

Abstract
Nitration of tyrosine residues by nitric oxide (NO)-derived species results in the accumulation of 3-nitrotyrosine in proteins, a hallmark of nitrosative stress in cells and tissues. Tyrosine nitration is recognized as one of the multiple signaling modalities used by NO-derived species for the regulation of protein structure and function in health and disease. Various methods have been described for the quantification of protein 3-nitrotyrosine residues, and several strategies have been presented toward the goal of proteome-wide identification of protein tyrosine modification sites. This chapter details a useful protocol for the quantification of 3-nitrotyrosine in cells and tissues using high-pressure liquid chromatography with electrochemical detection. Additionally, this chapter describes a novel biotin-tagging strategy for specific enrichment of 3-nitrotyrosine-containing peptides. Application of this strategy, in conjunction with high-throughput MS/MS-based peptide sequencing, is anticipated to fuel efforts in developing comprehensive inventories of nitrosative stress-induced protein-tyrosine modification sites in cells and tissues.
1. Introduction
A large and growing body of evidence has associated the accumulation of 3-nitrotyrosine (3-NT) in proteins with major neurological (Alzheimer's, Parkinson's, multiple sclerosis, and stroke) and cardiovascular (atherosclerosis, myocardial infarction, coronary artery disease, hypertension, and diabetic vasculopathy) diseases that share inflammation as a contributor to pathogenesis. While inflammation-associated protein-3-NT forms predominantly via the reaction of proteinaceous tyrosine residues with peroxynitrite (ONOO−), the latter arising from the near diffusion-limited reaction of nitric oxide (•NO) with superoxide (O2•−)(Beckmann et al., 1994; Pacher et al., 2007), other nitration mechanisms have also been implicated (Eiserich et al., 1998). Regardless of the mechanism of formation in vivo, protein 3-NT levels provide a useful measure of the severity of tissue exposure to reactive nitrogen species and a telling indicator of inflammatory disease severity and progression Brodsky et al., 2004; Halejcio-Delophont et al., 2001; Skinner et al., 1997; Upmacis et al., 2007). Semiquantitative immunological methods (Ischiropoulos, 1998), as well as quantitative high-pressure liquid chromatography (HPLC)-based methods that utilize ultraviolet-visible (UV/VIS) absorption, electrochemistry, and mass spectrometry (MS) for detection (Frost et al., 2000; Maruyama et al., 1996; Schwedhelm et al., 1999; Shigenaga et al., 1997; Yi et al., 2000), have all been employed for the quantification of protein 3-NT residues; each method offers its own particular strengths and limitations. Importantly, these alternative 3-NT detection methods differ widely with regard to their relative sensitivity, specificity, throughput, and accessibility (because of differing requirements for expensive and/or specialized instrumentation). The reader is referred to an excellent survey of proteomic and MS-based 3-NT assays by Kanski and Schoneich (2005), as well as chapters in this volume containing complementary information.
The goal of this chapter is threefold: (1) detail a useful protocol for assay of protein 3-NT in cells and tissues, (2) consider approaches for unbiased identification of protein 3-NT modification sites, and (3) describe a novel biotin-tagging approach to selectively enrich 3-NT-containing peptides for more comprehensive 3-NT site identification by liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS).
2. Quantification of 3-NT in Proteins Using HPLC Separation and Electrochemical (EC) Detection
This section provides procedural details for the quantification of protein-bound 3-NT using isocratic reverse-phased HPLC paired with EC detection. This method is essentially as described previously by Maruyama and colleagues (Maruyama et al., 1996; Skinner et al., 1997), but with some recommendations and implemented modifications. Strengths of this method are its relatively low cost and sufficient sensitivity for reliable measurement of protein 3-NT at the relatively low basal levels found in most “healthy” tissues. It is notable that the use of EC detection provides ≈100 times greater sensitivity than that which can be obtained using UV/VIS absorption. Protein hydrolysis for the release of free 3-NT from proteins is performed under neutral conditions, minimizing the potential for artifactual nitration that may occur in acidified nitrite-containing solutions (Oldreive et al., 1998). When performed as described, the efficiency of protein 3-NT retrieval from crude extracts of cells and tissues routinely exceeds 85%.
3. Protocol for Quantification of 3-NT in Hydrolyzed Proteins Using HPLC-EC
3.1. Materials
3-Nitrotyrosine, sodium octanesulfonate, acetonitrile, proteinase K, sodium hydrosulfite, and all other reagents are purchased from Sigma-Aldrich in the best available grade (minimum 95% purity; HPLC grade where available). An isocratic HPLC system with autosampler, pump, tubings, and fittings, as well as a multichannel electrochemical CoulArray detector and EC cell, are from ESA, Inc. HPLC mobile phase and all buffers and standard solutions are prepared using 18 MΩ resistance water, either purchased or prepared using a Milli-Q water purification system (Waters Inc.) or equivalent. The HPLC mobile phase is vacuum degassed and filtered through a 0.2-μm nylon membrane (Whatman) to reduce background electrochemical noise, prolonging the electrochemical cell lifetime and enhancing the 3-NT detection of sensitivity. 3-NT detection sensitivity can be enhanced progressively by continuous recirculation of the mobile phase through the EC cell to attenuate background oxidizable species. Centrifugal molecular weight cutoff filters (Microcon Ultracel YM-10; 10 kDa) are from Millipore Corporation. Polypropylene microcentrifuge tubes (2 ml) are from Sorenson Biosciences, ultracentrifuge tubes (1.5 ml) are from Beckman Instruments, and autosampler vials are from Fisher Scientific. Any reversed-phase C18 column may be used for 3-NT analysis; however, columns with smaller particle sizes (3 μm, or less) and longer column lengths (>100 mm) will enhance resolution, except at the expense of analysis time. We often use a relatively inexpensive Microsorb-MV 100 mm C18 (5-μm particle size) HPLC column from Varian Instruments. Also required for this protocol is a handheld homogenizer (Branson Scientific), microultracentrifuge (e.g., Beckman Optima TLX and TLA100.3 rotor), and a Speed-Vac concentrator (Savant).
