

Summary
Background
In neurones, release of neurotransmitter occurs through the fusion of synaptic vesicles with the plasma membrane. Many proteins required for this process have been identified, with the SNAREs syntaxin 1, SNAP-25 and synaptobrevin thought to constitute the core fusion machinery. However, there is still a large gap between our understanding of individual protein-protein interactions and the functions of these proteins revealed by perturbations in intact synaptic preparations. To bridge this gap, we have used purified synaptic vesicles, together with artificial membranes containing co-reconstituted SNAREs as reaction partners, in fusion assays.
Results
Using complementary experimental approaches, we show that synaptic vesicles fuse constitutively, and with high efficiency, with proteoliposomes containing the plasma membrane proteins syntaxin 1 and SNAP-25. Fusion is inhibited by clostridial neurotoxins and involves the formation of SNARE complexes. Despite the presence of endogenous synaptotagmin, Ca2+ does not enhance fusion, even if phosphatidylinositol 4,5-bisphosphate is present in the liposome membrane. Rather, fusion kinetics are dominated by the availability of free syntaxin 1/SNAP-25 acceptor sites for synaptobrevin.
Conclusions
Synaptic vesicles are constitutively active fusion machines, needing only synaptobrevin for activity. Apparently, the final step in fusion does not involve the regulatory activities of other vesicle constituents, although these may be involved in regulating earlier processes. This is particularly relevant for the calcium-dependent regulation of exocytosis, which, in addition to synaptotagmin, requires other factors not present in the vesicle membrane. The in-vitro system described here provides an ideal starting point for unravelling the molecular details of such regulatory events.
Introduction
Synaptic release of neurotransmitters is mediated by the Ca2+-dependent exocytosis of synaptic vesicles. In recent years, an increasingly complex network of interacting proteins has been identified, which is responsible for the regulation and execution of vesicle docking and fusion. Examples of proteins participating in this network include the SNAREs synaptobrevin, syntaxin 1 and SNAP-25 that catalyse fusion; synaptotagmin, complexin and Munc-13 that are all involved in the Ca2+-dependent control of exocytosis; the SM-protein Munc-18 that probably controls SNARE assembly via interaction with other proteins, such as Mints; and an array of protein kinases that phosphorylate proteins such as SNAP-25, providing additional layers of regulation [1].
In contrast to the increasing genetic and physiological evidence linking these proteins to synaptic function, it has been much more difficult to unravel the underlying molecular mechanisms. In fact, there are many cases in which the properties of individual biochemical features are difficult to reconcile with the physiological function of these proteins as studied, for instance, by ‘knock-out’ and ‘knock-in’ approaches. The reason is that there is still a large gap in complexity between perturbing an individual protein in an otherwise intact presynaptic preparation and the study of protein-protein interactions in the test tube using purified components.
Biochemical analysis of supramolecular structures under cell-free conditions can bridge the ‘gap in complexity’ between these approaches, by reconstituting such functional complexes from individual purified components. However, unlike other supramolecular machines, the molecular complexes involved in vesicle docking and fusion are ‘dirty’ nanostructures, in that they involve membranes with highly complex and variable compositions, which recruit proteins only temporarily from the cytoplasm, dissociating once the task is completed [1].
In the synapse, a large body of evidence supports the idea that fusion is catalysed by the SNARE proteins. According to the ‘zipper hypothesis’ of SNARE function, the vesicular SNARE synaptobrevin assembles with the SNAREs syntaxin 1 and SNAP-25 in the plasma membrane, forming a molecular bridge between the membranes. Assembly involves the SNARE motifs, conserved stretches of 60–70 amino acids that are unstructured in solution but which form a bundle containing four parallel α-helices after assembly – the so-called ‘core-complex’. Nucleation occurs at the N-terminal end of the SNARE motifs and progresses towards the C-terminal membrane anchors, thus tying the membranes together and probably exerting force on the membranes. As a result, repulsion between the membranes is overcome and fusion is induced [1]. Indeed, when appropriate SNAREs are reconstituted in artificial vesicles they readily fuse in a SNARE-dependent manner [2]. However, other features of exocytosis have not been reproduced in the liposome system. Furthermore, the relevance of such reconstituted systems for biological fusion reactions has been challenged. Liposomes become leaky and fusion-prone at high protein:phospholipid ratios and they may contain residual detergent that could potentially act as a non-specific fusion catalyst. Furthermore, liposome fusion can be induced solely by the SNARE trans-membrane domains, and vesicles containing synaptobrevin fuse with planar membranes containing syntaxin in the absence of SNAP-25, giving rise to concerns about whether these fusions do indeed represent the molecular intermediates of synaptic exocytosis (reviewed in [3]).
In the present study, we have taken the in vitro approach towards reconstituting synaptic exocytosis one step further, by investigating the fusion of synaptic vesicles isolated from rat brain with liposomes containing the SNAREs syntaxin 1 and SNAP-25. By using a native membrane as one of the fusion partners we hope to avoid potential artefacts, caused by leaky or fusion-prone liposomes [3]. Furthermore, purified synaptic vesicles should serve as a benchmark for studying the function of the SNARE synaptobrevin in its native membrane (which, in contrast to liposomes, is loaded with additional proteins which may affect exocytosis). Finally, our recent analysis of synaptic vesicle structure has provided quantitative data for all the major constituents, including synaptobrevin, thus allowing us to match liposomes to their native counterparts [4].
Our results show that synaptic vesicles fuse with such liposomes in a highly efficient and almost quantitative manner. Fusion is mediated by synaptobrevin interacting with syntaxin and SNAP-25 in the membrane and its kinetics are controlled by the availability of the syntaxin-SNAP-25 acceptor complex. Surprisingly, fusion is not accelerated but rather slowed by Ca2+. We conclude that synaptic vesicles are constitutively active fusion machines, which are dependent on additional docking and priming factors in order to undergo Ca2+-dependent exocytosis at the synapse.
Results
Synaptic Vesicles are Constitutively Active Fusion Machines
During initial experiments we were interested in establishing whether synaptobrevin on synaptic vesicles is free to engage in synaptic core-complexes and, if so, whether core-complex formation could lead to membrane fusion. To avoid potential bias resulting from the method of preparation, synaptic vesicles were purified by two different procedures, involving either size-exclusion chromatography (‘Method A’) or Optiprep® floatation gradients (‘Method B’) as the final purification step [4, 5]. SNAREs were reconstituted into liposomes from cholate micellar solutions, using a phospholipid composition resembling that of synaptic vesicles and a physiologically relevant SNARE:phospholipid molar ratio of 1:300 [4].
