

Abstract
The mammalian renal toxicant tetrafluoroethylcysteine (TFEC) is metabolized to a reactive intermediate that covalently modifies the lysine residues of a select group of mitochondrial proteins, forming difluorothioamidyl lysine protein adducts. Cellular damage is initiated by this process and cell death ensues. NH2-terminal sequence analysis of purified mitochondrial proteins containing difluorothioamidyl lysine adducts identified the lipoamide succinyltransferase and dihydrolipoamide dehydrogenase subunits of the α-ketoglutarate dehydrogenase complex (αKGDH), a key regulatory component of oxidative metabolism, as targets for TFEC action. Adduct formation resulted in marked inhibition of αKGDH enzymatic activity, whereas the related pyruvate dehydrogenase complex was unmodified by TFEC and its activity was not inhibited in vivo. Covalent modification of αKGDH subunits also resulted in interactions with mitochondrial chaperonin HSP60 in vivo and with HSP60 and mitochondrial HSP70 in vitro. These observations confirm the role of mammalian stress proteins in the recognition of abnormal proteins and provide supporting evidence for reactive metabolite-induced cell death by modification of critical protein targets.
Mitochondrial stress proteins perform many vital functions including the import, folding, and assembly of newly synthesized polypeptides (1–3). Genetic studies have clearly indicated that the 60-kDa heat shock protein (HSP60) and the mitochondrial 70-kDa heat shock protein (mtHSP70) are essential for yeast viability (4, 5). Crystal structures of prokaryotic homologs, GroEL and DnaK/peptide-binding domain, have recently been determined, largely defining their structure and function (6, 7). It is anticipated that eukaryotic HSP60 and mtHSP70 will resemble the prokaryotic forms and also function in abnormal fold state recognition, prevention of aggregation, and other “downstream” processes of organellar removal and degradation (8–10). To date, detailed functional studies of HSP60 and mtHSP70 in mammalian systems have been limited and information is only now beginning to emerge (11).
The nephrotoxicant S-(1,1,2,2-tetrafluoroethyl)-l-cysteine (TFEC) is a metabolite of the industrial gas tetrafluoroethene and is representative of several structurally related halogen-substituted gases with broad economic and environmental interest. These include inhalational anesthetics (e.g., halothane and sevoflurane), ozone-depleting chlorofluorocarbon substitutes, and other industrial gases (12–14). According to the covalent binding hypothesis of cell damage, first proposed more than 20 years ago (15, 16), the bioactivation of an otherwise chemically inert compound will result in generation of reactive intermediates that covalently modify critical cellular targets. Such proteotoxic processes are believed to result in an inactivation of target protein function and, ultimately, cell death and organ damage (17). Consequently, the presence of covalently bound protein adducts after drug or chemical exposure is used extensively to indicate a potential for toxic reactions, although the nature of critical protein target(s) has yet to be defined for any compound. TFEC represents a prototypical compound for examining such processes with enzymatic bioactivation that results in the formation of a reactive acyl fluoride species that covalently modifies lysine ɛ-amino groups to form difluorothioamidyl lysine (DFTAL) protein adducts (14).
Nephrotoxic doses of TFEC reproducibly modify five major proteins in rat kidney mitochondria with approximate molecular masses of 99, 84, 66, 52, and 48 kDa (18). We have previously shown that the prominently adducted 66-kDa and 84-kDa proteins are, respectively, chaperonin HSP60 and mtHSP70 (also known as the 74-kDa peptide-binding protein or mortalin; refs. 19–21). In addition, we identified a minor 42-kDa DFTAL-modified protein as the mitochondrial isoform of aspartate aminotransferase. Independent studies have subsequently shown that aspartate aminotransferase activity is decreased after DFTAL formation (22). We report herein the identification of additional TFEC target proteins, further studies outlining the molecular events associated with this form of cell death, and discuss implications for the covalent binding hypothesis and the actions of mammalian stress proteins.
MATERIALS AND METHODS
Materials.
Unless otherwise indicated chemicals were obtained from Sigma. TFEC and 35S-radiolabeled TFEC were prepared exactly as described from [35S]cysteine (Amersham) (23). Antiserum initially raised to halothane protein adducts (a gift from Lance Pohl, National Institutes of Health) was previously characterized as specific for difluorothioamidyl lysine residues (24). mAbs to HSP60 was purchased from StressGen Biotechnologies (Sidney, Canada).
DFTAL Protein Identification.