3.2. Sample preparation
Tissues and cells to be analyzed (wet weight of ≈50−100 mg) are rinsed of blood or media, respectively, by multiple washes in iced phosphate-buffered saline (PBS) buffer (pH 7.2−7.4). Tissue samples are placed in a weigh boat containing a volume of up to 500 μl buffer A, comprising 50 mM Tris-HCl, 150 mM NaCl, 0.1 mM EDTA, and 20 mM CHAPS (pH 7.4), and minced with a fine scissors. The minced tissue is transferred to a 1.5-ml ultracentrifugation tube and disrupted on ice using three 10-s bursts of a small handheld homogenizer. Cells in culture are scrape harvested (after washing in iced PBS), transferred quantitatively to a 15-ml conical tube, pelleted at 800 g, transferred in up to 500 μl of buffer A to an ultracentrifugation tube, and subjected to three 10-s bursts of a handheld homogenizer. Tissue and cell homogenates are ultracentrifuged at 100,000 g for 60 min in a Beckman TLA100.3 rotor, and supernatants are retained for isolation of 3-NT. A 20-μl aliquot of the supernatant should be preserved for the analysis of total protein content using the Bio-Rad DC assay (or other comparable protein assay) using bovine serum albumin as a standard. Note that assay of total protein in each sample is essential, as this determines the amount of protease to be added to the supernatant for complete protein digestion and additionally provides the normalization factor for final determination of protein 3-NT (i.e., protein 3-NT content is typically expressed on a pmol/mg protein basis). If a pure protein is the subject of 3-NT quantification, then 50 μg is recommended as a convenient starting quantity of protein. For analysis of 3-NT in pure proteins, the homogenization and ultracentrifugation steps can be bypassed.
Supernatants obtained after homogenization and ultracentrifugation (or known quantities of pure proteins) are treated with a 3:1 v/v of ice-cold acid precipitation buffer (0.1 M phosphoric acid, 0.23 M TCA). The pellet is resuspended in Buffer A and subjected to complete proteolytic digestion with proteinase K (10 U/mg protein) in a total volume of up to 500 μl. The digestion mixture should be allowed to incubate for 8 h at 55°C in 2-ml capacity polypropylene microcentrifuge tubes (note that these tubes accommodate the subsequently added volume of acetonitrile used for 3-NT extraction, described later). It was shown previously that proteinase K is more efficient than either pronase or trypsin for complete digestion of both plasma and tissue samples (Skinner et al., 1997). Using these digestion conditions, 30% of the proteinase K is self-hydrolyzed (determined by UV/VIS spectroscopy) and thus contributes to the total pool of amino acids analyzed by HPLC-EC. Nevertheless, proteinase K autolysis poses no interference for 3-NT detection in samples. Indeed, Fig. 1.1 (chromatogram B) affirms that HPLC-EC signals derived from the proteinase K autolysis are well separated from the 3-NT signal in a typical chromatogram and would not result in false 3-NT quantification. In addition, Fig. 1.1 (chromatogram A) demonstrates that proteinase K itself does not contribute a detectable level of 3-NT to a study sample.
Figure 1.1.
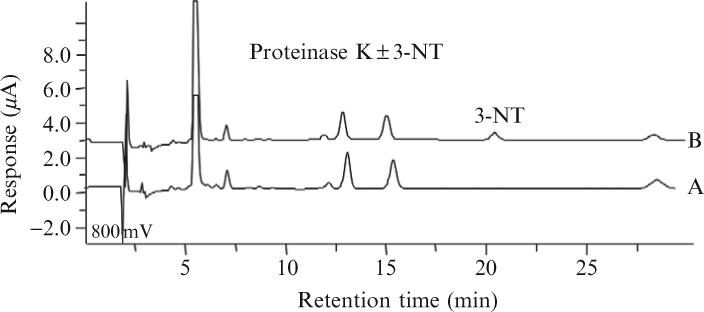
HPLC-EC analysis of 10 units proteinase K inthe presence (chromatogram B) and absence (chromatogram A) of (200 pmol) 3-NT. Proteinase K (±3-NT) was treated to the full procedure of heating, extraction, and isolation as for analysis of 3-NT in an unknown sample.
