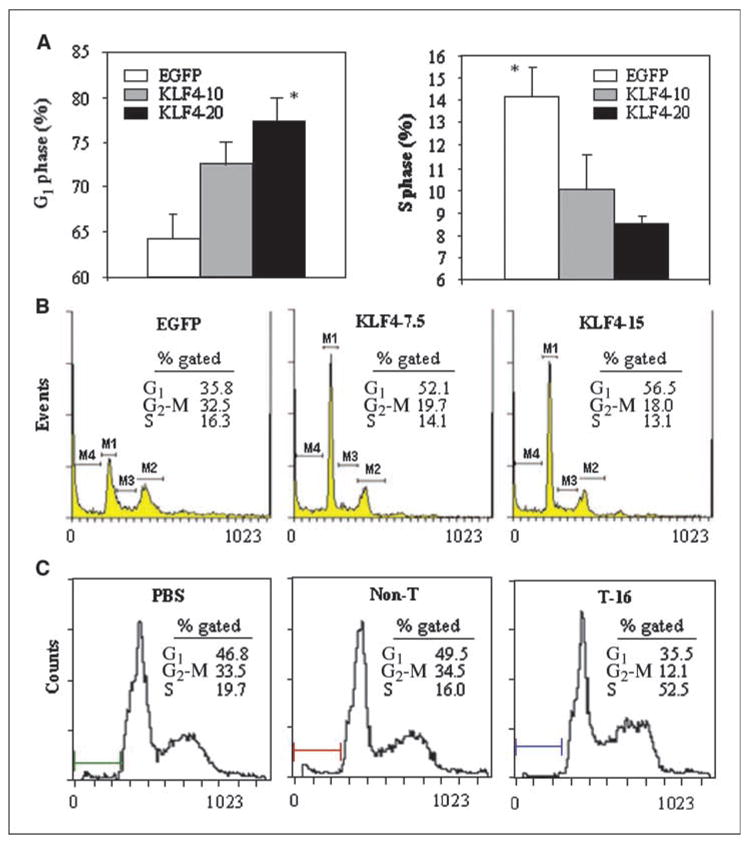
Altered KLF4 expressions affect cell cycle progression of pancreatic cancer cells. FG cells (A) or BxPC-3 cells (B) were transduced with Ad-KLF4 at the indicated MOI; Ad-EGFP was used as a control or to adjust the total MOI to be equal in each treatment condition. Cells were harvested at 40 h after transduction, fixed in 70% ethanol, and then stained with propidium iodide for FACS analysis. C, parental BxPC-3 cells and BxPC-3 cells stably carrying nontarget (Non-T) or specific target (T-16) shRNAs were cultured in DMEM containing 1% colony-stimulating factor for 24 h; cells were harvested and processed for FACS analysis. Bars, SE. *, P < 0.05 versus Ad-EGFP control.
KLF4 up-regulates p27Kip1 expression in pancreatic cancer cells
Next, to investigate the mechanism by which KLF4 induced cell cycle arrest, we assessed the expression of various cell cycle proteins. Following the expression of exogenous KLF4, the expression of p27Kip1 was significantly increased (Fig. 3A). Over-expression of KLF4 by FG cells reduced the expression of cyclin D1 and S-phase kinase-associated protein 2 (Skp2) but increased that of p21CIP1. Then, we undertook dose-response experiments and found that overexpression of KLF4 increased the expression of p27kip1 in both cell types (Fig. 3B), with the extent of the increase paralleling the MOI of Ad-KLF4. To determine whether endogenous KLF4 specifically regulates p27Kip1 expression, we used siRNA-mediated gene knockdown in the relatively high KLF4-expressing BxPC-3 cells and found corresponding dose-dependent reductions in both KLF4 and p27Kip1 expression (Fig. 3C). Because the ability of p27Kip1 to regulate cell cycling requires that it be in the nucleus, we next considered whether KLF4 could induce the nuclear accumulation of p27Kip1 protein. We found that the EGFP-KLF4 fusion protein was located mainly in the nuclei of FG cells, where it colocalized with p27Kip1 (Fig. 3D). Collectively, these findings indicate that KLF4 positively regulated p27Kip1 expression in pancreatic cancer cells.
Figure 3.
