

Abstract
Streptococcus pneumoniae is the main causal agent of pathologies that are increasingly resistant to antibiotic treatment. Clinical resistance of S. pneumoniae to β-lactam antibiotics is linked to multiple mutations of high molecular mass penicillin-binding proteins (H-PBPs), essential enzymes involved in the final steps of bacterial cell wall synthesis. H-PBPs from resistant bacteria have a reduced affinity for β-lactam and a decreased hydrolytic activity on substrate analogues. In S. pneumoniae, the gene coding for one of these H-PBPs, PBP2x, is located in the cell division cluster (DCW). We present here structural evidence linking multiple β-lactam resistance to amino acid substitutions in PBP2x within a buried cavity near the catalytic site that contains a structural water molecule. Site-directed mutation of amino acids in contact with this water molecule in the “sensitive” form of PBP2x produces mutants similar, in terms of β-lactam affinity and substrate hydrolysis, to altered PBP2x produced in resistant clinical isolates. A reverse mutation in a PBP2x variant from a clinically important resistant clone increases the acylation efficiency for β-lactams and substrate analogues. Furthermore, amino acid residues in contact with the structural water molecule are conserved in the equivalent H-PBPs of pathogenic Gram-positive cocci. We suggest that, probably via a local structural modification, the partial or complete loss of this water molecule reduces the acylation efficiency of PBP2x substrates to a point at which cell wall synthesis still occurs, but the sensitivity to therapeutic concentrations of β-lactam antibiotics is lost.
β-lactam compounds are the most frequently used antibiotics. Their targets, known as penicillin-binding proteins (PBPs), are involved in the final stages of peptidoglycan synthesis. Bacteria have between four and eight PBPs, depending on the species. They can be divided into three groups (1), one of low molecular mass and two of high molecular mass. The low molecular mass forms (≈30–40 kDa) are mainly d-alanyl d-alanine carboxypeptidases and are supposed to be responsible for the degree of cross-linking of the peptidoglycan. The high molecular mass forms (≈50–100 kDa) consist of class A forms, with two enzymatic activities (transglycosylation and transpeptidation), and class B forms, with only one well-defined function (transpeptidation). Since the 1960s, because of the intensive use of β-lactam compounds, β-lactam resistance has appeared in many species (2). In Gram-negative bacteria, the main mechanism of resistance is the production of β-lactamases that are highly effective in opening the β-lactam ring, yielding an inactive compound, whereas most Gram-positive bacteria gain resistance by either changing the specificity of their PBPs or acquiring new PBPs with lower affinity for antibiotics. Alteration of affinity is linked to multiple mutations of high molecular mass PBPs (3–5). At the present time, the therapeutic dose of penicillin, although 600-fold higher than that used in 1941, is still not always able to save patients infected with resistant strains of Streptococcus pneumoniae.
S. pneumoniae contains five high molecular mass PBPs, three of class A and two of class B; one of the latter, PBP2x, is an essential enzyme for the bacterium (6, 7). The determination of the x-ray structure of PBP2x has revealed a three-domain protein: two domains of unknown function and a central 350-residue domain with transpeptidase activity showing a low structural homology with class A β-lactamases (8). Mutations in PBP2x are a prerequisite for high-level resistance that is mediated by additional changes in up to five PBPs (9). PBP2x variants produced by resistant clinical isolates are encoded by mosaic genes that are believed to be the result of genetic exchange and recombinational events (10, 11). The resistant forms of PBP2x produced by clinical isolates have a reduced affinity for β-lactams and, in the case of the “resistant” form of Sp328 PBP2x used in this study, a lower enzymatic activity on thiolester substrate analogues (12).
Deciphering the molecular mechanism of resistance is particularly difficult because of multiple amino acid differences between sensitive and resistant PBP2x. Based on high-resolution structure of PBP2x and multiple sequence alignments, we have identified a restricted set of amino acids, lining a small water-containing cavity, putatively involved in PBP2x resistance to β-lactam antibiotics. We have confirmed the role of these residues by using site-directed mutagenesis and functional analysis.
METHODS
Site-Directed Mutagenesis.
Site-directed mutagenesis was performed by using the expression vector pGEX-4T1 (Pharmacia) previously modified by introducing the replication origin of phage f1 (ori f1) (13). The mutations were checked by sequencing the double-stranded DNA, using the dideoxy-chain termination method (14).