Digested protein solutions are cooled to room temperature, followed by extraction in the same tubes with ice-cold acetonitrile (3:1 v/v; acetonitrile: sample). Following the addition of acetonitrile, samples are vortexed, incubated on ice for 5 min, and then centrifuged at 12,000 g for 15 min at 4°C. Note that during the extraction step, precipitation is anticipated; however, the supernatant (acetonitrile extract) retains the hydrolyzed amino acids, including 3-NT. Next, 500-μl volumes of the supernatants are transferred to new microcentrifuge tubes and concentrated to dryness at room temperature in a Speed-Vac (Savant Instruments; note that limiting the supernatant volume to 500 μl/tube expedites the drying process). Depending on protein concentration and sample volume, this step typically takes ≈4 to 6 h. After drying, samples may be stored indefinitely at −80° prior to analysis for 3-NT content.
For analysis of 3-NT, dried samples are resuspended in HPLC mobile phase and pooled (when desired) to give a final combined volume of 250 μl. Prior to HPLC injection, the resuspended samples are then filtered through a 10-kDa cutoff centrifugal concentrator to remove residual high molecular weight species. The HPLC mobile phase buffer is as described previously (Crabtree et al., 2003), containing 90 mM sodium acetate, 35 mM citric acid, 130 μM EDTA, and 460 μM sodium octane sulfonate (pH 4.35) in Milli-Q water (vacuum filtered as described earlier).
3.3. Reverse-phase HPLC-EC analysis of 3-NT in samples
HPLC analysis is performed isocratically using a C18 reversed-phase column and mobile phase (see earlier), composed as described previously. Using a flow rate of 0.75 ml/min and on an optimally prepared C18 column, the retention time of 3-NT may range from 10 to 20 min, depending on the choice of column length, particle size, length and bore of tubing connections, and operating temperature. Confirmation of the authenticity of a putative 3-NT signal can be performed as described later. The optimal potential for complete oxidation of 3-NT and hence electrochemical detection is 800 mV. To eliminate interfering signals that arise from species that are oxidized more readily than 3-NT, and additionally affirm complete oxidation of 3-NT at 800 mV, the multichannel EC detector is additionally set to flanking voltages of 700 and 900 mV, respectively (Crabtree et al., 2003).
The reproducibility of HPLC-EC chromatogram signal intensity and the retention time of 3-NT are determined by the quality of the C18 column used and the proper maintenance of the electrochemical cell. Optimal reproducibility is ensured by (1) routine washing of the column between uses, with methanol (with the electrochemical cell disconnected), (2) safeguarding the electrochemical cell by ensuring that the potentials are not applied when the mobile phase is not flowing or when the system is being flushed with organic solvent (>10%), and (3) periodic reconditioning of the electrode when a performance loss is observed (briefly, this involves the repeated application of alternating potentials between 1000 and −450 mV, during perfusion with the mobile phase).
3.4. Interpretation of HPLC chromatograms for quantification of 3-NT
Quantification of 3-NT is performed by comparing the peak area of an unknown sample to the peak area of an external standard comprising a known concentration of pure 3-NT. External standards may range from 1 to 1000 pmol, although the expected range for biological samples is typically 1 to 400 pmol per mg protein. The limit of 3-NT detection by HPLC-EC with a pristine EC cell is ≈500 fmol, rising to ≈4 pmol with a 4-year-old extensively used but otherwise well-maintained EC cell.
The identity of a peak in a complex chromatogram as arising from 3-NT is evidenced by the fulfillment of multiple expectations: (1) retention time of the peak is indistinguishable from authentic 3-NT; (2) electrochemical properties of the peak are identical to authentic 3-NT, that is, minimal oxidation at 700 mV and complete oxidation at 800 mV; (3) the peak coelutes with authentic 3-NT in a 3-NT-“spiked” sample; and (4) treatment of the sample with a final concentration of 10 mM sodium hydrosulfite (dithionite) prior to HPLC-EC analysis reduces 3-nitrotyrosine to 3-aminotyrosine (Riordan and Sokolovsky, 1971), silencing the electrochemical signal of bona fide 3-NT (see note later on the dithionite reaction). Generally, it is desirable to employ more than one approach to confirm the identity of the 3-NT peak—nonetheless, sample availability may limit validation. Along with the retention time and electrochemical properties of the 3-NT peak (approaches 1 and 2 given earlier), chemical reduction of the 3-NT signal provides a convincing method for 3-NT confirmation (approach 5). It is notable that the addition of sodium dithionite typically results in a cloudy precipitate immediately after addition to test samples. Therefore, it is necessary to filter dithionite-treated samples with a syringe filter (0.22-μm Millex GV filters; Millipore Corp.) prior to sample injection.
Figure 1.2 shows a chromatogram (trace A) for ONOO−-treated bovine serum albumin (BSA), revealing multiple peaks and a complex array of preexisting oxidizable species and ONOO−-derived oxidation products. Trace B in Figure 1.2 demonstrates that the 3-NT peak in ONOO−-treated BSA can be identified by its retention time, relative to a 3-NT standard, and its disappearance following exposure of the sample to 10 mM dithionite.
Figure 1.2.

(A) HPLC-EC chromatogram of 3-NT in bovine serum albumin, following reaction of BSAwith 500 μM peroxynitrite (ONOO−). The retention time for 3-NT is denoted by the asterisk. (B) HPLC-EC chromatogram for ONOO−-treated BSA is as in chromatogram A, but following chemical reduction of 3-NT using sodium dithionite. As highlighted by the arrow, dithionite treatment silences the 3-NT signal by reducing the 3-nitro moiety to 3-amino, providing confirmation of the identity of the presumed 3-NT peak.
