

Abstract
The pathognomonic plaques of Alzheimer’s disease are composed primarily of the 39- to 43-aa β-amyloid (Aβ) peptide. Crosslinking of Aβ peptides by tissue transglutaminase (tTg) indicates that Gln15 of one peptide is proximate to Lys16 of another in aggregated Aβ. Here we report how the fibril structure is resolved by mapping interstrand distances in this core region of the Aβ peptide chain with solid-state NMR. Isotopic substitution provides the source points for measuring distances in aggregated Aβ. Peptides containing a single carbonyl 13C label at Gln15, Lys16, Leu17, or Val18 were synthesized and evaluated by NMR dipolar recoupling methods for the measurement of interpeptide distances to a resolution of 0.2 Å. Analysis of these data establish that this central core of Aβ consists of a parallel β-sheet structure in which identical residues on adjacent chains are aligned directly, i.e., in register. Our data, in conjunction with existing structural data, establish that the Aβ fibril is a hydrogen-bonded, parallel β-sheet defining the long axis of the Aβ fibril propagation.
The primary component of the amyloid plaques of Alzheimer’s disease is the 39- to 43-aa peptide β-amyloid (Aβ), a proteolytic product of the β-amyloid precursor protein (1–4). Mutations in Aβ and the presenilin have been found to increase the levels of both soluble Aβ peptides and of amyloid plaques and are associated with an earlier onset and more severe phenotype of Alzheimer’s disease (5–10). Furthermore, the number of amyloid plaques correlates directly with the severity of the disease (11). It has been shown, by electron microscopy and x-ray scattering, that Alzheimer’s amyloid plaques consist of a highly ordered, fibrillar structure which can be replicated in vitro by using synthetic peptides (12–20). The basic secondary structure of these fibrils is considered to be a β-strand (12–20), and the overall structure is often referred to as a “cross-β fibril.” However, the precise arrangement of the individual peptides in these fibrils has been elusive. The term “cross-β fibril” is an historical one and has been applied to Aβ on the basis of data obtained from low-resolution x-ray diffraction (21–23). Although such data are compatible with either a parallel or antiparallel conformation, the term “cross-β” often has been assumed to betoken an antiparallel conformation, and all of the prevailing models for β-amyloid fibrils incorporate this assumption to some degree (24–31). The experimental support for this contention, however, is limited to infrared spectroscopy, which does not yield high precision data and which cannot, furthermore, distinguish unambiguously between parallel and antiparallel β-conformations.
The elucidation of the structure of Aβ fibrils would be an important step toward understanding the pathogenesis of Alzheimer’s disease and ultimately toward understanding the propagating structure of the many fibrillar proteins involved in amyloid diseases. Efforts toward these goals have been hindered by the insoluble, noncrystalline nature of Aβ fibrils, which renders them unsuitable for high resolution techniques of protein structure determination such as solution-phase NMR and x-ray crystallography. In this paper, we use solid-state NMR to demonstrate that the core structure of Aβ contains β-strands in a parallel arrangement. Using this approach, we have determined the specific conformation and alignment of the core domain of Aβ peptide, which is critical for self-association and fibril formation (32–39).
EXPERIMENTAL PROCEDURES
Peptide Synthesis.
1-13C-l-Lys, 1-13C-l-Gln, 1-13C-l-Leu, 1-13C-l-Phe, and 1-13C-l-Val were obtained from Cambridge Isotope Laboratories (Cambridge, MA). Isotopic purities, from individual lot numbers, were 99.2% for 1-13C-l-Lys; 97.4% for 1-13C-l-Gln; and 99.0% for 1-13C-l-Leu, 1-13C-l-Phe, and 1-13C-l-Val. Protection of the 13C-labeled amino acids was performed by Midwest Biotech (Indianapolis). Aβ(10–35) with an amidated carboxy terminus was synthesized by using standard 9-fluoromethoxycarbonyl chemistry on an Applied Biosystems model 431A peptide synthesizer. Peptides were purified as described (40). Molecular mass was verified by matrix-assisted laser desorption ionization–time-of-flight mass spectroscopy. Peptide purity was >96% by RP-HPLC.
Formation of Aβ Fibrils for Solid-State NMR Studies.
Aβ(10–35) in 100% trifuoroacetic acid was precipitated into diethyl ether at 0°C, and residual trifuoroacetic acid was extracted with diethyl ether. Peptide (50 mg) was dissolved in distilled water to ≈0.6 mg/ml (≈0.2 mM); exact concentrations were determined by amino acid analysis. The pH of this solution was 2.9 because of the presence of traces of residual trifuoroacetic acid; at this point, there was no precipitate by centrifugation at 17,000 × g, and no fibrils or amorphous precipitate were seen by electron microscopy. The pH was adjusted to 7.40 by addition of 2-μl aliquots of 0.1 M NaOH every 2 min. The solution then was swirled gently at 0.5 rotations/s. The course of the reaction was monitored by disappearance of monomeric peptide peak on analytical RP-HPLC. At the end of this procedure, a precipitate had formed, which was entirely fibrillar by electron microscopy; that is, no amorphous precipitate was visible. Furthermore, as published elsewhere (41), the fibrils could be stained by Congo Red stain, and when viewed under doubly polarized light, they displayed classical apple-green birefringence. The suspension of fibrils then was flash-frozen, lyophilized, and stored at −20°C for solid-state NMR studies. As control experiments, we tested several other procedures for sample preparation, notably the use of flash-freezing at −80°C or −196°C, with or without subsequent lyophilization. In no case was any difference in the NMR spectra observed to result from these variations in sample preparation, so long as the pH was 7.40.
