

Abstract
Cerebral vasodilatory responses evoked by activation of NMDA receptors and by hypercapnia are important factors in the integrated vascular response to perinatal cerebral ischemia. Cerebral vasodilation to NMDA is mediated by nitric oxide in adult and newborn animals, whereas vasodilation to hypercapnia is thought to become modulated by nitric oxide, at least in swine, after the newborn period. The developmental role of nitric oxide in the cerebral blood flow response to NMDA and hypercapnia was investigated at mid- and late gestation in fetal sheep. Superfusion of 300 μM NMDA over the cerebral cortex through a closed cranial window on the exteriorized head of an anesthetized fetus increased laser-Doppler flow by 41 ± 7% (± S.E.) at 0.65 gestation. The increase was reduced by superfusion of a nitric oxide synthase inhibitor (18 ± 8%). At 0.9 gestation, the response to NMDA was augmented (85 ± 24%) compared to that at 0.65 gestation and was reduced by a nitric oxide synthase inhibitor (32 ± 6%). In unanesthetized fetal sheep, hypercapnic reactivity of microsphere-determined cerebral blood flow was not significantly attenuated by nitric oxide synthase inhibition at 0.65 gestation (4.6 ± 0.7 to 3.7 ± 1.0% change/mmHg pCO2) or at 0.9 gestation (4.0 ± 0.7 to 3.5 ± 0.9% change/mmHg pCO2). Therefore, nitric oxide-dependent cerebrovascular dilation to NMDA-receptor activation is present as early as 0.65 gestation in fetal sheep and increases further during the last trimester, whereas vasodilation to hypercapnia remains unchanged and independent of nitric oxide during the last trimester. Hence, cerebrovascular reactivities to different stimuli do not mature concurrently.
Keywords: Cerebral blood flow, Fetal sheep, Hypercapnia, NMDA receptor, Neurovascular coupling, Nitric oxide
1. Introduction
Hypoxia-ischemia in neonates born prematurely can result in white matter injury with secondary disturbances in gray matter (Volpe, 2005), whereas hypoxia-ischemia in those born near term can result in primary injury to peri-Rolandic cortical gray matter and basal ganglia (Miller et al., 2005). In fetal sheep, cortical gray matter is also less susceptible to ischemic injury at 0.65 gestation compared to 0.9 gestation (Reddy et al., 1998). This change in susceptibility is presumably related to maturation of synaptic connections, expression of excitatory neurotransmitter receptors, increased spontaneous electrical activity, and associated increased oxidative metabolism. In postnatal rats, the increased susceptibility to hypoxia-ischemia seen at postnatal day 7 corresponds to an increased susceptibility to excitotoxic injury by N-methyl-D-aspartate (NMDA) (McDonald et al., 1988; Ikonomidou et al., 1989).
Excitotoxic injury is largely dependent on stimulation of nitric oxide synthase (NOS) in neurons. Expression of NOS is developmentally regulated differentially among regions. In fetal sheep with 145 days of gestation, expression of NOS in the neuropil of the cortical plate reaches a peak at 71 days, whereas neuronal somatodendritic expression progressively increases between 71 and 110 days (Northington et al., 1996). This change in cortical cell localization corresponded with fourfold increase in NOS catalytic activity and threefold increase in cerebral blood flow (CBF) and cerebral O2 consumption between 92 days and 135 days (Northington et al., 1997). Co-localization of NOS in postsynaptic density proteins is important for activation by localized calcium influx through NMDA-receptor channels. Thus, the temporal localization of NOS parallels vulnerability to hypoxic-ischemic cortical injury in fetal sheep.
Coupling of cerebral vasodilation to activation of NMDA receptors is dependent on nitric oxide (NO) production (Faraci and Breese, 1993; Pelligrino et al., 1996). Cerebrovascular coupling dependent on NO has also been demonstrated in immature animals such as newborn piglets (Meng et al., 1995) and lambs (Northington et al., 1995). However, maturation of this vascular coupling mechanism has not been investigated during early development prenatally under conditions of intrauterine fetal blood gases and blood pressure.
In addition to NMDA, cerebral vasodilation to hypercapnia can require the presence of NO. However, in this case, NO is thought to act as a permissive enabler rather than a mediator of vasodilation (Iadecola and Zhang, 1996). The magnitude of NO dependency of the vascular reactivity to CO2 varies among adult species and appears to be greater in rat (Iadecola and Zhang, 1994) and cat (Sandor et al., 1994) than in monkey (McPherson et al., 1995) and human (White et al., 1998). In swine, NO assumes a greater role in juveniles than in newborns (Zuckerman et al., 1996). Thus, species and developmental differences exist in the contribution of NO to cerebrovascular reactivity. Whether developmental changes in the role of NO in CO2 reactivity exist in species other than pig and whether these changes occur prenatally in a species with precocious offspring are not known. Substantial cortical development occurs prenatally in fetal sheep, which have been used extensively to study development of cerebrovascular regulation. Baseline CBF increases threefold between 0.65 and 0.9 gestation in fetal sheep to match robust increases in oxidative metabolism (Gleason et al., 1989). However, the relative increase in CBF during hypercapnia is unchanged (Helou et al., 1991) and is similar to that in newborns (Rosenberg et al., 1982).
Regulation of the cerebral vasodilatory responses to NMDA and CO2 is presumed to be important in CBF regulation during perinatal hypoxia-ischemia, where NMDA receptors are activated and where carbonic acidosis occurs. Thus, the role of NO in these vascular responses during fetal development is of interest. We tested the hypotheses (1) that the percent increase in cortical CBF during NMDA administration in fetal sheep at 0.9 gestaion is greater than at 0.65 gestation, (2) that the increase in cortical CBF evoked by NMDA is reduced by an NO synthase inhibitor, and (3) that NO synthase inhibition reduces CBF during normocapnia and hypercapnia but does not reduce the percent change in CBF during hypercapnia at either gestational age.
2. Methods
All procedures were approved by the Johns Hopkins University Animal Care and Use Committee and conformed to the National Institutes of Health Guidelines.
