

Abstract
Pulmonary hypertension (PHT) develops in sickle cell disease (SCD) and is associated with high mortality. We previously showed that erythroid cells produce placenta growth factor (PlGF), which activates monocytes to induce proinflammatory cytochemokines, contributing to the baseline inflammation and severity in SCD. In this study, we observed that PlGF increased expression of endothelin-1 (ET-1) and endothelin-B receptor (ET-BR) from human pulmonary microvascular endothelial cells (HPMVECs) and monocytes, respectively. PlGF-mediated ET-1 and ET-BR expression occurred via activation of PI-3 kinase, reactive oxygen species and hypoxia inducible factor-1α (HIF-1α). PlGF increased binding of HIF-1α to the ET-1 and ET-BR promoters; this effect was abrogated with mutation of hypoxia response elements in the promoter regions and HIF-1α siRNA and confirmed by chromatin immunoprecipitation analysis. Furthermore, PlGF-mediated ET-1 release from HPMVECs and ET-BR expression in monocytes creates a PlGF–ET-1–ET-BR loop, leading to increased expression of MCP-1 and IL-8. Our studies show that PlGF-induced expression of the potent vasoconstrictor ET-1 and its cognate ET-BR receptor occur via activation of HIF-1α, independent of hypoxia. PlGF levels are intrinsically elevated from the increased red cell turnover in SCD and in other chronic anemia (eg, thalassemia) and may contribute to inflammation and PHT seen in these diseases.
Introduction
The clinical manifestations of sickle cell disease (SCD) include chronic hemolytic anemia, frequent infections, and intermittent episodes of vascular occlusion.1–6 Pulmonary disease, both acute and chronic, is the second most common cause of hospitalization and a leading cause of both morbidity and mortality in adults with SCD.1,7–9 The most frequent form of acute pulmonary disease is the acute chest syndrome (ACS), which occurs in 15% to 40% of patients with SCD.10 Pulmonary hypertension (PHT) occurs in both adults and children with SCD: it develops with increasing age and portends an extremely poor prognosis. PHT is a significant risk factor for early mortality in SCD.9,11–13 Studies have shown that there is a clinical syndrome of hemolysis-associated PHT in SCD9 that results from global impairment in nitric oxide (NO) bioavailability from its quenching by free heme.14 It is known that vascular tone is also modulated by vasoconstrictors such as endothelin-1 (ET-1). Studies have shown increased plasma levels of ET-1 in patients with SCD and ACS.15,16 Tissue hypoxemia due to microvascular occlusion and chronic mild-moderate desaturations in SCD may contribute to increased levels of ET-1, which is released from endothelial cells in response to hypoxia.17 Increased levels of ET-1, if sustained as a result of the underlying SCD pathophysiology, can contribute to the development of PHT.
SCD is also characterized by presence of a chronic inflammatory state manifested by leukocytosis and monocytosis and increased circulating levels of proinflammatory cytochemokines seen at steady state that occurs in the absence of acute infection or an acute vaso-occlusive event.18,19 Monocytes isolated from the blood of patients with SCD are in an activated state.18 The levels of interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α),20,21 and IL-822 are elevated in plasma of patients with SCD. However, the nature of the stimuli that cause the baseline inflammation in SCD is relatively less understood. Our previous studies showed that plasma levels of placenta growth factor (PlGF) are higher in patients with SCD compared with healthy control subjects and correlate with increased incidence of vaso-occlusive events.23 We have shown that PlGF significantly increased expression of proinflammatory cytochemokines from mononuclear cells (MNCs) in healthy subjects.23,24 Expression of these same cytochemokines was significantly elevated in MNCs from subjects with SCD at steady state compared with healthy control subjects.23 PlGF was initially found to be secreted by placental trophoblasts and umbilical vein endothelial cells.25 However, recent studies show that PlGF is also produced by erythroid cells but not by other hematopoietic cells.26 Because erythropoiesis is expanded in persons with with SCD, resulting in higher levels of PlGF in plasma,23 we hypothesized that PlGF may be a key mediator in activation of endothelial cells to promote expression of molecules that mediate vasoconstriction and inflammation.
In this report, we show that PlGF increased mRNA and protein expression of ET-1 from cultured human pulmonary microvascular endothelial cells (HPMVECs) via activation of hypoxia-inducible factor-1α (HIF-1α). In monocytes, PlGF increased mRNA and protein expression of endothelin-B receptor (ET-BR), which was also mediated by HIF-1α. Furthermore, PlGF-treated monocytic cells, which led to increased expression of ET-BR, when treated with ET-1 showed additive effect in monocyte chemoattractant protein-1 (MCP-1) and interleukin-8 (IL-8) mRNA expression, indicating that PlGF-primed cells are more responsive to ET-1 in cytochemokine expression. Because ET-1 release is also mediated by PlGF, this creates a PlGF–ET-1–ET-BR loop that augments inflammation from monocytes and may promote PHT. ET-1 mediated signaling in monocytic cells led to increased expression of inflammatory cytochemokines, augmenting the inflammatory effect of PlGF through vascular endothelial growth factor receptor-1 (VEGFR-1) binding.24 To the best of our knowledge, this is the first report of activation of HIF-1α via PlGF in a hypoxia-independent manner that results in up-regulation of a potent pulmonary vasoconstrictor, ET-1. The induction of ET-1 and ET-BR by chronically elevated levels of PlGF could contribute to PHT and further exaggerate the pro-inflammatory state in SCD.
Methods
Cell culture and reagents
Human pulmonary microvascular endothelial cells (HPMVECs) were obtained and cultured as described previously.27 Unless otherwise indicated, HPMVECs were kept overnight in EBM-2 containing 2% serum, followed by serum-free media for 3 hours, before treatment with PlGF or other experimental conditions. THP-1, a promonocytic cell line, was obtained from the ATCC (Manassas, VA) and cultured in RPMI-1640 medium with 10% heat-inactivated fetal bovine serum (FBS).24 Unless otherwise indicated, THP-1 cells were cultured overnight in RPMI-1640 medium without serum for treatment with PlGF or other experimental conditions. Peripheral blood monocytes (PBM) were isolated and purified from blood obtained from healthy volunteers after obtaining informed consent according to a protocol approved by the Institutional Review Board at University of Southern California-Los Angeles County Hospital.24
Recombinant human PlGF was purchased from R&D Systems (Minneapolis, MN). LY294002, PD98059, SB203580, and SP600125 were obtained from Tocris Bioscience (Ellisville, MO). Primary antibodies against HIF-1α, HIF-1β, ET-BR, prolyl hydroxylase-2 (PHD-2), and secondary antibodies conjugated to horseradish peroxidase (HRP) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Unless otherwise specified, all other reagents were purchased from Sigma-Aldrich (St Louis, MO).