To monitor fusion, we utilised a standard fluorescence dequenching assay, which reports fusion based on lipid mixing [2]. First, we investigated whether synaptic vesicles (purified by ‘Method A’) fuse with proteoliposomes containing an N-terminally truncated variant of syntaxin 1A (syntaxinH3) and SNAP-25 in a preformed binary complex (see Supplementary Data for further details). When synaptic vesicles and liposomes were mixed, a robust dequenching signal was observed (Figure 1A, left hand panel), that developed rapidly and saturated after 10–20 min. To examine whether this dequenching was reporting SNARE-mediated membrane fusion, we used clostridial neurotoxin light chains (which are zinc-dependent proteases) to specifically target the neuronal SNARE proteins [6]. Preincubation with either Botulinum neurotoxin C1 (BoNT/C1) to cleave syntaxin 1A, or Tetanus toxin (TeNT) to cleave synaptobrevin, resulted in an inhibition of fusion (Figure 1A; see also Supplementary Data Figure S1). No inhibition of fusion was observed when toxin mutants were used that had been rendered inactive by an amino acid substitution in the Zn2+-coordination site. This demonstrates that the fusion reaction requires both intact syntaxin 1 (on liposomes) and synaptobrevin (on synaptic vesicles).
Figure 1. Synaptic vesicles fuse constitutively with syntaxin 1/SNAP-25 liposomes, in a SNARE-dependent manner, irrespective of purification method.

(A) Purified synaptic vesicles (‘Method A’) were added to NBD/Rhodamine labelled liposomes, containing syntaxin 1/SNAP-25. Synaptic vesicles fused constitutively with liposomes, as shown by the robust dequenching signal (No Addition). Preincubation of the membranes (60 min, 37°C) with the light chain of Botulinum neurotoxin C1 (BoNT/C1-WT; cleaving syntaxin 1) or Tetanus toxin (TeNT-WT; cleaving synaptobrevin) largely inhibited fusion. Parallel incubations with light chain mutants, inactivated by a point mutation in the Zn2+-coordination site (BoNT/C1-Mut, TeNT-Mut), did not result in any inhibition (left hand panel). Fusion also resulted in the formation of SDS-resistant, heat sensitive SNARE core-complexes. Increasing the amount of syntaxin/SNAP-25 proteoliposomes (5x and 10x the standard amount) in the reaction mixture increased the amount of synaptobrevin engaged in core-complexes. Synaptic vesicles also contain low amounts of endogenous SDS-resistant complexes that were only detectable at longer exposure times (not shown). An estimate of the maximum number of SDS-resistant complexes that could form was obtained by incubating proteoliposomes and synaptic vesicles in the presence of 2% (vol:vol) of Triton X-100 (TX100) (right hand panel).
(B) Robust fusion was also seen between proteoliposomes and synaptic vesicles prepared using ‘Method B’. However, some fusion persisted after incubation with TeNT, despite quantitative cleavage of synaptobrevin (syb). Synaptophysin (syp) is shown as a loading control. Traces are averages of 8 experiments ± SEMs.
(C) Fusion was observed for all concentrations of liposomes and synaptic vesicles (‘Method A’ left hand panel; ‘Method B’ right hand panel) tested. Figures refer to the final concentration of proteoliposomes (recombinant protein in nM) and amount of synaptic vesicles (total protein in µg) in the reaction mixture.
In addition to toxin sensitivity, SNARE-mediated fusion should result in the formation of ‘cis’-SNARE complexes in the membrane, which are detectable as distinct, heat-sensitive bands of high molecular mass by SDS-PAGE. This was indeed the case. Interestingly, vesicular synaptobrevin was almost quantitatively shifted into core-complexes when an excess of liposomes was included in the reaction mixture, which is evident by a major loss of the monomeric form (approximately 70% in this case when comparing non-boiled with boiled samples). When the membranes were preincubated with either BoNT/C1 or TeNT, complex formation was largely inhibited (Figure 1A, right hand panel and data not shown).
Several control experiments were carried out to confirm that fusion between synaptic vesicles and proteoliposomes is dependent on the formation of SNARE complexes. First, we tested whether the fusion kinetics are altered when full-length syntaxin 1 is used, instead of syntaxinH3. The N-terminal domain of syntaxin is known to interact with the SNARE motif, down-regulating its capability to enter SNARE complexes [7]. However, as shown Figure 2A, the fusion rate of liposomes containing the full-length protein was only slightly slower than that of liposomes containing the truncated version, suggesting that the N-terminal domain is not rate-limiting once SNAP-25 is bound to syntaxin. Lowering the concentration of vesicles approximately 6-fold reduced both the rate and extent of fusion by about half, with a similar reduction being observed when the liposome concentration was reduced (Figure 1C, left hand panel). By way of comparison, reliable fusion was also observed with synaptic vesicles prepared according to ‘Method B’ (Figure 1B and Figure 1C, right hand panel). In addition, fusion of these vesicles was concomitant with the appearance of SDS resistant core-complexes, with the majority of synaptobrevin being free to engage in core-complexes when an excess of liposomes was included in the reaction mixture (see Supplementary Data Figure S4). However, some fusion persisted after incubation with TeNT despite quantitative cleavage of synaptobrevin (Figure 1B).
Figure 2. Characterisation of fusion between purified synaptic vesicles and proteoliposomes containing syntaxin 1 and SNAP-25, using fluorescence dequenching.

(A) Fusion of synaptic vesicles with liposomes containing SNAP-25 and either full-length syntaxin 1 (SyxFL) or a truncated syntaxin 1 variant (SyxH3), which lacks the autoinhibitory N-terminal Habc domain, showing that full-length syntaxin is only slightly less active in the lipid dequenching assay. (Traces are averages of at least eight experiments ± SEMs). Fusion is inhibited when liposomes and synaptic vesicles are preincubated for 60 min at room temperature with the cytoplasmic fragments of either synaptobrevin (1x molar excess) or syntaxinH3/SNAP-25 (5x molar excess), respectively.
(B) Fusion of synaptic vesicles to proteoliposomes requires the presence of all three neuronal SNAREs with correct topology. Proteoliposomes produced with a preformed syntaxin/SNAP-25 complex fuse with synaptic vesicles. No fusion was observed with syntaxinH3 proteoliposomes alone. Fusion was partially restored by either incubating proteoliposomes with an 8x molar excess of SNAP-25 relative to syntaxin for 60 min prior to the reaction (SN25 preinc), or by adding SNAP-25 during the reaction (arrow). No ‘homotypic’ fusion between syntaxinH3/SNAP-25 proteoliposomes or synaptobrevin proteoliposomes and synaptic vesicles was observed.
(C) Addition of lysophosphatidylcholine (LPC) results in an inhibition of fusion.