NH2-terminal sequences for the 48-kDa and 52-kDa DFTAL-modified proteins were obtained from two independent F344 rat mitochondrial preparations. Renal tissue protein was isolated 6 h after TFEC (30 mg/kg) was administered i.p. and purified DFTAL proteins were subject to replicate NH2-terminal sequence analyses, as described (18), before confirmation of target protein adduction using immunoprecipitation. Homology searches were as described (18). Amino acid residues are numbered from the translational initiation codon for the published lipoamide succinyltransferase (E2o) or dihydrolipoamide dehydrogenase (E3o) sequences (25–27).
α-Ketoglutarate Dehydrogenase (αKGDH) and Pyruvate Dehydrogenase (PDH) Complex Activities.
αKGDH activities were determined from homogenized kidney tissue by measuring the formation of NADH by monitoring absorbance at 340 nm as described (28). Potential inhibition of αKGDH activity by TFEC was assessed with porcine heart αKGDH and found not to be inhibited directly (data not presented). PDH activities were determined by coupling the generation of NADH to the oxidation of 2-(p-iodophenyl)-3-p-nitrophenyl-5-phenyltetrazolium chloride (INT) (29).
Assessment of Renal Function.
Urinary glucose and blood urea nitrogen levels were determined at regular intervals after administration of TFEC (30 mg/kg, i.p.) by using commercially available diagnostic kits.
Immunohistochemistry.
Rats were treated with TFEC (30 mg/kg, i.p.) and sacrificed at regular intervals after dosing. Kidneys were decapsulated, fixed in neutral buffered formalin, and embedded in paraffin. After deparaffination, sections were stained immunohistochemically for DFTAL-modified proteins as described (24).
Immunoprecipitation and Immunoblots.
Immunoprecipitations from tissue homogenates or in vitro-generated samples were performed as described by using 5 μl of HSP60 mAb, 20–40 μl of anti-α-ketoglutarate decarboxylase (E1o) antiserum, 20–40 μl of anti-E2o antiserum, 20–40 μl of anti-E3o antiserum, 20–40 μl of anti-PDH antiserum, 20–40 μl of normal rabbit serum, or 20–40 μl of an unrelated mAb (18). Immunoprecipitates were separated on denaturing polyacrylamide gels (SDS/PAGE) and subjected to fluorography and autoradiography to detect radiolabeled proteins or to enhanced chemiluminescence after transfer to nitrocellulose (ECL, Amersham).
Isolation of Mitochondria and in Vitro Incubation Conditions.
Kidney mitochondria were isolated from untreated F344 rats as described (18). Tissue homogenates from control or TFEC-treated animals (30 mg/kg, i.p.; 6 h) were used immediately for immunoprecipitation (see Fig. 2) or for the preparation of mitochondria (control animals only) (see Figs. 3 and 4). Cytosolic contamination of mitochondria, as judged by residual lactate dehydrogenase activity, was typically 10–15%. Isolated mitochondria were incubated with either unlabeled TFEC (100 or 500 μM) or [35S]TFEC (80 or 400 μM) for 1 h at 25°C before examination by immunoblot analysis or autoradiography. Mitochondria were incubated in isolation buffer not containing ADP in the absence or the presence of 10 mM aminooxyacetic acid. For studies examining the effect of ATP on complex formation, mitochondria were incubated in isolation buffer containing 1 mM ATP.
Figure 2.

HSP60 interacts with damaged αKGDH subunits in vivo after TFEC treatment. (A and B) Specific interactions between HSP60 and TFEC target proteins were detected by immunoprecipitation from control or TFEC-treated (30 mg/kg, i.p.; 6 h) renal homogenates. HSP60 immune complexes were immunoblotted with antiserum to the E2o (A) or E3o (B) subunits of αKGDH. The presence of mature E2o or E3o subunits complexed with HSP60 only after TFEC treatment are indicated to the right (arrowhead). (C) Detection of αKGDH subunit-associated HSP60 by immunoprecipitation of control or TFEC-treated homogenates with E1o-, E2o-, and E3o-specific antiserum or with normal rabbit sera (NS). Immune complexes were immunoblotted for the presence of HSP60 (Blot ab). HSP60 association with αKGDH subunits was increased after TFEC treatment. (D and E) Confirmation of αKGDH E2o and E3o antisera specificity by immunoprecipitation from control and TFEC-treated homogenates with subunit-directed antiserum or with normal rabbit serum (NS). Blots were incubated with antiserum identical to either E2o (D) or E3o (E). The presence of either E2o or E3o (Blot ab) only in anticipated immunoprecipitates is shown to the right in D and E (arrowhead). Treatments and precipitating antibodies (IP ab) are indicated at the top. The positions of molecular weight standards are shown to the left. Nonspecific interactions between secondary antibody and the heavy chain of IgG (H-chain IgG) were observed in A, C, and E.
Figure 3.