4. Beyond Quantification: Specification of 3-NT Sites in Proteins
While quantification of total protein 3-NT levels is extremely useful for identifying changing levels of exposure to nitrosative stress in cells and tissues and can be accomplished effectively using the HPLC-EC method detailed earlier, mechanistic questions regarding nitrosative signaling are not answered by knowledge of the total protein 3-NT content. For example, in neurodegenerative illnesses such as Alzheimer's disease, it is established that 3-NT levels increase in the brain, especially in disease-associated plaques (Smith et al., 1997; Tohgi et al., 1999), yet it is uncertain whether protein nitration is a cause or effect of the disease process. To understand how tyrosine nitration may contribute to Alzheimer's disease pathogenesis, one would need to identify the particular proteins that become nitrated, specify their sites of nitration, and elucidate the functional consequences of this modification. Further, one would want to know the degree of nitration at these sites (i.e., quantity of nitrated vs non-nitrated tyrosines) to infer the extent of nitration-evoked change in the function of a given protein. Unfortunately, methods that enable an unbiased identification and quantification of protein nitration sites in biologically complex protein mixtures have been elusive. To date, discovery of the majority of currently recognized biologically relevant protein nitration sites have been arduously identified on a protein-by-protein basis, using an iterative approach involving mass spectrometry-based peptide sequencing, Tyr mutagenesis, and protein functional analysis. Notwithstanding, various strategies have been described in the literature for unbiased protein tyrosine-nitration site identification, although none can be applied comprehensively to discover low endogenous modification levels, physiologically present in protein extracts from tissues. Furthermore, no proteomic strategy has yet been devised to enable the simultaneous, high-throughput and unbiased quantification of changes in 3-NT content in what is likely to be thousands of individual 3-NT-containing proteins present in tissues following exposure to acute or chronic nitrosative stress.
Among the strategies that have been presented for unbiased identification of protein nitration sites, bottom-up mass spectrometry (MS; i.e., analysis of protein digests) seems to be uniquely suited for 3-NT site specification (Kanski et al., 2005). However, given the enormous complexity of protein biological tissues, coupled with a relatively low abundance of protein tyrosine nitration, peptide prefractionation and enrichment steps are likely to be crucial for broad specification of 3-NT modification sites. A recent step toward success in this endeavor is described in a report by Smith and colleagues, who were able to identify 29 nitrated proteins from healthy mouse brain extracts using first-dimension HPLC prefractionation, followed by nano-LC-MS/MS analysis (Sacksteder et al., 2006). It is notable that this relative success was achieved using extensive sample prefractionation and MS instrument time, sufficient to enable the identification of 7792 proteins in total, culminating in the discovery of a mere 29 nitration sites. Although this represents a significant advance, it is reasonable to assume that a far larger number of nitrated proteins and their cognate sites went undiscovered.
Because most researchers who seek to identify endogenously nitrated proteins do not have access to the powerful mass spectrometry resources employed by Smith and co-workers for the study just described (Sacksteder et al., 2006), other investigators have attempted enrichment of nitrated proteins prior to analysis by MS (Kanski and Schoneich, 2005). For example, some investigators have used anti-nitrotyrosine antibodies to immunoprecipitate nitrated proteins prior to further purification and analysis by MS or MS/MS (Kanski et al., 2005). Although immunoprecipitation can be used for effective enrichment of 3-NT-containing proteins, it is notable that a significant number of nonnitrated proteins coimmunoprecipitate. This apparent lack of specificity may arise from undesired interactions of anti-3-NT antibodies with 3-NT-free proteins (Aulak et al., 2004), such as those which contain 5-hydroxynitrotryptophan (Rebrin et al., 2007), and the predicted pull down of proteins that associate with 3-NT-containing proteins.
Perhaps the most commonly used unbiased approach for identifying 3-NT-containing proteins has been two-dimensional (2D) gel electrophoresis, wherein protein spots that are specifically recognized by Western blotting with an anti-3-NT antibody are cored for subsequent in-gel trypsinolysis and protein identification by MS or MS/MS (Castegna et al., 2003; Turko et al., 2003). Although this method has been used successfully to identify novel nitrated proteins, it suffers from several shortcomings (Kanski and Schoneich, 2005). Principal among these shortcomings is that, only in the odd case where a specific site of nitration can be assigned by MS/MS to a peptide ion (e.g., by the observed addition of 45 Da as nitrate, or loss of 181 Da as a hallmark immonium product ion), the identity of a putative 3-NT-containing protein can only be considered as tentative. An additional weakness of the approach is that the labor involved in individually extracting and processing proteins from the 2D gel limits the number of proteins that can be analyzed practically by MS. Furthermore, the resolution of proteins on 2D gels is biased against hydrophobic proteins and proteins at the extremes of the pI and molecular mass ranges. Moreover, low-abundance 3-NT-containing proteins will be challenging to detect by Western blotting and hence, will tend to not be selected for MS analysis. Finally, the 2D gel approach can result in numerous false positives, resulting from either the unintentional extraction of overlapping proteins or the identification of proteins that were detected nonspecifically by the 3-NT antibody.