Solid-State NMR.
Cross-polarization/magic-angle spinning 13C NMR experiments were performed on a Bruker Avance DSX spectrometer (Bruker, Bellerica, MA) tuned to a frequency of 50.3 MHz. The dipolar recoupling in a windowless sequence (DRAWS) pulse sequence was implemented (42). Spectra were acquired with 272 scans for the 50-mg, 13C-labeled samples (1-13C-Gln15-Aβ, 1-13C-Leu17-Aβ, and 1-13C-Val18-Aβ) and 600 scans for unlabeled or 15-mg samples (1-13C-Lys16-Aβ). Therefore, to acquire all of the points for a single DRAWS curve, with a 5-fold repetition of points, typically took 5–21 h. All samples contained a small amount of hexamethylbenzene (8–12 mg) as an internal control. Samples were spun at 4,525 (±3) Hz. The 13C radio frequency power level was set to 38.5 kHz. The 1H decoupling level was 120 kHz. Spectra from DRAWS experiments were processed and analyzed as described (42).
Simulated data were created by numerical calculation by using a density matrix approach (58). The input parameters to the numerical calculation program included the chemical shift tensor elements for the spin-1/2 nuclei, the dipolar coupling strengths, Euler angles that rotate the chemical shift tensors from the molecular frame to their respective principle axis systems, an initial density matrix ρ(0), an observable, and any relevant relaxation parameters. In this work, the Euler angles were set to zero because it was determined that they had a negligible effect on the simulated curves. The chemical shift tensor parameters were taken from the data of Ye et al. (44). The initial density matrix and the observable are given by either
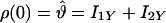 |
or
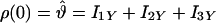 |
for two spin and three spin systems, respectively. This was propagated under the DRAWS Hamiltonian (44). The calculated “signal” was found by taking the trace
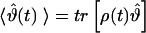 |
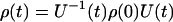 |
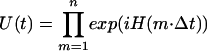 |
and
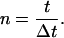 |
The time increment was typically 2–3 ms. The program also performed a powder average of 2,000 randomly selected crystallite orientations.
Relaxation effects were modeled by multiplying the single-quantum density matrix elements by an exponential factor at the end of each time increment. Data from the unlabeled sample were used to determine the appropriate single-quantum relaxation time constant. Moreover, this decay rate was reproduced in several other samples, including unfibrilized Aβ containing single and multiple 13C labels. Aβ fibril samples containing a single 13C label diluted to 10% in unlabeled material also showed in a decay rate similar to that measured for unlabeled Aβ samples.
In other work, we performed several additional experiments, confirming that modeling of other relaxation processes as an additional, single-exponential decay was reasonable. First, DRAWS was performed on 13C-labeled model compounds (succinic and adipic acid) with carbonyl–carbonyl carbon distances of 3.8 Å and 6.3 Å, respectively. Accordingly, measured DRAWS distances were found to be 3.8 + 0.1 Å and 6.0 + 0.5 Å, validating the accuracy of the measurements over distances >4 Å. Furthermore, only single-quantum relaxation effects were taken into account in the calculations. Results from numerical simulations showed that effects of zero-quantum relaxation were negligible on the DRAWS results. On the other hand, simulations showed that double-quantum relaxation can have a measurable effect on the results. However, for unprotonated carbonyl carbons, simulations were found to fit the data better when effects from double-quantum relaxation were ignored; this included distances from 3.4 to 6.0 Å measured for the Aβ fibrils. Therefore, double-quantum relaxation appeared to have a negligible effect on our experiments.
Given the inherent resolution of solid-state NMR spectra, virtually all β-sheet carbonyl chemical shifts are superimposible at δ ≈ 171 ppm (45). It was necessary, therefore, to subtract the natural abundance signal derived from other amino acid residues. For ST equaling total experimentally observed signal, one obtains
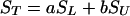 |
where SL equals normalized signal attributable to the labeled carbonyl carbon, and SU equals normalized aggregated signal caused by natural abundance of 13C at all 25 other carbonyl carbon atoms, and a + b = 1. This treatment assumes that the 13C abundance at the labeled position is 100% and is 1.1% for each of the other 25 amino acids in the peptide. Thus, a = 0.78 and
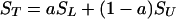 |
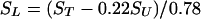 |
Structural Modeling.
The distances tabulated in Fig. 2 are for crystallgraphic structures of 1BNH (46) and 2DLH (47). Individual distances for β-sheets varied by as much as 0.5 Å for a given position, especially when approaching a turn or the end of a sheet; however, in each of the structures examined, the mean measurement for a given position fell within the standard error for the structures represented in the table. The DRAWS experiment gives a single result, representing the average distance for that contact in the molecule; hence, standard error and not standard deviation is presented in Fig. 2.
Figure 2.