2.1. NMDA experiment
To measure CBF responses to NMDA, NMDA was applied topically in a cranial window on the fetal skull and the local perfusion was measured by laser-Doppler flowmetry (LDF) on the cortical surface in nonsurvival experiments on 12 fetuses at 0.65 gestation and 8 fetuses at 0.9 gestation. Cranial windows have been used by others in fetal sheep to assess developmental vasoreactivity in vivo (Kurth and Wagerle, 1992). LDF monitors red blood cell flux in approximately 1 mm3 of underlying cortex. Use of LDF is well suited for capturing the CBF time course and peak response, which can vary among animals. The ewe was anesthetized with halothane and mechanically ventilated through a tracheostomy with halothane and supplemental O2. End-tidal CO2 monitoring was used to adjust ventilation. The maternal femoral vessels were catheterized for pressure monitoring and infusion of lactated Ringer solution. Rectal temperature was maintained at 39.5 °C with a heating blanket. Through a midline laparotomy, an incision was made in the uterus to expose the foreleg for catheterizing the axillary artery. After inserting the foreleg back into the uterus, the head of the fetus was exteriorized, wrapped in cellophane to reduce evaporative cooling, and supported in a molded plaster cast connected to a metal frame to reduce head movement. The rest of the body of the fetus remained in the uterus to minimize disturbance of the placental circulation. A closed cranial window was constructed over the parietal cortex by cementing a 1-cm plastic ring around a craniotomy and gently dissecting the dura. The window was filled with artificial cerebrospinal fluid and closed by gluing a coverslip to the ring, which contained ports for superfusing fluid and for measuring pressure and fluid temperature in the window. Fluid temperature was maintained by external heating. The LDF probe was secured to the glass coverslip, and a drop of mineral oil was used to provide optical continuity. The window was covered to reduce background light scattering.
Artificial cerebrospinal fluid containing 300 μM NMDA was superfused through the cranial window at a rate of 0.2 ml/min for 10 min. The percent change in LDF was measured at 1-min intervals for 40 min. After 40 min of observation, the window was flushed with artificial cerebrospinal fluid. The window was superfused with 1 mM of the NOS inhibitor Nω-nitro-L-arginine (L-NNA) in artificial cerebrospinal fluid at a rate of 0.1 ml/min for 20 min and 0.05 ml/min for an additional 40 min. The total 60-min duration of L-NNA superfusion was based on studies that showed more complete inhibition of NOS activity and vascular responses in tissue underlying an LDF probe when 60 min rather than 30 min was allowed for tissue penetration (Irikura et al., 1994). The LDF response to superfusion of NMDA + L-NNA was then measured. Measurements of blood gases, hemoglobin concentration, and O2 saturation were made on axillary arterial samples obtained at baseline and at 1 h of L-NNA superfusion (before starting each NMDA superfusion). Arterial pressure was continuously monitored.
2.2. Hypercapnia experiment
To measure the CBF response to hypercapnia, the radioactive microsphere technique was used in chronically catheterized fetuses of time-dated pregnant sheep at 0.65 gestation (92–94 days; n = 6) and 0.9 gestation (132–135 days; n = 7), as previously described (Harris et al., 2001; Harris et al., 1989). Because the CBF response to hypercapnia is more stable than the transient response to NMDA, microspheres injected at a single time point can be used to obtain regional CBF measurements during steady state hypercapnia. The sheep were anesthetized with halothane, intubated, and mechanically ventilated with 1.5% halothane, 30% N2O, and balance O2. Under aseptic conditions, the uterus was exposed through a midline abdominal incision. Through a single incision in the uterus, catheters were placed into the fetal axillary arteries, pedal vein, and sagittal sinus. Incisions were closed with suture, and the catheters were exteriorized to the flank of the ewe. Antibiotic administration included 1,200,000 U of procaine penicillin, i.m., and 500 mg of ampicillin into the amniotic fluid.
Regional CBF was measured by injection of 1–1.5 million microspheres into the inferior vena cava (Harris et al., 2001). The microspheres had a nominal diameter of 15 μm and were labeled with either 153Gd, 114mIn, 113Sn, 103Ru, 95Nb, or 46Sc. An arterial blood reference sample was obtained by withdrawal of blood from the axillary artery at a rate of 1.17 ml/min in 93-day fetuses and 2.55 ml/min in 133-day fetuses starting 0.5 min before the microsphere injection and ending 1 min after the end of the injection. The brain was sectioned into cerebrum (cortical grey plus white matter), diencephalon, brainstem, and cerebellum. Cervical spinal cord samples were also obtained. Tissue and blood samples were counted for radioactivity, with correction for spectral overlap of isotopes. Blood flow was calculated as the product of microsphere counts in the tissue and the arterial blood reference withdrawal rate divided by microsphere counts in the arterial reference sample (Harris et al., 1989). Cerebrovascular resistance (CVR) was calculated using the difference between arterial and sagittal sinus pressure as cerebral perfusion pressure.
Fetal blood that was withdrawn during the microsphere injections and sampling for blood gas determination was replaced with blood having a similar O2 affinity. On the day of the experiment (two days after surgery), blood was withdrawn from the ewe. The maternal blood with low O2 affinity was exchange-transfused with the fetus 1 h before the start of the experiment. The blood withdrawn from the fetus during the exchange transfusion had a high O2 affinity and was later transfused back to the fetus to replace blood withdrawn during the experiment.
Baseline measurements were made of fetal arterial blood pressure, pH, pCO2, pO2, hemoglobin, O2 saturation, O2 content, and CBF. Sagittal sinus O2 content was also measured to permit the calculation of cerebral O2 consumption (Harris et al., 2001). Hypercapnia was induced by increasing the CO2 in a gas mixture flowing through a plastic bag that was secured over the head of the ewe, which was standing in a cart. Measurements were made at 8–10 min of hypercapnia. After return to normocapnia, the NOS inhibitor Nω-nitro-L-arginine methyl ester (L-NAME) was infused intravenously into the fetus at a nominal dose of 60 mg/kg based on an estimate of the weight of the fetus during surgery. This dose was previously shown to inhibit brain NOS activity by 82% at 92 days and by 89% at 135 days of gestation (Northington et al., 1997). After 30 min, a third set of measurements was made. Hypercapnia was then repeated and a fourth set of measurements was obtained.