The relatively low abundance of 3-NT compared to non-3-NT-containing proteins in cells and tissues, combined with the limitations of 2D gel electrophoresis and Western blotting for recognition of 3-NT-containing proteins, evidences the need for a more effective approach to enrich endogenously nitrated proteins and/or peptides for high-throughput sequence analysis by MS. One solution would be to develop an affinity-tagging strategy for the selective purification of 3-NT-containing peptides in digests of biologically complex protein mixtures. To address an analogous methodological need, our laboratory previously developed the SNOSID method for isolation and unbiased specification of S-nitrosylation sites on cysteine residues in complex protein mixtures (Hao et al., 2006). The SNOSID method builds on the biotin-switch technique (Jaffrey et al., 2001) for introduction of a chemically cleavable biotin-containing “tag” at sites of protein S-nitrosylation. The biotin tag enables highly selective capture of peptides on immobilized avidin and allows facile peptide release upon cleavage of the tag. Thus, just as the SNOSID method enabled the identification of endogenously S-nitrosylated proteins, we hypothesize that a similar biotin-based affinity purification strategy may be applied to broadly identify sites of endogenous nitration on peptides in complex mixtures and specify their proteins of origin.
A review of the protein 3-NT literature finds that two such affinity-purification strategies have previously been proposed, both of which take significant steps toward the goal of unbiased identification of modification sites in endogenously nitrated proteins. In 2003, Nikov et al. proposed a strategy that takes advantage of the ability of sodium dithionite to selectively convert 3-NT to 3-aminotyrosine, thus providing a chemical means to introduce a new amino group into a peptide that can be targeted for modification with an amine-reactive affinity tag. To test their approach, the authors nitrated human serum albumin and then treated it with a 200-fold molar excess of sodium dithionite in 50 mM ammonium bicarbonate buffer. After removal of the dithionite on a desalting column, the protein was reconstituted in 50 mM sodium acetate buffer, pH 5.0, in 0.1% sodium dodecyl sulfate and treated for 30 min with a 2-fold molar excess of an amine-reactive biotin reagent, sulfo-NHS-SS-biotin. The resulting biotinylated proteins were affinity captured on immobilized streptavidin, digested with trypsin, and then analyzed by MS-based sequencing—details of this approach are schematized in Fig. 1.3.
Figure 1.3.

A strategy described by Tannenbaum and colleagues (Nikov et al., 2003) for immunoprecipitation of 3-NT-containing proteins and enrichment of 3-NT peptides for potential unbiased identification (modified from Nikov et al., 2003).
The Nikov et al. (2003) strategy represents an important conceptual advance and enabled the investigators to identify an in vitro nitrated protein that had been spiked into the complex milieu of mouse plasma. However, the authors were unable to accomplish the practical goal of discovering endogenously nitrated proteins and specifying their modification sites. This shortcoming can be partly explained by the authors' reliance on pH to guide the specificity of the sulfo-NHS-SS-biotin reagent. Because sulfo-NHS-SS-biotin reacts only with nonprotonated amines and the amino group of 3-aminotyrosine has a pKa of ≈5 (more than three orders of magnitude less than the pKa of ε-lysine and N-terminal amino groups; 10.5 and 8.5, respectively), the authors reasoned that aminotyrosines would be selectively biotinylated at pH 5.0. However, this selectivity was apparently not achieved, likely a consequence of the far greater number of ε-lysine and N-terminal amines in the samples (relative to aminotyrosine) and the progressive reaction of sulfo-NHS-SS-biotin with all amine groups over time.
This limitation was clearly appreciated by Zhang et al. (2007), who modified the Nikov et al. strategy and thereby enabled the identification of a number of protein nitration sites in a peroxynitrite-treated brain extract. The Zhang et al. (2007) protocol involves protein digestion with trypsin as a first step, followed by reduction and alkylation of all free cysteine thiols with 5 mM dithiothreitol and 20 mM iodoacetamide. In this procedure, all free amines are then blocked by acetylation, using a 200-fold molar excess of acetic anhydride in ammonium bicarbonate buffer, pH 8.5. (Note: undesired acetylation of hydroxyl residues is reversed by treatment with 20 mM hydroxylamine.) After removal of excess acetic anhydride with a desalting column, peptide 3-NT residues are reduced to 3-aminotyrosine with sodium dithionite treatment. However, instead of biotinylating the newly formed 3-aminotyrosine groups with an amine-reactive biotinylating reagent, as in the Nikov et al. (2003) strategy, the peptides are treated with a 50-fold molar excess of N-succinimidyl S-acetylthioacetate (SATA), followed by treatment with 0.5 M hydroxylamine. This SATA/hydroxylamine treatment results in replacement of the amino group on 3-aminotyrosine with a sulfhydryl group on a chemical linker. The authors then employ a thiol-enrichment technique, pioneered in their laboratory (Liu et al., 2004), and the resulting thiol-enriched peptides are subjected to MS/MS-based amino acid sequence analysis—procedural details are schematized in Fig. 1.4.
Figure 1.4.