Further experiments were carried out using synaptic vesicles prepared using ‘Method A’, showing that addition of a 1x molar excess of soluble synaptobrevin, or of soluble syntaxinH3/SNAP-25 (both in 5x molar excess), effectively inhibited fusion (Figure 2A), as expected for competitive inhibition. Second, no fusion was observed when syntaxin alone was incorporated into liposomes without SNAP-25. Addition of an 8x molar excess of SNAP-25, either 60 min before or immediately after the start of the reaction, partially restored fusion (Figure 2B). Thus, fusion depends on the formation of SNARE acceptor complexes containing both SNAP-25 and syntaxin 1. No ‘homotypic’ fusion was observed between syntaxinH3 proteoliposomes, or between synaptobrevin-containing proteoliposomes and synaptic vesicles, regardless of whether SNAP-25 was present (Figure 2B). Finally, no non-specific fusion with protein-free liposomes was detected (see Figure 4B).
Figure 4. Synaptic vesicles undergo multiple rounds of fusion.
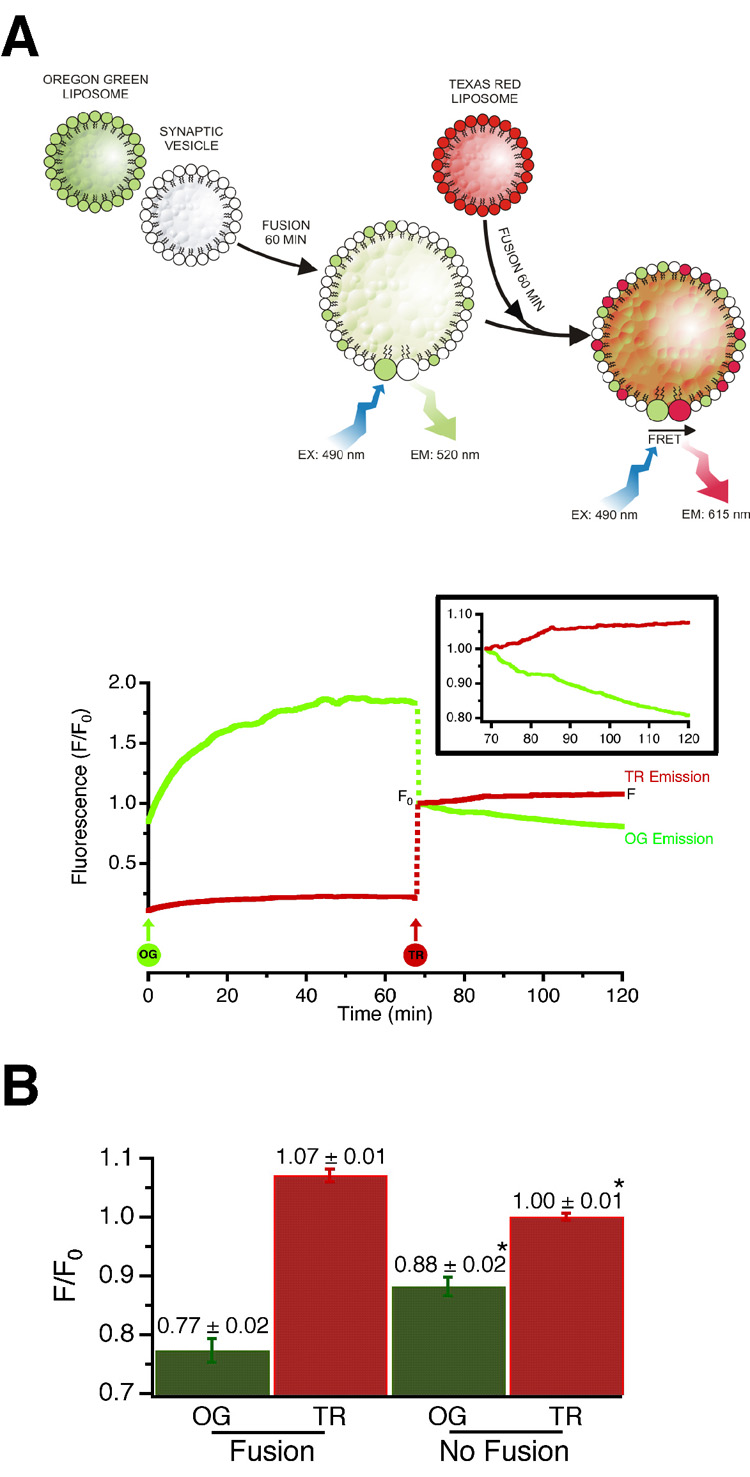
(A) Synaptic vesicles were first fused with syntaxinH3/SNAP-25 proteoliposomes containing Oregon Green labelled lipid, followed by proteoliposomes containing Texas Red labelled lipid. Because fusion is strictly dependent on one of the fusion partners containing synaptobrevin, FRET only develops if a Texas Red liposome fuses with a synaptic vesicle that has acquired Oregon Green labelled lipids in a preceding fusion step. FRET was monitored fluorometrically using excitation at 490 nm and emission at both 520 nm (Oregon Green donor, green trace on graph) and 615 nm (Texas Red acceptor, red trace on graph).
SyntaxinH3/SNAP-25 proteoliposomes, labelled with Oregon Green or Texas Red, were added to synaptic vesicles at the indicated time points (circles OG and TR). The increase in OG fluorescence at the beginning of the experiment is due to dequenching of the dye on fusion. Both OG and TR fluorescence signals were normalised to an arbitrary value of 1 after the TR liposomes were added, to account for the dilution effect (dashed lines; F0). The inlay shows a portion of the graph expanded after addition of TR liposomes, to show the decrease in OG (donor) and increase in TR (acceptor) signals typical of FRET.
(B) Donor and acceptor emission signals at the end of the reaction (F) normalised to the point at which the TR proteoliposomes were added (F0). Addition of TR proteoliposomes to the reaction mixture caused a decrease in the OG signal and a concomitant increase in the TR signal (Fusion; n=12, average ± SEMs), when compared to protein free liposomes (No Fusion; n=11, average ± SEMs) (*p<0.001).
SNARE assembly is thought to overcome the repulsive forces existing between membranes, resulting in fusion. Fusion is thought to proceed via a transient ‘stalk’ that connects the two membranes. Such ‘stalk-like’ intermediates possess a negative overall curvature and can be destabilised by amphiphiles possessing an inverted cone shape. Indeed, such lipids have inhibited all fusion reactions examined to date, with lysophosphatidylcholine (LPC) being most commonly used. Addition of increasing amounts of LPC, up to a concentration of 400 µM (which is still below its critical micellar concentration [8]), led to a progressive decrease in fusion (Figure 2C).