2.3. Statistical analysis
The percent change in LDF after topical application of NMDA was compared over time between groups by two-way repeated measures analysis of variance (ANOVA). The maximum percent change in the response was compared between age groups by unpaired t-test. The effect of L-NNA on the maximum LDF response and other physiologic parameters was compared with and without L-NNA by unpaired t-tests. Reactivity to CO2 was calculated as the percent change in CBF or CVR divided by the change in arterial pCO2. Reactivity was compared before and after L-NAME administration by paired t-tests. Other measurements in the hypercapnia experiment were compared by two-way repeated measures ANOVA, where L-NAME was one factor and CO2 was a second factor. If one of these factors had a significant overall effect or a significant interaction with the other factor, contrasts were performed by paired t-tests with the false-discovery-rate procedure for multiple comparisons (Curran-Everett, 2000). Data are presented as mean ± S.E. with p < 0.05 considered significant.
3. Results
3.1. Responses to NMDA
During 10 min of superfusion of 300 μM NMDA in fetal cranial windows, LDF gradually increased and remained elevated before gradually decreasing at 25–40 min (Fig. 1A). Two-way ANOVA indicated a significant effect of time and of L-NNA treatment in each age group. When the percent responses were averaged at individual time-points, the increase in LDF was blocked at 0.65 gestation (Fig. 1B) and markedly blunted at 0.9 gestation (Fig. 1C).
Fig. 1.

Cortical perfusion measured by laser-Doppler flowmetry and expressed as a percent of baseline values (± S.E.) during and after 10 min of superfusion of a closed cranial window with 300 μM NMDA in fetal sheep at 0.65 (n = 12) and 0.9 (n = 8) gestation (A). The time course of the control response to NMDA is compared with the response to NMDA after superfusion of 1 mM L-NNA in fetal sheep at 0.65 (B) and 0.9 (C) gestation. ANOVA indicated an overall effect of L-NNA treatment on the flow response in both age groups.
The maximum change in LDF at any particular time point was used for individual group comparisons. The maximum response to NMDA was significantly greater in the 0.9-gestation group than in the 0.65-gestation group (Fig. 2). Treatment with L-NNA significantly attenuated the maximum increase in each age group.
Fig. 2.
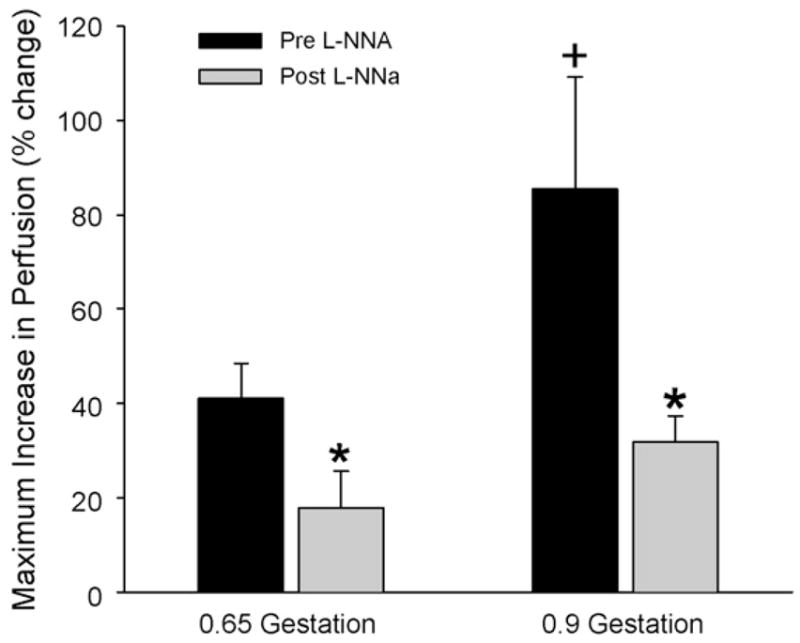
Maximum increase in laser-Doppler flow response (± S.E.) to NMDA over the 40-min observation period before and after L-NNA superfusion over cerebral cortex of fetal sheep at 0.65 and 0.9 gestation. *p < 0.05 from pre-L-NNA response; +p < 0.05 between pre-L-NNA responses of 0.65- and 0.9-gestation groups.
After the 40-min observation period for the NMDA response and the subsequent 60-min period of L-NNA superfusion, fetal arterial O2 content decreased moderately as a result of decreased O2 saturation without a change in hemoglobin concentration (Table 1). This decrease in fetal O2 saturation was assumed to be due to effects of surgery and prolonged anesthesia on the placental circulation. Other physiologic variables such as mean arterial blood pressure and arterial pCO2 were unchanged for the NMDA exposure after L-NNA administration.
Table 1.
Physiologic variables in NMDA experiments
| 0.65 Gestation | 0.9 Gestation | ||
|---|---|---|---|
| MABP (mmHg) | Control | 36 ± 1 | 44 ± 1 |
| L-NNA | 34 ± 1 | 42 ± 1 | |
| PaCO2 (mmHg) | Control | 45 ± 1 | 46 ± 2 |
| L-NNA | 51 ± 2 | 49 ± 3 | |
| Hemoglobin (g/dl) | Control | 12.8 ± 0.4 | 15.6 ± 0.8 |
| L-NNA | 12.7 ± 0.4 | 15.3 ± 0.5 | |
| O2 Saturation (%) | Control | 69 ± 4 | 62 ± 2 |
| L-NNA | 49 ± 4* | 46 ± 4* | |
| Arterial O2 content (ml O2/dl) | Control | 11.9 ± 0.6 | 13.2 ± 0.6 |
| L-NNA | 8.6 ± 0.5* | 9.7 ± 0.8* |
Values are means ± S.E. MABP, mean arterial blood pressure; PaCO2, arterial pCO2.
p < 0.05 from control.