Modeling of β-strands indicating that orientation and alignment of Aβ can be determined by peptides containing single 1-13C labels in Gln15 or Lys16. (A) Top pair: the chemical and schematic views of a single alignment of two parallel β strands. The “pleat” of the β-pleated sheet is denoted by the offsets of the amino acids in the schematic view (right). For this single alignment, there are two possible offsets of the carbonyls, i.e., one where the carbonyls representing Gln15 (pink arrows) point out of the β-sheet (top pair) and one where the carbonyls of Gln15 point into the β-sheet (bottom pair). Interstrand carbonyl–carbonyl distances for parallel and antiparallel β-sheets were measured for >50 structures from the Brookhaven National Laboratory Protein Data Base, including isolated pairs of β-strands, sheets, and barrels. Two representative structures were chosen (46, 47). Shown in B are the measurements obtained for one antiparallel structure, in Å ± SE (n = 23–29), correlated with possible interstrand alignments of singly 1-13C labeled (Gln15 or Lys16) Aβ peptides. In C are the measurements obtained for one parallel structure, in Å ± SE (n = 34–38), correlated with possible interstrand alignments of singly 1-13C labeled (Gln15 or Lys16) Aβ peptides. The DRAWS experiment has a sensitivity limit of 6 Å and a precision of 0.1–0.2 Å; hence, it was clear at the outset of the experiment that if the Gln15 was indeed “near” the Lys16 of an adjacent peptide, combined distance data for the 1-13C-Gln15 and 1-13C-Lys16 peptides could be used to predict the form (parallel or antiparallel) and alignment of the peptides as they self-associate in the fibrils.
RESULTS AND DISCUSSION
In recent years, solid-state NMR methods have emerged that are capable of determining the local structures of biomolecules. These methods have allowed the measurement of internuclear distances between specific labels to an accuracy of ±0.2 Å (42, 48–65). Until now, solid-state NMR had not been used successfully to determine the structures of complex, self-associating proteins such as the amyloids. This lack of success is caused largely by two particular barriers. First, the experiments require the incorporation of specific nuclear labels at discrete positions; the decision of where to place these labels, in the context of a large molecule, is nontrivial. Thus, most experiments have been limited to small peptides or oligonucleotide sequences, often those with known crystallographic structures. Second, the usual approach is to place two specific labels within a single molecule and measure the distance between them. In the study of self-associating amyloids and prion proteins, however, untangling intramolecular interactions in the presence of stronger intermolecular interactions has proven to be complex, and the resulting structural data have been limited to the tentative assignment of conformational structure (43). Thus, an approach that examines only intermolecular associations is highly desirable.
The decision of where to place these labels was aided by studies of tissue transglutaminase (tTg) crosslinking of Aβ (66–70). This enzyme specifically crosslinks Gln15 and Lys16 of Aβ but never involves Lys28 (70), suggesting that Gln15 and Lys16 are proximate in an aggregated form of Aβ. For our experiments, we synthesized a 26-aa peptide, comprising residues 10–35 of the Aβ peptide (Aβ(10–35), Fig. 1A). For Aβ(10–35), the reaction of tTg was self-limiting, as only products up to the size of a hexamer were observed (Fig. 1B). This specificity of the tTg reaction suggested initial sites of Gln15 and Lys16 for the placement of 13C labels in Aβ(10–35) for the NMR experiments.
Figure 1.

Crosslinking by tTg suggests design of NMR experiments. (A) Amino acid sequence of Aβ(10–35). Peptides containing a 13C label at the carbonyl carbon of either Gln15 or Lys16 (underlined) were synthesized for use in solid-state NMR experiments. (B) Unlabeled Aβ(10–35) was crosslinked by tTg (69, 70) and was analyzed on Tris⋅Tricine SDS/PAGE (83). Reaction mixtures contained 1 mg/ml Aβ(10–35) peptide, 0.04–0.20 mg/ml tTg (Sigma), and 0.2–0.4 mM CaCl2 in 40 mM Tris (pH 8.6). After 4 h at 23°C, the experiments were stopped by addition of excess EDTA. Crosslinked peptide species ranged from dimeric to hexameric. The proportion of higher molecular weight species depended on the amount of tTg included; no higher order oligomers were observed. Specific crosslinking of Gln15 of one peptide to Lys16 of another, in a regular, ordered fashion, suggested placing 13C labels at these positions.
For structural studies, a biologically relevant peptide was required, which could be prepared in a homogeneous fibrillar form. Our selection of Aβ(10–35) was based on the following considerations: (i) Model peptide studies have demonstrated that truncated peptides form fibrils (12–20); (ii) neurotoxicity is linked with a peptide’s ability to self-associate into filamentous β-strand aggregates (71–76); (iii) Aβ(10–35) incorporates the core region, point mutations of which significantly obstruct fibril formation and have been used to generate inhibitors of fibrillogenesis (33–39); (iv) Aβ(10–35) retains the ability to add to bona fide Alzheimer’s plaques, in contrast to other truncated peptides (77–81), and forms fibrils morphologically similar to those of the full length peptide; (v) and of most importance, the full length peptide, Aβ(1–42), is intractable for the controlled formation of fibrils from aqueous media because at the earliest time points, some of the peptide exists as an amorphous precipitate. In contrast, the use of Aβ(10–35) allowed the reproducible and controlled formation of fibrils from aqueous solutions, under defined conditions of pH, ionic strength, and peptide concentration and thus yielded the required homogeneous fibrils.