In vitro formation of DFTAL and [35S]DFTAL-modified proteins by using isolated mitochondria. Isolated mitochondria from naïve F344 rat kidneys were incubated with either unlabeled TFEC or high-specific-activity [35S]TFEC to detect formation of DFTAL-modified proteins by immunoblot analysis (lanes DFTAL epitope) or autoradiography (lanes Total 35S-DFTAL). The β-lyase inhibitor aminooxyacetic acid (lanes +AOA; 10 mM) was added to some incubations to block TFEC metabolism and to determine the specificity of DFTAL antiserum detection and 35S-radiolabel incorporation (B and C Right). (A) Immunoblot of DFTAL-modified mitochondrial proteins formed in vivo after 6 h as detected with adduct-specific antisera (18). The positions of major adducted proteins are shown to the left as the unknown 99-kDa protein (unk), mitochondrial HSP70 (mtHSP70), HSP60, and αKGDH complex subunits (E2o and E3o). Positions of molecular weight markers are indicated to the right and are also shown to the left of B and C. DFTAL-protein formation in vitro was detected by either anti-DFTAL antiserum or [35S]DFTAL radiolabel: (B Left) Immunoblot of DFTAL-modified mitochondrial proteins formed in the presence of 100 μM unlabeled TFEC. (B Middle) Autoradiograph of [35S]DFTAL proteins formed after incubation of mitochondria with 80 μM [35S]TFEC. (C Left) DFTAL-protein immunoblot from mitochondria incubated in the presence of 500 μM unlabeled TFEC. (Middle) Autoradiograph of [35S]DFTAL proteins observed after incubation of mitochondria with 400 μM [35S]TFEC. (Right) The positions of the major adducted proteins formed are shown in addition to a minor DFTAL protein [aspartate aminotransferase (AAT)].
Figure 4.

Confirmation of TFEC target protein identities by immunoprecipitation of [35S]DFTAL-E2o and [35S]DFTAL-E3o and interactions with mitochondrial stress proteins in vitro. In vitro-generated [35S]DFTAL proteins were immunoprecipitated from isolated mitochondria after incubation with 80 or 400 μM [35S]TFEC. The precipitating antibodies are indicated above each lane. Nonspecific immunoprecipitations with normal rabbit serum (NRS) are indicated to the left. Lanes marked with 80/400 indicate that the results from nonspecific immunoprecipitations were the same regardless of the concentration of [35S]TFEC. (A) The 48-kDa [35S]DFTAL-E2o was observed as the predominant band in immunoprecipitates and coprecipitated with other known [35S]DFTAL proteins (indicated on the right). (B) The 55-kDa [35S]DFTAL-E3o was observed in antibody complexes with [35S]DFTAL-E2o and other known DFTAL-modified proteins in 80 μM (Middle) or 400 μM (Right) [35S]TFEC incubations (indicated on the right). (C) Confirmation of [35S]DFTAL formation on HSP60 and coprecipitation with other [35S]DFTAL labeled proteins. Less total 35S-derived radiolabel was observed at 400 μM compared with 80 μM [35S]TFEC incubations and further reduced by incubation of mitochondria with ATP (see text). Migrations of prestained molecular mass standards (in kDa) are shown to the left.
Statistical Analysis.
Analysis of variance used the Bonferroni/Dunn post hoc test with statview (Abacus Concepts, San Francisco, CA). Statistical significance was assumed when the P value was <0.01. All data are presented as mean ± SEM.
RESULTS AND DISCUSSION
To identify the 48-kDa and 52-kDa DFTAL proteins, we partially purified these proteins while monitoring for adduct localization (18). NH2-terminal Edman degradation sequence analyses for the 48-kDa DFTAL protein gave N57DVITVQtPAXaESVtEGDV76 confirming it as the mature form of the E2 (lipoamide succinyltransferase) subunit of αKGDH (E2o; ref. 25). Replicate NH2-terminal sequence determinations for the 52-kDa adducted protein yielded A36DQPIDADVTViGsGPGgY54, indicating complete identity with the human and porcine forms of mature E3 subunit (lipoamide dehydrogenase) of αKGDH (E3o; refs. 26 and 27). Assignments are as follows: x represents an equivocal residue, and lowercase letters indicate reduced or variable yields. Identification of the mature forms of E2o and E3o as targets for reactive metabolites of TFEC is consistent with attack on assembled αKGDH complex after transport, maturation, and assembly. Interestingly, the E1 subunit of αKGDH (E1o) was not detected as a DFTAL-modified protein, indicating differential susceptibilities among αKGDH subunits with regard to adduct formation. After we identified αKGDH E2o and E3o subunits as putative targets, we examined the effect of covalent modification on αKGDH in vivo activity and independently confirmed the status of E2o and E3o as adducted proteins (see below).