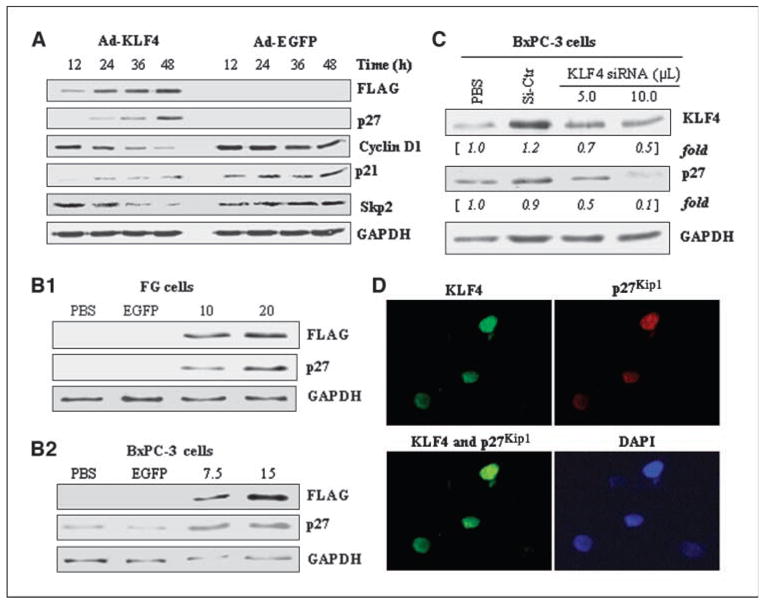
KLF4 up-regulates p27Kip1 expression in human pancreatic cancer cells. A, FG cells were transduced with adenoviral KLF4 or EGFP (control) at a MOI of 20 and expression of KLF4 and cell cycle–related proteins was assessed at the indicated times by Western blotting. B, p27Kip1 expression was determined in FG and BxPC-3 cells at 48 h after transduction with Ad-KLF4 at indicated MOI as noted for Fig. 2. C, BxPC-3 cells were treated with control (scrambled) siRNA or the indicated volumes of KLF4 siRNA (with control siRNA used to adjust the total volume to 10.0 μL/well); p27Kip1 expression was determined 48 h later by Western blotting. D, transduction of FG cells with an EGFP-KLF4 fusion protein followed by immunofluorescence showed colocalization of KLF4 and p27Kip1 in the nuclei. DAPI, 4′,6-diamidino-2-phenylindole.
KLF4 transcriptionally activates p27Kip1 promoter
Because p27Kip1 protein levels can be regulated by several mechanisms (28, 30, 31), we next sought to determine if KLF4 transcriptionally regulates p27Kip1 expression. Indeed, transduction with Ad-KLF4 induced the expression of p27Kip1 RNA as determined by Northern blotting (Fig. 4A) and induced p27Kip1 promoter activity in luciferase reporter analyses (Fig. 4B). These findings indicate that KLF4 can transcriptionally regulate p27Kip1 expression. Next, we identified the region in the p27Kip1 promoter responsible for transcriptional activation by KLF4 by transfecting HEK 293 cells with a series of 5′ deletion mutants based on the 3,568–bp p27Kip1 promoter, along with a KLF4 expression vector or a control vector, and testing the effect on promoter activity. Loss of the region spanning −3,358 to −462 nucleotides in p27Kip1 promoter construct did not substantially affect the induction of p27Kip1 promoter activity by KLF4. However, further deletion of the 5′ promoter region up to −435 nucleotides significantly increased the induction of p27Kip1 promoter activity, and further deletion of the nucleotides to −60 completely abolished the induction of p27Kip1 promoter activity, suggesting that the −435/−60 region contains the regulatory elements that are required for KLF4 to induce p27Kip1 promoter activity.
Figure 4.
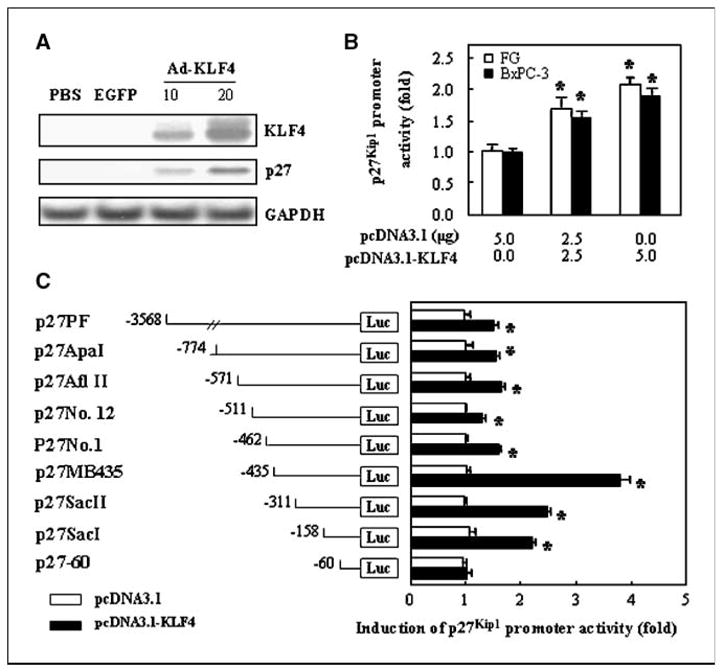
KLF4 transcriptionally activates p27Kip1 promoter. A, FG cells were transduced with adenoviral KLF4 or EGFP as described for Fig. 2, and KLF4 and p27Kip1 expression was measured 36 h later by Northern blotting. B, a full-length p27Kip1 promoter reporter (p27PF) was transfected into FG and BxPC-3 cells with pcDNA3.1-KLF4 or pcDNA3.1 (control) as shown and p27Kip1 promoter activity was determined 48 h later by dual luciferase assay. Results are expressed as the “fold” changes of the RR of pcDNA3.1-KLF4 vector versus pcDNA3.1 control vector. C, a series of 5′ deletion mutants based on the 3,568-bp p27Kip1 promoter were transfected into HEK 293 cells by using a KLF4 expression vector or control vector, and promoter activities were measured with a dual luciferase assay. Results are expressed as fold changes of the RR of KLF4 expression vector versus pcDNA3.1 control vector. Bars, SE. *, P < 0.05 versus the value of pcDNA3.1 control vector.