Protein Expression and Purification.
The water-soluble PBP2x* (corresponding to amino acids 49–750) from S. pneumoniae R6 (minimum inhibitory concentration of penicillin = 0.006 μg/ml) was expressed as a fusion protein to glutathione S-transferase with a thrombin-cleavable linker (pGEX-4T1 plasmid, Pharmacia) in the host strain Escherichia coli MC1061. The cell extract (obtained from 1 liter of culture) was loaded onto a glutathione-Sepharose 4B affinity column (Pharmacia) (1 × 10 cm), previously equilibrated with 50 mM Tris⋅HCl, pH 8, 100 mM NaCl, 1 mM EDTA buffer. One hundred units of human thrombin (Sigma) was applied onto the column at room temperature, to elute the water-soluble PBP2x*. The thrombin activity then was inhibited by 1 mM phenylmethylsulfonyl fluoride. All purification steps were performed at 4°C except when specified. Analysis of purity by SDS/polyacrylamide gels showed a single band with an electrophoretic mobility corresponding to an apparent molecular mass of 77 kDa. Mass spectrometry analysis gave a value of 77,256 ± 7 Da consistent with that predicted from the amino acids sequence (77,254 Da). R-PBP2x* [a water-soluble form of clinically produced resistant Sp 328 PBP2x (residues 49–750)] from S. pneumoniae 328 (minimum inhibitory concentration of penicillin = 4 μg/ml), a penicillin-resistant clinical isolate, and all mutants described in this paper were purified to homogeneity by the same method. The yield range was 20–50 mg of pure protein per liter of culture medium depending on the protein.
Acylation Efficiency Determination.
High-Mr PBPs interact with β-lactam antibiotics according to the three-step scheme (15):
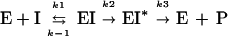 |
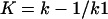 |
where E is the PBP enzyme; I, the β-lactam antibiotic; EI, the Michaelis-Menten complex; EI*, the acyl-enzyme covalent complex; and P, the product of the reaction (degraded β-lactam antibiotic).
The k2/K parameter, accounting for the acylation step efficiency, was determined by following the decrease of the intrinsic fluorescence of the protein at 37°C by using spectrofluorometric measurements coupled with a Biologic SFM3 stopped-flow apparatus (16). The tryptophan-specific excitation wavelength was 280 nm, and the emission was measured in the 305–360 nm range. S-PBP2x* [the water-soluble form of R6 PBP2x (residues 49–750)], R-PBP2x*, and mutants of PBP2x* at a concentration of 0.6 μM in 10 mM phosphate buffer, pH 7 were incubated with concentrations of β-lactam antibiotic ranging from 5 μM to 2 mM.
Hydrolysis Activity Determination.
Hydrolysis activity was measured by using N-benzoyl-d-alanylmercaptoacetic thiolester (S2d) and carboxymethyl benzoylaminothioacetate, synthetic thiolesters analogues of the cell wall stem peptide, according to the method described in ref. 12. Assays were performed at 37°C.
k3 Determination by Electrospray-Ionization Mass Spectrometry (ESI-MS).
Mass spectrometry analysis of the proteins was carried out on a PE Sciex (Toronto, Canada) API III+ triple quadrupole mass spectrometer equipped with an ionspray source. The single spectra and the kinetic studies were recorded in the 900–2,400 and 1,500–1,750 (charges from 46+ to 56+) range of mass-to-charge (m/z) ratios in steps of 0.6 and 0.4 m/z, with a 2- and 6-ms dwell time, respectively. The signal was averaged over 2–4 scans. The electrospray probe tip was held at 5 kV, and the declustering voltage was set at 90 V. The reconstructed molecular mass profiles were obtained by using a deconvolution algorithm (PE Sciex). S-PBP2x*, R-PBP2x*, and mutants of PBP2x* (100 μM) were incubated at 37°C in ammonium acetate, pH 7 with a small excess of antibiotic during 5 min, to obtain complete formation of the acyl-enzyme covalent complex. β-lactamase was added to remove the excess of free antibiotic. The addition of β-lactamase corresponds to initial time. Samples were withdrawn after various periods of time and analyzed by ESI-MS.
RESULTS
Structure-Function Studies of PBP2x Variants.