Dominant-negative (Dn) mutant of phosphatidylinositol 3-kinase (PI-3 kinase) (Dn-Δp85)24 (Dr David Ann), PTEN plasmid (Dr Debbie Johnson) and HIF-1α and HIF-1β expression plasmids (Dr Michael Stallcup) were obtained from the specified investigators at University of Southern California (USC) Keck School of Medicine (Los Angeles, CA). HIF-1α siRNA, HIF-1α scRNA, PHD-2 siRNA, and PHD-2 scRNA were synthesized at the Microchemical core facility of USC Comprehensive Cancer Center (CCC) as described previously.27 ET-1 promoter-luciferase plasmids (−669 and −176 base pairs [bp]) with mutation in the HIF-1α binding site were generously provided by Keith Webster (Miami, FL).28
Generation of ET-BR promoter luciferase constructs
A 1.752-kilobase segment (−1259/+493, wild type) of an ET-BR promoter (Gene Accession no. D13168) was cloned from genomic DNA isolated from THP-1 cells using polymerase chain reaction (PCR). All the primer sequences used to generate deletion constructs of ET-BR promoter are listed in Table 1. All the forward primers and a common reverse primer contained the recognition sequence for restriction enzymes KpnI and BglI, respectively. Forty 2 cycles of PCR were performed using 100 ng of wild type ET-BR promoter as a template. PCR conditions were: 95°C for 30 seconds, 61°C for 60 seconds, and 72°C for 120 seconds for 30 cycles. PCR products were cloned into pGL3 vector (Promega, Madison, WI). Mutations of HIF-1α binding sites located at −155 to −152 bp and −371 to −368 bp in −392/+493 bp ET-BR promoter were carried out using the primers shown in Table 1 according to the QuikChange site-directed mutagenesis protocol (Stratagene, La Jolla, CA). DNA sequences of all the constructs were confirmed by sequencing (Microchemical Core Facility, USC Norris Comprehensive Cancer Center).
Table 1.
Oligonucleotide primers used in this study
| Gene/fragment location | Method | Forward sequence | Reverse sequence |
|---|---|---|---|
| ET-BR promoter (−1259/+493) | PCR | aatcatggtaccacatggtgcgtgataacttg | acgtacagatctcgccctttaccttgtaga |
| ET-BR promoter (−867/+493) | PCR | tagtcgtggtacctgtctgtattcctcccgttac | acgtacagatctcgccctttaccttgtaga |
| ET-BR promoter (−555/+493) | PCR | tagtcgtggtaccgtctccattcgtcgctctg | acgtacagatctcgccctttaccttgtaga |
| ET-BR promoter (−392/+493) | PCR | tagtcgtggtaccgtctccattcgtcgctctg | acgtacagatctcgccctttaccttgtaga |
| ET-BR promoter (−392/+493)-HIF-1α-M1 | SDM | cttgagttacttttgagcagggatactggcgaagaggctg | cagcctcttcgccagtatccctgctcaaaagtaactcaag |
| ET-BR promoter (−392/+493)-HIF-1α-M2 | SDM | ctgtattcctcccgttacaggaaagagttcggagctttg | caaagctccgaactctttcctgtaacgggaggaatacag |
| HIF-1α consensus oligo | EMSA | tctgtacgtgaccacactcacctc | gaggtgagtgtggtcacgtacaga |
| ET-1 promoter (−606/+5) | ChIP | cctcacggatctttctccgat | gtctgacttggacagctctct |
| ET-BR promoter (−619/+2) | ChIP | tgagaagtctctgcgggtttc | atgctgctacctgctccagaa |
Bold and underlined sequences highlight the specific mutations.
ChIP indicates chromatin immunoprecipitation; EMSA, electrophoretic mobility shift assay; PCR, polymerase chain reaction; and SDM, site-directed mutagenesis.
Transient transfection
THP-1 cells (106) were suspended in 100 μL of Nucleofector Solution (Nucleofector kit V; Amaxa Biosystems, Cologne, Germany) containing 0.5 μg of expression plasmids and a pCMV-β-galactosidase vector (0.5 μg) followed by nucleofection according to the manufacturer's instructions using program V-001 (Amaxa Nucleofector II). The cells were then transferred to 6-well plates and incubated overnight in complete medium containing 10% FBS. For transfection of HPMVECs, cells were grown to 80% confluence in EBM-2 containing 5% FBS and nucleofected in 100 μL of Nucleofector Solution (Amaxa Kit-T) containing 1 μg of expression plasmid and 0.5 μg of β-gal reporter plasmid using program S-005. Cells were then transferred overnight to 6-well plates containing EBM-2 complete media, followed by incubation in serum-free medium for 3 hours, before stimulation with PlGF for the indicated time (6 hours). After treatment, cells were rinsed once with phosphate-buffered saline (PBS), lysed with reporter lysis buffer (Promega, Madison, WI), and centrifuged at 8000 rpm for 5 minutes. The supernatants were collected and assayed for luciferase activity using a luminometer (Lumat LB 9501; Berthold, Bad Wildbach, Germany); β-galactosidase activity was measured using the Promega kit. Firefly luciferase values were normalized to β-galactosidase values for transfection efficiency. Data were expressed as values relative to the promoterless pGL3 basic vector.
RNase protection assay
Total RNA from PlGF treated THP-1 monocytes and HPMVECs was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA). RNase protection assay (RPA) was performed with a custom-made Riboquant Multi-probe (ET-1, HIF-1α, ET-BR, MCP-1, IL-8, and glyceraldehyde-3-phosphate dehydrogenase [GAPDH]) obtained from BD Biosciences (San Jose, CA) as described previously.29 The intensity of bands was analyzed using Spot-Denso software on Alpha Imager 2000 gel documentation system (Alpha Innotech, San Leandro, CA). The mRNA levels were normalized to housekeeping GAPDH mRNA. The data are expressed as percentage change relative to untreated cells.
Quantification of ET-1
HPMVECs (106 cells/mL) in serum free-medium were treated with PlGF (250 ng/mL) in the presence or absence of inhibitors for 6 hours. The supernatants were assayed for ET-1 release using an enzyme-linked immunosorbent assay (ELISA) kit (American Research Products, Belmont, MA).
Preparation of nuclear extracts
Nuclear extracts were prepared from HPMVECs and THP-1 cells according to the modified procedure of Dignam et al.30 HPMVECs or THP-1 (5 × 106 cells) were treated with PlGF (250 ng/mL) for indicated time periods (4-12 hours) in the absence and presence of SiRNA, followed by a wash with cold PBS. Cells were suspended in 400 μL of cell lysis buffer (100 mM KCl, 1.5 mM MgCl2, 0.1 mM ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 0.5 mM dithiothreitol (DTT), 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), pH 8.0, 0.5% Nonidet P-40, and 1 μL/mL protease inhibitor cocktail) and allowed to swell on ice for 30 minutes. The homogenate was centrifuged at 10 000g for 1 minute, and the cytosolic supernatants were collected. The nuclear pellet was resuspended in 50 μL of nuclear extraction buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 420 mM NaCl, 0.1 mM EGTA, 0.5 mM DTT, 5% glycerol, 0.5mM PMSF, and 1 μL/mL protease inhibitor cocktail) and mixed intermittently for 60 minutes. The nuclear extracts were obtained by centrifugation at 10 000g for 10 minutes at 4°C. The protein content was quantified by Bradford method (Bio-Rad Laboratories, Hercules, CA).