KLF4 binds to the p27Kip1 promoter in vivo
Next, we sought to determine if KLF4 binds to the promoter region of the p27Kip1 gene in vivo using ChIP assays. The PCR forward and reverse primers flanking the essential regulatory region are shown in Fig. 5A. First, to determine whether endogenous KLF4 binds to the p27Kip1 promoter, we immunoprecipitated chromatin fragments from BxPC-3 cells with control IgG or a specific anti-KLF4 antibody and found that the DNA fragment of the p27Kip1 promoter was immunoprecipitated by the specific anti-KLF4 antibody (Fig. 5B1, lane 6), but the DNA fragment of p27Kip1 3′-UTR was not immunoprecipitated by the specific anti-KLF4 antibody (Fig. 5B1, lane 7). Next, to determine whether exogenous KLF4 (i.e., that produced via gene transfer) binds to the p27Kip1 promoter, we transduced FG with Ad-KLF4 or Ad-EGFP and immunoprecipitated chromatin fragments with specific anti-FLAG antibody. Compared with EGFP-transduced cells or control FG cells, exogenous KLF4 clearly bound to the p27Kip1 promoter (Fig. 5B2, lane 12) but not to 3′-UTR of p27Kip1 gene (Fig. 5B2, lane 15). Thus, both exogenous and endogenous KLF4 bound to the p27Kip1 promoter in pancreatic cancer cells in vivo.
Figure 5.
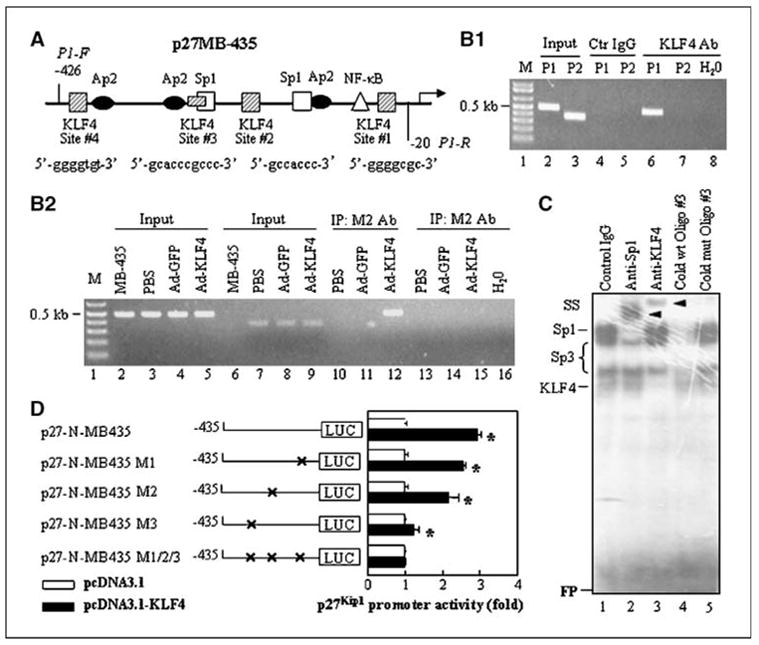
Binding of KLF4 to the p27Kip1 promoter in vivo and in vitro. A, schematic structure of p27Kip1 promoter. The locations and sequences of putative KLF4-binding sites and forward (P1-F) and reverse (P1-R) PCR primers flanking the p27Kip1 promoter were shown. B1, binding of endogenous KLF4 to p27Kip1 promoter. Chromatin fragments were prepared from BxPC-3 cells and ChIP assay was performed using a control IgG or anti-KLF4 antibody. Chromatin fragments without IgG or antibody were used as input controls. The PCR was performed using two sets of primers (P1, for p27Kip1 promoter; P2, for p27Kip1 3′-UTR as a control) as described in Materials and Methods. B2, binding of exogenous KLF4 to p27Kip1 promoter. Chromatin fragments were prepared from FG cells (PBS), FG cells transduced with Ad-EGFP (Ad-EGFP), or Ad-KLF4 harboring FLAG-tagged KLF4 gene (Ad-KLF4) and ChIP assay was performed using a specific anti-FLAG antibody. The PCR was performed using two sets of primers (P1, for lanes 2–5 and 10–12; P2, for lanes 6–9 and 13–16). C, EMSA. Nuclear protein was extracted from BxPC-3 cells transduced with Ad-KLF4. Radioisotope-labeled oligo #3 corresponding to #3 KLF4-binding site on p27 promoter was used as a probe. The probe was incubated with nuclear protein in the presence of control IgG (lane 1), anti-Sp1 (lane 2), anti-KLF4 (lane 3), cold wild-type oligo #3 (1:50 ratio; lane 4), or mutant oligo #3 (1:50 ratio; lane 5). Shifted bands were marked as “Sp1,” “Sp3,” or “KLF4”; supershifted bands “SS” were indicated by arrows; and free probes were marked as “FP.” D, p27 promoter activity. The p27 promoter reporter constructs with site-directed mutagenesis of three putative KLF4-binding sites were transfected into BxPC-3 cells in triplicate with KLF4 expression vector or control pcDNA3 vector and incubated for 48 h. The relative p27 promoter activities were determined and the activities in KLF4 groups were expressed as fold changes of that in their respective controls. This was a representative experiment of three with similar results. *, statistical significance (P < 0.01) compared with respective control groups.