We implemented the DRAWS pulse sequence to measure distances between 13C nuclei (42) in fibrillar peptides containing a single label in one amino acid. The DRAWS pulse sequence selectively restores the dipolar coupling interaction that is eliminated as a result of magic-angle spinning. Only interactions between proximal 13C labels in aggregated Aβ fibrils were observed as an accelerated rate of decay of the NMR signal with increasing DRAWS mixing time. Moreover, one of the distinct advantages of this method is its ability to measure distances between two sites containing an identical 13C-label with degenerate chemical shifts, e.g., two 13C carbonyl labels of a single amino acid. Because of the sensitivity of the inverse cube relationship of dipolar coupling to internuclear separation, the DRAWS approach allows precise measurement of 13C-13C internuclear separations, up to a maximum distance of ≈6 Å.
To analyze the distance measurements obtained in the DRAWS experiments, we obtained interstrand carbonyl–carbonyl distances for parallel and antiparallel alignments of β-strand peptide conformations based on data from structures in the Brookhaven National Laboratory Protein Data Bank, and these distances then were superimposed on sliding alignment models of Aβ peptides (Fig. 2). For peptides containing a single 13C backbone carbonyl, the predicted distances for different interstrand alignments indicated that, if the Gln15 of one peptide was indeed proximate to the Lys16 of an adjacent peptide, then a clear solution to the structure of this domain could be obtained given the precision of the NMR measurements. Furthermore, by labeling only one position per peptide, only intermolecular interactions between nuclei were measured.
Three sets of fibrils were prepared initially: 1-13C-Gln15-Aβ, 1-13C-Lys16-Aβ, and unlabeled-Aβ. To obtain distances between labels, DRAWS experiments were performed on the fibrils (Fig. 3). The signal of the labeled carbonyl peak (δ ≈ 171 ppm) was integrated for each mixing time, and the normalized signal intensity was plotted as a function of mixing time for both the labeled and unlabeled samples. The plots then were compared with theoretical curves obtained from numerical simulations. As shown in Fig. 3, distances measured for the 1-13C-Gln15-Aβ and 1-13C-Lys16-Aβ samples were 5.1 Å (±0.2) and 4.9 Å (±0.2), respectively. These distances were obtained for both repeated measurements on the same sample and for repeated sample preparations at pH 7.4, including control experiments as summarized briefly in Experimental Procedures. These aspects of the experiments are discussed elsewhere (82).
Figure 3.

DRAWS data demonstrating a 5-Å, multiple-contact model for both 1-13C-Gln15-Aβ and 1-13C-Lys16-Aβ fibrils. (A) Conventional cross polarization/magic angle spinning experiment (DRAWS mixing time = 0) for 50 mg of 1-13C-Gln15-Aβ (δ = 171 ppm) mixed with 10 mg of unlabeled hexamethylbenzene (internal control). (B) For each peptide, the DRAWS experiment was performed by using a series of mixing times from 0 to 22 ms. At each mixing time, the carbonyl peak was integrated and normalized to the first data point (mixing time = 0) to allow comparison between samples. Data also were adjusted for natural abundance signal as described in Experimental Procedures. Values shown are the mean ± 1 SD for 50 mg of 1-13C-Gln15-Aβ lyophilized fibrils (red squares, n = 5), 15 mg of 1-13C-Lys16-Aβ (blue circles, n = 6), and 50 mg of unlabeled fibrils (black triangles, n = 5) compared with numerical simulations of no interaction (dotted line), a 5.2-Å interaction (long dashed line), a 5.0-Å interaction (solid line), or a 4.8-Å interaction (short dashed line). Error bars for experimental data are shown only when they exceed the symbol size. C shows the same data for 1-13C-Gln15-Aβ plotted against simulations for two 13C labels interacting in isolation (i.e., a single, two-spin contact) at 5.0 Å (solid line), 4.8 Å (short dashed line), or 4.6 Å (dot-dash line). D shows the 1-13C-Gln15-Aβ data plotted against simulations for three or more 13C labels interacting simultaneously (multiple spin, i.e., each 13C label has at least two contacts) at 5.2 Å (long dashed line), 5.0 Å (solid line), or 4.8 Å (short dashed line). At mixing times beyond 16 ms, the experimental data clearly fit a multiple-interaction model (D) better than an isolated-interaction model (C).
Close analysis of the DRAWS results revealed yet another structural constraint. The contacts under observation did not fit a simple “two-spin” numerical simulation. As shown in Fig. 3C, at mixing times of 16 ms and greater, the experimental data deviate from a simulation designed to examine a pair of 13C carbonyls interacting in isolation. However, if the simulations are adjusted to model a condition under which every 13C carbonyl interacts with two, equidistant 13C carbonyls (multiple spin model, Fig. 3D), the experimental data conform more closely to the simulation curves. Additional simulated DRAWS curves using a model of four spins resulted in the same measured distance. Thus, it is reasonable to assume that any model in which each spin is coupled to several other spins will give similar results.
