

Abstract
Organic vinylidene species have found limited use in organic synthesis due to their inaccessibility. In contrast, metal vinylidenes are much more stable, and may be readily accessed through transition metal activation of terminal alkynes. These electrophilic species may be trapped by a number of nucleophiles. Additionally, metal vinylidenes can participate in pericyclic reactions and processes involving migration of a metal ligand to the vinylidene species. This review addresses the reactions and applications of metal vinylidenes in organic synthesis.
Keywords: atom economy, catalysis, chemoselectivity, organic synthesis, vinylidene
1. Introduction
Organic vinylidene species (2) are tautomers of the corresponding alkynes (1), but they are thermodynamically much less stable (Scheme 1). There is also a significant activation energy for the interconversion of the two species. Since harsh conditions are required for the generation of organic vinylidenes their application in organic synthesis has been severely limited.
Scheme 1.
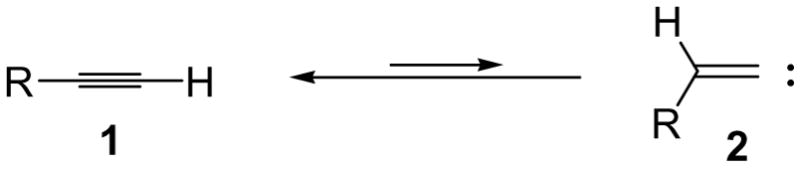
Terminal Alkyne and the Corresponding Vinylidene
A rare example of an organic vinylidene used in a synthetic context is displayed in Scheme 2. In a synthesis of (+/−)-isoptychanolide, Dreiding and coworkers reported that flash vacuum thermolysis of ynone 3 leads to cyclopentenone 5, presumably via generation of the transient vinylidene species 4 and its insertion into the proximal C-H bond.[1]
Scheme 2.
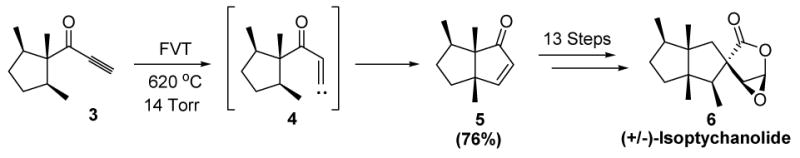
Synthetic Use of an Organic Vinylidene: Dreiding’s Synthesis of (+/−)-Isoptychanolide. FVT=flash vacuum thermolysis.
A significant advance was the observation that metal vinylidenes are much more stable than organic (free) vinylidenes.[2] The most straightforward route to metal vinylidenes is by the transition metal activation of terminal alkynes (Scheme 3). This process is reversible, and the relative stabilities of the metal-coordinated alkyne (7) and the metal vinylidene (8) depend on the electron configuration of the metal (M), the nature of the ligands (Ln), and the alkyne substituents.
Scheme 3.
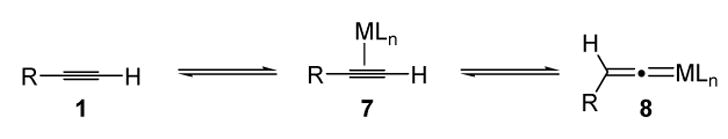
Formation of Metal Vinylidenes from Terminal Alkynes
There are two proposed mechanisms for the conversion of metal-coordinated terminal alkynes to metal vinylidenes. The first pathway involves oxidative addition of the metal into the alkyne C-H bond, followed by a concerted [1,3]-shift of the hydride. The second mechanism consists of a direct [1,2]-migration of a hydride over the alkyne (Scheme 4).
Scheme 4.
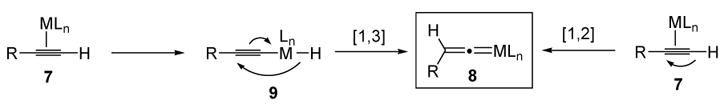
Mechanisms of Metal Vinylidene Formation
There has been experimental support for the first mechanism in some cases (ie. observation or isolation of the alkynyl metal hydride intermediate 9). This is the case for reactions involving RhCl(iPr3)2,[3] although even in this case it is not clear whether the [1,3]-migration step is a unimolecular[4] or a bimolecular[5] process. In the majority of reactions, however, it is not known which pathway is operative, and both experimental and computational studies have been carried out in order to delineate the mechanism.[6]
Vinylidenes behave as electron-withdrawing ligands on the metal, and thus electron-donating ligands (Ln) will stabilize this species (8) relative to the metal-complexed alkyne (7, Scheme 3). However, there must be a balance, as there is a danger of making the species too stable such that it does not undergo further reactions. Traditionally, nucleophiles (such as alcohols) have been added to metal vinylidenes in stoichiometric reactions to generate the corresponding Fischer carbene complexes. This mode of reactivity reflects the inherent electrophilicity of the vinylidene α carbon.
More recently, there has been a large number of publications involving conversion of terminal alkynes to various products in which metal vinylidenes serve as catalytic species.[7] The three types of transformations most commonly investigated are: (a) nucleophilic addition to the α carbon, (b) [1,2]-alkyl migration from the metal center to the α carbon, and (c) pericyclic reactions.[8] In this review, the various reactions involving catalytic metal vinylidene species are analyzed, with an emphasis on those that synthetic chemists may find useful. In most cases, a proposed mechanism (catalytic cycle) is presented in order to facilitate an understanding of the transformation at hand.
2. Heteroatom Nucleophile Reactions with Vinylidenes
2.1. Carbamate Nucleophiles
Dixneuf reported the first report of a metal vinylidene complex as a catalytic species in an organic reaction over 20 years ago.[9] In an interesting three component coupling reaction, Ru3(CO)12 catalyzed the addition of N,N-diethylcarbamate to n-hexyne to generate both the anti-Markovnikov (14 and 15) and Markovnikov (16) vinyl carbamates in low yield (Scheme 6). Such a transformation is noteworthy from the standpoint of atom economy, as well as the fact that it involves carbon dioxide, a highly stable molecule that is not easily activated in organic synthesis.[10]
Scheme 6.
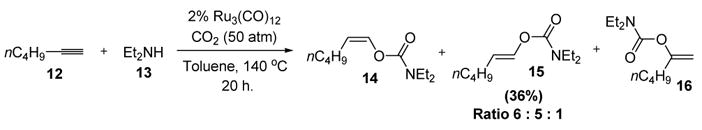
Vinylcarbamates from Terminal Alkynes, Secondary Amines, and Carbon Dioxide
The proposed mechanism for the formation of the anti-Markovnikov product involves vinylidene formation between the ruthenium catalyst (I) and the terminal alkyne, followed by nucleophilic attack of the carbamate (generated in situ) to give III (Scheme 7). Protonation of the metal, followed by reductive elimination, then releases both the anti-Markovnikov products 14 and 15 and regenerates the active catalyst. The Markovnikov product 16, however, is believed to form by nucleophilic attack on the more electrophilic (internal) carbon of the metal-coordinated alkyne (V).
Scheme 7.
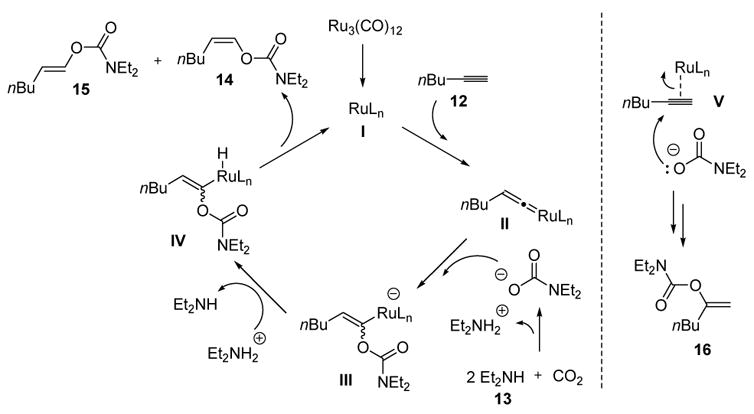
Mechanism of Vinylcarbamate Formation
Although the selectivities and yields of the process were low, the results spurred further generations of catalyst modification to improve the transformation. Thus by changing the catalyst and solvent phenylacetylene (17) was reacted with diethylamine to provide the corresponding vinylcarbamate in moderate yield and selectivity (equation 1, Scheme 8).[11] Acetylene (21) was also a viable coupling partner with pyrrolidine, although an excess of alkyne was required due to competing polymerization of the alkyne (equation 2).[12,13] Interestingly, 2-methyl-1-buten-3-yne (24) could be used as a substrate, reacting with morpholine to give diene products that are activated Diels-Alder reaction partners (equation 3).[14]
Scheme 8.
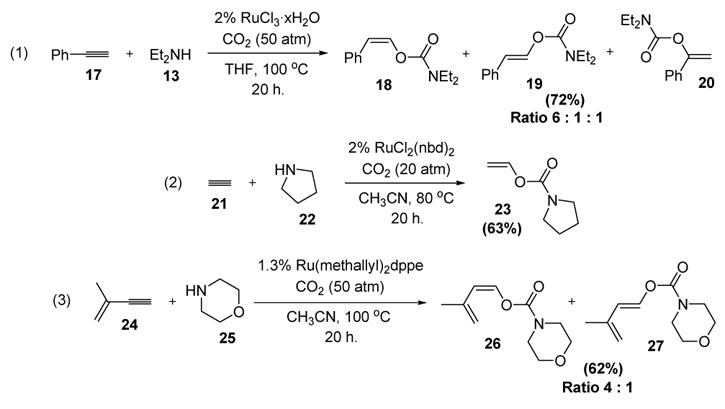
Synthesis of Vinylcarbamates: Scope of Alkynes and Secondary Amines. dppe=bis(diphenylphosphino)ethane, nbd=norbornadiene, THF=tetrahydrofuran.