KLF4 binds to the p27Kip1 promoter in vitro
To identify KLF4-specific binding sites in the proximal promoter region of p27Kip1, we analyzed previously reported KLF4-binding motifs (32, 33) and found four putative KLF4-binding sites in the essential region of p27Kip1 promoter. We then used EMSA to determine if KLF4 binds to these sites in vitro using synthesized oligonucleotides containing the core sequences shown in Fig. 5A. First, the putative KLF4-binding site #3 was radiolabeled and used as a hot probe in EMSA, resulting in four major shifted DNA-protein complexes (Fig. 5C). The shifted complexes were then competed with cold wild-type site #3 oligonucleotides (lane 4) but not with mutant oligonucleotides (lane 5). In supershift assays, the putative KLF4 band was further shifted by anti-KLF4 antibody (lane 3). Similarly, we showed that KLF4 also bound to putative KLF4-binding site #1 and #2 of p27Kip1 promoter in vitro (Supplementary Fig. S4). Thus, we were able to identify three putative KLF4-binding sites in the proximal region of the p27Kip1 promoter.
Finally, to provide direct evidence that these putative KLF4-binding sites within the proximal p27Kip1 promoter contribute to the activity of the p27Kip1 promoter, we performed site-directed mutagenesis of the three putative KLF4-binding sites (or the combination of all three of them). Disruption of the putative KLF4-binding sites #1 and #2 led to slight decreases in the induction of p27Kip1 promoter activity by KLF4 (Fig. 5D); disruption of site #3 led to a substantial reduction. Mutation of all three sites totally eliminated the induction of p27Kip1 promoter activity by KLF4 (Fig. 5D). These findings suggest that all three putative KLF4-binding sites must be intact for the full induction of p27Kip1 promoter activity by KLF4. Finally, because KLF4 can positively or negatively regulate the expression of several genes, we selected proximal promoters of Sp1 and p27Kip1 and cotransfected them with control or KLF4 expression vectors into HEK 293 cells. In these experiments, KLF4 positively regulated p27Kip1 promoter activity but negatively regulated Sp1 promoter activity (Supplementary Fig. S5). This result is consistent with our present and previous report (34) and indicates that the effect of KLF4 on gene expression (positive or negative) is specific to the gene promoters.
Discussion
In the present study, we found that KLF4 was expressed at various levels in human pancreatic cancer cell lines and that ectopic overexpression of KLF4 significantly inhibited pancreatic cancer cell growth in vitro and tumorigenicity in animal models, whereas knocking down of KLF4 expression promoted tumor growth in vivo. Our mechanistic investigations suggested that this antitumor activity resulted from cell cycle arrest, that ectopic overexpression of KLF4 by pancreatic cancer cells led to significant induction of p27Kip1 mRNA and protein, that KLF4 bound to the promoter region of p27Kip1, and that the intact of three KLF4-binding sites in the p27Kip1 promoter region is required for the full induction of p27Kip1 promoter activity by KLF4. Collectively, these findings are the first report of the mechanism of action of KLF4 in pancreatic cancer, supporting the notion that KLF4 acts as a tumor suppressor (23).