The functional consequence of covalent modification was assessed by determining the in vivo activity of the αKGDH complex after a nephrotoxic dose of TFEC. As an internal control for mitochondrial damage, the activity of the functionally related PDH multienzyme complex was also examined. The data confirmed that in vivo αKGDH activity was significantly inhibited, whereas PDH activity was not inhibited (Fig. 1A). Maximal αKGDH inhibition was observed 12 h after TFEC administration and declined to 45% of the activity of untreated controls (P < 0.001). The time course and extent of αKGDH inhibition was consistent with the relative number of immunohistochemically labeled target cells (i.e., protein covalent binding) (Fig. 1C). Maximal inhibition of αKGDH activity preceded peak renal damage at 24 h, as assessed by other biochemical and morphological indices (Fig. 1 B and C). In addition, loss of αKGDH activity was marginally significant (P = 0.016) as early as 6 h after TFEC administration. Subsequent time points indicated a return of αKGDH activity and, although not significant, still represented an approximately 25% inhibition of control levels (Fig. 1A).
Figure 1.

Inhibition of in vivo αKGDH activity and onset of cell death and kidney damage in response to TFEC. (A) Kidney homogenate αKGDH (■) or PDH (⧫) activities after TFEC administration from three determinations (n = 2 to 4 animals per point) except at 36 h (two determinations). Activities are expressed as a percentage of control (untreated) activity at each time point. αKGDH activity in control animals ranged from 12.2 to 18.9 nmol of NADH produced per min per mg of protein. αKGDH activity in TFEC-treated animals ranged from 6.1 to 14.5 nmol of NADH produced per min per mg of protein. PDH activity in control animals ranged from 1.48 to 1.73 μmol of INT reduced per min per mg and in TFEC-treated animals ranged from 1.44 to 1.74 μmol of INT reduced per min per mg of protein. Significant differences between treatment and control groups are indicated as follows: ∗, 0.05 < P > 0.01; ∗∗, P < 0.001. (B) Renal function was assessed for up to 72 h after TFEC was administered. Increases in urinary glucose concentration (□), an early indicator of renal damage, paralleled the onset of cell death (C) and preceded increased blood urea nitrogen levels (•) (n = 4 or 5, from one determination). Variance is less than symbol width when not visible. (C) Immunohistochemical localization of protein adducts in renal tissue using DFTAL-specific antiserum after TFEC treatment. Immunoreactivity was evident as early as 6 h after the dose was administered as punctate staining of mostly S3C proximal tubule epithelial cells (arrows). Staining was qualitatively maximal at 12 h in both S3C and S3M proximal tubule segments. Medullary ray (MR) and glomerular (G) structures remained unstained. Proximal tubule cell death, visualized as an exfoliation into the tubule lumen, was maximal between 18 and 48 h after the dose was administered. Immunoreactive cell debris was cleared progressively and was localized exclusively to the lumen by 72 h. (Relative magnifications: ×100, 0, 12, 48, and 72 h; ×160, 6 and 24 h.)
Our data confirm that DFTAL formation on the E2o and E3o subunits of αKGDH in vivo resulted in a pronounced inhibition of αKGDH activity. Earlier studies have shown an inhibition of the αKGDH complex after TFEC treatment but it has not been possible to determine from these studies whether this was a direct or an indirect effect (30, 31). Furthermore, we did not observe any decreases in PDH activity after TFEC treatment, indicating that the functionally and structurally related PDH complex was not a primary target for TFEC in vivo.
An expected repercussion of αKGDH covalent modification and inactivation would be the perturbation of subunit structure resulting in mitochondrial stress protein recognition (6, 8, 9). To test this, HSP60 and associated proteins were immunoprecipitated directly from untreated or TFEC-treated tissue homogenates (Fig. 2). HSP60 complexes were then immunoblotted with monospecific antiserum for each αKGDH subunit (Fig. 2 A and B and ref. 32). Similarly, αKGDH subunit-directed immune complexes were immunoblotted with HSP60 mAb (Fig. 2C). In both cases HSP60 was only found associated with αKGDH subunits after TFEC treatment, confirming that chaperonin recognition of αKGDH subunits occurs after the proteotoxic event (Fig. 2 A–C). HSP60 localization with the E1o subunit suggested that E1o is released from the αKGDH complex, thereby revealing normally inaccessible hydrophobic stretches required for HSP60 binding (Fig. 2C and ref. 6). We also examined potential HSP60 interactions with PDH subunits and found no comparable association (data not presented).