As shown above, the majority of vesicular synaptobrevin is driven into SNARE complexes when an excess of liposomes is used, indicating that all synaptic vesicles isolated by our procedure are essentially fusion-competent. To confirm this, two types of experiments were carried out. First, we asked whether fusion results in a detectable size increase. We determined the size-distribution of vesicles in a fusion mixture at the end of the reaction and compared it with the size distribution of a fusion reaction that was blocked by preincubation with TeNT. As shown in Figure 3, fusion resulted in a size-shift towards larger diameters, although this shift was not as pronounced as that previously observed for synaptobrevin proteoliposomes [9]. However, it needs to be remembered that a single fusion event between vesicles of a similar size only results in a diameter increase of 3√2, which is close to the detection limit of electron microscopy. The absence of vesicle clusters in the micrographs suggests that when liposomes and synaptic vesicles meet, fusion is fast with no prolonged docking state preceding fusion (see also Supplementary Data Figure S7).
Figure 3. Fusion between synaptic vesicles and liposomes results in a size-increase.
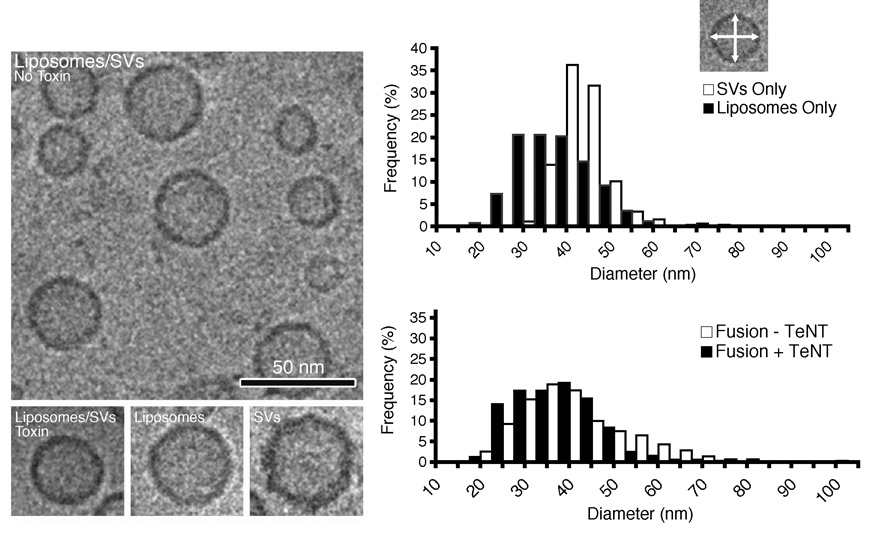
Cryo-electron micrographs of proteoliposomes, synaptic vesicles and particles resulting from a fusion reaction (left). Quantification of particle sizes, at the end of a 30 min fusion reaction (right). The longest and the shortest diameters (outer rim) of each particle were measured and the average diameter calculated (arrows on inset). The size distributions of the individual proteoliposome and synaptic vesicle populations are shown (top histogram). Fusion resulted in a shift to larger diameters (bottom histogram). As a control, a parallel reaction was made with Tetanus toxin treated synaptic vesicles, in which no size shift was seen. Interestingly, few particle clusters were seen, indicating that fusion is fast and complete. The experiment was repeated twice and the numbers of particles counted for each condition were pooled. Particle numbers (and the associated standard deviations) are: liposomes 688 (8.81); synaptic vesicles 410 (6.37); liposomes/synaptic vesicles−TeNT 647 (14.02); liposomes/synaptic vesicles+TeNT 741 (10.36).
Together, these data show not only that isolated synaptic vesicles effectively fuse with proteoliposomes containing SNAP-25 and syntaxin 1 but also that fusion is SNARE-mediated. Fusion is almost quantitative, in good agreement with our previous observation showing that vesicular synaptobrevin is not restricted in its ability to engage in SNARE complexes with soluble partner SNAREs [10].
Synaptic Vesicles Undergo Multiple Fusion Events
As shown in Figure 3, fusion leads to the generation of larger vesicles, with diameters ranging between 60 and 80 nm, suggesting that these vesicles may result from multiple fusion events. To investigate this the following fluorescence resonance energy transfer (FRET) based strategy was used. First, we fused synaptic vesicles with non-saturating amounts of syntaxin/SNAP-25 proteoliposomes containing Oregon Green labelled phospholipids. Next, an excess amount of syntaxin/SNAP-25 proteoliposomes containing Texas Red labelled phospholipids was added. Since fusion is dependent on synaptobrevin, a FRET signal can only develop if a Texas Red labelled liposome fuses with a synaptic vesicle that has acquired Oregon Green labelled lipids in a preceding fusion step (see Figure 4A). Indeed, a FRET signal was observed which increased over the course of the experiment. The signal was small, probably due to the fact that only a small amount of Oregon Green labelled proteoliposomes was used in the initial fusion step. To confirm that the FRET signal was caused by SNARE-mediated fusion, we used protein free Texas Red liposomes. As shown in Figure 4B, there was no increase in acceptor fluorescence. Donor fluorescence decreased somewhat, albeit less than under fusion conditions, which is probably attributable to photobleaching. We conclude that under these in vitro conditions, synaptic vesicles are capable of undergoing multiple rounds of fusion. Synaptobrevin molecules that were not involved in the first fusion reaction retain their ability to undergo subsequent rounds of fusion (see Figure 1A, right hand panel and Discussion).
Synaptic Vesicle Fusion is Independent of Ca2+
The data described so far show that synaptic vesicles effectively fuse with liposomes containing syntaxin and SNAP-25. At the synapse, exocytosis is triggered by a rise in intracellular Ca2+ during stimulation. Synaptic vesicles contain at least 10 copies of the Ca2+-sensor synaptotagmin 1, so we wondered whether fusion is accelerated by the addition of Ca2+ ions. Surprisingly, no increase in the fusion rate was observable (Figure 5A). Rather, a minor inhibition was seen, even at concentrations known to stimulate exocytosis in neurones. A similar inhibition was also observed when the Ca2+ concentration was raised to (non-physiological) 1000 µM [11]. This inhibition was partially prevented when phosphatidylinositol 4,5-bisphosphate (PIP2) was included in the membrane of the syntaxin/SNAP-25 liposomes (Figure 5B), although the effects of PIP2 inclusion were rather minor, even when it was systematically varied over a large range (see Supplementary Data Figure S6). Finally, addition of the cytoplasmic domain of synaptotagmin to the fusion reaction produced only minor effects on the fusion rate, regardless of whether Ca2+ was present or not (data not shown). The inability of Ca2+ to stimulate fusion, despite the presence of endogenous synaptotagmin, shows that regulated exocytosis requires additional, hitherto unknown, control proteins (see Discussion).