This multiplicity constraint had an important bearing on the structural modeling which followed. As indicated by the data in Fig. 2 and illustrated in Fig. 4, distance constraints of 5 Å for both Gln15-Gln15 and Lys16-Lys16 could fit either a parallel or an antiparallel β-strand structure. In the parallel structure, every 13C carbonyl interacts with two, equidistant 13C carbonyls (“multiple spins”; Fig. 4A). In the antiparallel structure, however, each 13C carbonyl interacts with only one other 13C carbonyl at a distance of 4.8 Å; the second 13C carbonyl interaction of 8.1 Å would not be detectable in the DRAWS experiment. Hence, in the antiparallel model, each 13C interacts effectively with one spin in isolation (two spins; Fig. 4B). Thus, from the DRAWS experiments, there were two 5-Å distance constraints and a multiplicity constraint, which made the parallel, directly aligned structure (Fig. 4A) the clearly preferable solution.
Figure 4.

Further DRAWS experiments confirm parallel, directly aligned (“in register”) orientation of Aβ. Contacts (5 Å) for both Gln15 (pink) and Lys16 (blue) are consistent with either a parallel (A) or antiparallel (B) β-strand arrangement. In A, each 13C-labeled carbonyl has multiple interactions whereas in B, each 13C-labeled carbonyl has only paired interactions. The solid lines are contacts of ≈5 Å, and the dotted lines (B) are >8 Å, which would not be detectable in DRAWS experiments. Thus, the preferable fit of the data shown in Fig. 3D is consistent only with the parallel arrangement and not with the antiparallel arrangement. To validate this proposal, peptides containing 1-13C at Leu17 and Val18 were synthesized. A parallel arrangement (A) predicts 5-Å contacts for both peptides. An antiparallel arrangement (B) predicts that no measurable interaction at either Leu17 or Val18 would be found. (C) DRAWS experiments for 1-13C-Leu17-Aβ demonstrate a 5.1-Å (± 0.2) interaction at this position. Data shown are mean (± SD of mean) for two 50-mg samples, each run five times. (D) DRAWS experiments for 1-13C-Val18-Aβ demonstrate a 5.0-Å (± 0.2) interaction. Data shown are for one 50 mg-sample, run five times per data point. For both C and D, error bars are shown only when they exceed the size of the data point. Simulations shown are for multiple interactions (i.e., multiple spins) at 5.2-Å (long dashed line), 5.0-Å (solid line), or 4.8-Å (short dashed line).
To test the validity of this structure, two additional peptides were synthesized: 1-13C-Leu17-Aβ and 1-13C-Val18-Aβ. For both peptides, the parallel, directly aligned structure (Fig. 4A) predicts distances of 5 Å. The antiparallel structure (Fig. 4B) predicts distances of ≈8 Å and ≈13 Å for 1-13C-Leu17-Aβ and 1-13C-Val18-Aβ contacts, respectively. The increased interstrand distances predicted for Leu17 and Val18 in the antiparallel structure follow from the fact that, for antiparallel β-sheets, the internuclear distances increase continuously as the antiparallel chains proceed from the point of Lys15-Gln16 proximity. As can be seen in Fig. 4 C and D, DRAWS curves for 1-13C-Leu17-Aβ and 1-13C-Val18-Aβ demonstrate 5.1 Å (± 0.2) and 5.0 Å (± 0.2), respectively, which is consistent with only the parallel, directly aligned structure for the core of the Aβ fibril (Fig. 4A).
Our data, establishing that the Aβ peptide stacks as a directly aligned parallel β-sheet, now can explain important previous observations. x-ray diffraction studies have identified 5- and 10-Å reflections of Aβ fibrils. The intensity of the 5-Å reflection was attributed to the repeating unit occurring along the long axis of the fibril (32–39), and our data now can account for this 5-Å reflection by positing the parallel β-sheet oriented with H-bonds along the fibril axis. The weaker 10-Å reflection then would be attributable to the lamination of multiple β-sheets through side chain interactions, allowing for lateral thickening of the fibril. Therefore, our data provide an atomic resolution structure for an Aβ fibril, and the outlined approach is completely generalizable, providing a strategy for characterizing the fibrillar structures critical to amyloid diseases.
Acknowledgments
We are grateful to H. A. Fozzard, J. Fried, and G. S. Getz for valuable advice in preparing the manuscript. We acknowledge support from the Argonne National Laboratory (to R.E.B., D.G.L., and S.C.M.), the Division of Chemical Sciences, Office of Basic Energy Sciences, Department of Energy (contract W-31-109-ENG-38, to R.E.B.), the National Institutes of Health (Grant R21 RR 12723 to D.G.L., Grant 5 T32 HL07327 to T.S.B. and Grant 5 T32 GM07281 to T.L.S.B.), the American Federation for Aging Research (to T.L.S.B. and S.C.M.), and the American Foundation for Aging Research (to T.L.S.B.).