KLF4 has been implicated in regulation of the differentiation of pancreatic ductal cells (35, 36). A recent microarray analysis indicated that KLF4 was up-regulated in pancreatic intraepithelial neoplasia (PanIN) lesions (37), but whether the alteration of KLF4 expression plays a constraining or promoting role in pancreatic cancer remains unknown. Given that the effects of oncogenic KRAS in isolated pancreatic ductal cells derived from keratin 19 promoter-driven K-rasG12V transgenic mice were constrained by up-regulation of certain cell cycle regulators, such as p16INK4A/p19Arf and p27Kip1, resulting in controlled cellular proliferation (38); p16INK4A has been found to inhibit the progression of pancreatic adenocarcinoma in mice (2, 39); and homozygous deletion of the CDKN2A locus where p16INK4A resides resulted in significant down-regulation of KLF4 in melanoma cell lines (40), it is reasonable to speculate that up-regulation of KLF4 in the early premalignant lesions of PanIN may represent a defense mechanism against oncogene activation. In the present study, an ectopic overexpression of KLF4 significantly inhibited tumor cell growth in vitro and suppressed tumor growth and metastasis in vivo, whereas lentiviral-mediated knocking down of KLF4 expression promoted tumor growth in animal model. These results suggest that KLF4 has a tumor-suppressive effect. However, many questions remain about whether KLF4 is a bona fide tumor suppressor in pancreatic cancer, including whether the expression of KLF4 is different at various disease stages or whether genetic alterations of KLF4 are common in pancreatic cancer.
Several lines of evidence suggest that KLF4 is a negative regulator of cellular proliferation and an inducer of epithelial terminal differentiation (23). Among the cell cycle genes regulated by KLF4, many of those that are up-regulated are inhibitors of proliferation, whereas genes that promote proliferation are repressed (41). In the current study, transduction of KLF4 caused significant cell growth inhibition owing to blockage of G1-S and G2-M cell cycle progression. Importantly, we found that KLF4 up-regulated p27Kip1 and, to a lesser extent, p21CIP1 but down-regulated cyclin D1 expression. Previous studies have established the importance of KLF4 in regulation of p21CIP1 expression (7, 11, 42, 43). Mechanistically, KLF4 activates p21CIP1 through a specific Sp1-like cis-element in the p21CIP1 proximal promoter; interestingly, the same element is also required for p53 to activate the p21CIP1 promoter, although p53 does not bind to it (44). Similarly, KLF4 negatively regulates cyclin D1 expression through binding to the Sp1 motif in the proximal region of the cyclin D1 promoter (45).
p27Kip1 is a universal cyclin-dependent kinase inhibitor that directly inhibits the enzymatic activity of cyclin-CDK complexes, resulting in cell cycle arrest at G1. In the present study, we established that KLF4 positively regulated p27Kip1 expression. Our results are consistent with those from a previous study of redox-controlled vascular smooth muscle cell proliferation, in which KLF4 was found to be involved in the expression of p21CIP1, p53, and p27Kip1 induced by H-Fe (46). Moreover, our study provides a molecular mechanism by which KLF4 regulates p27Kip1 expression.
p27Kip1 protein levels are regulated by several mechanisms. Although transcriptional regulation is the primary mechanism, cellular abundance of p27Kip1 was previously thought to be regulated predominantly by ubiquitin/proteasome-mediated proteolysis (47) in a process that requires p27Kip1 to associate with cyclin/cyclin-dependent kinase complexes and be phosphorylated at Thr187, which is then recognized by SCFSkp2, a member of the F-box family of the specific substrate recognition subunit of Skp1/Cull/F-box ubiquitin-protein ligase complexes (30). However, more recently, loss of p27Kip1 was shown to promote tumorigenesis in the lung independently of SCFSkp2-mediated p27Kip1 degradation, with loss of p27Kip1 protein expression resulting from reduced expression of p27Kip1 mRNA in the tumors (31). These findings were consistent with the observation that down-regulation of p27Kip1 protein was caused by decreased abundance of p27Kip1 mRNA in more than half of human breast cancers tested (31). Therefore, in the present study, we focused on how KLF4 directly regulated transcription of p27Kip1, although it is possible that KLF4 may indirectly affect p27Kip1 stability and hence p27Kip1 protein levels through regulating Skp2 expression. Our data strongly suggested that KLF4 transcriptionally regulates p27Kip1 expression by directly binding to the proximal region of p27Kip1 promoter. Although this region has been reported to have promoter activity (48), the physiologic role of this region in the regulation of p27Kip1 expression remained to be defined. Therefore, our study provides novel insight into the functional role of the proximal region of p27Kip1 promoter in regulation of p27Kip1 gene expression. Interestingly, it is possible that KLF4 has an inhibitory effect on the other binding sites by recruiting other molecules to the promoter region of p27, which has elegantly been shown by a recent report (49). Thus, transcriptional regulation of p27Kip1 by KLF4 is complex and warrants further investigation.