Figure 5. Ca2+ does not accelerate fusion between synaptic vesicles and SNARE containing liposomes.
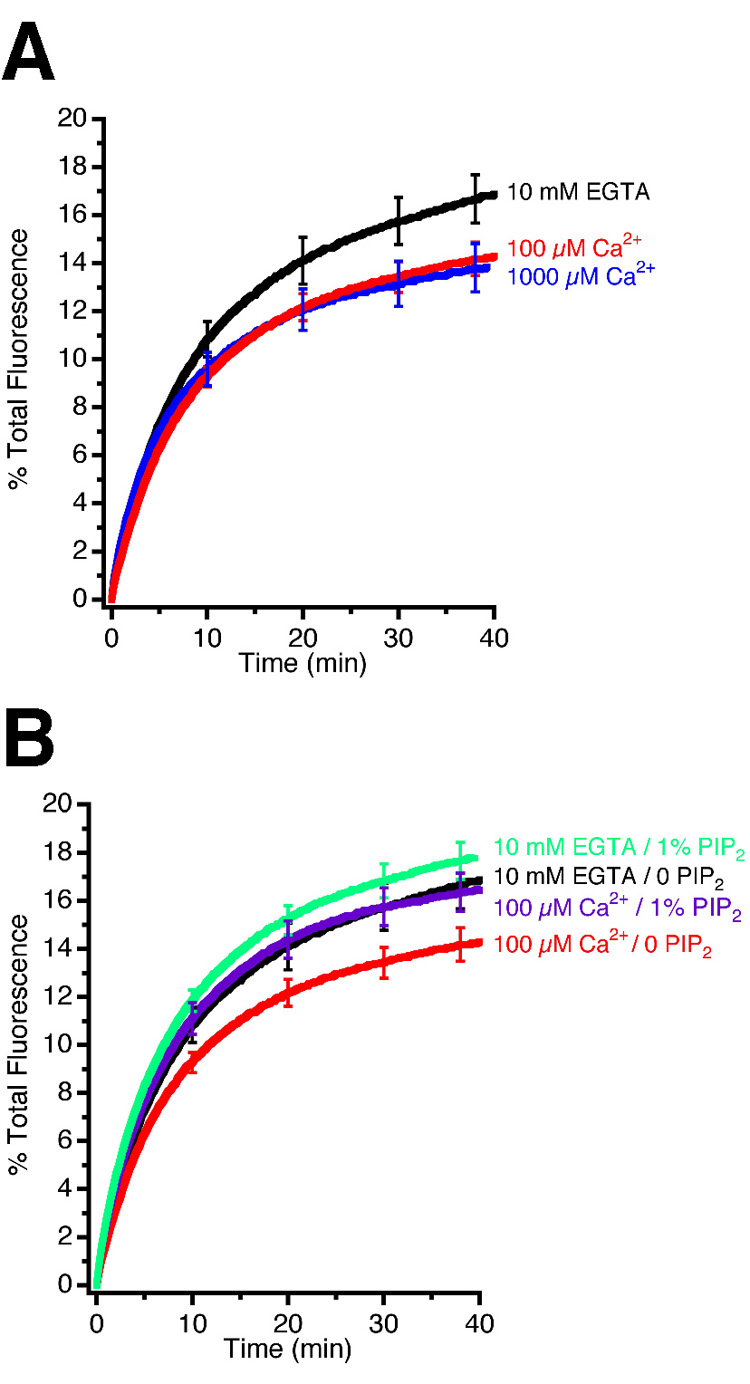
(A) Fusion of synaptic vesicles with syntaxin/SNAP-25 proteoliposomes, as measured by lipid dequenching, is not enhanced by calcium. In fact, a very reproducible inhibition was observable with both 100 µM and 1000 µM Ca2+. n ≥ 6 experiments ± SEMs.
(B) Inclusion of a synaptotagmin effector lipid (phosphatidylinositol 4,5-bisphosphate – PIP2) in the liposomal membrane did not produce a significant increase in the rate of fusion, as measured by lipid dequenching. However, the inhibition of fusion seen upon the addition of Ca2+ was partially prevented by the incorporation of 1% PIP2 in the proteoliposome membrane. n ≥ 9 experiments ± SEMs.
The Rate of Synaptic Vesicle Fusion is Determined by the Availability of Synaptobrevin Acceptor Sites
In the final set of experiments, we investigated whether the rate of vesicle-liposome fusion is dependent on the status of the acceptor SNAREs in the liposome membrane. When SNAP-25 and syntaxin 1 are mixed in vitro an unstable 1:1 complex is formed, that readily recruits a second syntaxin molecule with the equilibrium being strongly on the side of the 2:1 complex. Since the rate of SNARE assembly and SNARE-mediated liposome fusion depends on the concentration of free binding sites for synaptobrevin, the reaction can only proceed if the second syntaxin molecule (that blocks the synaptobrevin binding site) dissociates from the binary complex [12].
Two approaches were used to increase the concentration of synaptobrevin acceptor sites on the syntaxin/SNAP-25 proteoliposomes. First, complexes were preformed using increasing concentrations of SNAP-25, with the goal of increasing the concentration of 1:1 complexes by mass-action. As shown in Figure 6A, a moderate acceleration was observed when the SNAP-25 concentration was doubled, with no further increase at higher concentrations. Second, we took advantage of the recent observation that a free N-terminal acceptor site for synaptobrevin can be stabilised if a SNARE complex is formed containing N-terminally truncated synaptobrevin (referred to as ΔN complex), which greatly enhances liposome-liposome fusion [13]. Therefore, we asked whether the fusion of synaptic vesicles with liposomes containing ΔN complexes is also accelerated. As shown in Figure 6B, this is indeed the case, with the degree of acceleration comparable to that previously observed when synaptobrevin-containing liposomes were used. As expected, fusion was associated with the displacement of the N-terminally truncated peptide (see Supplementary Figure S7). We conclude that isolated synaptic vesicles are highly efficient fusion machines, whose fusion kinetics are determined by the availability of SNARE acceptor sites for synaptobrevin.
Figure 6. The rate of synaptic vesicle fusion is determined by the availability of free synaptobrevin acceptor sites.