ABBREVIATIONS
- Aβ
beta amyloid
- tTg
tissue transglutaminase
- DRAWS
dipolar recoupling in a windowless sequence
References
- 1.Glenner G G, Wong C W. Biochem Biophys Res Commun. 1984;122:1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 2.Masters C L, Simms G, Weinman N A, Multhaup G, McDonald B L, Beyreuther K. Proc Natl Acad Sci USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang J, Lemaire H G, Unterbeck A, Salbaum J M, Masters C L, Grzeschik K H, Multhaup G, Beyreuther K, Muller-Hill B. Nature (London) 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 4.Prelli F, Castaño E, Glenner G G, Frangione B. J Neurochem. 1988;51:648–651. doi: 10.1111/j.1471-4159.1988.tb01087.x. [DOI] [PubMed] [Google Scholar]
- 5.Levy E, Carman M D, Fernandez-Madrid I J, Power M D, Lieberburg I, van Duinen S G, Bots G T, Luyendijk W, Frangione B. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 6.Goate A, Chartier-Harlin M C, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L. Nature (London) 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 7.Murrell J, Farlow M, Ghetti B, Benson M D. Science. 1991;254:97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- 8.Schellenberg G D. Proc Natl Acad Sci USA. 1995;92:8552–8559. doi: 10.1073/pnas.92.19.8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selkoe D J. J Biol Chem. 1996;271:18295–18298. doi: 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- 10.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird T D, Hardy J, Hutton M, Kukull W, et al. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 11.Selkoe D J. J Neuropathol Exp Neurol. 1994;53:438–447. doi: 10.1097/00005072-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 12.Castaño E M, Ghiso J, Prelli F, Gorevic P D, Migheli A, Frangione B. Biochem Biophys Res Commun. 1986;141:782–789. doi: 10.1016/s0006-291x(86)80241-8. [DOI] [PubMed] [Google Scholar]
- 13.Gorevic P D, Castaño E M, Sarma R, Frangione B. Biochem Biophys Res Commun. 1987;147:854–862. doi: 10.1016/0006-291x(87)91008-4. [DOI] [PubMed] [Google Scholar]
- 14.Halverson K, Fraser P E, Kirschner D A, Lansbury P T., Jr Biochemistry. 1990;29:2639–2644. doi: 10.1021/bi00463a003. [DOI] [PubMed] [Google Scholar]
- 15.Fraser P E, Duffy L K, O’Malley M B, Nguyen J, Inouye H, Kirschner D A. J Neurosci Res. 1991;28:474–485. doi: 10.1002/jnr.490280404. [DOI] [PubMed] [Google Scholar]
- 16.Fraser P E, Nguyen J T, Surewicz W, Kirschner D A. Biophys J. 1991;60:1190–1201. doi: 10.1016/S0006-3495(91)82154-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraser P E, Nguyen J T, Inouye H, Surewicz W K, Selkoe D J, Podlisny M B, Kirschner D A. Biochemistry. 1992;31:10716–10723. doi: 10.1021/bi00159a011. [DOI] [PubMed] [Google Scholar]
- 18.Barrow C J, Zagorski M G. Science. 1991;253:179–182. doi: 10.1126/science.1853202. [DOI] [PubMed] [Google Scholar]
- 19.Barrow C J, Yasuda A, Kenny P T, Zagorski M G. J Mol Biol. 1992;225:1075–1093. doi: 10.1016/0022-2836(92)90106-t. [DOI] [PubMed] [Google Scholar]
- 20.Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman C, Glabe C. J Biol Chem. 1992;267:546–554. [PubMed] [Google Scholar]
- 21.Pauling L, Corey R. Proc Natl Acad Sci USA. 1951;37:729–740. doi: 10.1073/pnas.37.11.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonar L, Cohen A S, Skinner M. Proc Soc Exp Biol Med. 1967;131:1373–1375. doi: 10.3181/00379727-131-34110. [DOI] [PubMed] [Google Scholar]
- 23.Eanes E D, Glenner G G. J Histochem Cytochem. 1968;16:673–677. doi: 10.1177/16.11.673. [DOI] [PubMed] [Google Scholar]
- 24.Fabian H, Naumann D, Otbos L, Jr, Schulz C, Backmann J, Szendrei G I, Hahn U, Saenger W, Mantsch H H. J Mol Struct. 1995;348:5–8. [Google Scholar]
- 25.Kelly J W. Structure. 1997;5:595–600. doi: 10.1016/s0969-2126(97)00215-3. [DOI] [PubMed] [Google Scholar]
- 26.Kay C J. FEBS Lett. 1997;403:230–235. doi: 10.1016/s0014-5793(97)00076-8. [DOI] [PubMed] [Google Scholar]
- 27.Lazo N D, Downing D T. Biochemistry. 1998;37:1732. doi: 10.1021/bi971016d. [DOI] [PubMed] [Google Scholar]