Western blot analysis
Nuclear extracts from HPMVECs were subjected to SDS gel electrophoresis followed by transfer to nitrocellulose membrane. Membranes were probed with antibody to HIF-1α (1:250) as described previously.27 Blots were stripped and reprobed with HIF-1β antibody (1:250) to monitor protein loading. The protein bands were detected using Immunobilon Western reagents (Millipore, Billerica, MA).
Electrophoretic mobility shift assay for transcription factor HIF-α
The double stranded HIF-1α consensus oligonucleotide obtained from Santa Cruz Biotechnology was labeled with biotin using a light shift chemiluminescent electrophoretic mobility shift assay (EMSA) kit (Pierce, Rockford, IL). The DNA binding reactions containing nuclear extract (10 μg), 5% glycerol, 5 mM MgCl2, 50 ng/μL poly(dI·dC), 0.05% Nonidet P-40 and 0.5 ng of biotinylated duplex DNA were incubated at room temperature for 20 minutes. The samples were subjected to nondenaturing 6% polyacrylamide gel electrophoresis at 100 V in 0.5 × Tris-borate/ethylenediaminetetraacetic acid (EDTA) buffer and transferred to Hybond-N+ nylon membrane (GE Healthcare, Little Chalfont, United Kingdom). The bands were detected with streptavidin-HRP/chemiluminescence (Pierce, Rockford, IL). The specificity of DNA-protein interaction was demonstrated using 50-fold excess of unlabeled probe. In super shift assays, nuclear extracts were pre-incubated for 20 minutes with specific HIF-1α antibody (2 μg).
Chromatin immunoprecipitation (ChIP) assays
HPMVECs (5 × 106 cells) or THP-1(10 × 106 cells) in serum-free medium were treated with PlGF (250 ng/mL) in the presence or absence of inhibitors for 2 and 4 hours, respectively. ChIP analysis was performed using antibody to HIF-1α as described previously.27 In brief, 5 μL of DNA sample was subjected to PCR amplification using primers for ET-1 and ET-BR promoter region (Table 1). PCR was performed for 30 cycles under the following conditions; 94°C for 60 seconds, 56°C for 60 seconds and 72°C for 45 seconds. The PCR products were subjected to 0.8% agarose gel electrophoresis and visualized by ethidium bromide staining. Densitometric quantification of chromatin immunoprecipitation (ChIP) products was performed using Spot-Denso software on Alpha Imager 2000 gel documentation system. The values were normalized to input DNA. The data are expressed as percentage changes relative to untreated cells.
Flow cytometric analysis
THP-1 cells or PBM were treated with PlGF (250 ng/mL) for 24 hours in the presence and absence of pharmacologic inhibitors. Cells were washed with chilled PBS, resuspended in PBS at a density of 106 cells/mL and incubated with antibody to ET-BR (5 μg/mL) at 4°C for 60 minutes. Cells were washed, incubated with fluorescein isothiocyanate-conjugated anti–goat IgG, washed again and analyzed for ET-BR surface expression on FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ). Monocytes were gated based on forward and side-scatter parameters.
Statistical analysis
Data are presented as means (± SE). The significance of difference in mean values between multiple groups was analyzed with parametric one-way ANOVA followed by a Turkey-Kramer test using Instat 2 software program. (GraphPad, San Diego, CA). PlGF-treated sample values were used for multiple comparisons. Student t test was used to evaluate the significance of difference between untreated and PlGF-treated samples. Values of P less than .05 were considered statistically significant.
Results
PlGF-induced expression of ET-1 in HPMVECs involves activation of PI-3 kinase and reactive oxygen species
To determine whether elevated plasma levels of ET-1 at baseline/steady state in SCD15 are secondary to increased production of PlGF, we exposed HPMVECs to PlGF and analyzed ET-1 mRNA expression. PlGF treatment of HPMVECs resulted in a time-dependent increase in ET-1 mRNA expression with maximal increase by 5.5-fold (557 ± 20.5%) at 6 hours (Figure 1A). Treatment of HPMVECs with pharmacologic inhibitors of PI-3 kinase (LY294002) and nicotinamide-adenine dinucleotide phosphate (NADPH)-oxidase (diphenylene iodonium [DPI]) reflective of reactive oxygen species (ROS), reduced ET-1 mRNA expression by 74% (± 4%) and 77% (± 4%), respectively (Figure 1B). However, inhibitors of mitogen-activated protein (MAP) kinase (PD98059) and p38 MAP kinase (SB203580) modestly reduced (17 ± 4%), whereas inhibitor of nuclear factor-κB (pyrrolidine dithiocarbamate [PDTC]) did not (< 5%) affect PlGF-mediated ET-1 mRNA expression (Figure 1B). Pharmacologic inhibitors can act in a nonspecific manner to cause this effect, we therefore transfected a Dn mutant of PI-3 kinase and phosphatase and tensin homolog (PTEN; a negative regulator of PI-3 kinase/Akt), both of which inhibited PlGF-mediated ET-1 mRNA expression by 89% (± 8%) and 105% (± 7%), respectively (Figure 1C). In addition, siRNA against p47phox, a subunit of NADPH-oxidase (95 ± 9%), but not scrambled p47phox siRNA (13 ± 11%), inhibited PlGF-mediated ET-1 expression (Figure 1C lane 3).
Figure 1.

PlGF-mediated ET-1 expression involves PI-3 kinase, ROS, and HIF-1α. HPMVECs were treated with PlGF (250 ng/mL) for 6 hours, unless otherwise indicated, in the absence and presence of pharmacologic inhibitors or transfected with plasmids. (A-C) RNase protection assay. Data are representative of 3 different experiments. (D,E) ET-1 release by ELISA. Data are expressed as percentage change relative to untreated cells and represent means (± SE) of 3 independent experiments. ***P < .001; **P < .01; ns, P > .05. Where indicated, the vertical lines show the repositioned gel lanes.
At the protein level, PlGF increased ET-1 release from HPMVECs by 2.5-fold (253 ± 7%), which was inhibited by the same pharmacologic inhibitors that inhibited ET-1 mRNA expression, LY294002 (70 ± 1%) and DPI (90 ± 10%), but not by PD98059 (25 ± 3%), SB203580 (25 ± 5%), and PDTC (15 ± 8%; Figure 1D). Moreover, transfection of HPMVECs with p47phox siRNA and PTEN plasmid resulted in 89% (± 14%) and 55% (± 10%) reduction in PlGF-mediated ET-1 release, respectively (Figure 1E). Taken together, these data indicate that PlGF-induced activation of both PI-3 kinase and NADPH-oxidase led to increase in ET-1 release from HPMVECs.