In summary, KLF4 was expressed at various levels in pancreatic cancer cells and that ectopic overexpression of KLF4 suppressed pancreatic cancer cell growth in vitro and in vivo via induction of p27Kip1 expression and cell cycle arrest, suggesting that KLF4 has tumor suppressor functions in pancreatic cancer. Our findings also provide new insights into the transcriptional regulation of p27Kip1 expression by KLF4 and may help to clarify the role of KLF4 in pancreatic cancer progression.
Acknowledgments
Grant support: National Cancer Institute Pancreatic Cancer Specialized Program of Research Excellence grants 5P20CA101936-01-PP4, 1R01CA093829, and 1R03CA124523; American Cancer Society Research Scholar Grant CSM-106640; and American Association of Cancer Research-PanCAN career development award.
We thank Christine Wogan for editorial assistance.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed.
References
- 1.American Cancer Society. Cancer facts and figures 2007. Atlanta: American Cancer Society; 2007. [Google Scholar]
- 2.Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–49. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- 3.Maitra A, Hruban RH. A new mouse model of pancreatic cancer: PTEN gets its Akt together. Cancer Cell. 2005;8:171–2. doi: 10.1016/j.ccr.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 4.Altomare DA, Tanno S, De Rienzo A, et al. Frequent activation of AKT2 kinase in human pancreatic carcinomas. J Cell Biochem. 2003;88:470–6. doi: 10.1002/jcb.10287. [DOI] [PubMed] [Google Scholar]
- 5.Segre JA, Bauer C, Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet. 1999;22:356–60. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- 6.Shields JM, Christy RJ, Yang VW. Identification and characterization of a gene encoding a gut-enriched Kruppel-like factor expressed during growth arrest. J Biol Chem. 1996;271:20009. doi: 10.1074/jbc.271.33.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoon HS, Yang VW. Requirement of Kruppel-like factor 4 in preventing entry into mitosis following DNA damage. J Biol Chem. 2004;279:5035–41. doi: 10.1074/jbc.M307631200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen ZY, Wang X, Zhou Y, Offner G, Tseng CC. Destabilization of Kruppel-like factor 4 protein in response to serum stimulation involves the ubiquitin-proteasome pathway. Cancer Res. 2005;65:10394–400. doi: 10.1158/0008-5472.CAN-05-2059. [DOI] [PubMed] [Google Scholar]
- 9.Yng Y, Goldstein BG, Chao HH, Katz JP. KLF4 and KLF5 regulate proliferation, apoptosis and invasion in esophageal cancer cells. Cancer Biol Ther. 2005;4:1216–21. doi: 10.4161/cbt.4.11.2090. [DOI] [PubMed] [Google Scholar]
- 10.Huang CC, Liu Z, Li X, et al. KLF4 and PCNA identify stages of tumor initiation in a conditional model of cutaneous squamous epithelial neoplasia. Cancer Biol Ther. 2005;4:1401–8. doi: 10.4161/cbt.4.12.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katz JP, Perreault N, Goldstein BG, et al. Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology. 2005;128:935–45. doi: 10.1053/j.gastro.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 12.Wei D, Gong W, Kanai M, et al. Drastic down-regulation of Kruppel-like factor 4 expression is critical in human gastric cancer development and progression. Cancer Res. 2005;65:2746–54. doi: 10.1158/0008-5472.CAN-04-3619. [DOI] [PubMed] [Google Scholar]
- 13.Dang DT, Chen X, Feng J, Torbenson M, Dang LH, Yang VW. Overexpression of Kruppel-like factor 4 in the human colon cancer cell line RKO leads to reduced tumorigenecity. Oncogene. 2003;22:3424–30. doi: 10.1038/sj.onc.1206413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohnishi S, Ohnami S, Laub F, et al. Downregulation and growth inhibitory effect of epithelial-type Kruppel-like transcription factor KLF4, but not KLF5, in bladder cancer. Biochem Biophys Res Commun. 2003;308:251–6. doi: 10.1016/s0006-291x(03)01356-1. [DOI] [PubMed] [Google Scholar]
- 15.Yasunaga J, Taniguchi Y, Nosaka K, et al. Identification of aberrantly methylated genes in association with adult T-cell leukemia. Cancer Res. 2004;64:6002–9. doi: 10.1158/0008-5472.CAN-04-1422. [DOI] [PubMed] [Google Scholar]