To exclude nonspecific HSP60 interactions with the intact complete αKGDH complex, we examined E1o, E2o, and E3o immunoprecipitations for other “contaminating” subunits (Fig. 2 D and E). Although we were unable to detect E1o by immunoprecipitation under these conditions, we were able to confirm that E2o and E3o immunoprecipitations were very selective in precipitating the subunit of interest (Fig. 2 D and E). Lack of E1o detection, in these experiments, may be due to the decreased immunoreactivity of E1o-directed antiserum to fragments of this proteolytically sensitive subunit. Overall, these data indicated that increased HSP60 binding to individual αKGDH subunits was related to toxicant-mediated changes to these subunits and the αKGDH complex.
We independently confirmed the in vivo data of αKGDH adduct localization by incubating normal rat mitochondria with [35S]TFEC (Fig. 3) and using subunit-specific antibodies to immunoprecipitate radiolabeled subunits (Fig. 4). Isolated mitochondria were incubated with [35S]TFEC at either 40% (80 μM) or 200% (400 μM) of the calculated IC50 value for inhibition of state 3 respiration (33). HSP60 was the most prominently radiolabeled protein with incubations at the moderately toxic 80 μM level (Fig. 3B Middle) confirming our previous observations (18). Other 35S-labeled proteins were also observed in the expected molecular mass range for E2o (48 kDa) and E3o (55 kDa, Fig. 3B). Although the pattern of radiolabeled proteins was similar to that found with DFTAL-directed antiserum, considerable differences in band intensity between these two techniques were observed (Fig. 3B). A lack of agreement between immunochemical and radiochemical procedures has been observed previously with other studies that have compared protein adduct localization by both techniques (34).
Mitochondrial incubations with higher concentrations of unlabeled TFEC (500 μM) generated a pattern similar to that observed in vivo (Fig. 3C Left, compare with Fig. 3A). Analogously, 35S-labeled proteins from incubations with 400 μM [35S]TFEC were nearly identical to those observed in vivo above 35-kDa molecular mass (Fig. 3C Middle, compare with Fig. 3A). The more complex pattern of radiolabeled proteins at lower mass is suggestive of protein degradation. In all cases, the addition of aminooxyacetic acid (35), a specific inhibitor of TFEC bioactivation by β-lyase, completely inhibited 35S-labeling or immunoreactivity, confirming that β-lyase was required for these protein modification events (Fig. 3 B Right and C Right). We concluded that in vitro incubations of normal isolated mitochondria with [35S]TFEC can model the protein modification events found in vivo.
The identity of E2o as a target protein was confirmed by immunoprecipitation of the radiolabeled subunit from in vitro mitochondrial incubations with E2o-specific antiserum (Fig. 4A). Similarly, adduct localization on E3o was confirmed by immunoprecipitation with E3o-specific antiserum (Fig. 4B). Although E2o and E3o subunits were observed as a prominent band in each immunoprecipitation, other DFTAL-labeled proteins also coprecipitated. The composition of E2o- and E3o-directed immune complexes varied with the concentration of [35S]TFEC, and increased stress protein binding at the more toxic concentrations suggested some specificity to these interactions (refer later). Radiolabeled E1o or PDH subunits were not detected in corresponding immunoprecipitations, indicating that neither are direct targets for TFEC, in agreement with the related studies of halothane-mediated hepatitis (36, 37).
Immunoprecipitation of radiolabeled HSP60 from in vitro mitochondrial incubations was in accord with the in vivo data showing that HSP60 was a primary target and that it coprecipitated with other DFTAL proteins (Fig. 4C and ref. 18). In contrast to E2o and E3o immunoprecipitates, however, the extent of radiolabel on HSP60 per se was consistently lower compared with other coprecipitating [35S]DFTAL proteins (Fig. 4C, compare with Fig. 4 A and B). This preferential enrichment of other [35S]DFTAL proteins would be expected if HSP60 acts to recognize damaged proteins as the functional form. Furthermore, impairment of HSP60 peptide binding was suggested by less immunoprecipitated radiolabel at higher concentrations of [35S]TFEC (Fig. 4C). Total radiolabeled proteins coprecipitating with HSP60 were further decreased, but not abolished, when mitochondria were incubated in the presence of added ATP and ATP-generating substrates (Fig. 4C Right). Thus, in agreement with the in vivo data of Fig. 2, functional HSP60 preferentially associated with adducted αKGDH subunits (as well as other modified mitochondrial proteins) and that a proportion of these proteins likely exist as kinetically trapped species.
Because the composition of immune complexes varied, these data argue against simple protein aggregation events for stress protein recognition of DFTAL-modified proteins. For example, HSP60 coprecipitated with E2o but not E3o, whereas mtHSP70 precipitated with both (Fig. 4). These data indicate that mammalian HSP60 and mtHSP70 may interact in the recognition of abnormal proteins and are themselves recognized when adducted. However, we have noted the formation of higher molecular weight complexes, both in vivo and in vitro, which are traditionally associated with protein aggregation events (Figs. 3C and 4). The possibility remains, therefore, that inactivation of mitochondrial stress proteins will result in the formation of insoluble aggregates that ultimately lead to cell death.