3.2. Hypercapnic reactivity
Baseline CBF in cerebrum of fetal sheep at 0.9 gestation was threefold that at 0.65 gestation. Increasing fetal arterial pCO2 by approximately 20–25 mm Hg increased CBF in both age groups (Fig. 3). Two-way ANOVA indicated a significant overall effect of L-NAME administration and an overall effect of pCO2 in both groups. After administration of L-NAME, CBF during normocapnia decreased by 28 ± 7% at 0.65 gestation and by 25 ± 6% at 0.9 gestation. Subsequent hypercapnia increased CBF in both groups from normocapnic levels. The level of CBF during hypercapnia after L-NAME administration was less than the level during hypercapnia before L-NAME administration. Thus, L-NAME shifted CBF in cerebrum downward during both normocapnia and hypercapnia.
Fig. 3.
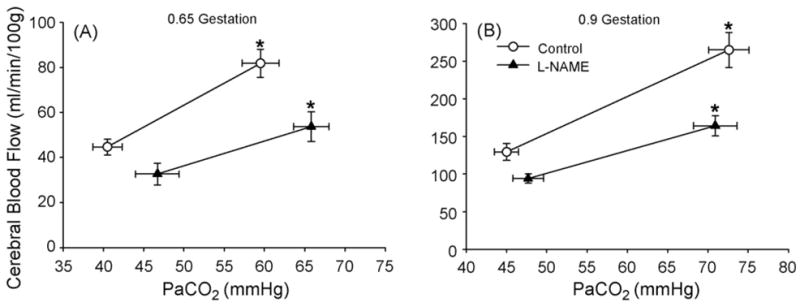
Cerebral blood flow (± S.E.) measured by radiolabeled microspheres in cerebrum vs. arterial pCO2 (PaCO2) before and after systemic administration of L-NAME in fetal sheep at 0.65 gestation (A, n = 6) and at 0.9 gestation (B, n = 7). *p < 0.05 from normocapnic blood flow.
An increase in fetal arterial pO2 during hypercapnia prevented a decrease in arterial O2 saturation or O2 content as a result of the Bohr effect induced by hypercapnia (Table 2). Hemoglobin concentration was elevated after L-NAME administration in both groups, possibly as a result of splenic contracture. Hemoglobin concentration did not change during hypercapnia. Arterial O2 content was statistically increased after L-NAME administration only in the case of hypercapnia in the 0.9-gestation group. Cerebral O2 consumption was not significantly changed by L-NAME administration or by hypercapnia. Mean arterial blood pressure increased after L-NAME administration in both age groups, but only minor changes occurred with induction of hypercapnia. Calculation of CVR in cerebrum indicated significant increases after L-NAME administration (Fig. 4). Decreases in CVR occurred during hypercapnia both before and after L-NAME administration in both groups. As with CBF, two-way ANOVA on CVR data indicated significant overall effects of L-NAME administration and of CO2 level.
Table 2.
Physiologic variables in hypercapnia experiments
| 0.65 Gestation
|
0.9 Gestation
|
||||
|---|---|---|---|---|---|
| Baseline | CO2 | Baseline | CO2 | ||
| MABP (mmHg) | Control | 44 ± 1 | 48 ± 1† | 54 ± 2 | 55 ± 2 |
| L-NAME | 55 ± 2* | 54 ± 1* | 75 ± 3* | 71 ± 3* | |
| PaO2 (g/dl) | Control | 24 ± 2 | 31 ± 1† | 22 ± 2 | 27 ± 2† |
| L-NAME | 21 ± 1* | 26 ± 2*,† | 24 ± 2 | 33 ± 2*,† | |
| Hemoglobin (g/dl) | Control | 9.7 ± 1.9 | 9.9 ± 2.2 | 11.6 ± 3.8 | 11.4 ± 2.8 |
| L-NAME | 12.5 ± 4.4* | 12.2 ± 3.8* | 12.9 ± 4.0* | 12.5 ± 4.6* | |
| O2 Saturation (%) | Control | 67 ± 3 | 67 ± 3 | 59 ± 4 | 61 ± 4 |
| L-NAME | 55 ± 4* | 56 ± 5* | 60 ± 4 | 69 ± 2† | |
| Arterial O2 content (ml O2/dl) | Control | 8.8 ± 0.5 | 9.0 ± 0.5 | 9.3 ± 0.5 | 9.5 ± 0.6 |
| L-NAME | 9.4 ± 0.9 | 9.4 ± 1.0 | 10.5 ± 0.7 | 11.7 ± 0.4*,† | |
| CMRO2 (ml/min/100 g) | Control | 1.3 ± 0.1 | 1.4 ± 0.2 | 3.7 ± 0.2 | 3.1 ± 0.6 |
| L-NAME | 1.4 ± 0.1 | 1.1 ± 0.1 | 3.9 ± 0.4 | 3.8 ± 0.3 | |
Values are means ± S.E. MABP, mean arterial blood pressure; PaO2, arterial pO2; CMRO2, cerebral metabolic rate of oxygen.
p < 0.05 from control.
p < 0.05 from baseline.
Fig. 4.
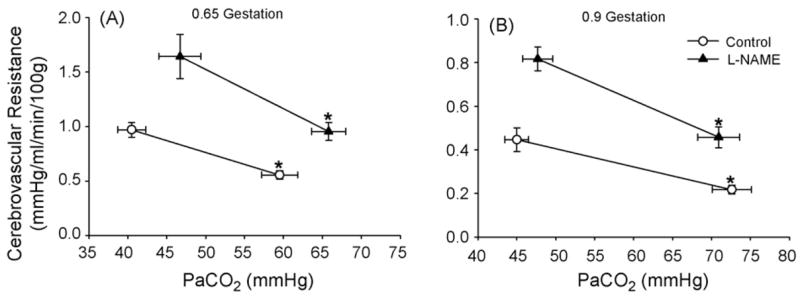
Cerebrovascular resistance in cerebrum (± S.E.) vs. arterial pCO2 (PaCO2) before and after systemic administration of L-NAME in fetal sheep at 0.65 gestation (A, n = 6) and at 0.9 gestation (B, n = 7). *p < 0.05 from normocapnic value.