The PBP2x transpeptidase domain sequence from a resistant clinical isolate contains an average of about 40-aa substitutions distributed throughout the domain when compared with a sensitive PBP2x (S-PBP2x) sequence. Fig. 1 shows the distribution, along the sequence, of the mutation probability in the transpeptidase domain of resistant PBP2x produced by clinical isolates. Analysis of 26 sequences of resistant PBP produced by clinical isolates highlights a few interesting amino acid positions. Because mutations in the active site are more likely to lead to changes in the specificity of the enzyme, mutation of residue 338 was considered. A Thr residue in S-PBP2x, it is mutated to Ala (in the majority of cases investigated), rarely to Pro or Gly. The determination of the high-resolution x-ray structure of S-PBP2x* showed that Thr-338 is located at the beginning of helix α2; its side chain points away from the catalytic residue Ser-337 and is buried in a small cavity shielded from the active site by the protein main chain (8). This cavity contains a structural water molecule (WAT) that is hydrogen-bonded to a carbonyl of the main chain (P335) and three side-chain hydroxyl groups (T338, S571, and Y586): none of the WAT ligand residues can be in contact with a bound antibiotic (Fig. 2). To confirm the correlation between the presence of this water molecule and resistance, we investigated the effect of mutations of WAT ligands on the reactivity to antibiotics.
Figure 1.
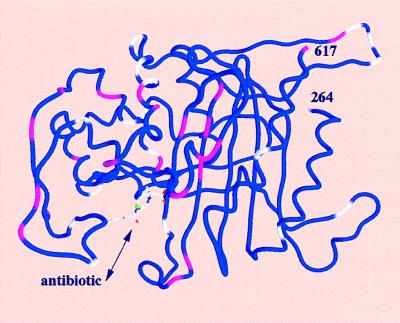
Worm representation of the transpeptidase domain of PBP2x (residues 264–617). The worm is color-coded with colors representing the mutation probability in clinically produced resistant sequences, with blue (0), white (between 0 and 0.75), and pink (higher than 0.75) at the Cα position. The mutation probability is based on the analysis of 26 sequences of resistant PBP2x produced by clinical isolates. Secondary structure elements are shown as in figure 3b of ref. 8. A cephalosporin molecule is modeled in the active site.
Figure 2.
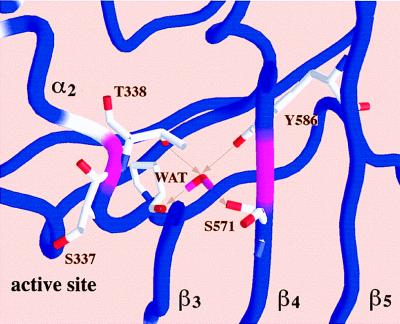
Closer view of the worm representation showing the side chains interacting with the buried water molecule (WAT) and the active-site serine S337. For clarity only a slice of the structure is represented. Arrows indicate hydrogen bonds between WAT and neighboring atoms. Figs. 1 and 2 were made with the program grasp (27).
S-PBP2x* and R-PBP2x*, derived from a sensitive strain and a resistant clinical isolate Sp328, have been expressed in E. coli as fusion proteins with glutathione S-transferase, and purified to homogeneity (8). Mutants, generated by site-directed mutagenesis (13), were produced and purified by the same method. All proteins were thermostable at 37°C and could be fully acylated by benzylpenicillin (Pen-G) or cefotaxime (two classical members of the β-lactam family), as shown by electrospray-ionization mass spectrometry (ESI-MS). The half-life of the acyl-enzyme, measured by ESI-MS, varied from 3.6 to 14.8 hr, depending on the mutation. Stopped-flow fluorimetry was used to measure acylation efficiencies of PBPs by both antibiotics (16).
The importance of the hydroxyl group of residue 338 is shown by the kinetic data of Table 1. The mutation T338A reduces the acylation efficiency of S-PBP2x* by cefotaxime by a factor of 2; conversely the sole mutation A338T in the resistant R-PBP2x* increases this rate by a factor of 6 (Fig. 3). Similar results are obtained for Pen-G. Thus, the absence of a hydroxyl group at position 338 appears to lower the k2/K value of S-PBP2x* for β-lactams, even though this group is not in direct contact with the antibiotic (8). Similarly, the addition of a hydroxyl group at position 338 in R-PBP2x* increases the acylation efficiency for β-lactams. The k2/K reduction is even stronger for mutants T338G and T338P of S-PBP2x* (8- and 5-fold, respectively) than for T338A. In agreement with this result is the fact that substitutions T338G or T338P are found in clinically produced PBP2x with unusually high levels of β-lactam resistance (17).