2.2. Carboxylic Acid Nucleophiles
2.2.1 Intermolecular Reactions
Contemporaneous with his work on vinylcarbamate formation, Dixneuf disclosed the use of carboxylic acid nucleophiles to capture metal vinylidenes in a catalytic synthesis of enol esters.[15,16] Thus phenylacetylene (17) and benzoic acid (28) react, in the presence of a catalytic amount of ruthenium trichloride trihydrate, to give a mixture of anti-Markovnikov (29 and 30) and Markovnikov (31) addition products (Scheme 9).
Scheme 9.
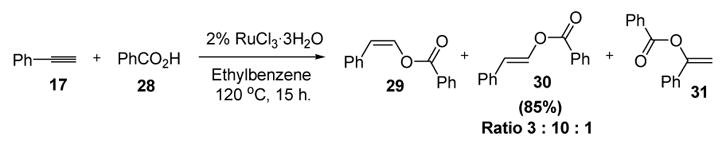
Formation of Enol Esters from Carboxylic Acids and Alkynes
Similar to the aforementioned formation of vinylcarbamates, the anti-Markovnikov enol ester products can be accounted for by invoking a vinylidene mechanism (Scheme 10). Thus vinylidene formation between the ruthenium catalyst and the terminal alkyne 17, followed by nucleophilic attack of the carboxylate, protonation of the metal, and reductive elimination can give products 29 and 30. The regioisomeric Markovnikov product 31 likely forms by electrophilic activation of the alkyne followed by nucleophilic attack.
Scheme 10.
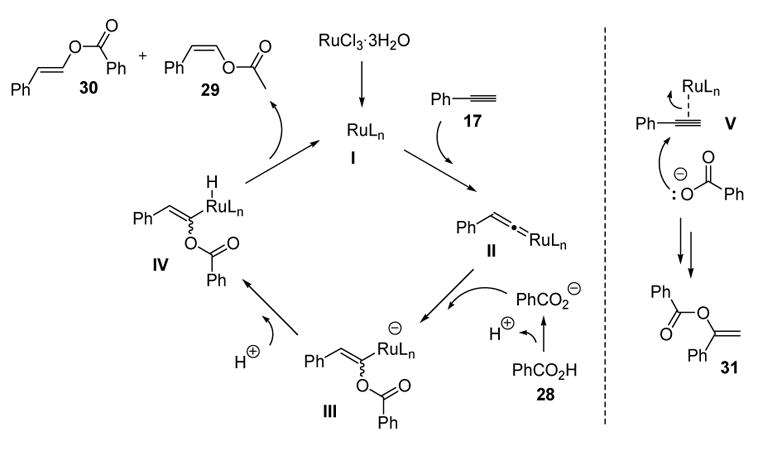
Mechanism of Enol Ester Formation
Subsequent studies showed that the complex Ru(methallyl)2dppb was a better catalyst for a variety of alkynes and carboxylic acids. For example, benzoic acid added in a regio- and stereoselective manner to phenylacetylene (equation 1),[17] n-hexyne (equation 2),[18] and 2-methyl-1-buten-3-yne (equation 3)[19] to give the corresponding enol esters in excellent yields (Scheme 11).
Scheme 11.
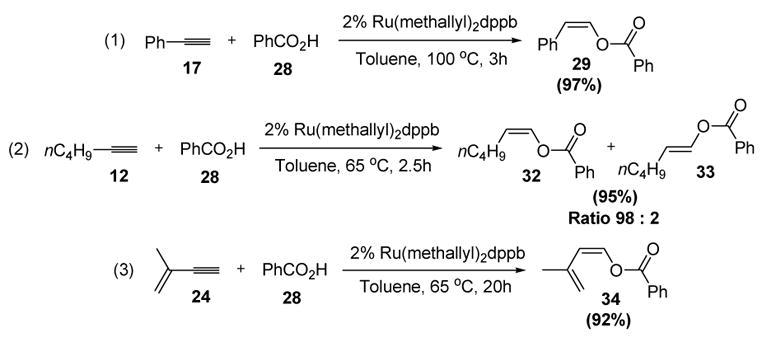
Ruthenium-Catalyzed Formation of Enol Esters: Scope of Alkynes. dppb=bis(diphenylphosphino)butane.
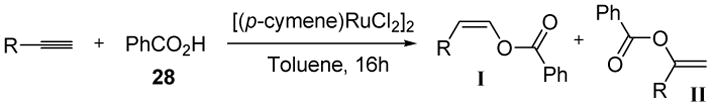
A number of ruthenium catalysts have since been found to catalyze the regioselective, anti-Markovnikov formation of enol esters from various alkynes and carboxylic acids.[20,21] One of the most interesting reports showed that changing the ligand and base, with the same ruthenium catalyst, allowed access to either the anti-Markovnikov or Markovnikov products in excellent yield (Table 1).[22]
Table 1.
Ligand Control in Regioselectivity of Enol Ester Formation
 | ||||||
|---|---|---|---|---|---|---|
| Alkyne (R) | % Catalyst | Ligand | Base | Temp (°C) | Yield (%) | Ratio (I/II) |
| nBu | 1 | 3% P(4-Cl-C6H4)3 | 4% DMAP | 60 | 89 | 50:1 |
| nBu | 0.4 | 0.8% P(furyl)3 | 1.6% Na2CO3 | 50 | 90 | 1:30 |
| Ph | 1 | 3% P(4-Cl-C6H4)3 | 4% DMAP | 60 | 99 | 50:1 |
| Ph | 0.4 | 0.8% P(furyl)3 | 1.6% Na2CO3 | 70 | 88 | 1:1.5 |
| tBu | 1 | 3% P(4-Cl-C6H4)3 | 4% DMAP | 80 | 68[a] | 50:1 |
| tBu | 0.4 | 0.8% P(furyl)3 | 1.6% Na2CO3 | 50 | 80 | 1:10 |
Solvent: 1,2-dichloroethane. DMAP=4-dimethylaminopyridine.
To highlight the utility of this type of reaction, Dixneuf and coworkers have investigated a ruthenium-catalyzed isomerization process (Scheme 12). In this transformation, benzoic acid adds regioselectively to putative metal vinylidene complexes, and the resulting Z-enol esters then fragment at higher temperatures to give enal products in good yields.[23] Alternatively, the intermediate enol esters may be treated with a catalytic amount of p-toluenesulfonic acid at room temperature to effect the elimination reaction.[24]
Scheme 12.
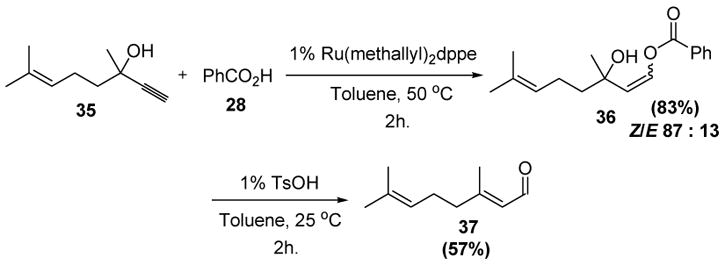
Dixneuf’s Isomerization of Propargyl Alcohols to Enals. dppe=bis(diphenylphosphino)ethane, TsOH=p-toluenesulfonic acid.
This process constitutes the synthetic equivalent of a Meyer-Schuster rearrangement,[25] but has the advantage of taking place under much milder reaction conditions. Although this reaction has not yet been used in target-oriented synthesis, one can imagine that it might find use as an alternative to the Horner-Wadsworth-Emmons or Wittig olefinations of ketones.
2.2.2 Intramolecular Reactions
Valerga and coworkers have found that carboxylic acids can add to terminal alkynes in an intramolecular fashion.[26] Thus α,ω-alkynoic acids (38) may be converted to the corresponding enol lactones 39, in one case even forming a macrocycle, in good to excellent yield by heating in the presence of a ruthenium catalyst (Scheme 13). The exclusive formation of the endocyclic enol lactones is consistent with vinylidene formation, followed by intramolecular trapping by the tethered carboxylic acids. Verpoort and colleagues later reported that ruthenium complexes of the form RuClx(p-cymene)(triazol-5-ylidene) were able to catalyze the cyclization of 4-pentynoic acid in excellent yield and regioselectivity.[27]
Scheme 13.

Valerga’s Cyclization of α,ω-Alkynoic Acids
2.3. Alcohol Nucleophiles
2.3.1 Intermolecular Reactions
Despite the success of carboxylic acids, the use of alcohols to intercept metal vinylidene intermediates in intermolecular catalytic reactions has been problematic. For the most part, such reactions occur only stoichiometrically to provide the corresponding Fischer carbenes.[28] One notable exception, however, has been the use of allylic alcohols. In what is termed the ruthenium-catalyzed reconstitutive condensation reaction, Trost has reported that treatment of alkynes 40 and excess allylic alcohol 41 with a ruthenium catalyst and ammonium hexafluorophosphate leads to β,γ-unsaturated ketone 43 (Scheme 14).[29] This in fact represents the first use of vinylidenes to form C-C bonds in a catalytic reaction.
Scheme 14.