Because of the shift in baseline CBF and CVR in cerebrum after L-NAME administration, the lower CBF during hypercapnia after L-NAME administration could be attributed to a change in the normocapnic baseline rather than a change in CO2 reactivity. Moreover, the change in arterial pCO2 during hypercapnia after L-NAME administration could not be precisely matched to that before L-NAME administration. The relationship of CBF to arterial pCO2 is approximately linear over a range of 30–80 mmHg (Reivich, 1964; Rosenberg et al., 1982). Therefore, the slopes of the percent responses to CO2 alterations were analyzed as a measure of CO2 reactivity. The percent change in CBF per change in pCO2 during hypercapnia after L-NAME was not different from the percent change before L-NAME at either 0.65 or 0.9 gestation (Fig. 5). Likewise, the percent change in CVR per change in pCO2 during hypercapnia after L-NAME was not different from the percent change before L-NAME. Moreover, hypercapnic reactivity of the CBF and CVR in cerebrum was similar at 0.65 and 0.9 gestation.
Fig. 5.

Cerebral blood flow (CBF) reactivity in cerebrum (A) and cerebrovascular resistance (CVR) reactivity in cerebrum (B) to hypercapnia, expressed as a percent change per mmHg increase in arterial pCO2 (± S.E.) before and after systemic administration of L-NAME in fetal sheep at 0.65 and 0.9 gestation. L-NAME had no significant effect on reactivity.
In addition to cerebrum, analysis of blood flow in cervical spinal cord, cerebellum, brainstem, and diencephalon indicated overall effects of L-NAME and CO2. Two-way ANOVA on percent reactivity of CBF to hypercapnia indicated an overall effect of brain region in each age group but no overall effect of L-NAME administration and no interaction of L-NAME with brain region. Thus, administration of L-NAME did not have a consistent effect on CO2 reactivity in any region at either 0.65 or 0.9 gestation (Fig. 6). However, calculated reactivity was somewhat more variable in subcortical regions among fetuses than in cerebrum.
Fig. 6.
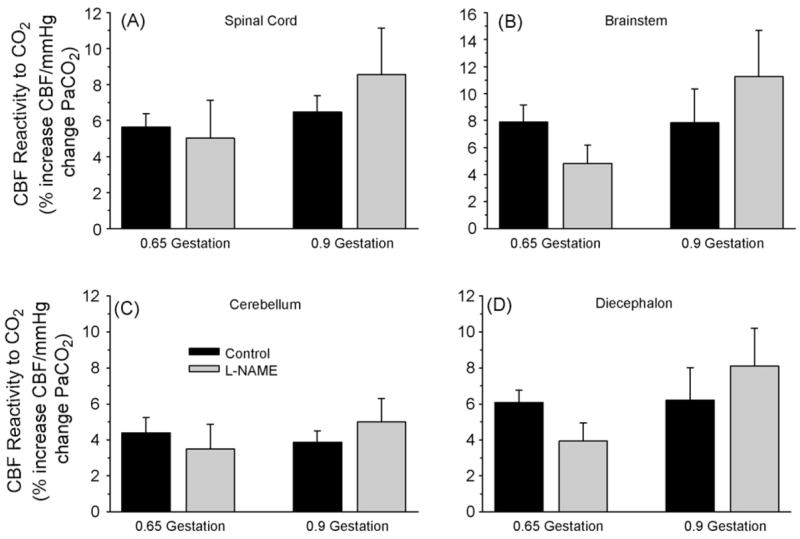
Regional blood flow (CBF) reactivity to hypercapnia, expressed as a percent change per mmHg increase in arterial pCO2 (± S.E.), in cervical spinal cord (A), brainstem (B), cerebellum (C), and diencephalon (D) before and after systemic administration of L-NAME in fetal sheep at 0.65 and 0.9 gestation. L-NAME had no overall significant effect on regional flow reactivity.
4. Discussion
The major findings of this study are (1) that neurovascular coupling to activation of NMDA receptors in neocortex increases during the last trimester in fetal sheep, (2) that NO contributes to this neurovascular coupling as early as 0.65 gestation, and (3) that NO contributes to basal blood flow but is not required for cerebrovascular reactivity to hypercapnia at either 0.65 or 0.9 gestation. Therefore, developmental increases in the role of NO in neurovascular regulation are specific for NMDA-receptor activation.
4.1. NMDA response
The maturation in neurovascular coupling in neocortex corresponds to enrichment of cortical synaptic connections and various glutamate receptors (Furuta and Martin, 1999), a threefold increase in cerebral O2 consumption (Gleason et al., 1989), and a fourfold increase in cortical NOS catalytic activity (Northington et al., 1997). Coupling of Ca2+ currents by NMDA receptors to NOS activity normally requires anchoring by PSD95 (Christopherson et al., 1999). While NOS is present in neurons in the cortical plate at 0.65 gestation, subcellular localization in synaptic processes coincident with NMDA receptors is presumably critical for evoking an increase in NO sufficient for relaxing smooth muscle. In contrast, endothelial NOS is already present in vessels at 0.4 gestation (Northington et al., 1996). Decreases in baseline CBF with systemic L-NAME administration at 0.65 gestation indicate that vascular smooth muscle is already responsive to tonic NO production derived from endothelium and/or neurons. Therefore, increases in the blood flow response to NMDA between 0.65 and 0.9 gestation are likely to be related to maturation of the synaptic molecular machinery that links NMDA receptors with NOS, rather than to an inability of the vascular smooth muscle to respond to NO.
The increase in CBF in response to hypoxia is greater at 0.9 gestation than at 0.65 gestation (Harris et al., 2001). In this case, however, the percent change in CBF was not diminished by L-NAME. In contrast, the CBF response to NMDA was reduced by NOS inhibition at both gestational ages. Thus, vascular responses to both NO-dependent and NO-independent stimuli change during this developmental period.