- 28.Halverson K, Fraser P E, Kirschner D A, Lansbury P T., Jr Biochemistry. 1990;29:2639–2644. doi: 10.1021/bi00463a003. [DOI] [PubMed] [Google Scholar]
- 29.Choo L P, Wetzel D L, Halliday W C, Jackson M, LeVine S M, Mantsch H H. Biophys J. 1996;71:1672–1679. doi: 10.1016/S0006-3495(96)79411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hilbich C, Kisters-Woike B, Reed J, Masters C L, Beyreuther K. J Mol Biol. 1991;218:149–163. doi: 10.1016/0022-2836(91)90881-6. [DOI] [PubMed] [Google Scholar]
- 31.Blake C, Serpell L. Structure. 1996;4:989–998. doi: 10.1016/s0969-2126(96)00104-9. [DOI] [PubMed] [Google Scholar]
- 32.Kirschner D A, Inouye H, Duffy L K, Sinclair A, Lind M, Selkoe D J. Proc Natl Acad Sci USA. 1987;84:6953–6957. doi: 10.1073/pnas.84.19.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hilbich C, Kisters-Woike B, Reed J, Masters C L, Beyreuther K. J Mol Biol. 1992;228:460–473. doi: 10.1016/0022-2836(92)90835-8. [DOI] [PubMed] [Google Scholar]
- 34.Fraser P E, McLachlan D R, Surewicz W K, Mizzen C A, Snow A D, Nguyen J T, Kirschner D A. J Mol Biol. 1994;244:64–73. doi: 10.1006/jmbi.1994.1704. [DOI] [PubMed] [Google Scholar]
- 35.Boland K, Manias K, Perlmutter D H. J Biol Chem. 1995;270:28022–28028. doi: 10.1074/jbc.270.47.28022. [DOI] [PubMed] [Google Scholar]
- 36.Soto C, Castaño E M, Frangione B, Inestrosa N C. J Biol Chem. 1995;270:3063–3067. doi: 10.1074/jbc.270.7.3063. [DOI] [PubMed] [Google Scholar]
- 37.Esler W P, Stimson E R, Ghilardi J R, Lu Y A, Felix A M, Vinters H V, Mantyh P W, Lee J P, Maggio J E. Biochemistry. 1996;35:13914–13921. doi: 10.1021/bi961302+. [DOI] [PubMed] [Google Scholar]
- 38.Wood S J, Wetzel R, Martin J D, Hurle M R. Biochemistry. 1995;34:724–728. doi: 10.1021/bi00003a003. [DOI] [PubMed] [Google Scholar]
- 39.Maggio J E, Mantyh P W. Brain Pathol. 1996;6:147–162. doi: 10.1111/j.1750-3639.1996.tb00797.x. [DOI] [PubMed] [Google Scholar]
- 40.Braddock D T, Mercurius K O, Subramanian R M, Dominguez S R, Davies P F, Meredith S C. Biochemistry. 1996;35:13975–13984. doi: 10.1021/bi960006u. [DOI] [PubMed] [Google Scholar]
- 41.Burkoth T S, Benzinger T L S, Jones D N M, Hallenga K, Meredith S C, Lynn D G. J Am Chem Soc. 1998;120:7655–7656. [Google Scholar]
- 42.Gregory D M, Mehta M A, Shiels J C, Drobny G P. J Chem Phys. 1997;107:28–42. [Google Scholar]
- 43.Lansbury P T, Jr, Costa P R, Griffiths J M, Simon E J, Auger M, Halverson K J, Kocisko D A, Hendsch Z S, Ashburn T T, Spencer R G. Nat Struct Biol. 1995;2:990–998. doi: 10.1038/nsb1195-990. [DOI] [PubMed] [Google Scholar]
- 44.Ye C, Fu R, Hu J, Hou L, Ding S. Magn Reson Chem. 1993;31:699–704. [Google Scholar]
- 45.Saitô H. Magn Reson Chem. 1986;24:835–852. [Google Scholar]
- 46.Kobe B, Deisenhofer J. J Mol Biol. 1993;231:137–140. doi: 10.1006/jmbi.1993.1263. [DOI] [PubMed] [Google Scholar]
- 47.Stern L J, Brown J H, Jardetzky T S, Gorga J C, Urban R G, Strominger J L, Wiley D C. Nature (London) 1994;368:215–221. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 48.Gullion T, Schaefer J. Adv Magn Reson. 1989;13:57–83. [Google Scholar]
- 49.Levitt M H, Raleigh D P, Creuzet F, Griffin R G. J Chem Phys. 1990;92:6347–6364. [Google Scholar]
- 50.Tycko R, Dabbagh G. J Am Chem Soc. 1991;113:9444–9448. [Google Scholar]
- 51.Griffiths J M, Griffin R G. Anal Chim Acta. 1993;283:1081–1101. [Google Scholar]
- 52.Griffiths J M, Lakshmi K V, Bennett A E, Raap J, van der Wielen C M, Lugtenburg J, Herzfeld J, Griffin R G. J Am Chem Soc. 1994;116:10178–10181. [Google Scholar]
- 53.McDermott A E, Creuzet F, Gebhard R, van der Hoef K, Levitt M H, Herzfeld J, Lugtenburg J, Griffin R G. Biochemistry. 1994;33:6129–6136. doi: 10.1021/bi00186a012. [DOI] [PubMed] [Google Scholar]
- 54.Smith S O, Jonas R, Braiman M, Bormann B J. Biochemistry. 1994;33:6334–6341. doi: 10.1021/bi00186a037. [DOI] [PubMed] [Google Scholar]
- 55.McDowell L M, Klug C A, Beusen D D, Schaefer J. Biochemistry. 1996;35:5395–5403. doi: 10.1021/bi9529059. [DOI] [PubMed] [Google Scholar]
- 56.McDowell L M, Schaefer J. Curr Opin Struct Biol. 1996;6:624–629. doi: 10.1016/s0959-440x(96)80028-5. [DOI] [PubMed] [Google Scholar]