The studies presented herein and our earlier work (18) on the submitochondrial distribution of DFTAL proteins suggest the sequence of events represented in Fig. 5. Reactive metabolites (Rδ+) from TFEC attack E2o (which forms the structural inner core of the αKGDH complex) and E3o subunits. This disrupts αKGDH quaternary structure, releasing component subunits into differing submitochondrial compartments. Mitochondrial stress protein HSP60 and/or mtHSP70 recognition follows, thereby determining the subsequent cellular fate of adducted proteins (38, 39).
Figure 5.
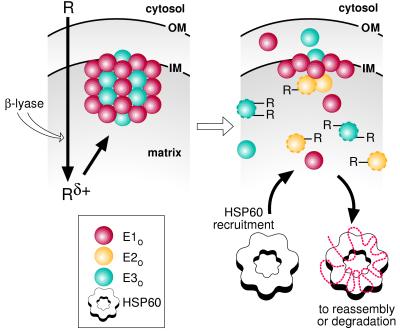
Model for the events leading to TFEC-mediated cell death in vivo. In the first step, the toxicant (R) is converted to a reactive intermediate (Rδ+) by the action of β-lyase within the mitochondrion. Attack on the α-KGDH complex (Left), located on the matrix face of the inner mitochondrial membrane (IM), results in altered protein structure (subunit-R) and disassociation of the enzyme complex (Right). Finally, disassembly of α-KGDH subunits and altered subunit structure increases recognition and binding of mitochondrial stress proteins (Lower Right). The relative positions of the mitochondrial outer membrane (OM) and the cytosol are also represented.
Current evidence indicates that the E3 subunit of all multienzyme ketoacid dehydrogenase complexes are identical (40, 41). Because the reactive acylhalides produced from TFEC metabolism should react indiscriminately with all Nɛ-lysine residues in close proximity, it is expected that the E3 subunit of PDH (E3p) represents an equally likely target for DFTAL modification. Our observations, however, indicate that E3p is not adducted and that its enzymatic activity is not inhibited. These results suggest that E3p was spatially removed from the site of reactive metabolite formation within the mitochondrion.
These studies have indicated that TFEC toxicity is associated with altered mitochondrial α-ketoglutarate utilization thereby disrupting the tricarboxylic acid cycle and mitochondrial function. Indeed, congenital defects in the αKGDH complex are fatal (42, 43) and E3o-null mice die at 7.5 days postcoitum as aberrant embryos (41). Although cross-species extrapolation should be viewed with caution, the relative degree of αKGDH inhibition reported herein is comparable to that seen in fibroblasts taken from αKGDH-deficient patients (43). To conclude, TFEC-mediated cell death and organ damage can be accounted for by perturbations to oxidative metabolism, which is fundamental for organism viability.
For more than 20 years, a central tenet in toxicology has been that biologically benign foreign compounds may be converted to reactive metabolites that can then modify cellular macromolecules and alter the structure and/or function of the target (15–17). The covalent binding hypothesis has, consequently, guided the majority of mechanistic studies in the toxicology of reactive intermediates during this time despite a lack of data defining the relative contribution of critical versus inconsequential protein targets. Concurrently, an expansion of knowledge regarding the functions of stress proteins has indicated that this class of proteins is characterized by their selective affinity for nonnative proteins (6–10). These results support the conclusion that in situ covalent modifications of a critical cellular target (i.e., the αKGDH complex by TFEC) results in an inhibition of the function of the target. Of equal importance is that the modified target protein is bound by stress proteins that function in the recognition of abnormal fold states. Thus, the physiological roles of mammalian HSP60 and mtHSP70 can now include the recognition of postmaturation reactive-intermediate-modified protein conformations.
Acknowledgments
We thank Dr. Lance Pohl for providing antisera to DFTAL protein adducts, Dr. Richard Hallberg for stimulating conversations, J. North for synthesis of [35S]TFEC, Dr. T. Ichimura for immunohistochemistry, Karen West for assistance with NH2-terminal sequence analyses, and Drs. Sid Nelson and Bill Atkins for critical review of the manuscript. Also, Katrina and Lauren for inspiration and support. The work was supported by National Institutes of Health Grant GM51916 to S.B.