In newborn lambs NMDA increased CBF, and the response was inhibited by 1 mM of the methyl ester form of L-NNA (Northington et al., 1995). Although perfusion of microdialysis probes in cerebral cortex with the 1-mM concentration almost completely suppressed basal NOS catalytic activity in vivo, a small increase in NOS activity and CBF persisted when NMDA was added to the dialysis perfusion. Thus, the 1-mM dose of L-NNA used in the present study may not have completely blocked NOS activity when NMDA was superfused, and incomplete inhibition may have accounted for the residual LDF response to NMDA seen in the 0.9-gestation group. Alternatively, other mediators may make a small contribution to the vascular response to NMDA by 0.9 gestation. For example, pial arteriolar dilation to NMDA is dependent on both NO (Meng et al., 1995) and on carbon monoxide derived from heme oxygenase activity in piglets (Robinson et al., 2002). Carbon monoxide and NO signaling can interact in a complex manner in piglet pial arteries (Leffler et al., 2005b,a). Upregulation of heme oxygenase function may be important in protecting the brain from oxidative stress at birth. Thus, carbon monoxide or other mediators might also contribute to the residual LDF response to NMDA in near-term fetal sheep.
Sheep were anesthetized with halothane in this experiment. Halothane and other inhalational anesthetics are capable of decreasing NMDA receptor-mediated currents (Criswell et al., 2004) by an interaction with transmembrane segment 3 of NR1 (Ogata et al., 2006) and lead to an attenuation of evoked increases in cGMP (Zuo et al., 1999). An underlying assumption in the present study is that halothane’s suppressive effect on NMDA currents was equivalent in the two gestational age groups of fetuses.
4.2. Hypercapnic response
In agreement with previous work (Harris et al., 2001), administration of L-NAME increased fetal arterial pressure at both ages. The increase in arterial pressure at 0.9 gestation was attributed to increases in vascular resistance throughout the major fetal organs and placenta (Harris et al., 2001). In the present study, administration of L-NAME increased CVR proportionately more than it increased arterial pressure, and it resulted in a decrease in CBF. Nevertheless, hypercapnic reactivity, whether based on the percent change in CBF or the percent change in CVR from the post-L-NAME baseline, was unchanged from the control hypercapnic reactivity. Other physiologic variables did not provide an explanation for the maintained hypercapnic reactivity after L-NAME administration. Oxidative metabolism was not significantly altered by L-NAME or hypercapnia. Arterial O2 content increased approximately 10% during hypercapnia after L-NAME administration in the 0.9-gestation group, but this increase would have acted to suppress hypercapnic reactivity rather than to maintain it (Massik et al., 1989). Thus, the effect of the systemically administered NOS inhibitor was to shift CBF and CVR during both normocapnia and hypercapnia but not to change the relative response to a hypercapnic challenge.
Inhibition of NOS decreases cerebrovascular CO2 reactivity in adult rat and cat (Iadecola and Zhang, 1994; Sandor et al., 1994). In monkeys, the decrease in CO2 reactivity after NOS inhibition was modest and selective for cortex (McPherson et al., 1995). In humans, the role of NO in CO2 reactivity is uncertain (White et al., 1998). Thus, the role of NO in CO2 reactivity appears to vary among species. In swine, the role of NO varies with development. Pial arteriolar reactivity to CO2 was not reduced by NOS inhibition in newborn pigs, whereas reactivity became impaired after NOS inhibition in juvenile pigs (Zuckerman et al., 1996; Willis and Leffler, 1999). Cerebrovascular reactivity to CO2 is primarily dependent on changes in extracellular pH (Kontos et al., 1977; Koehler and Traystman, 1982). In adult rats, the role of NO/cGMP is one of a permissive enabler rather than a mediator (Iadecola and Zhang, 1996; Okamoto et al., 1997), whereas in piglets, prostacyclin/cAMP serves the role of permissive enabler to hypercapnic dilation (Leffler et al., 1994). Results in the present study that showed no effect on the CBF and CVR percent reactivity by L-NAME in either age group of fetuses are consistent with the findings in piglet pial arteries. Thus, data from both sheep and pig indicate that any contribution of NO to CO2 reactivity occurs relatively late in development. Furthermore, L-NAME did not reduce CO2 reactivity at 0.9 gestation in subcortical regions where neural maturation generally precedes neocortex and where CO2 reactivity was somewhat greater than in neocortex. Moreover, CO2 reactivity in newborn lambs is similar to that in 0.9-gestation fetuses when normalized for postnatal increases in cerebral O2 consumption (Rosenberg et al., 1982). Interestingly, further increases in CO2 reactivity were detected between the newborn lambs and adult sheep (Rosenberg et al., 1982). Perhaps postnatal maturation of an NO-dependent vascular mechanism permits additional vasodilation to hypercapnia in adults.
Although absolute CBF increases threefold during the last trimester in fetal sheep, the relative increase in CBF during hypercapnia is similar over this period (Helou et al., 1991). The present work, which showed similar CBF and CVR reactivity to the change in arterial pCO2 on a percentage basis under control conditions at 0.65 and 0.9 gestation, confirms this finding. Other cerebrovascular responses, such as the pial arteriolar dilatory responses to acetylcholine (Wagerle et al., 1992) and adenosine analogs (Kurth and Wagerle, 1992), are also similar at these two gestational ages. Fetuses at 0.65 gestation also exhibit a modest intrinsic ability to autoregulate CBF to decreases in perfusion pressure, but the range of autoregulation is limited (Helou et al., 1994). Thus, some vascular responses, such as autoregulation, hypoxic vasodilation and neurovascular coupling to NMDA, continue to develop between 0.65 and 0.9 gestation, whereas vascular responsivity to CO2, acetylcholine, and adenosine remain unchanged during this period. Taken together with previous work, the present results support the view that selected aspects of cerebrovascular reactivity mature over different time courses in prenatal development rather than in concert.
Acknowledgments
This work was supported by a grant from the National Institutes of Health NS20020.
The authors thank Debra Flock and George Kuck for their technical assistance and Tzipora Sofare for her editorial assistance.