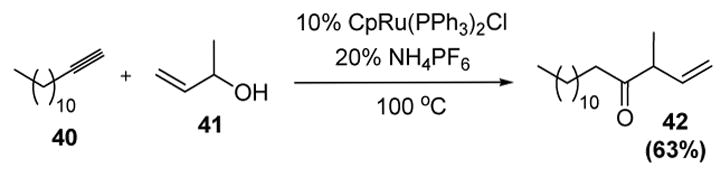
Ruthenium-Catalyzed Reconstitutive Condensation. Cp=η5-cyclopentadienyl.
Evidence for vinylidene intermediates in this process was gathered through exploration of substrate scope and labeling studies; a mechanism consistent with the results was then proposed (Scheme 15).[30] Loss of chloride ion from the ruthenium complex is expected to lead to the active cationic catalyst I. Vinylidene formation with alkyne 40 leads to II. Coordination of olefin 41, with loss of phosphine, can lead to III, and subsequent nucleophilic attack by the pendant alcohol can give IV. Ionization of the resulting allyl enol ether then provides acyl ruthenium species V, which can be represented as σ complex VI. Finally, reductive elimination releases the product (42) and regenerates the active catalyst.
Scheme 15.
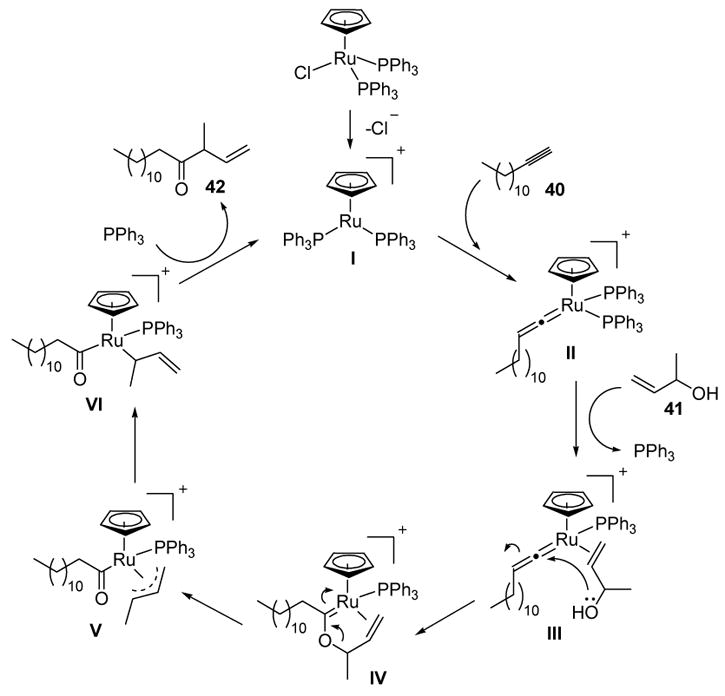
Proposed Mechanism of Ruthenium-Catalyzed Reconstitutive Condensation
The power of this reaction was demonstrated in a concise synthesis of the fully functionalized side chain of the steroid ganoderic acid (48, Scheme 16).[31] The terminal alkyne substrate 44 was prepared by Corey-Fuchs homologation of the commercially available 3-oxopregn-4-ene-20β-carboxaldehyde (43).[32] In this transformation, the enone present in the substrate was effectively protected from side reactions by conversion to the corresponding enolate prior to the reaction. The ruthenium-catalyzed reconstitutive condensation with allyl alcohol (46) then proceeded in good yield to give a mixture of 46 and 47; treatment of this mixture with catalytic rhodium trichloride in aqueous tetrahydrofuran accomplished complete conversion to the α,β-unsaturated ketone 47. Conjugate addition of cyanide, followed by nitrile hydrolysis, then gave 48.
Scheme 16.
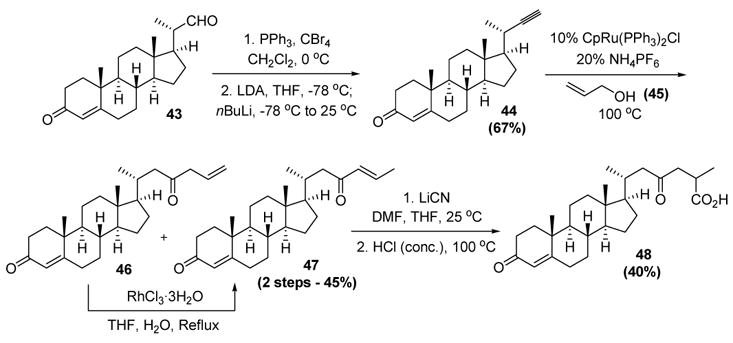
Trost’s Synthesis of a Functionalized Steroid Side Chain. Cp=η5-cyclopentadienyl., DMF=N,N-dimethylformamide, LDA=lithium diisopropylamide, THF=tetrahydrofuran.
The utility of this transformation was further demonstrated in a short synthesis of the fragrance rosefuran (53, Scheme 17).[33] Thus addition of propargylmagnesium bromide to acetone, followed by acylation of the resulting tertiary alcohol, gave alkyne 50 in good yield. Reconstitutive condensation with allylic alcohol 41 then gave 51, a molecule containing all of the carbon atoms present in the target molecule. A tandem dihydroxylation-cyclization provided furan 52. Ester hydrolysis, followed by thermal dehydration, then gave rosefuran.
Scheme 17.
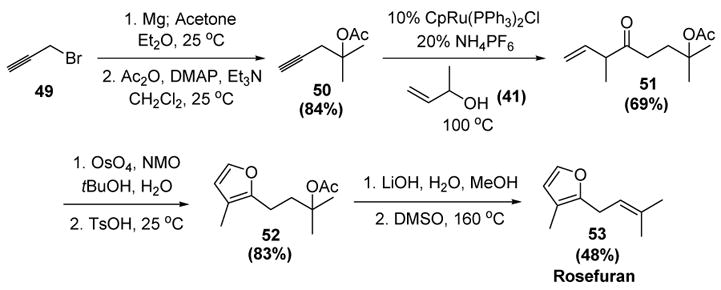
Trost’s Synthesis of Rosefuran. Cp=η5-cyclopentadienyl., DMSO=dimethyl sulfoxide, NMO=N-methylmorpholine-N-oxide, TsOH=p-toluenesulfonic acid.
Terminal alkynes bearing propargyl alcohols have the ability to form allenylidene species. These complexes are electrophilic, and thus offer opportunities for reactions with nucleophilic functional groups. In this context, Trost has reported the catalytic reaction of propargyl alcohol substrates bearing pendant alcohol groups to form tetrahydrofurans and tetrahydropyrans (Scheme 18).[34]
Scheme 18.
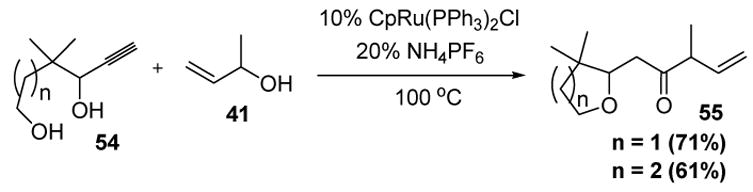
Trost’s Tandem Cyclization-Reconstitutive Condensation. Cp=η5-cyclopentadienyl.
The mechanism of this tandem reaction is depicted in Scheme 19. The active catalyst is generated by loss of chloride ion to give I. Insertion of the metal into the alkyne C-H bond then forms II, whereby loss of water leads to allenylidene III. The pendant alcohol then attacks this electrophilic species to give vinylidene IV. The mechanism then follows the same sequence of events as shown in Scheme 15 to give the β,γ-unsaturated ketone products.
Scheme 19.
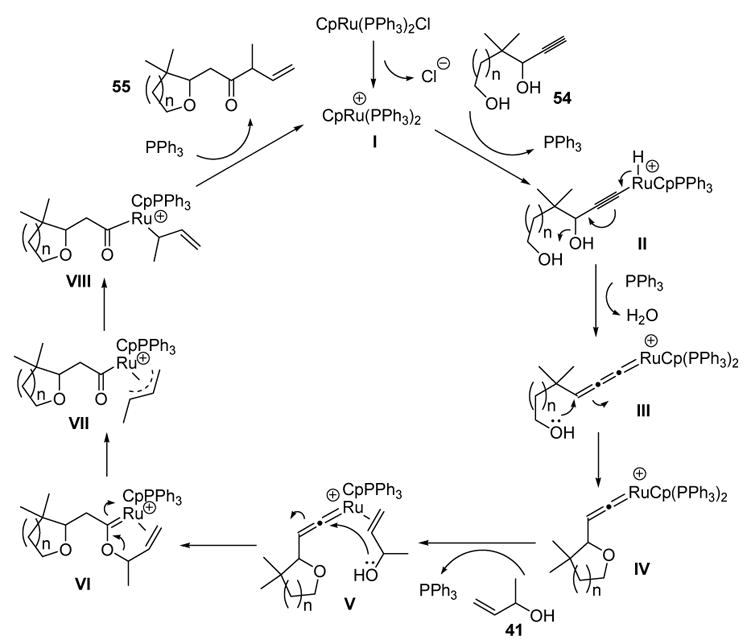
Mechanism of Trost’s Tandem Cyclization-Reconstitutive Condensation. Cp=η5-cyclopentadienyl.
This reaction has been used in a synthesis of the spiroketal subunit of the phosphatase inhibitor (−)-calyculin A (Scheme 20).[35] The sequence began with reduction of (R)-pantolactone (56) to the corresponding lactol. Addition of vinylmagnesium bromide, followed by acetonide formation, then afforded 57. Oxidation and subsequent acetonide cleavage-lactonization then led to 58. Ozonolysis of this intermediate with reductive workup then gave a diol that was ketalized with acetone to give 59. Reduction of the lactone to the lactol, followed by addition of lithium acetylide, then provided a mixture of epimeric propargyl alcohols. Ultimately, the stereochemistry was inconsequential, as the ruthenium-catalyzed tandem cyclization-reconstitutive condensation gave 61 as a single diastereomer with respect to the tetrahydrofuran ring. Asymmetric dihydroxylation, followed by selective protection of the primary alcohol, led to 62. Finally, oxidative cyclization gave the spirocyclic core of (−)-calyculin A (63).