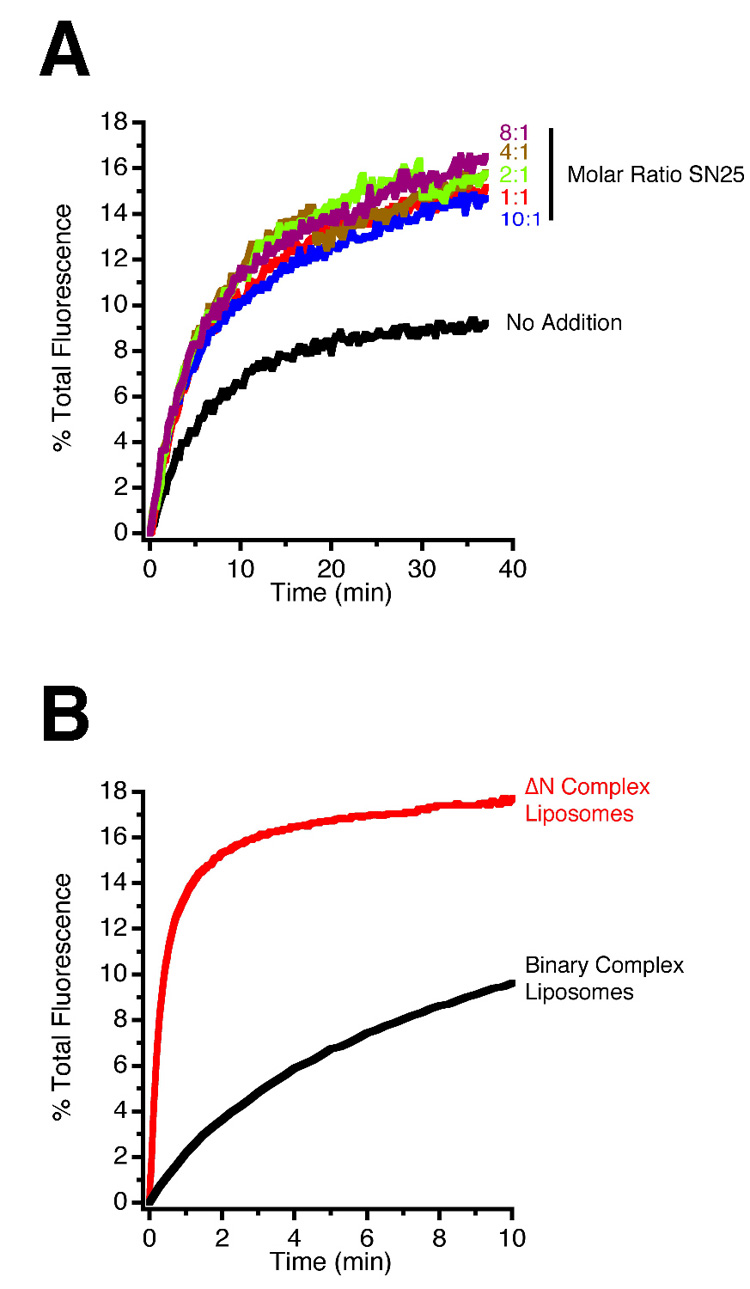
(A) Increasing the concentration of free SNAP-25 accelerates vesicle-liposome fusion by shifting the equilibrium by mass action from the energetically favoured 2:1 syntaxin/SNAP-25 complex to the unstable 1:1 syntaxin/SNAP-25 complex, which serves as an acceptor for synaptobrevin.
(B) Fusion of liposomes with synaptic vesicles, as measured by lipid dequenching, is greatly accelerated when a syntaxin/SNAP-25 acceptor complex is used that contains N-terminally truncated synaptobrevin (amino acids 49–96, ΔN complex), which leaves a stabilised, freely accessible N-terminal binding site for synaptobrevin. Note that the time-scale in B is expanded in comparison to A.
Discussion
We have demonstrated that isolated synaptic vesicles effectively fuse with liposomes containing the cognate neuronal SNAREs syntaxin 1 and SNAP-25. Fusion is dependent on the formation of SNARE complexes, but is not accelerated by calcium or PIP2, despite the presence of native and intact synaptotagmin in the vesicle membrane. Fusion is highly efficient, with most vesicles capable of fusing at least once, and fusion kinetics are predominately governed by the availability of an appropriately configured acceptor complex of syntaxin 1 and SNAP-25.
Our data lend strong support to the notion that SNAREs function as the minimal fusion machinery. Our results support the view that synaptic vesicle exocytosis requires all three neuronal SNAREs in correct topology, with at least 2 SNAREs containing trans-membrane domains. Our data also suggest that vesicular synaptobrevin does not need to be activated in order to engage in fusion-competent SNARE complexes with syntaxin 1 and SNAP-25. This conclusion disagrees with several previous studies reporting that synaptobrevin – either in native synaptic vesicles or after reconstitution of the recombinant protein into liposomes – is inactive with respect to SNARE interactions [5, 14]. Upon re-investigation of this issue, we have been unable to reproduce these findings. Rather, we observed that synaptobrevin anchored in native or artificial membranes can be quantitatively driven into SNARE complexes by addition of exogenous SNAP-25 and syntaxin 1 [2, 10]. Our present findings are fully consistent with these data and confirm that synaptobrevin is constitutively active on the vesicle membrane. Furthermore, several lines of evidence including lipid mixing (coupled to the degree of SNARE complex formation) and size-increase measured by electron microscopy conclusively document that a large majority, if not all, purified vesicles are competent for SNARE-mediated fusion. The reasons for these discrepancies are unclear at present, possibly being caused by differences in the purification of the SNARE proteins, which, in some studies, involved harsh denaturing steps; or the use of a high protein:lipid ratio (1:10) during proteoliposome preparation which may result, amongst other things, in steric hindrance of the proteins [5].
Our data also show that only a fraction of the synaptobrevin molecules on each vesicle are engaged in SNARE complexes during each fusion event. This is not surprising, as each vesicle contains 70 copies of synaptobrevin [4], many more than are thought needed for fusion [15]. This observation explains why vesicles can undergo multiple rounds of fusion, and is in agreement with previous findings on a NSF mutant in Drosophila, showing that synaptic vesicles can still undergo several rounds of exo-endocytotic cycling when the disassembly of SNAREs is inhibited [16].