PlGF-mediated ET-1 expression requires HIF-1α
Previous studies have shown that ET-1 expression was induced by hypoxia in human umbilical vein endothelial cells through HIF-1α.28,31 We therefore analyzed whether PlGF-induced expression of ET-1 also involved HIF-1α. As shown in Figure 1A middle panel, PlGF increased HIF-1α mRNA expression by 3.5-fold (358 ± 22.5%) at 6 hours. This increase was inhibited in response to LY294002 (Figure 1B middle panel), transfection with Dn PI-3 kinase and PTEN (Figure 1C middle panel) by 77% (± 6%), 103% (± 7%), and 90% (± 8%), respectively. However, DPI and p47phox siRNA transfections did not change PlGF-induced HIF-1α mRNA levels (Figure 1B,C middle panels). These data indicate that PlGF-mediated up-regulation of HIF-1α mRNA required activation of PI-3 kinase but not the ROS pathway.
Figure 2A demonstrates that PlGF-induced ET-1 and HIF-1α mRNA expression were both reduced by 125% (± 6%) upon HIF-1α siRNA transfections (lane 3). However, transfection with scHIF-1α siRNA (lane 4) had no effect on the levels of ET-1 or HIF-1α mRNA. Furthermore, overexpression of HIF-1α (lane 5) but not of HIF-1β (lane 6) increased ET-1 mRNA expression by 2.7-fold (270 ± 13%) in the absence of PlGF. The role of HIF-1α was further substantiated by transfections with PHD-2 siRNA, which showed 3.7-fold (371 ± 13%) increase in ET-1 mRNA expression compared with cells transfected with scPHD-2 siRNA (140 ± 8%) in response to PlGF (lane 7 and 8). It is pertinent to note that PlGF treatment of HPMVECs resulted in a time-dependent increase in HIF-1α protein levels (Figure 2B), with a maximal increase at 12 hours. Transfection with HIF-1α siRNA (Figure 2B lane 5) in PlGF-treated cells reduced HIF-1α protein levels, whereas transfection with PHD-2 siRNA (Figure 2B lane 7) resulted in an increase in HIF-1α protein levels. In addition, ascorbate, which causes degradation of HIF-1α protein,32 led to significant decrease (72 ± 4%) in ET-1 mRNA expression in response to PlGF (Figure 1B lane 8). Taken together, these data confirm that PlGF-mediated ET-1 expression involves activation of the transcription factor HIF-1α.
Figure 2.

The role of HIF-1α in PlGF-mediated ET-1 mRNA expression. HPMVECs were transfected with indicated plasmids followed by PlGF treatment for 6 hours. (A) RPA. (B) Western blot analysis of nuclear extracts (20 μg) using antibodies to HIF-1α and HIF-1β. Data are representative of 3 different experiments. Where indicated, the vertical lines show the repositioned gel lanes.
PlGF-mediated up-regulation of the ET-1 gene involves proximal HIF-1α binding motif
Next, we determined regions of the ET-1 promoter, which was responsible for ET-1 up-regulation. HPMVECs transfected with full-length (−669 bp) and truncated (−176 bp) ET-1 promoter-luciferase constructs resulted in approximately 6-fold increase in ET-1 promoter activity by the addition of PlGF, which was attenuated when HIF-1α binding site at −118 bp was mutated in full-length or truncated ET-1 promoter (Figure 3A). Next, we examined whether pharmacologic inhibitors, which had shown significant reduction of the PlGF-mediated ET-1 mRNA expression also affected the ET-1 promoter activity. As shown in Figure 3B, PlGF-induced ET-1 promoter activity was inhibited by inhibitors of PI-3 kinase and ROS by nearly 50%. To determine whether HIF-1α binds to the ET-1 promoter in the native chromatin of HPMVECs, we performed ChIP assays on chromatin obtained from HPMVEC cells treated with PlGF. Chromatin samples were immunoprecipitated with an antibody to either HIF-1α or normal rabbit IgG (control). HPMVEC cells treated with PlGF showed 3-fold (308 ± 8%) increase in expected 611-bp PCR product that corresponds to −606 to +5bp of the ET-1 promoter region containing HIF-1α binding sites. The binding to the promoter region was attenuated when HPMVECs were pretreated with ascorbate (76 ± 6%), LY294002 (78 ± 5%), and DP1 (73 ± 3%; Figure 3C top panel). No bands were evident in control IgG immunoprecipitates (Figure 3C bottom panel). The amplification of input DNA before immunoprecipitation was equal in all the samples (Figure 3C middle panel). Overall, these results indicate that PlGF promotes binding of HIF-1α to the proximal ET-1 promoter element in vivo to increase the transcription of ET-1.
Figure 3.

The regulation of ET-1 promoter by PlGF involves HIF-1α response elements. HPMVECs were transfected with indicated wild-type and mutant constructs followed by treatment with PlGF for 6 hours in the absence or presence of pharmacologic inhibitors. (A and B) relative luciferase activity. Data are expressed as means (± SE) of 3 independent experiments. ***P < .001. (C) HIF-1α binding to native chromatin was demonstrated by ChIP assay using HIF-1α or normal rabbit IgG antibodies. The ChIP products were amplified by PCR using primers flanking HIF-1α binding sites in the ET-1 promoter as shown in Table 1. Data are representative of 2 different experiments.
PlGF induces expression of the ET-1 cognate receptor ET-BR in THP-1 cells
ET-1 exerts its physiologic effect via activation of ET-A receptor (ET-AR) and ET-BR. We hypothesized that ET-1 and PlGF, both present at high levels in SCD, may act synergistically to augment the inflammatory response in monocytes. THP-1 cells and PBM express ET-BR, but not ET-AR,33 which was confirmed by quantitative real-time PCR (qRT-PCR; data not shown). Treatment of PBM with PlGF showed 2-fold increase in ET-BR mRNA expression as determined by qRT-PCR (data not shown). In THP-1 cells, PlGF caused a 4-fold (398 ± 11%) increase in the ET-BR mRNA, which was inhibited by LY294002 (73 ± 4%) and DPI (87 ± 4%; Figure 4A). However, PDTC and PD98059 did not affect PlGF− induced ET-BR expression. Ascorbate also reduced (71 ± 3%) PlGF-mediated ET-BR expression, suggesting that HIF-1α is involved in increased ET-BR expression. Indeed, PlGF-induced ET-BR expression was attenuated by HIF-1α siRNA (104 ± 8%) but not by scHIF-1α siRNA (Figure 4B lanes 3 and 4). In addition, overexpression of HIF-1α but not HIF-1β in THP-1 cells was able to increase by 2.8-fold (280 ± 13%) the expression of ET-BR in absence of PlGF (Figure 4B lanes 5 and 6), showing that ET-BR expression was specifically mediated by HIF-1α and not HIF-1β.
Figure 4.