Scheme 20.
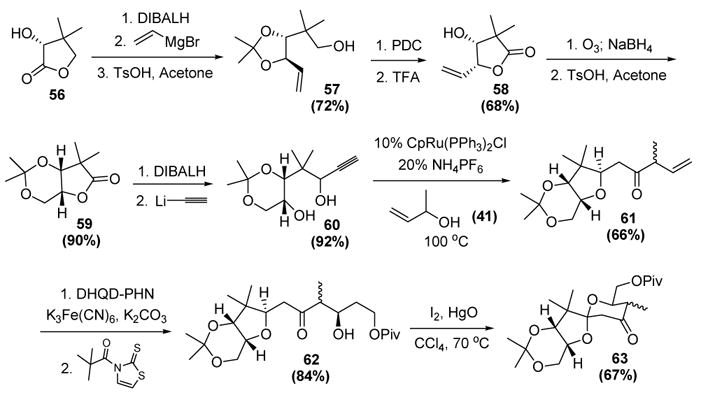
Trost’s Synthesis of The Spiroketal Subunit of (−)-Calyculin A. Cp=η5-cyclopentadienyl, (DHQD)PHN=dihydroquinidine 9-O-(9′-phenanthryl) ether, DIBALH=diisobutylaluminum hydride, PDC=pyridinium dichromate, TFA=trifluoroacetic acid, TsOH=p-toluenesulfonic acid.
2.3.2 Intramolecular Reactions
Contrary to the intermolecular reaction of alcohols with terminal alkynes through vinylidene intermediates, the endo cyclization of alkynols has found success.[36] The first report of such a transformation dates back almost 15 years (Scheme 21).[37] Although the yields were modest, chromium, tungsten, and molybdenum carbonyl complexes were found to promote the stoichiometric cyclization of homopropargyl alcohol 64 to the corresponding Fischer carbene complexes. In the latter case triethylamine was able to effect in situ demetalation to the corresponding dihydrofuran 67.
Scheme 21.
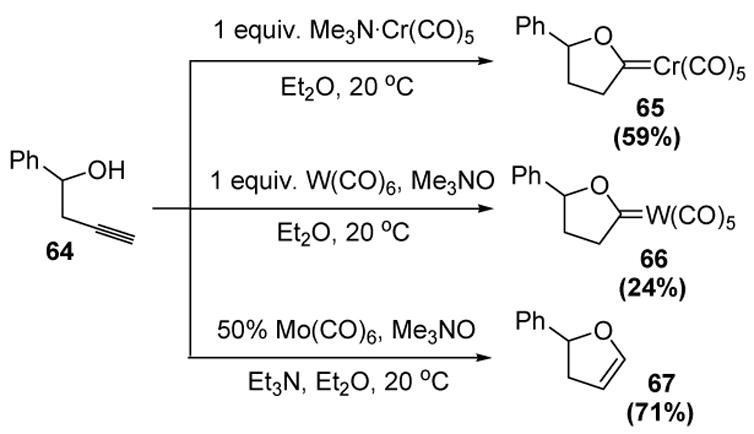
McDonald’s Seminal Alkynol Cycloisomerization Reaction
The products formed can be explained by considering the putative mechanism (Scheme 22). Molybdenum hexacarbonyl reacts with trimethylamine oxide to give the active catalyst I. Vinylidene formation with alkyne 64 gives II, which undergoes nucleophilic capture by the pendant alcohol to give Fischer carbene III. In the presence of triethylamine, deprotonation of this electrophilic oxacarbene can give anion IV. Protonation of the metal, followed by reductive elimination, gives 67 and releases the active catalyst. Although the catalyst loading was almost stoichiometric, this initial report paved the way for future investigations.
Scheme 22.
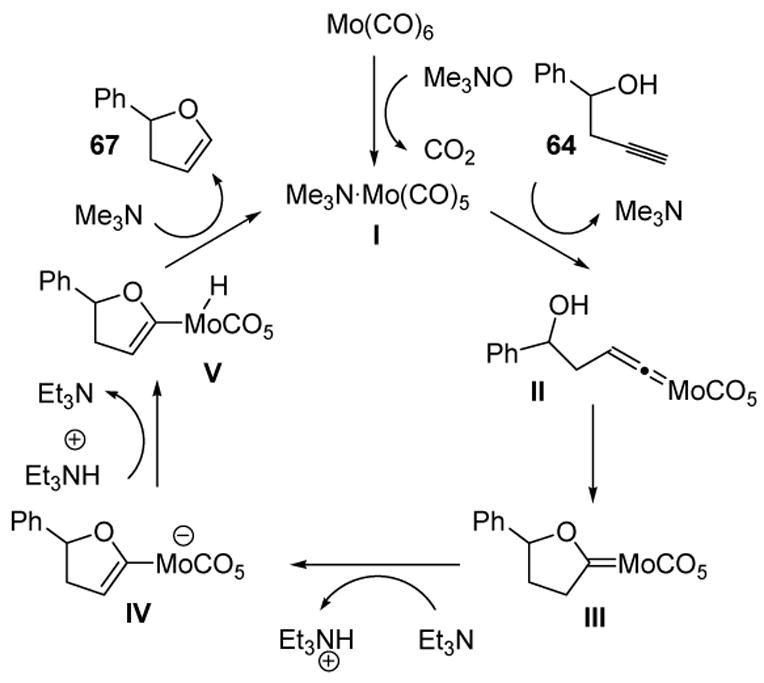
Mechanism of Molybdenum-Catalyzed Alkynol Cycloisomerization
Recognizing the utility of this transformation, synthetic applications followed soon after. McDonald first used this reaction in concise enantioselective syntheses of the deoxynucleosides stavudine and cordycepin (Scheme 23).[38] The starting point was a Katsuki-Sharpless epoxidation of allyl alcohol (45), followed by in situ protection of the alcohol, to provide (S)-glycidyl pivaloate 68. Regioselective epoxide opening with lithium acetylide then gave 69, which proved to be a capable substrate for the molybdenum-catalyzed cycloisomerization reaction, providing the key dihydrofuran intermediate 70 in good yield. Iodine-mediated introduction of thymine on to this molecule provided iodonucleoside 71. Pivaloate methanolysis and concomittant elimination of HI then gave stavudine (72), a substance possessing anti-HIV activity. Alternatively, dihydroxylation of 70 and acylation of the crude diol provided a mixture of four diastereomeric diacetates, the major product being 73. Lewis acid-catalyzed addition of a protected adenine derivative then gave 74, whereby methanolysis of the pivaloate, acetate, and benzoate protecting groups gave cordycepin (75), a substance possessing antibiotic activity.
Scheme 23.
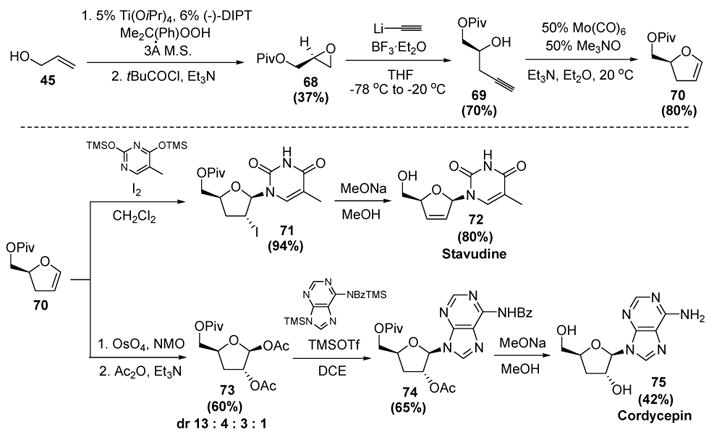
McDonald’s Syntheses of Stavudine and Cordycepin. DCE=1,2-dichloroethane, DIPT=diisopropyltartrate, M. S.=molecular sieves, NMO=N-methylmorpholine-N-oxide, THF=tetrahydrofuran, TMSOTf=trimethylsilyl trifluoromethanesulfonate.
This manner of deoxynucleotide synthesis was further extended to 3-aminofuranose glycals (Scheme 24).[39] Once again, asymmetric epoxidation provided the enantioenriched starting material. Regioselective opening of epoxide 77, followed by protection of the primary alcohol, gave 78 in moderate yield. Azide reduction, and protection of the resulting amine as either the acetamide or the trifluoroacetamide, gave substrates for cycloisomerization (79). In the event, the key molybdenum-promoted cycloisomerization provided the desired dihydrofurans (80) in excellent yield. Importantly, it was demonstrated that the presence of the amide in the propargylic position did not affect the reaction, unlike alcohol and azide functionalities (which are prone to elimination). Subsequent transformations finally allowed access to a variety of 3-aminofuranose glycals, including puromycin aminonucleoside (81).
Scheme 24.