A strategy described by Smith and colleagues (Zhang et al., 2007) for unbiased enrichment of nitrated peptides and sequencing by MS/MS (modified from Zhang et al., 2007). Peptide enrichment is performed by the reduction of peptidyl-3-NT to aminotyrosine (using dithionite), which is then reacted with SATA to introduce a sulfhydryl group for selective peptide capture and release on a thiol affinity matrix.
Using this strategy, the authors confidently identified 150 nitration sites on 102 unique proteins in a mouse brain homogenate that had been nitrated previously in vitro. While this is recognized as a significant advance over the Nikov et al. (2003) strategy, which was only used to identify a single nitrated protein spiked into plasma, it fell short of the goal of identifying endogenously nitrated proteins. Furthermore, the 150 protein nitration sites identified by Zhang et al. (2007) represented only 35% of the sequenced spectra—the other 65% of peptides that were “enriched” by the affinity protocol were nonnitrated. The relative inefficiency of the Zhang et al. (2007) strategy for the enrichment of in vitro nitrated peptides is likely to preclude the effective use of this approach to capture and sequence peptides in protein digests containing low endogenous levels of tyrosine nitration, estimated at <0.1% of total tyrosine residues. We predict that the high false-positive peptide capture rate with the Zhang et al. (2007) method lies in the choice of affinity tag. Notably, the efficiency and selectivity of thiol-peptide capture are likely to be many orders of magnitude less than that which can be achieved by taking advantage of the femtomolar Kd interaction of biotin for avidin, one of the strongest interactions known to occur in biological systems (Green, 1963). Incomplete blocking of cysteine thiols may have also contributed to the significant capture of nonnitrated peptides on thiol-Sepharose beads by Zhang et al. (2007).
To overcome some of the perceived shortcomings of the Zhang et al. (2007) protocol, moving it closer to the goal of unbiased identification of endogenous protein nitration sites, we suggest several key modifications. While it is essential that an approach is used that results in virtually complete blocking of all free amine groups (ε-lysine amino and N-terminal amino) prior to reducing peptide 3-NT to aminotyrosine residues, it is important that a positive charge is retained. Notably, a survey of the nitrated peptides discovered by Zhang et al. (2007) reveals that 109 of the 150 identified peptides contain a C-terminal arginine residue, despite the prediction of an approximately equal number of C-terminal lysine- and C-terminal arginine-containing tryptic peptides. This discrepancy is reconciled by the fact that acetic anhydride-mediated acetylation of peptides with a C-terminal lysine will neutralize the positive charge on this amine (in addition to neutralizing the positive charge on N termini), limiting detection by positive ion monitoring MS. In contrast, C-terminal arginine residues will not be acetylated and therefore will predictably retain a single positive charge, allowing them to be detected by positive ion monitoring MS (although sensitivity is likely to be diminished for not being doubly charged). Thus, the choice of acetic anhydride as an amino-blocking agent will result in an unacceptable loss of peptide ion signals by MS.
To overcome the problem of signal loss on MS, we are employing reductive dimethylation to block free amines on tryptic peptides, while retaining their positive charge (see Fig. 1.5). As has been shown previously, reductive dimethylation is a rapid, specific, irreversible, and, most importantly for the present application, allows the modified amines to retain their positive charge at acidic pH (Hsu et al., 2007). We found that 10 μg of a tryptic digest of bovine serum albumin is >99% dimethylated following treatment with a combination of 0.48% formaldehyde and 50 mM sodium cyanoborohydride for 1 h at 37° in 50 mM potassium phosphate buffer (pH 7) and results in peptide ions that can be detected readily in positive ion mode by MS/MS. Another significant advantage of reductive dimethylation is that excess formaldehyde can then be consumed efficiently by the addition of 100 mM glycine preventing the need for a desalting step and attendant peptide losses.
Figure 1.5.

A proposed proteomic strategy for efficient capture of 3-NT-containing peptides in tryptic digests of complex protein mixtures to enable broad proteomic identification of 3-NT modification sites using MS/MS (Nuriel and Gross, unpublished). The schematized approach is exemplified for a representative dipeptide, NO2-Tyr-Lys. The overall strategy is an extension of the biotin-switch method (Jaffrey and Snyder, 2001) adapted for high-throughput identification of S-nitrosylation sites on protein cysteine residues (Hao et al., 2006). After complete blocking of free amino groups (by dimethylation), peptides are treated with dithionite to chemically reduce NO2-Tyr to NH2-Tyr. This newly formed amine is then modified by reaction with sulfo-NHS-SS-biotin, a thiol-cleavable and amine-selective biotinylating agent (note that an acid-cleavable biotinylating agent can similarly be employed). The biotinylated peptides are affinity captured on streptavidin-Sepharose and washed extensively, and nonbiotinylated peptides are eluted by treatment with β-mercaptoethanol for LC-MS/MS-based amino acid sequence analysis. This approach is expected to provide a more confident and comprehensive identification of protein 3-NT-modification sites than obtained previously.