PlGF-mediated ET-BR expression involves PI-3kinase, ROS and HIF-1α. THP-1 monocytes were treated with PlGF for 24 hours in the absence and presence of inhibitors (A) and after transfection with siRNA constructs (B) and transfection with HIF expression plasmids (B). (A,B) RNase protection assay. (C) Schematics of ET-BR promoter showing the locations and sequence of HIF-1α binding sites and corresponding mutations as shown by asterisk. (D) PlGF-mediated ET-BR promoter luciferase activity of full-length and deletion constructs. (E) THP-1 cells were transfected with − 392/+493 ET-BR luciferase promoter construct, followed by treatment with PlGF in the absence and presence of LY294002 (15 μM) or DPI (10 μM). (F) PlGF-mediated − 392/+493 ET-BR luciferase promoter activity in THP-1 cells transfected with proximal HIF-1α binding site mutant (HIF-1α-MI) and distal HIF-1α binding site mutant (HIF-1α-M2) as shown in panel C. Data in panels D through F are expressed as means (± SE) of 3 independent experiments. ***P < .001; **P < .01; ns, P > .05. Where indicated, the vertical lines show the repositioned gel lanes.
PlGF-induced ET-BR expression requires HIF-1α motif in its promoter
Studies have shown that the 5′-flanking promoter region of the ET-BR gene lacks the conventional TATA and CCCAAT boxes but contains potential SP-1, GATA, acute phase regulatory elements, and E-box motifs.34 Examination of the promoter element (−1259 to −1 bp) of ET-BR (GenBank Accession number D13168) revealed the additional presence of several RCGTG, HIF-1α response elements (HRE) and inverted HRE (GCAC bases) as indicated in Figure 4C. Transfection of −1259/+493 bp ET-BR promoter luciferase plasmid into THP-1 cells, followed by treatment with PlGF resulted in a 6-fold increase in luciferase activity compared with untreated controls (Figure 4D). Analysis of truncated ET-BR promoter constructs showed that −392/+493 bp ET-BR luciferase plasmid was as effective as −1259/+493 bp plasmid (Figure 4D). Thus, we used this plasmid for further studies. As shown in Figure 4E, THP-1 cells transfected with −392/+493 bp ET-BR plasmid and pre-incubated with LY294002 or DPI showed reduced activity in response to PlGF. The region −392 to +493bp of the ET-BR promoter, as shown in Figure 4C, contains 2 putative HREs (−155 to −152 bp and −371 to −368 bp) along with AP-1 site (located at −92 to −87 bp), relative to transcriptional start site. We observed that mutation of HIF-1α site (−155 to −152 bp, indicated as HIF-1α-M1) but not the second HIF-1α site (−371 to −368 bp, indicated as HIF-1α-M2) resulted in an attenuation of PlGF-induced ET-BR promoter luciferase activity (Figure 4F). These results indicate that PlGF-induced ET-BR promoter activity requires HIF-1α motif (−155 to −152bp) in its promoter. Taken together, these results showed that PlGF up-regulated ET-BR expression in THP-1 cells via activation of PI-3kinase, NADPH-oxidase, and HIF-1α.
PlGF promotes binding of HIF-1α in vitro (EMSA) and in vivo (ChIP) to the promoter region of ET-BR
Next, we determined whether PlGF promotes the DNA binding activity of HIF-1α. As shown in Figure 5A, PlGF led to increase in HIF-1α binding to an HIF-1α bona fide probe in THP-1 nuclear extract as determined by EMSA. The specificity of HIF-1α binding was shown by competition with either 50-fold excess unlabeled probe or by super shift in HIF-DNA complex with antibody to HIF-1α. Next, we performed ChIP assay on chromatin samples obtained from THP-1 cells treated with PlGF for 4 hours. Chromatin samples immunoprecipitated with HIF-1α antibody showed a 2.7-fold (275 ± 6%) increase in expected PCR product of 621-bp size (Figure 5B top panel) corresponding to ET-BR promoter (−619 to +2 bp) containing both HIF binding sites. Ascorbate (77 ± 3%), LY294002 (74 ± 2%), and DPI (81 ± 2%) reduced the expected PCR product. Amplification of input DNA before immunoprecipitation was equal in all the samples (Figure 5B middle panel). Moreover, immunoprecipitation of chromatin samples with normal rabbit IgG showed no amplification of expected products (Figure 5B bottom panel). ChIP analysis confirmed that HIF-1α binds to the ET-BR promoter in native chromatin of THP-1 cells.
Figure 5.

PlGF promotes HIF-1α binding to DNA in vitro and in vivo. (A) EMSA demonstrating binding activity of HIF-1α in THP-1 nuclear extracts to its consensus DNA binding sequence. Where indicated, 50-fold excess of unlabeled probe or antibody to HIF-1α was added. Complex formation was visualized by autoradiography. (B) THP-1 cells were treated with PlGF for 4 hours in the absence and presence of pharmacologic inhibitors. The soluble chromatin was isolated and immunoprecipitated with either antibody to HIF-1α (top panel) or control rabbit IgG (bottom panel). The primers flanking HIF-1α binding sites in the ET-BR promoter as indicated in Table 1 were used to amplify the products by PCR. The bottom panel is amplification of input DNA before immunoprecipitation. Data are representative of 2 independent experiments. Where indicated, the vertical lines show the repositioned gel lanes.
PlGF induces surface expression of ET-BR in THP-1 cells and PBM
PlGF treatment of THP-1 cells and PBM exhibited 3- and 3.6-fold increase in the surface expression of ET-BR, respectively, as determined by flow cytometry (Figure 6A,C). Pretreatment with pharmacologic inhibitors ascorbate, DPI, and LY294002 of THP-1 and PBM reduced ET-BR surface expression by more than 90% and more than 75%, respectively (Figure 6B,D). These studies indicate that PlGF-induced signaling through activation of PI-3 kinase and NADPH-oxidase causes increased surface expression of ET-BR in THP-1 and PBM via HIF-1α.
Figure 6.

FACS analysis of ET-BR surface expression in THP-1 and PBM. (A,C) THP-1 cells and PBM were treated with PlGF for 24 hours and surface expression of ET-BR was analyzed by flow cytometry. (B,D) The mean fluorescence intensity of ET-BR in PlGF treated THP-1 cells and PBM in the presence or absence of indicated pharmacologic inhibitors is shown as a histogram. Data are representative of 3 independent experiments in duplicates.