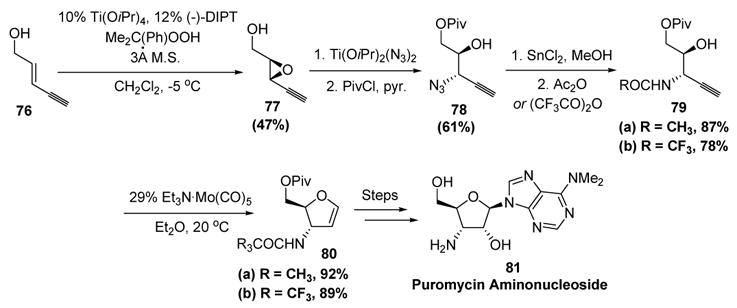
McDonald’s Syntheses of 3-Aminonucleosides
McDonald further demonstrated the utility of this transformation in a synthesis of digitoxin (82).[40] In this convergent approach, the trisaccharide moiety of the natural product was constructed by iterative cycloisomerizations,[41] and then connected to digitoxigenin (83) by glycosidation (Scheme 25).
Scheme 25.
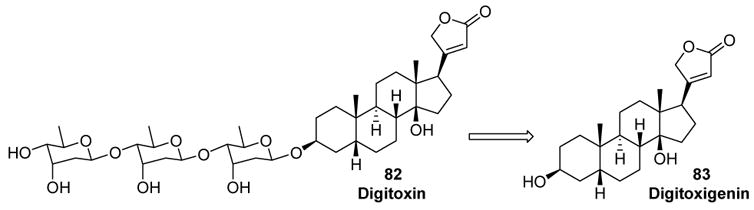
McDonald’s Retrosynthetic Analysis of Digitoxin
The chiral information of the nascent trisaccharide was installed by an enantioselective carbonyl reduction of enynone 84 and a diastereoselective epoxidation of the resultant allylic alcohol (Scheme 26). Regioselective epoxide opening with benzoic acid, followed by protodesilylation, then afforded compound 86. Diol protection followed by removal of the benzoate protecting group provided alkynol 87, which underwent a smooth cycloisomerization to give 6-deoxy-D-ribo glycal 88.[42] Glycosidation with a chiral alcohol proceeded in good yield and facial selectivity. Benzoate removal followed by cycloisomerization afforded disaccharide 90 in excellent yield. A series of transformations, including another cycloisomerization, provided 82. Finally, glycosidation with digitoxin, followed by protecting group removal, gave digitoxin.
Scheme 26.
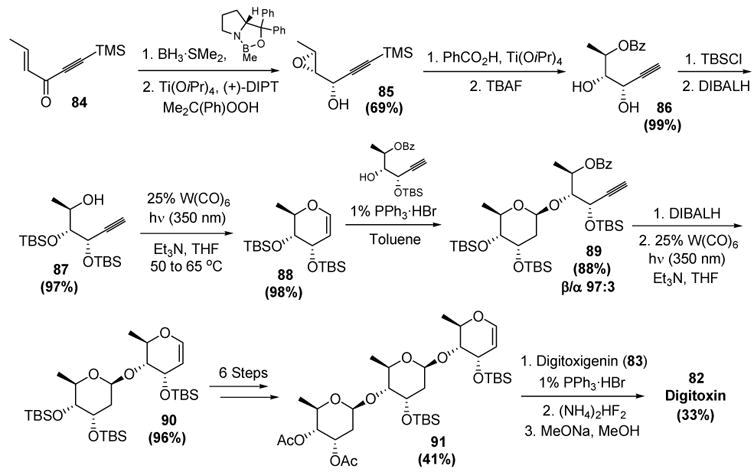
McDonald’s Iterative Cycloisomerization Approach to Digitoxin. DIBALH=diisobutylaluminum hydride, DIPT=diisopropyltartrate, TBAF=tetrabutylammonium fluoride, TBSCl=tert-butyldimethylsilyl chloride, THF=tetrahydrofuran.
More recently, McDonald has reported that use of DABCO as the base allows one to significantly lower the catalyst loading for the formation of dihydropyrans. This base may stabilize intermediates in the catalytic cycle more effectively than does triethylamine. Wipf has investigated these conditions with respect to the stereochemistry and nature of substituents on the chain linking the alkyne and the alcohol in the substrate.[43] McDonald has successfully applied these new conditions in syntheses of (L)-vancosamine (93)[44] and (D)-desosamine (95)[45] glycals (Scheme 27).
Scheme 27.
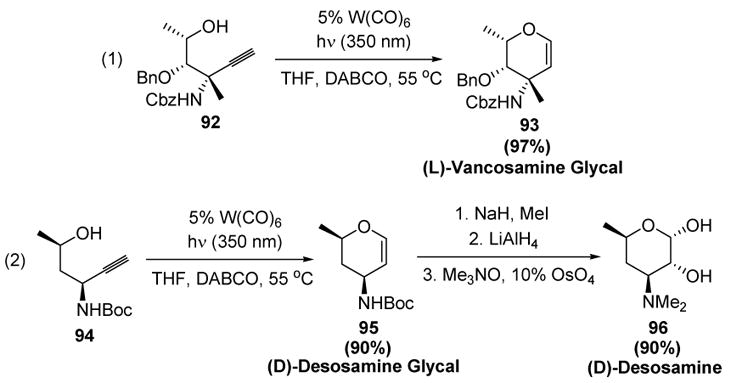
McDonald’s Syntheses of (L)-Vancosamine and (D)-Desosamine Glycals. DABCO=1,4-diazabicyclo[2.2.2]octane, THF=tetrahydrofuran.
McDonald and colleagues have also reported that α-stannyl vinyl ethers could be prepared from alkynols by either trapping the intermediate Fischer carbene anions with a tin electrophile in situ or through a stepwise process. For example, homopropargyl alcohol 64 could be converted to α-(tributylstannyl)dihydrofuran 97 in reasonable yield with a catalytic amount of a molybdenum pentacarbonyl complex and tributyltin triflate (equation 1, Scheme 28).[46] The bis-homopropargyl alcohol 98, on the other hand, could only be converted to a Fischer carbene by treatment with a tungsten pentacarbonyl complex. Upon treatment with the tin reagent, α-(tributylstannyl)dihydrofuran 100 was then obtained in quantitative yield (equation 2).[47] The vinyl tin moiety can undergo a variety of metal-catalyzed cross coupling reactions with alkyl halides, thus making these products useful building blocks for organic synthesis.
Scheme 28.
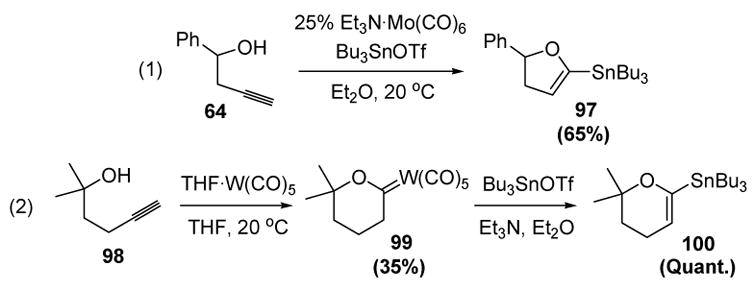
McDonald’s Formation of α-Stannyl Vinyl Ethers from Alkynols. THF=tetrahydrofuran.
Trost has reported that the ruthenium-catalyzed cycloisomerization of homopropargyl alcohols, such as 101, could be coupled to an oxidation reaction, thus providing γ-butyrolactones, such as 103 (Scheme 29).[48]
Scheme 29.
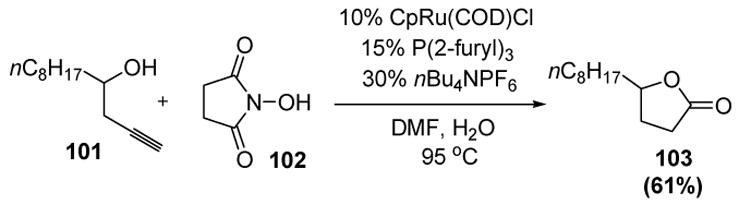
Ruthenium-Catalyzed Oxidative Cyclization of Homopropargyl Alcohols. COD=1,5-cyclooctadiene, Cp=η5-cyclopentadienyl, DMF=N,N-dimethylformamide.
Studies on the related reactions of bis-homopropargyl alcohols showed that employing the electron-donating ligand tris(4-methoxphenyl)phosphine gave δ-valerolactone products (Scheme 30). Interestingly, the electronics of the ligand could be tuned to give a different product. Thus use of the electron-withdrawing ligand tris(4-fluorophenyl) phosphine gave dihydropyrans.[49]
Scheme 30.
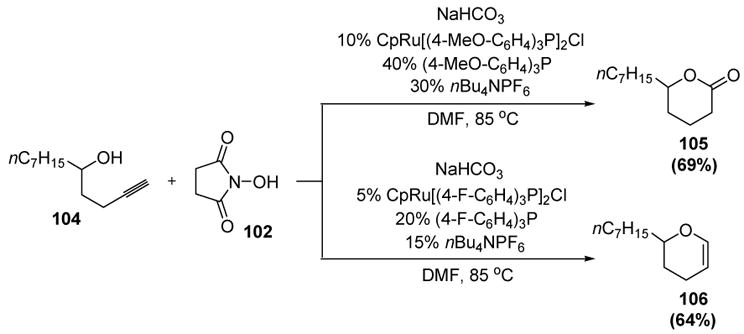
Oxidative Cyclization vs. Cycloisomerization of Bis-homopropargyl Alcohols. Cp=η5-cyclopentadienyl, DMF=N,N-dimethylformamide.