References
- Christopherson KS, Hillier BJ, Lim WA, Bredt DS. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J Biol Chem. 1999;274:27467–27473. doi: 10.1074/jbc.274.39.27467. [DOI] [PubMed] [Google Scholar]
- Criswell HE, Ming Z, Pleasant N, Griffith BL, Mueller RA, Breese GR. Macrokinetic analysis of blockade of NMDA-gated currents by substituted alcohols, alkanes and ethers. Brain Res. 2004;1015:107–113. doi: 10.1016/j.brainres.2004.04.050. [DOI] [PubMed] [Google Scholar]
- Curran-Everett D. Multiple comparisons: philosophies and illustrations. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1–R8. doi: 10.1152/ajpregu.2000.279.1.R1. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Breese KR. Nitric oxide mediates vasodilation in response to activation of N-methyl-D-aspartate receptors in brain. Circ Res. 1993;72:476–480. doi: 10.1161/01.res.72.2.476. [DOI] [PubMed] [Google Scholar]
- Furuta A, Martin LJ. Laminar segregation of the cortical plate during corticogenesis is accompanied by changes in glutamate receptor expression. J Neurobiol. 1999;39:67–80. [PubMed] [Google Scholar]
- Gleason CA, Hamm C, Jones MD., Jr Cerebral blood flow, oxygenation, and carbohydrate metabolism in immature fetal sheep in utero. Am J Physiol Regul Integr Comp Physiol. 1989;256:R1264–R1268. doi: 10.1152/ajpregu.1989.256.6.R1264. [DOI] [PubMed] [Google Scholar]
- Harris AP, Helou S, Gleason CA, Traystman RJ, Koehler RC. Fetal cerebral and peripheral circulatory responses to hypoxia after nitric oxide synthase inhibition. Am J Physiol Regul Integr Comp Physiol. 2001;281:R381–R390. doi: 10.1152/ajpregu.2001.281.2.R381. [DOI] [PubMed] [Google Scholar]
- Harris AP, Koehler RC, Gleason CA, Jones MD, Jr, Traystman RJ. Cerebral and peripheral circulatory responses to intracranial hypertension in fetal sheep. Circ Res. 1989;64:991–1000. doi: 10.1161/01.res.64.5.991. [DOI] [PubMed] [Google Scholar]
- Helou SM, Hudak ML, Jones MD., Jr Cerebral blood flow response to hypercapnia in immature fetal sheep. Am J Physiol Heart Circ Physiol. 1991;261:H1366–H1370. doi: 10.1152/ajpheart.1991.261.5.H1366. [DOI] [PubMed] [Google Scholar]
- Helou SM, Koehler RC, Gleason CA, Jones MD, Jr, Traystman RJ. Cerebrovascular autoregulation during fetal development in sheep. Am J Physiol Heart Circ Physiol. 1994;266:H1069–H1074. doi: 10.1152/ajpheart.1994.266.3.H1069. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F. Nitric oxide-dependent and -independent components of cerebrovasodilation elicited by hypercapnia. Am J Physiol. 1994;266:R546–R552. doi: 10.1152/ajpregu.1994.266.2.R546. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F. Permissive and obligatory roles of NO in cerebrovascular responses to hypercapnia and acetylcholine. Am J Physiol Regul Integr Comp Physiol. 1996;271:R990–R1001. doi: 10.1152/ajpregu.1996.271.4.R990. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Mosinger JL, Shahid Salles K, Labruyere J, Olney JW. Sensitivity of the developing rat brain to hypobaric/ischemic damage parallels sensitivity to N-methyl-aspartate neurotoxicity. J Neurosci. 1989;9:2809–2818. doi: 10.1523/JNEUROSCI.09-08-02809.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irikura K, Maynard KI, Moskowitz MA. Importance of nitric oxide synthase inhibition to the attenuated vascular responses induced by topical L-nitroarginine during vibrissal stimulation. J Cereb Blood Flow Metab. 1994;14:45–48. doi: 10.1038/jcbfm.1994.7. [DOI] [PubMed] [Google Scholar]
- Koehler RC, Traystman RJ. Bicarbonate ion modulation of cerebral blood flow during hypoxia and hypercapnia. Am J Physiol Heart Circ Physiol. 1982;243:H33–H40. doi: 10.1152/ajpheart.1982.243.1.H33. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Raper AJ, Patterson JL. Analysis of vasoactivity of local pH, PCO2 and bicarbonate on pial vessels. Stroke. 1977;8:358–360. doi: 10.1161/01.str.8.3.358. [DOI] [PubMed] [Google Scholar]
- Kurth CD, Wagerle LC. Cerebrovascular reactivity to adenosine analogues in 0.6-0.7 gestation and near-term fetal sheep. Am J Physiol Heart Circ Physiol. 1992;262:H1338–H1342. doi: 10.1152/ajpheart.1992.262.5.H1338. [DOI] [PubMed] [Google Scholar]
- Leffler CW, Balabanova L, Fedinec AL, Parfenova H. Nitric oxide increases carbon monoxide production by piglet cerebral microvessels. Am J Physiol Heart Circ Physiol. 2005a;289:H1442–H1447. doi: 10.1152/ajpheart.00464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler CW, Fedinec AL, Parfenova H, Jaggar JH. Permissive contributions of NO and prostacyclin in CO-induced cerebrovascular dilation in piglets. Am J Physiol Heart Circ Physiol. 2005b;289:H432–H438. doi: 10.1152/ajpheart.01195.2004. [DOI] [PubMed] [Google Scholar]
- Leffler CW, Mirro R, Pharris LJ, Shibata M. Permissive role of prostacyclin in cerebral vasodilation to hypercapnia in newborn pigs. Am J Physiol. 1994;267:H285–H291. doi: 10.1152/ajpheart.1994.267.1.H285. [DOI] [PubMed] [Google Scholar]
- Massik J, Jones MD, Jr, Miyabe M, Tang YL, Hudak ML, Koehler RC, Traystman RJ. Hypercapnia and response of cerebral blood flow to hypoxia in newborn lambs. J Appl Physiol. 1989;66:1065–1070. doi: 10.1152/jappl.1989.66.3.1065. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Silverstein FS, Johnston MV. Neurotoxicity of N-methyl-D-aspartate is markedly enchanced in developing rat central nervous system. Brain Res. 1988:200–203. doi: 10.1016/0006-8993(88)90306-x. [DOI] [PubMed] [Google Scholar]