PlGF-ET-1-ETBR loop in HIF-1α mediated cytochemokine expression in THP-1 cells
Our previous studies showed that PlGF increases the expression of MCP-1 and IL-8 in THP-1 and PBM.23,24 Because PlGF caused an increase in ET-1 release from HPMVECs and ET-BR expression in monocytes, we hypothesized that ET-1 and PlGF in circulation may act synergistically to increase cytochemokine expression and hence contribute to inflammatory state of SCD. As shown in Figure 7, we observed that ET-1 (250 nM) treatment for 30 minutes caused an increase in MCP-1 and IL-8 mRNA expression, with no change in ET-BR mRNA expression. PlGF treatment alone for 24 hours increased MCP-1, IL-8, and ET-BR mRNA expression in THP-1 cells. However, PlGF-treated cells, which show increased expression of ET-BR, when treated with ET-1 showed 3-fold increase in MCP-1 and IL-8 mRNA expression, indicating that PlGF-primed cells are more responsive to ET-1 in cytochemokine expression. Thus, PlGF-mediated ET-1 release from HPMVECs and ET-BR expression in monocytes creates a PlGF–ET-1–T-BR loop that augments inflammation and may promote PHT.
Figure 7.
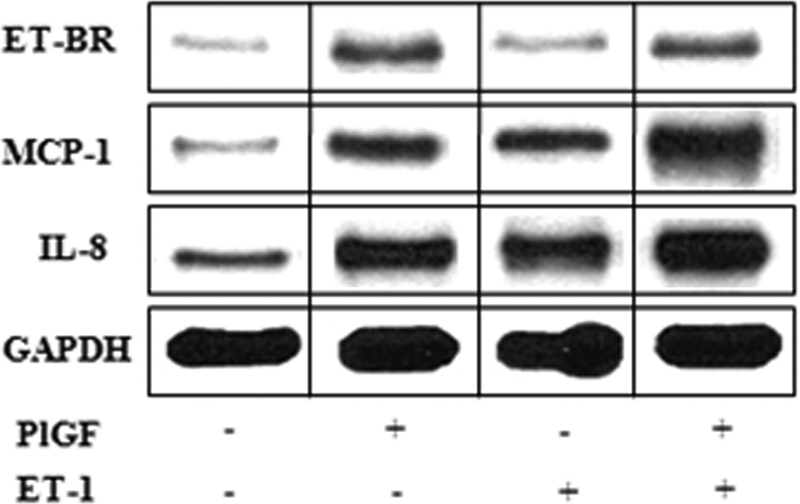
PlGF and ET-1 augment cytochemokine mRNA expression. THP-1 (106 cells/mL) were either treated with PlGF for 24 hours (250 ng/mL) or ET-1 (250 nmol/L) for 30 minutes. For priming, THP-1 cells were first treated with PlGF for 24 hours, washed with PBS and then treated with ET-1 for 30 minutes. Total mRNA was isolated and RPA was performed as described under “RNase protection assay.” The data are representative of 3 independent experiments. Where indicated, the vertical lines show the repositioned gel lanes.
Discussion
Pulmonary complications of SCD are now the most frequent cause of mortality. These include an increased incidence of airway hyper reactivity in children, ACS and cumulative lung damage. Restrictive lung disease develops with increasing age.11,35–37 In addition, increased incidence of PHT develops with increasing age.9,12,13 NO and ET-1 are opposing forces that determine the pulmonary vascular tone. Reduced bioavailability of NO due to increased hemolysis has been proposed to contribute to PHT in patients with SCD9,38 and in sickle transgenic mice.39 On the other hand, ET-1 levels are elevated in patients with SCD, further increased during vaso-occlusive crises, with the highest levels of ET-1 seen in sickle ACS.16 In this report, we show that an erythroid cell–derived angiogenic growth factor, PlGF, induced ET-1 expression in HPMVECs and up-regulated expression of its cognate receptor, ET-BR, in monocytes. Together with the proinflammatory response generated by PlGF, as previously shown,23,24 PlGF may also induce a potent pulmonary vasoconstrictive response via ET-1 and ET-BR up-regulation.40,41
Our studies show that PlGF-mediated up-regulation of ET-1 in HPMVECs involves activation of PI-3 kinase, NADPH-oxidase, and HIF-1α. Previous studies have shown that in response to hypoxia, HIF-1α binds to an inverted HRE (GCAC) at −118 bp upstream of the transcription start site of ET-1 promoter.28 Additional studies indicated that GATA-2, AP-1, and HIF-1α transcriptional factors formed a complex to regulate ET-1 gene expression in response to hypoxia.31 In our studies, the role of HIF-1α in PlGF-mediated expression of ET-1 was substantiated by transfections with HIF-1α siRNA, mutational analysis of HRE in ET-1 promoter, and ChIP analysis. Although hypoxia mediated ET-1 mRNA expression has been shown to be regulated by HIF-1α,28,31 this is the first report of a hypoxia-independent up-regulation of ET-1 by PlGF.
We have previously shown that monocytes are activated by PlGF via VEGFR-1 to generate cytochemokines.24 Herein, we observed that PlGF increased the expression of ET-1 cognate receptor ET-BR in THP-1 monocytic cells and PBM. Our studies show that PlGF regulates ET-BR expression via activation of PI-3 kinase, NADPH-oxidase, and HIF-1α, as was seen in the expression of ET-1 in HPMVECs. An in silico analysis of the ET-BR promoter element (−1259 to −1 bp)34 revealed the presence of several HRE. Our results show that PlGF-induced ET-BR promoter activity requires HIF-1α motif (−155 to −152 bp) in its promoter as determined by mutational analysis of 2 HRE sites. The role of HIF-1α in the up-regulation of ET-BR was further confirmed by EMSA, HIF-1α siRNA, and ChIP.
Our studies showed time-dependent increase in HIF-1α protein in response to PlGF, independent of hypoxia. Previous studies have shown that TGF-β1 induces HIF-1α stabilization through selective inhibition of prolyl-hydroxylase-2 (PHD-2) in HepG2 cells,42 whereas NO-induced degradation of HIF-1α involved PHD-2 induction.43 We showed that PHD-2 siRNA increases the HIF-1α protein level and concomitantly increases expression of ET-BR in response to PlGF, supporting the role of PHD-2 in HIF-1α degradation as shown in previous studies.42,43 It is pertinent to note that levels of PHD-2 enzyme remained unchanged in response to PlGF (data not shown), indicating that intracellular cofactors (eg, ascorbate, Fe+2, and succinate) affected by induction of ROS by PlGF may attenuate the PHD-2 enzyme activity44,45 and thus stabilize HIF-1α levels.
ET-1, ET-AR, and ET-BR transcripts are up regulated in the lungs and pulmonary artery in response to chronic hypoxia in rats.46 In addition, studies have shown increased expression of ET-1 in cultured endothelial cells in response to interaction with deoxygenated sickle red blood cells47 and hypoxia.28 The chronic hypoxia from the mild to moderate desaturations in patients with SCD at steady state and the high prevalence of nocturnal hypoxia that correlates with the degree of anemia48,49 may contribute significantly to increased ET-1 expression and PHT. It is noteworthy that PlGF levels are also chronically elevated in patients with SCD as a result of the increased erythroid cell turnover. Furthermore, PlGF concentrations in plasma correlate with increased incidence of vaso-occlusive admissions in SCD.23 Augmentation of ET-1 and ET-BR by PlGF can conceivably augment the PHT that is potentiated by the hypoxia in SCD, although this remains to be examined.