A mechanism that takes into account the ligand effects and explains the role of N-hydroxysuccinimide (102) is shown in Scheme 31. Vinylidene formation of active catalyst I with alkyne 104 gives II, and nucleophilic attack by the pendant alcohol then provides III. From this key intermediate C-protonation can give oxacarbene IV. Subsequent nucleophilic attack by the anion of N-hydroxysuccinimide, followed by protonation and reductive elimination, gives lactone product 105. Alternatively, ligand displacement of complex III by the anion of N-hydroxysuccinimide may give VI. Protonation and reductive elimination then can release dihydropyran product 106 and structure VII, which can undergo ligand displacement to regenerate the active catalyst. Electron-donating phosphines may promote protonation of intermediate III, thus leading to the preferential formation of lactone products. On the other hand, electron-withdrawing phosphines may enhance the electrophilicity of intermediate III, thus promoting nucleophilic attack by the anion of N-hydroxysuccinimide at ruthenium and leading to the formation of dihydropyrans. The large amount of phosphine ligands in both processes may serve to saturate the metal center. This would attenuate the electrophilicity of the metal, thus inhibiting competing processes while promoting vinylidene formation.
Scheme 31.
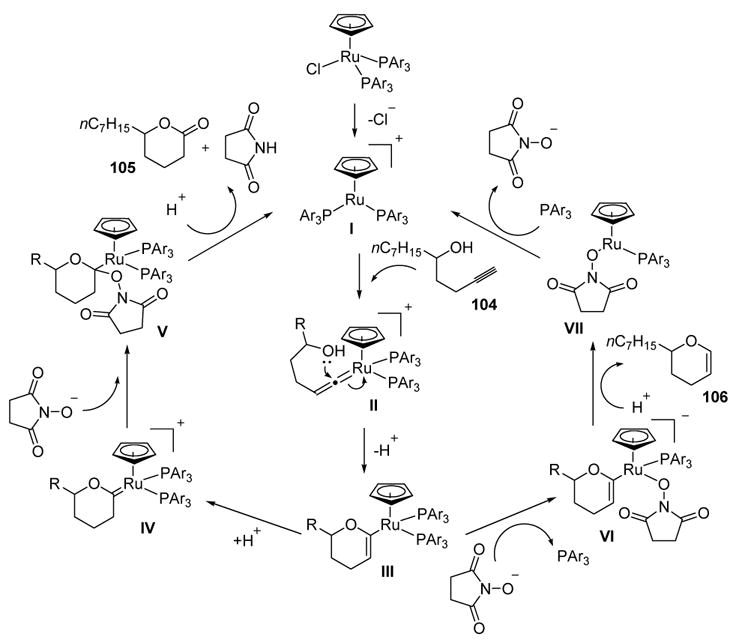
Proposed Mechanism of Ruthenium-Catalyzed Oxidative Cyclization and Cycloisomerization
Trost has shown the utility of these reactions in a novel iterative cycloisomerization approach to the marine ladder toxins prymnesin and yessotoxin (Scheme 32).[50] The sequence of reactions begins with a diastereoselective addition of propargyl zinc to the acetonide of glyceraldehyde (107). Protection of the resulting alcohol, followed by cleavage of the ketal, then gave diol 108. The ruthenium-catalyzed cycloisomerization reaction then gave dihydropyran 109 in good yield. Following protection of the alcohol, the enol ether was treated with dimethyl dioxirane, and the intermediate epoxide was regioselectively opened with allenylmagnesium bromide to give a mixture of diastereomeric bis-homopropargyl alcohols 112 and 113 (4: 1 ratio). The fact that a mixture was obtained is inconsequential though, as the desired isomer 113 could exclusively be obtained by a simple epimerization sequence. Exposure of this compound to the aforementioned ruthenium conditions led to the cycloisomerization product 114 in good yield. Repeating this sequence of events then gave the subunit of yessotoxin (115).
Scheme 32.
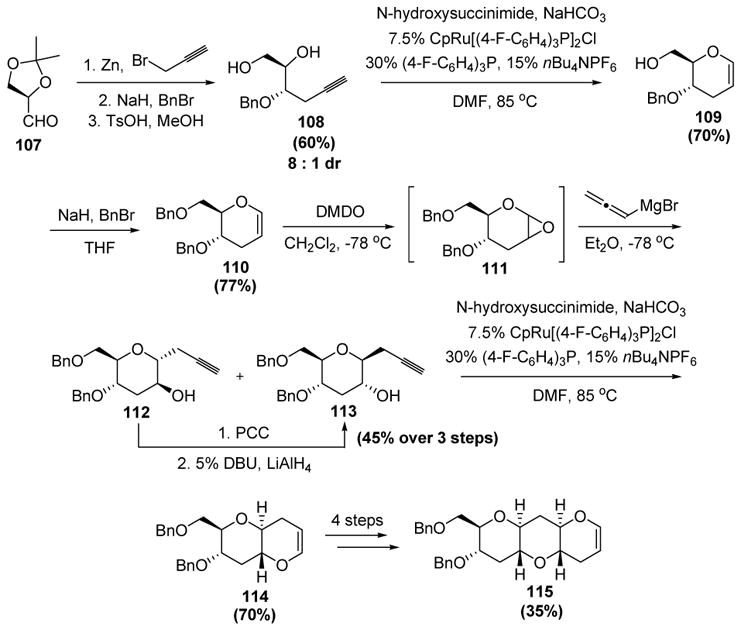
Trost’s Iterative Approach to Trans-Fused Polycyclic Ethers. BnBr=benzyl bromide, Cp=η5-cyclopentadienyl, DBU=1,8-diazabicyclo[5.4.0]undec-7-ene, DMDO=dimethyl dioxirane, DMF=N,N-dimethylformamide, PCC=pyridinium chlorochromate, THF=tetrahydrofuran, TsOH=p-toluenesulfonic acid.
Trost subsequently reported the cycloisomerization of homo- and bis-homopropargyl alcohols in the presence of rhodium complexes bearing electron-withdrawing phosphine ligands (Scheme 33).[51] Although relatively large amounts of ligand were still required, the reaction required less additives and showed higher catalyst turnover numbers (lower catalyst loadings) than the ruthenium-based system. Importantly, propargyl ethers were resistant to elimination (equation 2).
Scheme 33.
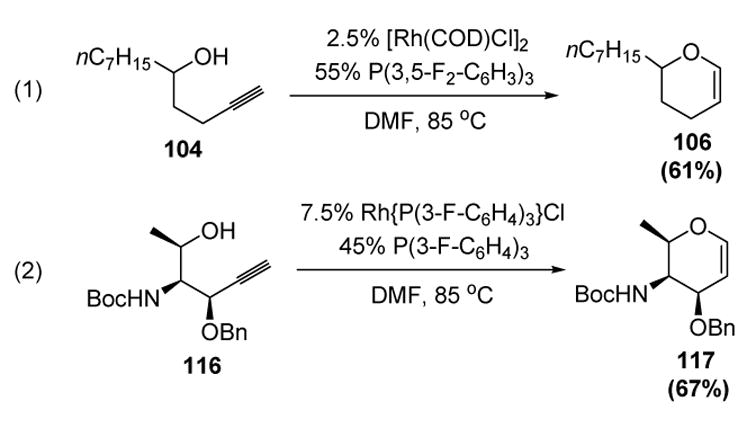
Rhodium-Catalyzed Cycloisomerization of Homo- and Bis-homopropargyl Alcohols. COD=1,5-cyclooctadiene, DMF=N,N-dimethylformamide.
2.4 Water as a Nucleophile
Whereas alcohols have failed to act as nucleophiles in catalytic intermolecular reactions with metal vinylidenes, water has been successful. This process accomplishes an atom economical conversion of terminal alkynes to aldehydes, a process that has been traditionally involved stoichiometric regioselective hydroboration and subsequent oxidation. This transformation thus holds considerable promise for organic synthesis, although it has not yet been utilized in target-oriented synthesis. Wakatsuki was the first to report this transformation; thus treatment of n-hexyne (12) with aqueous isopropanol and a ruthenium catalyst led to the corresponding aldehyde (118) as the major product (Scheme 34).[52] However, hindered alkynes such as phenylacetylene and t-butylacetylene failed to react.
Scheme 34.
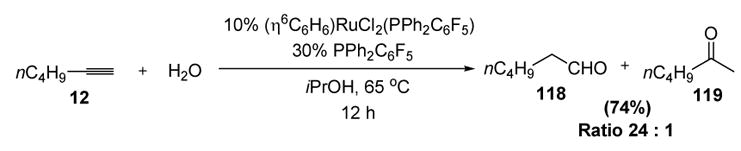
Wakatsuki’s Anti-Markovnikov Hydration of Terminal Acetylenes
A mechanism consistent with the observed regioselectivity is shown in Scheme 35. The terminal alkyne and the metal species I may form vinylidene species II, which is then trapped by water. Tautomerization and reductive elimination then can lead to the aldehyde product. The ketone (Markovnikov) product may be formed by electrophilic activation of the alkyne (V) and regioselective attack of water at the more electrophilic site. The excess phosphine ligand is thought to promote vinylidene formation (II) over simple alkyne activation (V).
Scheme 35.