For reduction of the 3-NT residues to 3-aminotyrosine, we are employing dithionite treatment (5 mM for < 1 min.). However, dithionite treatment has been shown to result in a small percentage of side-products. Therefore, we are currently investigating alternative means for reduction. After reduction, 3-Aminotyrosine residues can then be biotinylated using the thiol-cleavable amine-reactive reagent such as that employed by Nikov et al. (2003). Thus, by treating the dimethylated peptides with excess sulfo-NHS-SS-biotin in sodium acetate buffer (pH 5.0), biotinylation of 3-aminotyrosine becomes specific, as ε-lysine amines and N-terminal amines are unavailable for reaction. Moreover, even if dimethylation was incomplete, a low abundance of unblocked lysine and N-terminal amines would predictably be tolerated inasmuch as the biotinylation of these amines should be relatively inefficient under the acidic conditions employed. After removing the excess biotin reagent, the biotinylated proteins are affinity purified on streptavidin-Sepharose and eluted using β-mercaptoethanol. These eluted peptides are biotin free, but retain a thiol vestige on the aminotyrosine residue that can be utilized by MS analysis to verify their authenticity as formerly nitrated peptides.
The combination of amine blocking and biotin tagging is anticipated to greatly improve the purification efficiency of 3-NT-containing peptides, relative to the two previously reported strategies discussed earlier. Although the procedure awaits optimization, we expect a version of this strategy to significantly advance the goal of unbiased and comprehensive identification of endogenously nitrated proteins. Furthermore, because N termini of nitrated peptides are dimethylated during the course of discovery, it should be a simple matter to modify this protocol for isotopically encoded modifications (i.e., with H2/C13 vs H1/C12-labeled formaldehyde) to allow quantification of changes in endogenously nitrated protein expression with diseases and treatments.
ACKNOWLEDGMENT
This work was supported by NIH Grants HL46403, HL50656, and HL80702.
REFERENCES
- Aulak KS, Koeck T, Crabb JW, Stuehr DJ. Proteomic method for identification of tyrosine-nitrated proteins. Methods Mol. Biol. 2004;279:151–165. doi: 10.1385/1-59259-807-2:151. [DOI] [PubMed] [Google Scholar]
- Beckmann JS, Ye YZ, Anderson PG, Chen J, Accavitti MA, Tarpey MM, White CR. Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol. Chem. Hoppe Seyler. 1994;375:81–88. doi: 10.1515/bchm3.1994.375.2.81. [DOI] [PubMed] [Google Scholar]
- Brodsky SV, Gealekman O, Chen J, Zhang F, Togashi N, Crabtree M, Gross SS, Nasjletti A, Goligorsky MS. Prevention and reversal of premature endothelial cell senescence and vasculopathy in obesity-induced diabetes by ebselen. Circ. Res. 2004;94:377–384. doi: 10.1161/01.RES.0000111802.09964.EF. [DOI] [PubMed] [Google Scholar]
- Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer's disease brain. J. Neurochem. 2003;85:1394–13401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- Crabtree M, Hao G, Gross SS. Detection of cysteine S-nitrosylation and tyrosine 3-nitration in kidney proteins. Methods Mol. Med. 2003;86:373–384. doi: 10.1385/1-59259-392-5:373. [DOI] [PubMed] [Google Scholar]
- Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA, Halliwell B, van der Vliet A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- Frost MT, Halliwell B, Moore KP. Analysis of free and protein-bound nitrotyrosine in human plasma by a gas chromatography/mass spectrometry method that avoids nitration artifacts. Biochem. J. 2000;345:453–458. [PMC free article] [PubMed] [Google Scholar]
- Green NM. Avidin. 1. The use of (14-C)biotin for kinetic studies and for assay. Biochem. J. 1963;89:585–591. doi: 10.1042/bj0890585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halejcio-Delophont P, Hoshiai K, Fukuyama N, Nakazawa H. No evidence of NO-induced damage in potential donor organs after brain death. J. Heart Lung Transplant. 2001;20:71–79. doi: 10.1016/s1053-2498(00)00207-2. [DOI] [PubMed] [Google Scholar]
- Hao G, Derakhshan B, Shi L, Campagne F, Gross SS. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc. Natl. Acad. Sci. USA. 2006;103:1012–1017. doi: 10.1073/pnas.0508412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JL, Chen SH, Li DT, Shi FK. Enhanced a1 fragmentation for dimethylated proteins and its applications for N-terminal identification and comparative protein quantitation. J. Proteome Res. 2007;6:2376–2383. doi: 10.1021/pr060639n. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H. Biological tyrosine nitration: A pathophysiological function of nitric oxide and reactive oxygen species. Arch. Biochem. Biophys. 1998;356:1–11. doi: 10.1006/abbi.1998.0755. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein s-nitrosylation: A physiological signal for neuronal nitric oxide. Nat. Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci. STKE. 2001;2001:PL1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- Kanski J, Behring A, Pelling J, Schoneich C. Proteomic identification of 3-nitrotyrosine-containing rat cardiac proteins: Effects of biological aging. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H371–H381. doi: 10.1152/ajpheart.01030.2003. [DOI] [PubMed] [Google Scholar]