We were also interested in determining whether ET-1 mediated signaling via ET-BR leads to increased cytochemokine expression. Indeed, ET-1 via binding to ET-BR increased expression of MCP-1 and IL-8 in monocytes. Although we have shown previously that PlGF alone increases expression of MCP-1 and IL-8, herein, ET-1 mediated up-regulation of these chemokines is additive, not synergistic with the effect of PlGF alone.24 Chronic inflammation and additional up-regulation of factors that promote pulmonary vasoconstriction conceivably contribute to the complex vasculopathy in SCD, with progressive development of PHT over time.
Overall, we have demonstrated that PlGF activates pulmonary endothelium and monocytes to generate ET-1 and its cognate receptor ET-BR, respectively. In addition, PlGF elicits a direct proinflammatory response from monocytes and indirectly via ET-1 and ET-BR up-regulation. Chronic inflammation and pulmonary vasoconstriction can contribute to the development of PHT in SCD, and other chronic anemias, such as thalassemia. Indeed, PHT is a prominent feature in patients with thalassemia intermedia and inadequately transfused patients with thalassemia major. Because our studies show that PlGF increases ET-BR expression, ET-BR antagonist could be potentially useful in ameliorating PHT and inflammation in SCD. It is pertinent to note that Bosentan, a dual endothelin receptor antagonist, was in clinical trials for the treatment of PHT in SCD, although the study was stopped as a result of slow accrual (www.clinicaltrials.gov).
Acknowledgments
We thank Dr Keith Webster for kindly providing ET-1 promoter constructs. We thank Mike Xie for preparing ET-BR luciferase plasmid.
This work was supported by National Institutes of Health grants HL070595 (USC-CSCC) and R01-HL079916.
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: N.P. designed and performed most of the experiments, analyzed the data, and contributed to the writing of the manuscript. C.G. performed experiments on ET-1 release. P.M. contributed to the design of the study and to the writing of the manuscript. V.K.K. designed research and contributed to the preparation and writing of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Vijay Kalra, PhD, HMR 611, Department of Biochemistry and Molecular Biology USC Keck School of Medicine, Los Angeles, CA 90033; e-mail: vkalra@usc.edu; or Punam Malik, MD, Division of Experimental Hematology, Cincinnati Children's Hospital Medical Center, MLC 7013, 3333 Burnet Ave, Cincinnati, OH 45229-3039; e-mail: punam.malik@cchmc.org.
References
- 1.Francis RB, Jr., Johnson CS. Vascular occlusion in sickle cell disease: current concepts and unanswered questions. Blood. 1991;77:1405–1414. [PubMed] [Google Scholar]
- 2.Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med. 1991;325:11–16. doi: 10.1056/NEJM199107043250103. [DOI] [PubMed] [Google Scholar]
- 3.Buchanan G R. Infection. In: Embury SH, Hebbel RP, Mohandas N, Steinberg MH, editors. Sickle Cell Disease—Basic Principles and Clinical Practice. New York: Raven Press; 1994. pp. 567–587. [Google Scholar]
- 4.Wong WY, Powars DR, Chan L, et al. Polysaccharide encapsulated bacterial infection in sickle cell anemia: a thirty year epidemiologic experience. Am J Hematol. 1992;39:176–182. doi: 10.1002/ajh.2830390305. [DOI] [PubMed] [Google Scholar]
- 5.Embury S H, Hebbel R P, Steinberg M H, Mohandas N. Pathogenesis of vasoocclusion. In: Embury SH, Hebbel RP, Mohandas N, Steinberg MH, editors. Sickle Cell Disease—Basic Principles and Clinical Practice. New York: Raven Press; 1994. pp. 311–326. [Google Scholar]
- 6.Powars DR. Sickle cell anemia and major organ failure. Hemoglobin. 1990;14:573–598. doi: 10.3109/03630269009046967. [DOI] [PubMed] [Google Scholar]
- 7.Vichinsky EP, Styles LA, Colangelo LH, et al. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood. 1997;89:1787–1792. [PubMed] [Google Scholar]
- 8.Serjeant GR. Sickle-cell disease. Lancet. 1997;350:725–30. doi: 10.1016/S0140-6736(97)07330-3. [DOI] [PubMed] [Google Scholar]
- 9.Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350:886–895. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- 10.Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood. 1994;84:643–649. [PubMed] [Google Scholar]
- 11.Klings ES, Wyszynski DF, Nolan VG, Steinberg MH. Abnormal pulmonary function in adults with sickle cell anemia. Am J Respir Crit Care Med. 2006;173:1264–1269. doi: 10.1164/rccm.200601-125OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ataga KI, Sood N, De Gent G, et al. Pulmonary hypertension in sickle cell disease. Am J Med. 2004;117:665–669. doi: 10.1016/j.amjmed.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 13.De Castro LM, Jonassaint JC, Graham FL, Ashley-Koch A, Telen MJ. Pulmonary hypertension associated with sickle cell disease: clinical and laboratory endpoints and disease outcomes. Hematology. 2008;83:19–25. doi: 10.1002/ajh.21058. [DOI] [PubMed] [Google Scholar]
- 14.Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 15.Hammerman SI, Kourembanas S, Conca TJ, et al. Endothelin-1 production during the acute chest syndrome in sickle cell disease. Am J Respir Crit Care Med. 1997;156:280–285. doi: 10.1164/ajrccm.156.1.9611085. [DOI] [PubMed] [Google Scholar]
- 16.Graido-Gonzalez E, Doherty JC, Bergreen EW, et al. Plasma endothelin-1, cytokine, and prostaglandin E2 levels in sickle cell disease and acute vaso-occlusive sickle crisis. Blood. 1998;92:2551–2555. [PubMed] [Google Scholar]
- 17.Kourembanas S, Marsden PA, McQuillan LP, Faller DV. Hypoxia induces endothelin gene expression and secretion in cultured human endothelium. J Clin Invest. 1991;88:1054–1057. doi: 10.1172/JCI115367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood. 2000;96:2451–2459. [PubMed] [Google Scholar]
- 19.Platt OS. Sickle cell anemia as an inflammatory disease. J Clin Invest. 2000;106:337–338. doi: 10.1172/JCI10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Francis RB, Jr., Haywood LJ. Elevated immunoreactive tumor necrosis factor and interleukin-1 in sickle cell disease. J Natl Med Assoc. 1992;84:611–615. [PMC free article] [PubMed] [Google Scholar]