- 57.McDowell L M, Schmidt A, Cohen E R, Studelska D R, Schaefer J. J Mol Biol. 1996;256:160–171. doi: 10.1006/jmbi.1996.0074. [DOI] [PubMed] [Google Scholar]
- 58.Mehta M A, Gregry D M, Kiihne S, Mitchell D J, Hatcher M E, Shiels J C, Drobny G P. Solid State Nucl Magn Reson. 1996;7:211–218. doi: 10.1016/s0926-2040(96)01267-2. [DOI] [PubMed] [Google Scholar]
- 59.Smith S O. Magn Reson Rev. 1996;17:1–26. [Google Scholar]
- 60.Studelska D R, Klug C A, Beusen D D, McDowell L M, Schaefer J. J Am Chem Soc. 1996;118:5476–5477. [Google Scholar]
- 61.Tycko R, Weliky D P, Berger A E. J Chem Phys. 1996;105:7915–7930. [Google Scholar]
- 62.Weliky D P, Tycko R. J Am Chem Soc. 1996;118:8487–8488. [Google Scholar]
- 63.Heller J, Kolbert A C, Larsen R, Ernst M, Bekker T, Baldwin M, Prusiner S B, Pines A, Wemmer D E. Protein Sci. 1996;5:1655–1661. doi: 10.1002/pro.5560050819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Griffiths J M, Ashbirn T T, Auger M, Costa P R, Griffin R G, Lansbury P T., Jr J Am Chem Soc. 1995;117:3539–3946. [Google Scholar]
- 65.Costa P R, Kocisko D A, Sun B Q, Lansbury P T, Jr, Griffith R G. J Am Chem Soc. 1997;119:10487–10493. [Google Scholar]
- 66.Selkoe D J, Abraham C, Ihara Y. Proc Natl Acad Sci USA. 1982;79:6070–6074. doi: 10.1073/pnas.79.19.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rasmussen L K, Sørensen E S, Petersen T E, Gliemann J, Jensen P H. FEBS Lett. 1994;338:161–166. doi: 10.1016/0014-5793(94)80356-0. [DOI] [PubMed] [Google Scholar]
- 68.Ho G J, Gregory E J, Smirnova I V, Zoubine M N, Festoff B W. FEBS Lett. 1994;349:151–154. doi: 10.1016/0014-5793(94)00663-6. [DOI] [PubMed] [Google Scholar]
- 69.Dudek S M, Johnson G V W. Brain Res. 1994;651:129–133. doi: 10.1016/0006-8993(94)90688-2. [DOI] [PubMed] [Google Scholar]
- 70.Ikura K, Takahata K, Sasaki R. FEBS Lett. 1993;326:109–111. doi: 10.1016/0014-5793(93)81772-r. [DOI] [PubMed] [Google Scholar]
- 71.Pike C J, Walencewicz A J, Glabe C G, Cotman C W. Brain Res. 1991;563:311–314. doi: 10.1016/0006-8993(91)91553-d. [DOI] [PubMed] [Google Scholar]
- 72.Pike C J, Walencewicz A J, Glabe C G, Cotman C W. Eur J Pharmacol. 1991;207:367–368. doi: 10.1016/0922-4106(91)90014-9. [DOI] [PubMed] [Google Scholar]
- 73.Pike C J, Burdick D, Walencewicz A J, Glabe C G, Cotman C W. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Simmons L K, May P C, Tomaselli K J, Rydel R E, Fuson K S, Brigham E F, Wright S, Lieberburg I, Becker G W, Brems D N. Mol Pharmacol. 1993;45:373–379. [PubMed] [Google Scholar]
- 75.Pike C J, Walencewicz-Wasserman A J, Kosmoski J, Cribbs D H, Glabe C G, Cotman C W. J Neurochem. 1995;64:253–265. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- 76.Cribbs D H, Pike C J, Weinstein S L, Velazquez P, Cotman C W. J Biol Chem. 1997;272:7431–7436. doi: 10.1074/jbc.272.11.7431. [DOI] [PubMed] [Google Scholar]
- 77.Manyth P W, Stimson E R, Ghilardi J R, Allen C J, Dahl C E, Whitcomb D C, Vigna S R, Vinters H V, Labenski M E, Maggio J E. Bull Clin Neurosci. 1991;56:73–85. [Google Scholar]
- 78.Maggio J E, Stimson E R, Ghilardi J R, Allen C J, Dahl C E, Whitcomb D C, Vigna S R, Vinters H V, Labenski M E, Mantyh P W. Proc Natl Acad Sci USA. 1992;89:5462–5466. doi: 10.1073/pnas.89.12.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee J P, Stimson E R, Ghilardi J R, Mantyh P W, Lu Y A, Felix A M, Llanos W, Behbin A, Cummings M, Van Criekinge M, et al. Biochemistry. 1995;34:5191–5200. doi: 10.1021/bi00015a033. [DOI] [PubMed] [Google Scholar]
- 80.Esler W P, Stimson E R, Ghilardi J R, Vinters H V, Lee J P, Mantyh P W, Maggio J E. Biochemistry. 1996;35:749–757. doi: 10.1021/bi951685w. [DOI] [PubMed] [Google Scholar]
- 81.Esler W P, Stimson E R, Ghilardi J R, Felix A M, Lu Y A, Vinters H V, Mantyh P W, Maggio J E. Nat Biotechnol. 1997;15:258–263. doi: 10.1038/nbt0397-258. [DOI] [PubMed] [Google Scholar]
- 82.Gregory, D. M., Benzinger, T. L. S., Burkoth, T. S., Miller-Auer, H., Lynn, D. G., Meredith, S. C. & Botto, R. E. Solid State Nucl. Mgn. Reson., in press. [DOI] [PubMed]
- 83.Schägger H, von Jagow G. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]