Table 1.
Kinetic parameters of PBP2x variants with cefotaxime, Pen-G, and thiolester substrate S2d
| k2/K cefotaxime | k2/K Pen-G | kcat/Km S2d | |
|---|---|---|---|
| S-PBP2x* | 209,000 | 99,000 | 2,500 |
| T338G | 26,000 | ND | 500 |
| T338A | 114,000 | 36,700 | 500 |
| T338V | 110,000 | 51,700 | 700 |
| T338P | 41,000 | ND | 180 |
| S571A | 59,000 | 21,000 | 370 |
| S571P | 1,300 | ND | 10 |
| T338A/S571A | 33,000 | 3,900 | 100 |
| T338A/S571P | 230 | ND | 2 |
| Y586F | 252,000 | 71,500 | 1,300 |
| R-PBP2x* | 3,300 | ND | 20 |
| R-PBP2x*/A338T | 18,600 | 3,800 | 150 |
The k2/K parameters (M−1·s−1) account for the acylation step efficiency and were measured by following the decrease of the intrinsic fluorescence of the protein. They reflect the accumulation of the acylenzyme (22). The kcat/Km parameters (M−1·s−1) were determined by standard steady-state methods (12). SDs were calculated only when more than four measurements were made. They are of the order of 10% for cefotaxime and S2d, and of 20% for Pen-G. R-PBP2x* and S-PBP2x* sequences have 37 amino acid differences in the transpeptidase domain. ND, not determined, too weak to be measured accurately.
Figure 3.

Comparison of cefotaxime acylation efficiencies of various PBP2x variants. R-PBP2x* and S-PBP2x* sequences have 37 amino acid differences in the transpeptidase domain.
The mutant S-PBP2x*S571A exhibited a reduced acylation efficiency by cefotaxime and Pen-G comparable to that of the T338A mutant (Table 1). As predicted from their linkage to a common water molecule, the effects of mutations T338A and S571A were not independent according to the terminology of Carter et al. (18) (Table 1). Among the clinically produced “resistant” sequences, one contains a Pro at position 571; the conversion of the Ser-571 residue in S-PBP2x* to Pro resulted in a 160-fold reduction in acylation efficiency by the cefotaxime (Table 1). This marked reduction in the rate of β-lactam acylation may be attributed to a combination of a reduced conformational freedom of strand β4 (8) and the partial or complete loss of the water molecule, WAT. The Y586F mutation, at a distance of 10.82 Å from Ser-337 (OH to Oγ), was neutral in terms of cefotaxime and Pen-G acylation. Only a high-resolution structure of a resistant PBP2x* could give a clear structure-based interpretation of the observed changes in acylation efficiencies. However, the loss of WAT might affect the position of strand β3 lining the active site cleft (8) and consequently alter the enzyme machinery.
The hydrolytic activities (kcat/Km) of the parent molecules, S-PBP2x* and R-PBP2x*, and the mutants were measured by spectrophotometry using the substrate analogues, carboxymethyl benzoylaminothioacetate (S2a) and S2d (16, 19). The kcat/Km values for both S2d and S2a correlated with the k2/K values for cefotaxime and Pen-G, and a good correlation between the cefotaxime acylation efficiency and S2d/S2a hydrolytic activity was shown by all PBP2x variants (Fig. 4). The correlation between in vitro enzyme activity and cefotaxime sensitivity suggests that β-lactam resistance is caused by an equivalent reduction in acylation efficiency by both antibiotics and substrate analogues.
Figure 4.

Correlation between cefotaxime acylation efficiency and hydrolytic activity using the thiolester substrate, S2d. Best fit: (k2/K) = −6,630.0 + 189.0 (kcat/Km), r = 0.94. RC, clinical resistant Sp328 (R-PBP2x*); RC-A338T, R-PBP2x* A338T mutant.