- 16.Ghaleb AM, McConnell BB, Nandan MO, Katz JP, Kaestner KH, Yang VW. Haploinsufficiency of Kruppel-like factor 4 promotes adenomatous polyposis coli dependent intestinal tumorigenesis. Cancer Res. 2007;67:7147–54. doi: 10.1158/0008-5472.CAN-07-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang W, Chen X, Kato Y, et al. Novel cross talk of Kruppel-like factor 4 and β-catenin regulates normal intestinal homeostasis and tumor repression. Mol Cell Biol. 2006;26:2055–64. doi: 10.1128/MCB.26.6.2055-2064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao W, Hisamuddin IM, Nandan MO, Babbin BA, Lamb NE, Yang VW. Identification of Kruppel-like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene. 2004;23:395–402. doi: 10.1038/sj.onc.1207067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foster KW, Frost AR, McKie-Bell P, et al. Increase of GKLF messenger RNA and protein expression during progression of breast cancer. Cancer Res. 2000;60:6488–95. [PubMed] [Google Scholar]
- 20.Foster KW, Ren S, Louro ID, et al. Oncogene expression cloning by retroviral transduction of adeno-virus E1A-immortalized rat kidney RK3E cells: transformation of a host with epithelial features by c-MYC and the zinc finger protein GKLF. Cell Growth Differ. 1999;10:423–34. [PubMed] [Google Scholar]
- 21.Rowland BD, Peeper DS. KLF4, p21 and context-dependent opposing forces in cancer. Nat Rev Cancer. 2006;6:11–23. doi: 10.1038/nrc1780. [DOI] [PubMed] [Google Scholar]
- 22.Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol. 2005;7:1074–82. doi: 10.1038/ncb1314. [DOI] [PubMed] [Google Scholar]
- 23.Wei D, Kanai M, Huang S, Xie K. Emerging role of KLF4 in human gastrointestinal cancer. Carcinogenesis. 2006;27:23–31. doi: 10.1093/carcin/bgi243. [DOI] [PubMed] [Google Scholar]
- 24.Vezeridis MP, Tzanakakis GN, Meitner PA, Doremus CM, Tibbetts LM, Calabresi P. In vivo selection of a highly metastatic cell line from a human pancreatic carcinoma in the nude mouse. Cancer. 1992;69:2060–3. doi: 10.1002/1097-0142(19920415)69:8<2060::aid-cncr2820690810>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 25.Cullen JJ, Weydert C, Hinkhouse MM, et al. The role of manganese superoxide dismutase in the growth of pancreatic adenocarcinoma. Cancer Res. 2003;63:1297–303. [PubMed] [Google Scholar]
- 26.Wei D, Gong W, Oh SC, et al. Loss of RUNX3 expression significantly affects the clinical outcome of gastric cancer patients and its restoration causes drastic suppression of tumor growth and metastasis. Cancer Res. 2005;65:4809–16. doi: 10.1158/0008-5472.CAN-04-3741. [DOI] [PubMed] [Google Scholar]
- 27.Wei D, Wang L, He Y, Xiong HQ, Abbruzzese JL, Xie K. Celecoxib inhibits vascular endothelial growth factor expression in and reduces angiogenesis and metastasis of human pancreatic cancer via suppression of Sp1 transcription factor activity. Cancer Res. 2004;64:2030–8. doi: 10.1158/0008-5472.can-03-1945. [DOI] [PubMed] [Google Scholar]
- 28.Minami S, Ohtani-Fujita N, Igata E, Tamaki T, Sakai T. Molecular cloning and characterization of the human p27Kip1 gene promoter. FEBS Lett. 1997;411:1–6. doi: 10.1016/s0014-5793(97)00660-1. [DOI] [PubMed] [Google Scholar]
- 29.Nicolas M, Noe V, Jensen KB, Ciudad CJ. Cloning and characterization of the 5′-flanking region of the human transcription factor Sp1 gene. J Biol Chem. 2001;276:22126–32. doi: 10.1074/jbc.M010740200. [DOI] [PubMed] [Google Scholar]
- 30.Ganoth D, Bornstein G, Ko TK, et al. The cell-cycle regulatory protein Cks1 is required for SCF (Skp2)-mediated ubiquitinylation of p27. Nat Cell Biol. 2001;3:321–4. doi: 10.1038/35060126. [DOI] [PubMed] [Google Scholar]
- 31.Timmerbeul I, Garrett-Engele CM, Kossatz U, et al. Testing the importance of p27 degradation by the SCFskp2 pathway in murine models of lung and colon cancer. Proc Natl Acad Sci U S A. 2006;103:14009–14. doi: 10.1073/pnas.0606316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shields JM, Yang VW. Identification of the DNA sequence that interacts with the gut-enriched Kruppel-like factor. Nucleic Acids Res. 1998;26:796–802. doi: 10.1093/nar/26.3.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garrett-Sinha LA, Eberspaecher H, Seldin MF, de Crombrugghe B. A gene for a novel zinc-finger protein expressed in differentiated epithelial cells and transiently in certain mesenchymal cells. J Biol Chem. 1996;271:31384–90. doi: 10.1074/jbc.271.49.31384. [DOI] [PubMed] [Google Scholar]