ABBREVIATIONS
- DFTAL
difluorothioamidyl lysine
- E1o
α-ketoglutarate decarboxylase, E2o, lipoamide succinyltransferase
- E3o
dihydrolipoamide dehydrogenase
- HSP60
60-kDa heat shock protein
- INT
2-(p-iodophenyl)-3-p-nitrophenyl-5-phenyltetrazolium chloride
- αKGDH
α-ketoglutarate dehydrogenase
- mtHSP70
70-kDa mitochondrial heat shock protein
- PDH
pyruvate dehydrogenase
- TFEC
S-(1,1,2,2-tetrafluoroethyl)-l-cysteine
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Gething M J, Sambrook J. Nature (London) 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- 2.Glick B, Schatz G. Annu Rev Genet. 1991;25:21–44. doi: 10.1146/annurev.ge.25.120191.000321. [DOI] [PubMed] [Google Scholar]
- 3.Parsell D A, Lindquist S. Annu Rev Genet. 1993;27:437–496. doi: 10.1146/annurev.ge.27.120193.002253. [DOI] [PubMed] [Google Scholar]
- 4.Cheng M Y, Hartl F U, Martin J, Pollock R A, Kalousek F, Neupert W, Hallberg E M, Hallberg R L, Horwich A L. Nature (London) 1989;337:620–625. doi: 10.1038/337620a0. [DOI] [PubMed] [Google Scholar]
- 5.Craig E A, Kramer J, Shilling J, Werner-Washburne M, Holmes S, Kosic Smithers J, Nicolet C M. Mol Cell Biol. 1989;9:3000–3008. doi: 10.1128/mcb.9.7.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fenton W A, Horwich A L. Protein Sci. 1997;6:743–760. doi: 10.1002/pro.5560060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu X, Zhao X, Burkholder W F, Gragerov A, Ogata C M, Gottesman M E, Hendrickson W A. Science. 1996;272:1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin J, Horwich A L, Hartl F U. Science. 1992;258:995–998. doi: 10.1126/science.1359644. [DOI] [PubMed] [Google Scholar]
- 9.Kandror O, Busconi L, Sherman M, Goldberg A L. J Biol Chem. 1994;269:23575–23582. [PubMed] [Google Scholar]
- 10.Wagner I, Arlt H, van Dyck L, Langer T, Neupert W. EMBO J. 1994;13:5135–5145. doi: 10.1002/j.1460-2075.1994.tb06843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agsteribbe E, Huckriede A, Veenhuis M, Ruiters M H, Niezen Koning K E, Skjeldal O H, Skullerud K, Gupta R S, Hallberg R, et al. Biochem Biophys Res Commun. 1993;193:146–154. doi: 10.1006/bbrc.1993.1602. [DOI] [PubMed] [Google Scholar]
- 12.Harris J W, Pohl L R, Martin J L, Anders M W. Proc Natl Acad Sci USA. 1991;88:1407–1410. doi: 10.1073/pnas.88.4.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoet P, Graf M L, Bourdi M, Pohl L R, Duray P H, Chen W, Peter R M, Nelson S D, Verlinden N, Lison D. Lancet. 1997;350:556–559. doi: 10.1016/S0140-6736(97)03094-8. [DOI] [PubMed] [Google Scholar]
- 14.Pohl L R. Chem Res Toxicol. 1993;6:786–793. doi: 10.1021/tx00036a006. [DOI] [PubMed] [Google Scholar]
- 15.Brodie B B, Reid W D, Cho A K, Sipes G, Krishna G, Gillette J R. Proc Natl Acad Sci USA. 1971;68:160–164. doi: 10.1073/pnas.68.1.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gillette J R. Biochem Pharmacol. 1974;23:2785–2794. doi: 10.1016/0006-2952(74)90052-5. [DOI] [PubMed] [Google Scholar]
- 17.Nelson S D, Pearson P G. Annu Rev Pharmacol Toxicol. 1990;30:169–195. doi: 10.1146/annurev.pa.30.040190.001125. [DOI] [PubMed] [Google Scholar]
- 18.Bruschi S A, West K A, Crabb J W, Gupta R S, Stevens J L. J Biol Chem. 1993;268:23157–23161. [PubMed] [Google Scholar]
- 19.Bhattacharyya T, Karnezis A N, Murphy S P, Hoang T, Freeman B C, Phillips B, Morimoto R I. J Biol Chem. 1995;270:1705–1710. doi: 10.1074/jbc.270.4.1705. [DOI] [PubMed] [Google Scholar]