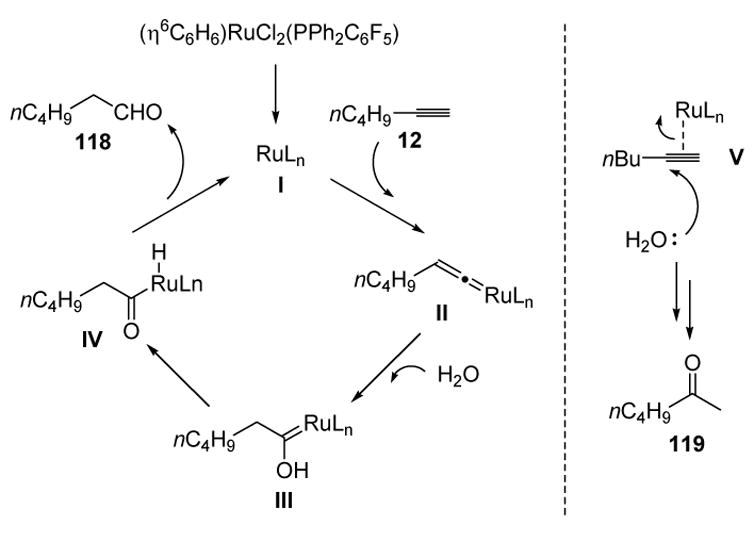
Mechanism of Anti-Markovnikov Alkyne Hydration
Wakatsuki subsequently reported that a variety of alkynes, including hindered and functionalized alkynes, could be effectively hydrated to the corresponding aldehydes with lower amounts of ruthenium complex 120 (Figure 1).[53] The bidentate nature of the dppm ligand was crucial to the success of this process. Other ruthenium complexes have also been used to catalyze this transformation. Bassetti and coworkers have reported that ruthenium complex 121 can catalyze the anti-Markovnikov hydration of alkynes in water containing surfactants such as sodium dodecylsulfate.[54] Grotjahn and colleagues utilized ruthenium complex 122 to hydrate a variety of alkynes, a catalyst that may deliver water in an intramolecular fashion to the vinylidene intermediate.[55] Breit has used ruthenium complex 123 to catalyze the transformation, a catalyst generated in situ based on the same principles of hydrogen bonding that guide the self-assembly of adenine and thymine in DNA.[56] More recently, Hintermann and colleagues reported the use of a catalyst generated from cationic ruthenium complex 124 and ligands such as 125. The benefits of this catalyst system include modification of the ligand through a modular synthesis as well as exceptional catalytic activity.[57]
Figure 1.
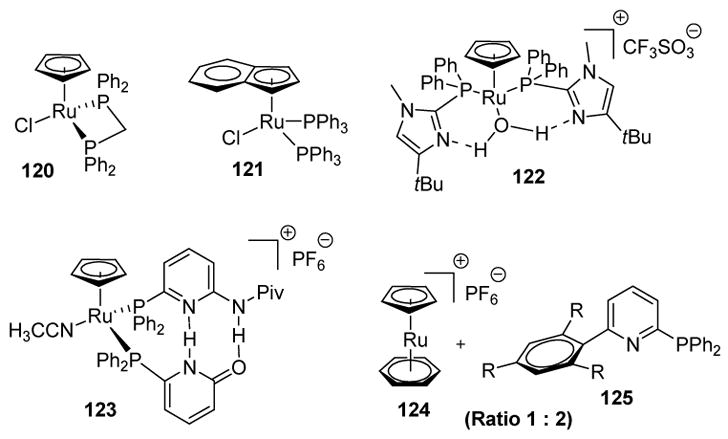
Ruthenium Catalysts for Anti-Markovnikov Alkyne Hydration.
Wakatsuki has also investigated the regioselective ruthenium-catalyzed hydration of propargyl alcohols to give enals. This reaction is a transition metal-catalyzed version of the Meyer-Schuster rearrangement, and thus provides a complimentary approach to the similar transformation involving benzoic acid developed by Dixneuf and coworkers (vide supra).[23,58] In an example of this reaction, propargyl alcohol 126 combines with water in the presence of a ruthenium catalyst to provide enal 127 in good yield and moderate Z/E stereoselectivity (Scheme 36).[59]
Scheme 36.
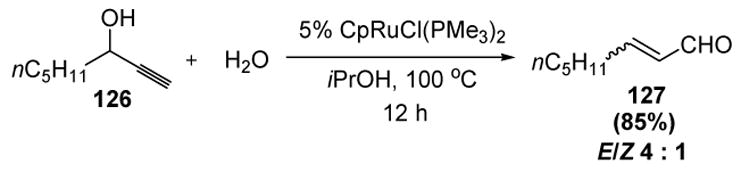
Wakatsuki’s Isomerization of Propargyl Alcohols. Cp=η5-cyclopentadienyl.
Labeling studies (H218O) suggest that the mechanism does not involve transposition of the alcohol oxygen to the carbonyl group. While it is not clear at which stage dehydration occurs, a plausible mechanism is presented in Scheme 37. The active catalyst I may insert into the alkyne C-H bond, and elimination of water then leads to allenylidene III. Addition of water to the electrophilic carbon then gives enol IV. Tautomerization, followed by reductive elimination, then gives enal 127 and regenerates the active catalyst.
Scheme 37.
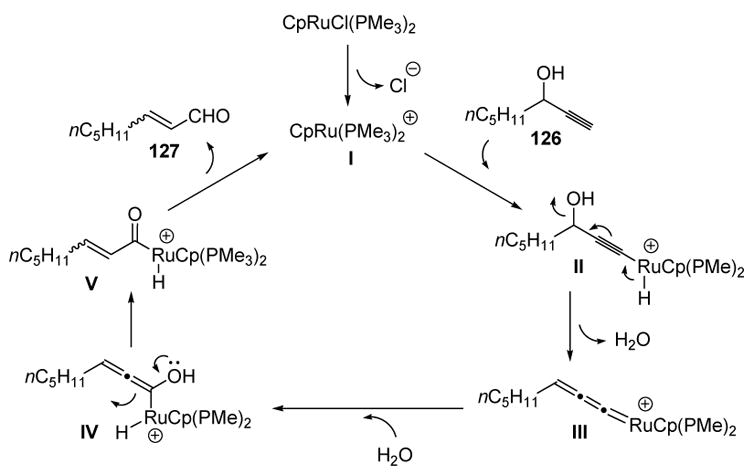
Proposed Mechanism of Propargyl Alcohol Isomerization. Cp=η5-cyclopentadienyl.
A novel tandem reaction involving water and a vinylidene intermediate has been recently reported by Lee and coworkers. In a process termed hydrative cyclization, a ruthenium complex catalyzes the formation of cyclopentanones from 1,5-enynes. An example of this reaction is depicted in Scheme 38.[60]
Scheme 38.
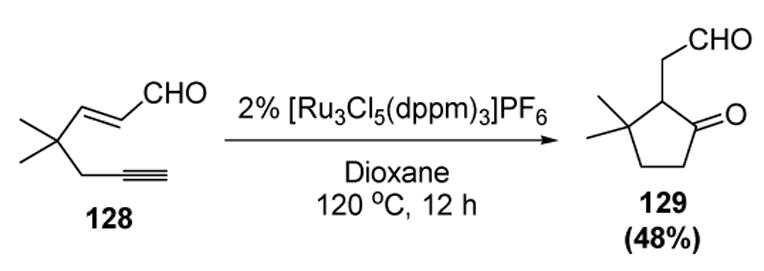
Lee’s Hydrative Cyclization of 1,5-Enynes. dppm=bis(diphenylphosphino)methane.
One mechanism that would explain this interesting domino reaction is depicted in Scheme 39. Thus nucleophilic capture of vinylidene II by water, followed by tautomerization, can give IV. Deprotonation of the metal and Michael addition of the acyl ruthenium species V leads to VI. Finally, protonation and reductive elimination regenerates the active catalyst and the cyclopentanone product 129.
Scheme 39.
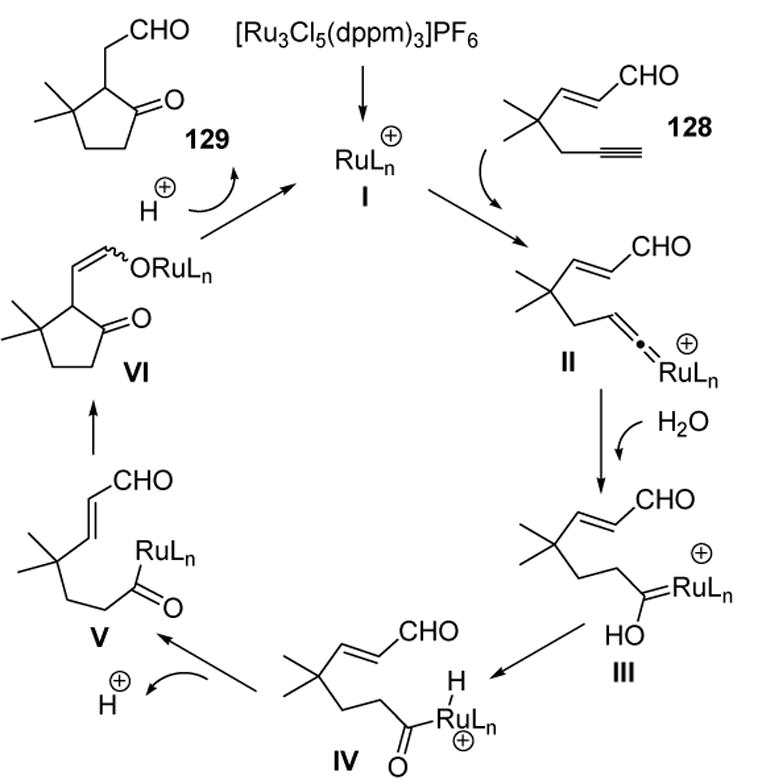
Proposed Mechanism for Lee’s Hydrative Cyclization of 1,5-Enynes. dppm=bis(diphenylphosphino)methane.