The laboratory-produced resistant PBP2xs differ from those found in clinically produced resistant strains. Selective pressure using a single antibiotic (cefotaxime) under laboratory conditions leads to a limited diversity of spontaneous point mutations, which are located mainly in the active site cleft and are liable to reduce the affinity for only the antibiotic used in their selection (20–23), whereas clinically produced resistant PBP2x exhibit multiple resistance to β-lactams and contain numerous amino acid substitutions. None of the laboratory mutants contained a mutated T338 residue, whereas most of the clinical isolates are altered at position 338, whose side chain is remote from the active site cleft.
We propose that mutation of T338 and/or S571 affects the reactivity of Ser-337 toward substrates and β-lactam antibiotics. Because most clinical isolates and sensitive strains are growing at the same rate the reduction of the transpeptidase activity has little effect on the metabolism of the bacteria, and biochemical changes in the peptidoglycan observed in some resistant isolates also appear to be independent of PBP alterations (24). Comparison of PBP2x and class A β-lactamase TEM1 structures reveals that three motifs are spatially conserved (8). One of these, SXXK, at the beginning of helix α2, contains the active site serine, S337 in PBP2x. The next residue is also a Thr in TEM1, but no water molecule is associated with it. Replacement of this Thr residue by all other amino acids (25) suggested that in TEM1 the Thr residue is not essential for substrate binding or catalysis but is important for protein stability, a conclusion that does not hold for PBP2x where activity is affected by mutations on Thr-338 but stability is conserved.
Generalization to Gram-Positive Bacteria.
The important role of the buried water molecule WAT is not unique to PBP2x. Interestingly, alignment of all available class B high molecular mass PBP (H-PBP) sequences shows that the amino acids related to WAT are conserved only in the H-PBP sequences whose genes belong to the cell division gene clusters (DCW) of Gram-positive bacteria: PBP2x (S. pneumoniae), PBPB and SP5D (Bacillus subtilis), PBPC (Enterococcus faecalis), PBP3s (E. hirae), and PBP1 (Staphylococcus aureus). Indeed, the amino acid side chains interacting with WAT are located in two conserved regions, YEPGST(338)XK, which includes the active serine, and S(571)-5X-P-3X-P-4X-Y(586) (nX indicating any n amino acids), downstream of the conserved motif K(547)(S/T)G. It is known that in E. coli PBP3 (whose gene is in a DCW cluster) plays a role in septum formation and cell division, and one can propose similar important functions for the PBPs in Gram-positive bacteria as well.
In streptococci, the introduction of the two mutations at positions 338 and 571 (e.g., T338A/S571P) into PBP2x are likely to result in a very low reactivity for any β-lactam antibiotic because on the basis of the present results we expect a 1,000-fold decrease in acylation efficiency; whereas, in other cocci, such as staphylococci and enterococci, in which genetic recombination is rare, resistance is acquired by the incorporation of an entire new gene coding for a PBP with low affinity for β-lactam antibiotics (26).
Acknowledgments
We thank Dr. E. Duée for helpful discussion. This work was supported by the French Ingénierie des Macromolécules Biologiques program, and the German Bundesmin sterium für Bildung und forschung Grant FKZ01K19703/2. This is publication no. 533 of the Institut de Biologie Structurale Jean-Pierre Ebel (Commissariat à l’Energie Atomique-Centre National de la Recherche Scientifique).