- McPherson RW, Kirsch JR, Ghaly RF, Traystman RJ. Effect of nitric oxide synthase inhibition on cerebral blood flow response to hypercapnia in primates. Stroke. 1995;26:682–687. doi: 10.1161/01.str.26.4.682. [DOI] [PubMed] [Google Scholar]
- Meng W, Tobin JR, Busija DW. Glutamate-induced cerebral vasodilation is mediated by nitric oxide through N-methyl-D-aspartate receptors. Stroke. 1995;26:857–863. doi: 10.1161/01.str.26.5.857. [DOI] [PubMed] [Google Scholar]
- Miller SP, Ramaswamy V, Michelson D, Barkovich AJ, Holshouser B, Wycliffe N, Glidden DV, Deming D, Partridge JC, Wu YW, Ashwal S, Ferriero DM. Patterns of brain injury in term neonatal encephalopathy. J Pediatr. 2005;146:453–460. doi: 10.1016/j.jpeds.2004.12.026. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Koehler RC, Traystman RJ, Martin LJ. Nitric oxide synthase 1 and nitric oxide synthase 3 protein expression is regionally and temporally regulated in fetal brain. Brain Res Dev Brain Res. 1996;95:1–14. doi: 10.1016/0165-3806(96)00051-x. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Tobin JR, Harris AP, Traystman RJ, Koehler RC. Developmental and regional differences in nitric oxide synthase activity and blood flow in the sheep brain. J Cereb Blood Flow Metab. 1997;17:109–115. doi: 10.1097/00004647-199701000-00014. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Tobin JR, Koehler RC, Traystman RJ. In vivo production of nitric oxide correlates with NMDA-induced cerebral hyperemia in newborn sheep. Am J Physiol Heart Circ Physiol. 1995;269:H215–H221. doi: 10.1152/ajpheart.1995.269.1.H215. [DOI] [PubMed] [Google Scholar]
- Ogata J, Shiraishi M, Namba T, Smothers CT, Woodward JJ, Harris RA. Effects of anesthetics on mutant N-methyl-D-aspartate receptors expressed in Xenopus oocytes. J Pharmacol Exp Ther. 2006;318:434–443. doi: 10.1124/jpet.106.101691. [DOI] [PubMed] [Google Scholar]
- Okamoto H, Hudetz AG, Roman RJ, Bosnjak ZJ, Kampine JP. Neuronal NOS-derived NO plays permissive role in cerebral blood flow response to hypercapnia. Am J Physiol. 1997;272:H559–H566. doi: 10.1152/ajpheart.1997.272.1.H559. [DOI] [PubMed] [Google Scholar]
- Pelligrino DA, Gay RL, III, Baughman VL, Wang Q. NO synthase inhibition modulates NMDA-induced changes in cerebral blood flow and EEG activity. Am J Physiol Heart Circ Physiol. 1996;271:H990–H995. doi: 10.1152/ajpheart.1996.271.3.H990. [DOI] [PubMed] [Google Scholar]
- Reddy K, Mallard C, Guan J, Marks K, Bennet L, Gunning M, Gunn A, Gluckman P, Williams C. Maturational change in the cortical response to hypoperfusion injury in the fetal sheep. Pediatr Res. 1998;43:674–682. doi: 10.1203/00006450-199805000-00017. [DOI] [PubMed] [Google Scholar]
- Reivich M. Arterial pCO2 and cerebral hemodynamics. Am J Physiol. 1964;206:25–35. doi: 10.1152/ajplegacy.1964.206.1.25. [DOI] [PubMed] [Google Scholar]
- Robinson JS, Fedinec AL, Leffler CW. Role of carbon monoxide in glutamate receptor-induced dilation of newborn pig pial arterioles. Am J Physiol Heart Circ Physiol. 2002;282:H2371–H2376. doi: 10.1152/ajpheart.00911.2001. [DOI] [PubMed] [Google Scholar]
- Rosenberg AA, Jones MD, Jr, Traystman RJ, Simmons MA, Molteni RA. Response of cerebral blood flow to changes in PCO2 in fetal, newborn, and adult sheep. Am J Physiol Heart Circ Physiol. 1982;242:H862–H866. doi: 10.1152/ajpheart.1982.242.5.H862. [DOI] [PubMed] [Google Scholar]
- Sandor P, Komjati K, Reivich M, Nyary I. Major role of nitric oxide in the mediation of regional CO2 responsiveness of the cerebral and spinal cord vessels of the cat. J Cereb Blood Flow Metab. 1994;14:49–58. doi: 10.1038/jcbfm.1994.8. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Encephalopathy of prematurity includes neuronal abnormalities. Pediatrics. 2005;116:221–225. doi: 10.1542/peds.2005-0191. [DOI] [PubMed] [Google Scholar]
- Wagerle LC, Kurth CD, Busija DW. Cholinergic reactivity of cerebral arteries in the developing fetal and newborn lamb. J Dev Physiol. 1992;17:51–54. [PubMed] [Google Scholar]
- White RP, Deane C, Vallance P, Markus HS. Nitric oxide synthase inhibition in humans reduces cerebral blood flow but not the hyperemic response to hypercapnia. Stroke. 1998;29:467–472. doi: 10.1161/01.str.29.2.467. [DOI] [PubMed] [Google Scholar]
- Willis AP, Leffler CW. NO and prostanoids: age dependence of hypercapnia and histamine-induced dilations of pig pial arterioles. Am J Physiol Heart Circ Physiol. 1999;277:H299–H307. doi: 10.1152/ajpheart.1999.277.1.H299. [DOI] [PubMed] [Google Scholar]
- Zuckerman SL, Armstead WM, Hsu P, Shibata M, Leffler CW. Age dependence of cerebrovascular response mechanisms in domestic pigs. Am J Physiol Heart Circ Physiol. 1996;271:H535–H540. doi: 10.1152/ajpheart.1996.271.2.H535. [DOI] [PubMed] [Google Scholar]
- Zuo Z, Tichotsky A, Johns RA. Inhibition of excitatory neurotransmitter-nitric oxide signaling pathway by inhalational anesthetics. Neuroscience. 1999;93:1167–1172. doi: 10.1016/s0306-4522(99)00194-3. [DOI] [PubMed] [Google Scholar]