- 34.Kanai M, Wei D, Li Q, et al. Loss of Kruppel-like factor 4 expression contributes to Sp1 overexpression and human gastric cancer development and progression. Clin Cancer Res. 2006;12:6395–402. doi: 10.1158/1078-0432.CCR-06-1034. [DOI] [PubMed] [Google Scholar]
- 35.Brembeck FH, Rustgi AK. The tissue-dependent keratin 19 gene transcription is regulated by GKLF/KLF4 and Sp1. J Biol Chem. 2000;275:28230–9. doi: 10.1074/jbc.M004013200. [DOI] [PubMed] [Google Scholar]
- 36.Brembeck FH, Moffett J, Wang TC, Rustgi AK. The keratin 19 promoter is potent for cell-specific targeting of genes in transgenic mice. Gastroenterology. 2001;120:1720–8. doi: 10.1053/gast.2001.24846. [DOI] [PubMed] [Google Scholar]
- 37.Prasad NB, Biankin AV, Fukushima N, et al. Gene expression profiles in pancreatic intraepithelial neoplasia reflect the effects of Hedgehog signaling on pancreatic ductal epithelial cells. Cancer Res. 2005;65:1619–26. doi: 10.1158/0008-5472.CAN-04-1413. [DOI] [PubMed] [Google Scholar]
- 38.Schreiber FS, Deramaudt TB, Brunner TB, et al. Successful growth and characterization of mouse pancreatic ductal cells: functional properties of the Ki-RAS(G12V) oncogene. Gastroenterology. 2004;127:250–60. doi: 10.1053/j.gastro.2004.03.058. [DOI] [PubMed] [Google Scholar]
- 39.Bardeesy N, Aguirre AJ, Chu GC, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA. 2006;103:5947–52. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bloethner S, Hemminki K, Thirumaran RK, et al. Differences in global gene expression in melanoma cell lines with and without homozygous deletion of the CDKN2A locus genes. Melanoma Res. 2006;16:297–307. doi: 10.1097/01.cmr.0000222597.50309.05. [DOI] [PubMed] [Google Scholar]
- 41.Chen X, Whitney EM, Gao SY, Yang VW. Transcriptional profiling of Kruppel-like factor 4 reveals a function in cell cycle regulation and epithelial differentiation. J Mol Biol. 2003;326:665–77. doi: 10.1016/S0022-2836(02)01449-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoon HS, Chen X, Yang VW. Kruppel-like factor 4 mediates p53-dependent G1/S cell cycle arrest in response to DNA damage. J Biol Chem. 2003;278:2101–5. doi: 10.1074/jbc.M211027200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghaleb AM, Katz JP, Kaestner KH, Du JX, Yang VW. Kruppel-like factor 4 exhibits antiapoptotic activity following γ-radiation-induced DNA damage. Oncogene. 2007;26:2365–73. doi: 10.1038/sj.onc.1210022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang W, Geiman DE, Shields JM, et al. The gut-enriched Kruppel-like factor (Kruppel-like factor 4) mediates the transactivating effect of p53 on the p21WAF1/Cip1 promoter. J Biol Chem. 2000;275:18391–8. doi: 10.1074/jbc.C000062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shie JL, Chen ZY, Fu M, Pestell RG, Tseng CC. Gut-enriched Kruppel-like factor represses cyclin D1 promoter activity through Sp1 motif. Nucleic Acids Res. 2000;28:2969–76. doi: 10.1093/nar/28.15.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nickenig G, Baudler S, Muller C, et al. Redox-sensitive vascular smooth muscle cell proliferation is mediated by GKLF and Id3 in vitro and in vivo. FASEB J. 2002;16:1077–86. doi: 10.1096/fj.01-0570com. [DOI] [PubMed] [Google Scholar]
- 47.Pagano M, Tam SW, Theodoras AM, et al. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–5. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- 48.Liu Z, Dong Z, Han B, Yang Y, Liu Y, Zhang JT. Regulation of expression by promoters versus internal ribosome entry site in the 5′-untranslated sequence of the human cyclin-dependent kinase inhibitor p27kip1. Nucleic Acids Res. 2005;33:3763–71. doi: 10.1093/nar/gki680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Evans PM, Zhang W, Chen X, Yang J, Bhakat KK, Liu C. Kruppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. J Biol Chem. 2007;282:33994–4002. doi: 10.1074/jbc.M701847200. [DOI] [PubMed] [Google Scholar]