One of our most surprising findings is that neither calcium nor PIP2 in the liposome membrane affects the fusion rate to a major degree, despite the fact that synaptic vesicles contain endogenous synaptotagmins [1]. In this respect, our findings disagree with the previous report by Hu and colleagues, in which fusion between vesicles and liposomes only took place when calcium was present [5]. In our hands, we could consistently and reliably see fusion, irrespective of the method used to prepare vesicles; and while we did see an enhancement of fusion in the presence of Ca2+ when using vesicles prepared using ‘Method B’, we found this enhancement is unrelated to fusion mediated by synaptic SNAREs (see Supplementary Data Figure S5). Furthermore, our data are consistent with previous reports in which no calcium-dependent acceleration was observed in SNARE-dependent liposome fusion when full-length synaptotagmin was co-reconstituted with synaptobrevin [17, 18]. In fact, in our previous study, we observed a Ca2+-dependent inhibition of proteoliposome fusion in the presence of synaptotagmin, which is reminiscent of the inhibition reported here [18]. More detailed analysis revealed that this inhibition is due to the Ca2+-dependent binding of synaptotagmin to its own membrane when acidic phospholipids are present (as they are in synaptic vesicles) [4]. This binding apparently inactivates synaptotagmin, preventing it from interacting in ‘trans’ with syntaxin/SNAP-25 and/or acidic phospholipids in the target membrane. In our hands, the effect of PIP2 in partially preventing this Ca2+-dependent inhibition suggests that increasing the amount of this phospholipid in the acceptor liposome is sufficient to reestablish some ‘trans’ interaction between liposome and synaptic vesicle, which is mediated by synaptotagmin [19].
Several conclusions can be drawn concerning the in vitro reconstitution of exocytotic membrane fusion using isolated native membranes or artificial vesicles. First, isolated and purified synaptic vesicles exhibit properties virtually identical to proteoliposomes containing comparable amounts of synaptobrevin and synaptotagmin, when liposomes containing syntaxin and SNAP-25 are used as fusion partners. In both cases, fusion proceeds with comparable kinetics (being primarily dependent on the availability of SNARE acceptor sites), is close to being quantitative (with multiple rounds of fusion occurring), and is moderately inhibited in the presence of calcium. These striking similarities are surprising, when considering that in contrast to artificially prepared proteoliposomes, synaptic vesicles are loaded with other proteins and have a highly complex, probably asymmetric, lipid composition. Apparently, the massive release of energy during SNARE complex formation is capable of driving fusion under a wide variety of conditions, being able to ‘plough’ through densely packed cohorts of membrane proteins, as well as accommodating a large variety of membrane lipids. The data also indicates that the in vitro fusion of liposomes is not compromised by an artificially high fusogenicity, as has recently been suggested [3].
Second, the data shows that the regulatory steps involved in controlling neuronal exocytosis have not yet been adequately reconstructed in artificial fusion systems. Most importantly, the fast triggering by Ca2+ of exocytotic membrane fusion, which has been unequivocally shown to be mediated by synaptotagmins 1 and 2 [1], has not been reproduced in such fusion assays. Apparently, Ca2+-triggering requires the synaptic vesicle to be docked and primed at the release site, a molecular configuration that is not yet understood (for a more detailed description see Stein et al. [18]).
Several additional proteins, such as Munc-18 and complexin, are also known to be crucial for neuronal exocytosis, even though their precise role is not understood. Our present understanding is that these proteins are recruited from the cytoplasm to the site of fusion, where they are known to bind to free and/or assembled SNAREs. Munc-18 has been suggested to regulate the availability of SNAREs in the target membrane, whilst complexin, which binds to assembled SNAREs, has been proposed to clamp the SNARE complex in a prefusion state ready for Ca2+-dependent exocytosis, suggesting a co-operative action with synaptotagmin [1]. These interactions clearly add yet another layer of regulation. Obviously, more work is needed in order to reconstruct these and other, as yet unknown, regulatory steps in the test tube. However, it seems that such regulation is mediated primarily by factors that are either located at the plasma membrane or which are recruited from the cytoplasm, whereas isolated synaptic vesicles lack endogenous control proteins and are constitutively active for fusion.
Experimental Procedures
Experimental procedures were all based on published protocols (see references). Brief outlines are given below, with fuller descriptions being found in the Supplementary Data).
Recombinant protein expression and purification [12, 18]
SNAREs were cloned using rat (Rattus norvegicus) sequences as templates. SNARE proteins were expressed in E. coli and purified by Ni2+-NTA affinity chromatography, followed by ion exchange chromatography on an Äkta system (GE Healthcare). Purification of proteins containing a trans-membrane domain was done in the presence of 15 mM CHAPS.
Tetanus toxin and Botulinum toxin (including respective mutants) were purified using a similar strategy.
The concentrations of all recombinant proteins were determined by absorption at 280 nm.
Preparation of artificial and native membranes
Proteoliposome preparation [9, 18]
Lipids were mixed in chloroform to give (in molar ratios): phosphatidylcholine (5), phosphatidylethanolamine (2), phosphatidylserine (1), phosphatidylinositol (1) and cholesterol (1). In liposomes containing PIP2, the level of PI was reduced accordingly. For use in fluorescence-based assays, unlabelled PE was substituted in part by NBD-PE and Rhodamine-PE (lipid dequenching), and Oregon Green-PE or Texas Red-PE (FRET assays). After drying, lipids were resuspended in HB100 buffer (in mM) (100 KCl, 1 DTT, 25 Hepes; pH 7.4 KOH) with 5% sodium cholate at a total lipid concentration of 13.5 mM. SNARE proteins were added (at a protein:lipid ratio of 1:300) and detergent removed by gel filtration chromatography, either on a SMART system with a PC 3.2/10 Fast Desalting column (GE Healthcare) or a Bio-Rad EconoColumn filled with Sephadex G-50 superfine. Columns were equilibrated in HB100.
Synaptic vesicle purification [4, 5]
Synaptic vesicles were isolated from rat brain, using two different procedures. However, both procedures utilise standard differential centrifugation to initially isolate synaptosomes, from which synaptic vesicles are released by hypo-osmotic shock. In ‘Method A’, synaptic vesicles are then enriched by differential centrifugation, prior to purification on continuous sucrose density gradients and a final step of size-exclusion chromatography using controlled pore glass beads. In ‘Method B’, by way of contrast, large membrane fragments produced during lysis are subsequently removed by differential centrifugation, before the separation of synaptic vesicles from any remaining cytosolic components by floatation on Optiprep® gradients.
Concentrations of membrane proteins were determined using a modified Lowry procedure.
Fluorescence assays [2, 9, 20]
Liposome fusion measured by lipid dequenching was carried out by incubating NBD/Rhodamine labelled syntaxin/SNAP-25 liposomes with synaptic vesicles at 37°C. Fusion was reported by the increase in NBD fluorescence (measured at 460 nm excitation and 538 nm emission in a Fluoromax-2 spectrometer) that occurs on lipid mixing. At the end of the reaction, Triton X-100 was added to completely solubilise the membranes and maximally dequench NBD fluorescence. To normalise the experiments, the lowest NBD florescence signal was set to 0% and the maximal signal reached after detergent addition was set to 100%.