- 21.Croizat H. Circulating cytokines in sickle cell patients during steady state. Br J Haematol. 1994;87:592–597. doi: 10.1111/j.1365-2141.1994.tb08318.x. [DOI] [PubMed] [Google Scholar]
- 22.Duits AJ, Schnog JB, Lard LR, Saleh AW, Rojer RA. Elevated IL-8 levels during sickle cell crisis. Eur J Haematol. 1998;61:302–305. doi: 10.1111/j.1600-0609.1998.tb01092.x. [DOI] [PubMed] [Google Scholar]
- 23.Perelman N, Selvaraj SK, Batra S, et al. Placenta growth factor activates monocytes and correlates with sickle cell disease severity. Blood. 2003;102:1506–1514. doi: 10.1182/blood-2002-11-3422. [DOI] [PubMed] [Google Scholar]
- 24.Selvaraj SK, Giri RK, Perelman N, et al. Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood. 2003;102:1515–1524. doi: 10.1182/blood-2002-11-3423. [DOI] [PubMed] [Google Scholar]
- 25.Hauser SWH. A heparin-binding form of placenta growth factor (PlGF-2) is expressed in human umbilical vein endothelial cells and in placenta. Growth Factors. 1993;9:259–268. doi: 10.3109/08977199308991586. [DOI] [PubMed] [Google Scholar]
- 26.Tordjman R, Delaire S, Plouet J, et al. Erythroblasts are a source of angiogenic factors. Blood. 2001;97:1968–1974. doi: 10.1182/blood.v97.7.1968. [DOI] [PubMed] [Google Scholar]
- 27.Kim KS, Rajagopal V, Gonsalves C, Johnson C, Kalra VK. A novel role of hypoxia-inducible factor in cobalt chloride and hypoxia-mediated expression of IL-8 chemokine in human endothelial cells. J Immunol. 2006;177:7211–7224. doi: 10.4049/jimmunol.177.10.7211. [DOI] [PubMed] [Google Scholar]
- 28.Hu J, Discher DJ, Bishopric NH, Webster KA. Hypoxia regulates expression of the endothelin-1 gene through a proximal hypoxia-inducible factor-1 binding site on the antisense strand. Biochem Biophys Res Commun. 1998;245:894–899. doi: 10.1006/bbrc.1998.8543. [DOI] [PubMed] [Google Scholar]
- 29.Giri RK, Selvaraj SK, Kalra VK. Amyloid peptide-induced cytokine and chemokine expression in THP-1 monocytes is blocked by small inhibitory RNA duplexes for early growth response-1 messenger RNA. J Immunol. 2003;170:5281–5294. doi: 10.4049/jimmunol.170.10.5281. [DOI] [PubMed] [Google Scholar]
- 30.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA. Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, AND p300/CBP. J Biol Chem. 2001;276:12645–12653. doi: 10.1074/jbc.M011344200. [DOI] [PubMed] [Google Scholar]
- 32.Knowles HJ, Raval RR, Harris AL, Ratcliffe PJ. Effect of ascorbate on the activity of hypoxia-inducible factor in cancer cells. Cancer Res. 2003;63:1764–1768. [PubMed] [Google Scholar]
- 33.King JM, Srivastava KD, Stefano GB, et al. Human monocyte adhesion is modulated by endothelin B receptor-coupled nitric oxide release. J Immunol. 1997;158:880–886. [PubMed] [Google Scholar]
- 34.Arai H, Nakao K, Takaya K, et al. The human endothelin-B receptor gene. Structural organization and chromosomal assignment. J Biol Chem. 1993;268:3463–3470. [PubMed] [Google Scholar]
- 35.Vendramini EC, Vianna EO, De Lucena Angulo I, De Castro FB, Martinez JA, Terra-Filho J. Lung function and airway hyperresponsiveness in adult patients with sickle cell disease. Am J Med Sci. 2006;332:68–72. doi: 10.1097/00000441-200608000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Sylvester KP, Patey RA, Milligan P, et al. Pulmonary function abnormalities in children with sickle cell disease. Thorax. 2004;59:67–70. [PMC free article] [PubMed] [Google Scholar]
- 37.Koumbourlis AC, Zar HJ, Hurlet-Jensen A, Goldberg MR. Prevalence and reversibility of lower airway obstruction in children with sickle cell disease. J Pediatr. 2001;138:188–192. doi: 10.1067/mpd.2001.111824. [DOI] [PubMed] [Google Scholar]
- 38.Jison ML, Gladwin MT. Hemolytic anemia-associated pulmonary hypertension of sickle cell disease and the nitric oxide/arginine pathway. Am J Respir Crit Care Med. 2003;168:3–4. doi: 10.1164/rccm.2304002. [DOI] [PubMed] [Google Scholar]
- 39.Hsu LL, Champion HC, Campbell-Lee SA, et al. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood. 2007;109:3088–3098. doi: 10.1182/blood-2006-08-039438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bauer M, Wilkens H, Langer F, et al. Selective upregulation of endothelin B receptor gene expression in severe pulmonary hypertension. Circulation. 2002;105:1034–1036. doi: 10.1161/hc0902.105719. [DOI] [PubMed] [Google Scholar]
- 41.Chen YFOS. Endothelin and pulmonary hypertension. J Cardiovasc Pharmacol. 2000;35:S49–S53. doi: 10.1097/00005344-200000002-00012. [DOI] [PubMed] [Google Scholar]
- 42.McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem. 2006;281:24171–24181. doi: 10.1074/jbc.M604507200. [DOI] [PubMed] [Google Scholar]
- 43.Berchner-Pfannschmidt U, Yamac H, Trinidad B, Fandrey J. Nitric oxide modulates oxygen sensing by hypoxia-inducible factor 1-dependent induction of prolyl hydroxylase 2. J Biol Chem. 2007;282:1788–1796. doi: 10.1074/jbc.M607065200. [DOI] [PubMed] [Google Scholar]
- 44.Gao P, Zhang H, Dinavahi R, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12:230–238. doi: 10.1016/j.ccr.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaelin J. ROS: really involved in oxygen sensing. Cell Metabolism. 2005;1:357–358. doi: 10.1016/j.cmet.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 46.Li H, Chen SJ, Chen YF, et al. Enhanced endothelin-1 and endothelin receptor gene expression in chronic hypoxia. J Appl Physiol. 1994;77:1451–1459. doi: 10.1152/jappl.1994.77.3.1451. [DOI] [PubMed] [Google Scholar]
- 47.Phelan M, Perrine SP, Brauer M, Faller DV. Sickle erythrocytes, after sickling, regulate the expression of the endothelin-1 gene and protein in human endothelial cells in culture. J Clin Invest. 1995;96:1145–1151. doi: 10.1172/JCI118102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Setty BNY, Stuart MJ, Dampier C, Brodecki D, Allen JL. Hypoxaemia in sickle cell disease: biomarker modulation and relevance to pathophysiology. Lancet. 2003;362:1450–1455. doi: 10.1016/S0140-6736(03)14689-2. [DOI] [PubMed] [Google Scholar]
- 49.Raj AB, O'Brien LM, Bertolone SJ, Gozal D, Edmunds HL., Jr Cerebral oximetry improves detection of sickle cell patients at risk for nocturnal cerebral hypoxia. Pediatr Pulmonol. 2006;41:1088–1094. doi: 10.1002/ppul.20502. [DOI] [PubMed] [Google Scholar]