ABBREVIATIONS
- PBP
penicillin-binding proteins
- S-PBP2x*
water-soluble form of R6 PBP2x (residues 49–750)
- R-PBP2x*
water-soluble form of clinically produced resistant Sp 328 PBP2x (residues 49–750)
- S2d
N-benzoyl-d-alanylmercaptoacetic thiolester
- Pen-G
benzylpenicillin
References
- 1.Ghuysen J M. Trends Microbiol. 1994;2:372–380. doi: 10.1016/0966-842x(94)90614-9. [DOI] [PubMed] [Google Scholar]
- 2.Neu H C. Science. 1992;257:1064–1073. doi: 10.1126/science.257.5073.1064. [DOI] [PubMed] [Google Scholar]
- 3.Smith J M, Dowson C G, Spratt B G. Nature (London) 1991;349:29–31. doi: 10.1038/349029a0. [DOI] [PubMed] [Google Scholar]
- 4.Spratt B G. Science. 1994;264:388–393. doi: 10.1126/science.8153626. [DOI] [PubMed] [Google Scholar]
- 5.Hakenbeck R. Biochem Pharmacol. 1995;50:1121–1127. doi: 10.1016/0006-2952(95)00158-v. [DOI] [PubMed] [Google Scholar]
- 6.Kell C M, Sharma U K, Dowson C G, Town C, Balganesh T S, Spratt B G. FEMS Microbiol Lett. 1993;106:171–175. doi: 10.1111/j.1574-6968.1993.tb05954.x. [DOI] [PubMed] [Google Scholar]
- 7.Pucci M J, Thanassi J A, Discotto L F, Kessler R E, Dougherty T J. J Bacteriol. 1997;179:5632–5635. doi: 10.1128/jb.179.17.5632-5635.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parés S, Mouz N, Petillot Y, Hakenbeck R, Dideberg O. Nat Struct Biol. 1996;3:284–289. doi: 10.1038/nsb0396-284. [DOI] [PubMed] [Google Scholar]
- 9.Hakenbeck R, Konig A, Kern I, van der Linden M, Keck W, Billot-Klein D, Legrand R, Schoot B, Gutmann L. J Bacteriol. 1998;180:1831–1840. doi: 10.1128/jb.180.7.1831-1840.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laible G, Spratt B G, Hakenbeck R. Mol Microbiol. 1991;5:1993–2002. doi: 10.1111/j.1365-2958.1991.tb00821.x. [DOI] [PubMed] [Google Scholar]
- 11.Dowson C G, Coffey T J, Spratt B G. Trends Microbiol. 1994;2:361–366. doi: 10.1016/0966-842x(94)90612-2. [DOI] [PubMed] [Google Scholar]
- 12.Zhao G, Yeh W K, Carnahan R H, Flokowitsch J, Meier T I, Alborn W E, Jr, Becker G W, Jaskunas S R. J Bacteriol. 1997;179:4901–4908. doi: 10.1128/jb.179.15.4901-4908.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kunkel T A. Proc Natl Acad Sci USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanger F, Nicklen S, Coulson A R. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frére J M, Joris B. Crit Rev Microbiol. 1985;11:299–396. doi: 10.3109/10408418409105906. [DOI] [PubMed] [Google Scholar]
- 16.Jamin M, Damblon C, Millier S, Hakenbeck R, Frére J-M. Biochem J. 1993;292:735–741. doi: 10.1042/bj2920735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reichmann P, König A, Liñares J, Alcaide F, Tenover F C, McDougal L, Swidsinski S, Hakenbeck R. J Infect Dis. 1997;176:1001–1012. doi: 10.1086/516532. [DOI] [PubMed] [Google Scholar]
- 18.Carter P J, Winter G, Wilkinson A J, Fersht A R. Cell. 1984;38:835–840. doi: 10.1016/0092-8674(84)90278-2. [DOI] [PubMed] [Google Scholar]
- 19.Adam M, Damblon C, Jamin M, Zorzi W, Dusart V, Galleni M, el Kharroubi A, Piras G, Spratt B G, Keck W, et al. Biochem J. 1991;279:601–604. doi: 10.1042/bj2790601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grebe T, Hakenbeck R. Antimicrob Agents Chemother. 1996;40:829–834. doi: 10.1128/aac.40.4.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laible G, Hakenbeck R. J Bacteriol. 1991;173:6986–6990. doi: 10.1128/jb.173.21.6986-6990.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jamin M, Hakenbeck R, Frére J-M. FEBS Lett. 1993;331:101–104. doi: 10.1016/0014-5793(93)80305-e. [DOI] [PubMed] [Google Scholar]
- 23.Sifaoui F, Kitzis M-D, Gutmann L. Antimicrob Agents Chemother. 1996;40:152–156. doi: 10.1128/aac.40.1.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Severin A, Figueiredo A M, Tomasz A. J Bacteriol. 1996;178:1788–1792. doi: 10.1128/jb.178.7.1788-1792.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schultz S C, Richards J H. Proc Natl Acad Sci USA. 1986;83:1588–1592. doi: 10.1073/pnas.83.6.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomasz A. Trends Microbiol. 1994;2:380–385. doi: 10.1016/0966-842x(94)90615-7. [DOI] [PubMed] [Google Scholar]
- 27.Nicholls A, Charp K A, Honing B. Proteins Struc Funct Genet. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]