When using FRET to study the fusion reaction, Oregon Green labelled syntaxin/SNAP-25 liposomes (donor) were incubated with synaptic vesicles for 60 min, at which time fusion, as measured by Oregon Green dequenching, had proceeded to completion. At this point, a saturating amount of Texas Red labelled syntaxin/SNAP-25 liposomes (acceptor) were added and the reaction left for a further 60 min. The donor signal (excitation 490 nm; emission 520 nm) and the acceptor/FRET signal (excitation 490 nm; emission 615 nm) were measured in a Fluoromax-2 at 37°C. The final donor and FRET signals (F) were then expressed relative to the point at which the Texas Red liposomes were first added (F0).
All data analysis was performed using Igor Pro (Wavemetrics).
Electron microscopy [9]
Reaction mixtures were applied to carbon coated grids, plunge frozen in liquid ethane and transferred to a Philips CM120 Biofilter electron microscope using a Gatan cryo-stage. Images were recorded on a Gatan slow scan CCD at a magnification of 31,000X using Digital Micrograph software, at a constant defocus. Micrographs were then digitised using a flat-bed scanner at a resolution of 1200 dpi and the shortest and longest diameters of each particle measured.
SDS-PAGE and immunoblotting [9, 21, 22]
SDS-PAGE was carried out using a glycine or tricine based buffer system to ensure maximum resolution in the molecular weight range of interest. Immunoblotting was carried out using standard semi-dry electrophoretic techniques. Detection was by enhanced chemiluminescence using a LAS reader (Fuji). Primary antibodies were from Synaptic Systems or Abcam. Peroxidase-labelled secondary antibodies were from Bio-Rad
Supplementary Material
Acknowledgements
This work was supported by US National Institutes of Health grant P01 GM072694 (to RJ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lang T, Jahn R. Core proteins of the secretory machinery. Handb Exp Pharmacol. 2008:107–127. doi: 10.1007/978-3-540-74805-2_5. [DOI] [PubMed] [Google Scholar]
- 2.Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Söllner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 3.Rizo J, Chen X, Araç D. Unraveling the mechanisms of synaptotagmin and SNARE function in neurotransmitter release. Trends Cell Biol. 2006;16:339–350. doi: 10.1016/j.tcb.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 5.Hu K, Carroll J, Fedorovich S, Rickman C, Sukhodub A, Davletov B. Vesicular restriction of synaptobrevin suggests a role for calcium in membrane fusion. Nature. 2002;415:646–650. doi: 10.1038/415646a. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi T, McMahon H, Yamasaki S, Binz T, Hata Y, Südhof TC, Niemann H. Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 1994;13:5051–5061. doi: 10.1002/j.1460-2075.1994.tb06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parlati F, Weber T, McNew JA, Westermann B, Söllner TH, Rothman JE. Rapid and efficient fusion of phospholipid vesicles by the alpha-helical core of a SNARE complex in the absence of an N-terminal regulatory domain. Proc Natl Acad Sci U S A. 1999;96:12565–12570. doi: 10.1073/pnas.96.22.12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chernomordik LV, Vogel SS, Sokoloff A, Onaran HO, Leikina EA, Zimmerberg J. Lysolipids reversibly inhibit Ca2+-, GTP- and pH-dependent fusion of biological membranes. FEBS Lett. 1993;318:71–76. doi: 10.1016/0014-5793(93)81330-3. [DOI] [PubMed] [Google Scholar]
- 9.Schuette CG, Hatsuzawa K, Margittai M, Stein A, Riedel D, Küster P, König M, Seidel C, Jahn R. Determinants of liposome fusion mediated by synaptic SNARE proteins. Proc Natl Acad Sci U S A. 2004;101:2858–2863. doi: 10.1073/pnas.0400044101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siddiqui TJ, Vites O, Stein A, Heintzmann R, Jahn R, Fasshauer D. Determinants of Synaptobrevin Regulation in Membranes. Mol Biol Cell. 2007;18:2037–2046. doi: 10.1091/mbc.E07-01-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schneggenburger R, Neher E. Presynaptic calcium and control of vesicle fusion. Curr Opin Neurobiol. 2005;15:266–274. doi: 10.1016/j.conb.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Fasshauer D, Margittai M. A transient N-terminal interaction of SNAP-25 and syntaxin nucleates SNARE assembly. J Biol Chem. 2004;279:7613–7621. doi: 10.1074/jbc.M312064200. [DOI] [PubMed] [Google Scholar]
- 13.Pobbati AV, Stein A, Fasshauer D. N- to C-terminal SNARE complex assembly promotes rapid membrane fusion. Science. 2006;313:673–676. doi: 10.1126/science.1129486. [DOI] [PubMed] [Google Scholar]
- 14.Kweon DH, Kim CS, Shin YK. Regulation of neuronal SNARE assembly by the membrane. Nat Struct Biol. 2003;10:440–447. doi: 10.1038/nsb928. [DOI] [PubMed] [Google Scholar]
- 15.Montecucco C, Schiavo G, Pantano S. SNARE complexes and neuroexocytosis: how many, how close? Trends Biochem Sci. 2005;30:367–372. doi: 10.1016/j.tibs.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Littleton JT, Chapman ER, Kreber R, Garment MB, Carlson SD, Ganetzky B. Temperature-sensitive paralytic mutations demonstrate that synaptic exocytosis requires SNARE complex assembly and disassembly. Neuron. 1998;21:401–413. doi: 10.1016/s0896-6273(00)80549-8. [DOI] [PubMed] [Google Scholar]
- 17.Mahal LK, Sequeira SM, Gureasko JM, Söllner TH. Calcium-independent stimulation of membrane fusion and SNAREpin formation by synaptotagmin I. J Cell Biol. 2002;158:273–282. doi: 10.1083/jcb.200203135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stein A, Radhakrishnan A, Riedel D, Fasshauer D, Jahn R. Synaptotagmin activates membrane fusion through a Ca2+-dependent trans interaction with phospholipids. Nat Struct Mol Biol. 2007;14:904–911. doi: 10.1038/nsmb1305. [DOI] [PubMed] [Google Scholar]
- 19.Bai J, Tucker WC, Chapman ER. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat Struct Mol Biol. 2004;11:36–44. doi: 10.1038/nsmb709. [DOI] [PubMed] [Google Scholar]
- 20.Zwilling D, Cypionka A, Pohl WH, Fasshauer D, Walla PJ, Wahl MC, Jahn R. Early endosomal SNAREs form a structurally conserved SNARE complex and fuse liposomes with multiple topologies. EMBO J. 2007;26:9–18. doi: 10.1038/sj.emboj.7601467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Otto H, Hanson PI, Jahn R. Assembly and disassembly of a ternary complex of synaptobrevin, syntaxin, and SNAP-25 in the membrane of synaptic vesicles. Proc Natl Acad Sci U S A. 1997;94:6197–6201. doi: 10.1073/pnas.94.12.6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.