- Kanski J, Schoneich C. Protein nitration in biological aging: Proteomic and tandem mass spectrometric characterization of nitrated sites. Methods Enzymol. 2005;396:160–171. doi: 10.1016/S0076-6879(05)96016-3. [DOI] [PubMed] [Google Scholar]
- Liu T, Qian WJ, Strittmatter EF, Camp DG, 2nd, Anderson GA, Thrall BD, Smith RD. High-throughput comparative proteome analysis using a quantitative cysteinyl-peptide enrichment technology. Anal. Chem. 2004;76:5345–5353. doi: 10.1021/ac049485q. [DOI] [PubMed] [Google Scholar]
- Maruyama W, Hashizume Y, Matsubara K, Naoi M. Identification of 3-nitro-L-tyrosine, a product of nitric oxide and superoxide, as an indicator of oxidative stress in the human brain. J. Chromatogr. B Biomed. Appl. 1996;676:153–158. doi: 10.1016/0378-4347(95)00400-9. [DOI] [PubMed] [Google Scholar]
- Nikov G, Bhat V, Wishnok JS, Tannenbaum SR. Analysis of nitrated proteins by nitrotyrosine-specific affinity probes and mass spectrometry. Anal. Biochem. 2003;320:214–222. doi: 10.1016/s0003-2697(03)00359-2. [DOI] [PubMed] [Google Scholar]
- Oldreive C, Zhao K, Paganga G, Halliwell B, Rice-Evans C. Inhibition of nitrous acid-dependent tyrosine nitration and DNA base deamination by flavonoids and other phenolic compounds. Chem. Res. Toxicol. 1998;11:1574–1579. doi: 10.1021/tx980163p. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebrin I, Bregere C, Kamzalov S, Gallaher TK, Sohal RS. Nitration of tryptophan 372 in succinyl-CoA:3-ketoacid CoA transferase during aging in rat heart mitochondria. Biochemistry. 2007;46:10130–10144. doi: 10.1021/bi7001482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JF, Sokolovsky M. Reduction of nitrotyrosyl residues in proteins. Biochim Biophys. Acta. 1971;236:161–163. doi: 10.1016/0005-2795(71)90160-7. [DOI] [PubMed] [Google Scholar]
- Sacksteder CA, Qian WJ, Knyushko TV, Wang H, Chin MH, Lacan G, Melega WP, Camp DG, 2nd, Smith RD, Smith DJ, Squier TC, Bigelow DJ. Endogenously nitrated proteins in mouse brain: Links to neurodegenerative disease. Biochemistry. 2006;45:8009–8022. doi: 10.1021/bi060474w. [DOI] [PubMed] [Google Scholar]
- Schwedhelm E, Tsikas D, Gutzki FM, Frolich JC. Gas chromatographic-tandem mass spectrometric quantification of free 3-nitrotyrosine in human plasma at the basal state. Anal. Biochem. 1999;276:195–203. doi: 10.1006/abio.1999.4361. [DOI] [PubMed] [Google Scholar]
- Shigenaga MK, Lee HH, Blount BC, Christen S, Shigeno ET, Yip H, Ames BN. Inflammation and NOx-induced nitration: Assay for 3-nitrotyrosine by HPLC with electrochemical detection. Proc. Natl. Acad. Sci. USA. 1997;94:3211–3216. doi: 10.1073/pnas.94.7.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner KA, Crow JP, Skinner HB, Chandler RT, Thompson JA, Parks DA. Free and protein-associated nitrotyrosine formation following rat liver preservation and transplantation. Arch. Biochem. Biophys. 1997;342:282–288. doi: 10.1006/abbi.1997.0114. [DOI] [PubMed] [Google Scholar]
- Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J. Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohgi H, Abe T, Yamazaki K, Murata T, Ishizaki E, Isobe C. Alterations of 3-nitrotyrosine concentration in the cerebrospinal fluid during aging and in patients with Alzheimer's disease. Neurosci. Lett. 1999;269:52–54. doi: 10.1016/s0304-3940(99)00406-1. [DOI] [PubMed] [Google Scholar]
- Turko IV, Li L, Aulak KS, Stuehr DJ, Chang JY, Murad F. Protein tyrosine nitration in the mitochondria from diabetic mouse heart: Implications to dysfunctional mitochondria in diabetes. J. Biol. Chem. 2003;278:33972–33977. doi: 10.1074/jbc.M303734200. [DOI] [PubMed] [Google Scholar]
- Upmacis RK, Crabtree MJ, Deeb RS, Shen H, Lane PB, Benguigui LE, Maeda N, Hajjar DP, Gross SS. Profound biopterin oxidation and protein tyrosine nitration in tissues of ApoE-null mice on an atherogenic diet: Contribution of inducible nitric oxide synhase. Am. J. Phys. Heart Circ. Phys. 2007;293:H2878–H2887. doi: 10.1152/ajpheart.01144.2006. [DOI] [PubMed] [Google Scholar]
- Yi DH, Ingelse BA, Duncan MW, Smythe GA. Quantification of 3-nitrotyrosine in biological tissues and fluids: Generating valid results by eliminating artifactual formation. J. Am. Soc. Mass Spectrom. 2000;11:578–586. doi: 10.1016/S1044-0305(00)00113-6. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Qian WJ, Knyushko TV, Clauss TR, Purvine SO, Moore RJ, Sacksteder CA, Chin MH, Smith DJ, Camp DG, 2nd, Bigelow DJ, Smith RD. A method for selective enrichment and analysis of nitrotyrosine-containing peptides in complex proteome samples. J. Proteome Res. 2007;6:2257–2268. doi: 10.1021/pr0606934. [DOI] [PubMed] [Google Scholar]