- 20.Domanico S Z, DeNagel D C, Dahlseid J N, Green J M, Pierce S K. Mol Cell Biol. 1993;13:3598–3610. doi: 10.1128/mcb.13.6.3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wadhwa R, Kaul S C, Ikawa Y, Sugimoto Y. J Biol Chem. 1993;268:6615–6621. [PubMed] [Google Scholar]
- 22.Kato Y, Asano Y, Cooper A J L. Dev Neurosci. 1996;18:505–514. doi: 10.1159/000111447. [DOI] [PubMed] [Google Scholar]
- 23.Hayden P, Schaeffer V H, Larsen G, Stevens J L. Methods Enzymol. 1987;143:228–234. doi: 10.1016/0076-6879(87)43043-7. [DOI] [PubMed] [Google Scholar]
- 24.Hayden P J, Ichimura T, McCann D J, Pohl L R, Stevens J L. J Biol Chem. 1991;266:18415–19418. [PubMed] [Google Scholar]
- 25.Nakano K, Matuda S, Yamanaka T, Tsubouchi H, Nakagawa S, Titani K, Ohta S, Miyata T. J Biol Chem. 1991;266:19013–19017. [PubMed] [Google Scholar]
- 26.Otulakowski G, Robinson B H. J Biol Chem. 1987;262:17313–17318. [PubMed] [Google Scholar]
- 27.Pons G, Raefsky-Estrin C, Carothers D J, Pepin R A, Javed A A, Jesse B W, Ganapathi M K, Samols D, Patel M S. Proc Natl Acad Sci USA. 1988;85:1422–1426. doi: 10.1073/pnas.85.5.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas A P, Denton R M. Biochem J. 1986;238:93–101. doi: 10.1042/bj2380093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinman L M, Blass J P. J Biol Chem. 1981;256:6583–6586. [PubMed] [Google Scholar]
- 30.Stonard M D, Parker V H. Biochem Pharmacol. 1971;20:2417–2427. doi: 10.1016/0006-2952(71)90242-5. [DOI] [PubMed] [Google Scholar]
- 31.Lock E A, Schnellmann R G. Toxicol Appl Pharmacol. 1990;104:180–190. doi: 10.1016/0041-008x(90)90293-4. [DOI] [PubMed] [Google Scholar]
- 32.Hunter A, Lindsay J G. Eur J Biochem. 1986;155:103–109. doi: 10.1111/j.1432-1033.1986.tb09464.x. [DOI] [PubMed] [Google Scholar]
- 33.Hayden P J, Stevens J L. Mol Pharmacol. 1990;37:468–776. [PubMed] [Google Scholar]
- 34.Myers T G, Dietz E C, Anderson N L, Khairallah E A, Cohen S D, Nelson S D. Chem Res Toxicol. 1995;8:403–413. doi: 10.1021/tx00045a012. [DOI] [PubMed] [Google Scholar]
- 35.Elfarra A A, Jakobson I, Anders M W. Biochem Pharmacol. 1986;35:283–288. doi: 10.1016/0006-2952(86)90527-7. [DOI] [PubMed] [Google Scholar]
- 36.Christen U, Quinn J, Yeaman S J, Kenna J G, Clarke J B, Gandolfi A J, Gut J. Eur J Biochem. 1994;223:1035–1047. doi: 10.1111/j.1432-1033.1994.tb19082.x. [DOI] [PubMed] [Google Scholar]
- 37.Frey N, Christen U, Jenö P, Yeaman S J, Shimomura Y, Kenna J G, Gandolfi A J, Ranek L, Gut J. Chem Res Toxicol. 1994;8:736–746. doi: 10.1021/tx00047a014. [DOI] [PubMed] [Google Scholar]
- 38.Taguchi H, Makino Y, Yoshida M. J Biol Chem. 1994;269:8529–8534. [PubMed] [Google Scholar]
- 39.Gottesman S, Wickner S, Maurizi M R. Genes Dev. 1997;11:815–823. doi: 10.1101/gad.11.7.815. [DOI] [PubMed] [Google Scholar]
- 40.Perham R N. Biochemistry. 1991;30:8501–8512. doi: 10.1021/bi00099a001. [DOI] [PubMed] [Google Scholar]
- 41.Johnson M T, Yang H-S, Magnuson T, Patel M S. Proc Natl Acad Sci USA. 1997;94:14512–14517. doi: 10.1073/pnas.94.26.14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonnefont J P, Chretien D, Rustin P, Robinson B, Vassault A, Aupetit J, Charpentier C, Rabier D, Saudubray J M, Munnich A. J Pediatr. 1992;121:255–258. doi: 10.1016/s0022-3476(05)81199-0. [DOI] [PubMed] [Google Scholar]
- 43.Kohlschutter A, Behbehani A, Langenbeck U, Albani M, Heidemann P, Hoffmann G, Kleineke J, Lehnert W, Wendel U. Eur J Pediatr. 1982;138:32–37. doi: 10.1007/BF00442325. [DOI] [PubMed] [Google Scholar]