2.5 Epoxide Nucleophiles
McDonald was the first to report the cyclization of epoxyalkynes to furans through a vinylidene mechanism.[61] For example, treatment of 130 with a catalytic amount of a molybdenum pentacarbonyl catalyst led to 131 (equation 1, Scheme 40). Liu and coworkers subsequently disclosed that the same type of transformation could be catalyzed by a lower loading of a ruthenium complex (equation 2).[62]
Scheme 40.
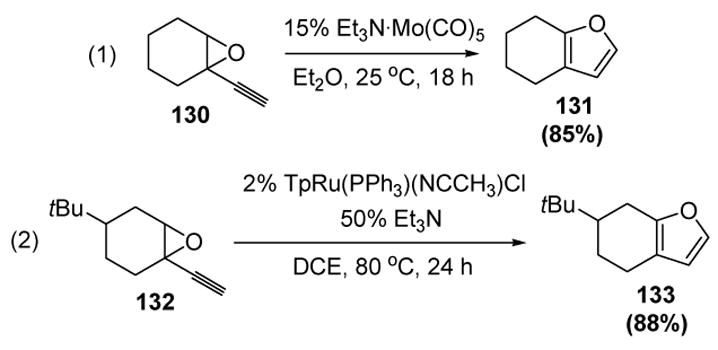
Formation of Furans from Epoxyalkynes. DCE=1,2-dichloroethane, Tp=tris(pyrazolyl)borate.
The mechanism of this transformation is rationalized by considering vinylidene intermediate II (Scheme 41). Attack by the epoxide oxygen can then lead to the rearranged Fischer carbene III. Vinylogous deprotonation, followed by protonation on the metal center, yields intermediate V, and reductive elimination then gives the product and regenerates the catalyst I.
Scheme 41.
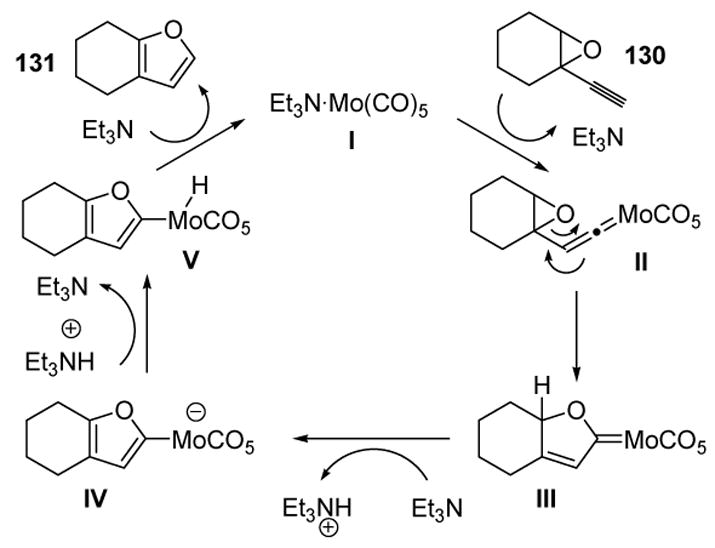
Mechanism of Furan Formation from Epoxyalkynes.
An interesting variation of the substrate structure can lead to different products.[63] Thus (o-ethynyl)styrene epoxides such as 134, when treated with catalytic amount of [TpRu(PPh3)(NCCH3)2]PF6, lead to 2-naphthol products such as 135 (equation 1, Scheme 42). Higher substituted epoxides (136), however, lead to 1-alkylidene-2-indanones 137 instead (equation 2).
Scheme 42.
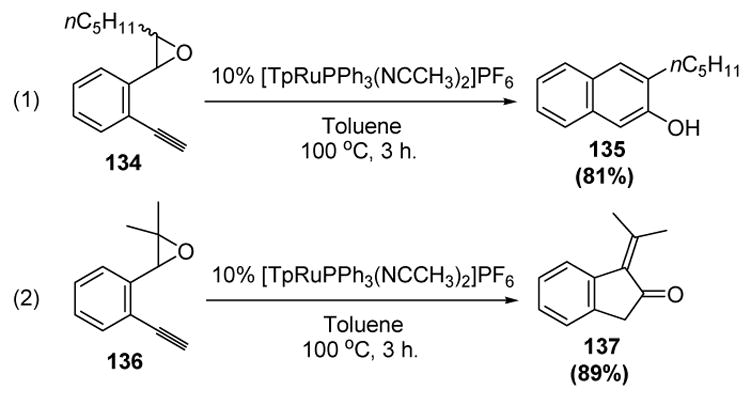
Liu’s Syntheses of 2-Naphthols and 1-Alkylidene-2-Indanones. Tp=tris(pyrazolyl)borate.
This dichotomy of products can be explained according to the mechanism depicted in Scheme 43. The loss of acetonitrile ligands opens up coordination spaces on the ruthenium complex to generate the active catalyst I. Vinylidene formation with either alkyne 134 or 136 then leads to II. Attack of the epoxide at the electrophilic carbon of the vinylidene species then leads to III, whereby cleavage gives the key ruthenium π-ketene intermediate IV. In the case of 1,2-disubstituted olefins (R1 = H, R2 = nC5H11), 6-endo-dig electrocyclization leads to benzylic carbocation V. Subsequent elimination provides VI, whereby protonation of the metal, followed by reductive elimination, gives 2-naphthol product 135 and liberates the active catalyst. Alternatively, in the case of trisubstituted olefins, IV undergoes 5-endo-dig cyclization to give the relatively stable tertiary carbocation VII. Elimination then gives VIII, and protonation at the metal, followed by reductive elimination, gives 1-alkylidene-2-indanone 137.
Scheme 43.
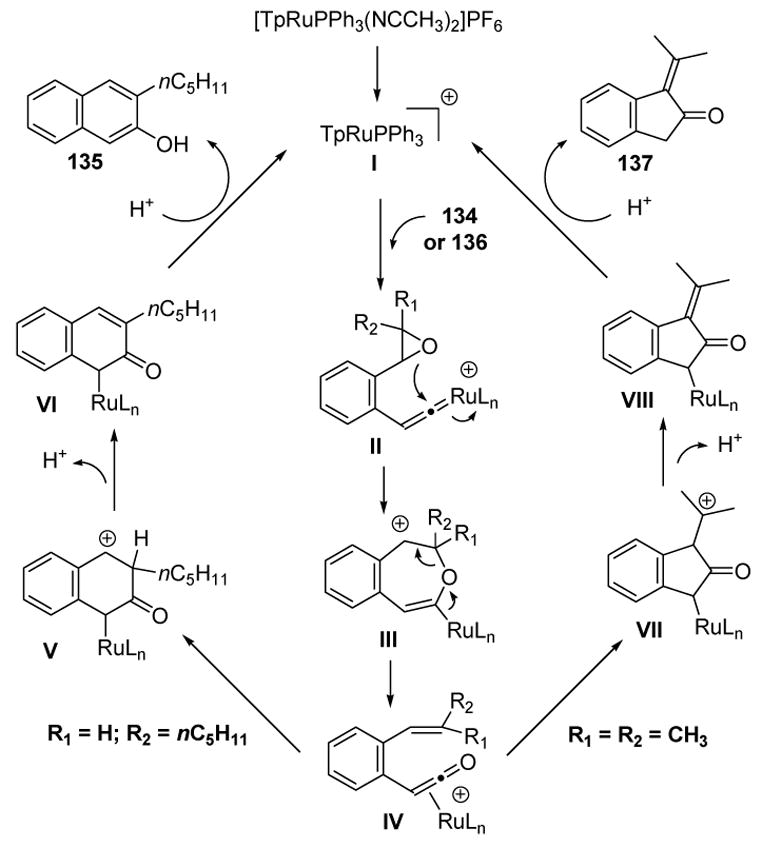
Mechanism of 2-Naphthol and 1-Alkylidene-2-Indanone Formation. Tp=tris(pyrazolyl)borate.
Recently, Liu and colleagues have disclosed that iodoalkynes, such as 138, can lead to 1-iodo-2-naphthol products, such as 139 (Scheme 44).[64] This result adds to the synthetic utility of the method, since aryl iodides can be further elaborated through a variety of metal-catalyzed cross-coupling reactions. Furthermore, the position of the iodide in the product lends credence to the proposed pathway of these reactions, as this group would migrate to the 1-position upon formation of the vinylidene intermediate (cf. Scheme 43).
Scheme 44.
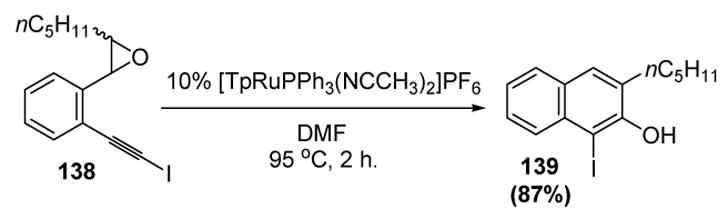
Liu’s Formation of 1-Iodo-2-Naphthols. Tp=tris(pyrazolyl)borate.
2.6 Carbonyl Nucleophiles
The interception of metal vinylidene intermediates by carbonyl groups has only been recently reported. In the first report, Uemura and coworkers disclosed that conjugated enyne esters (140) were transformed into pyranylidene metal complexes (142), presumably from vinylidene formation (141) followed by [3,3]-sigmatropic rearrangement (Scheme 45). These Fischer carbene products are analogs of α-pyrones, and thus were anticipated to participate in Diels-Alder reactions with dienophiles. In the event, dimethyl acetylenedicarboxylate added to pyranylidene 142 to generate tetrahydronaphthalene 144 in low yield, presumably via retrocycloaddition of intermediate 143.[65,66]
Scheme 45.
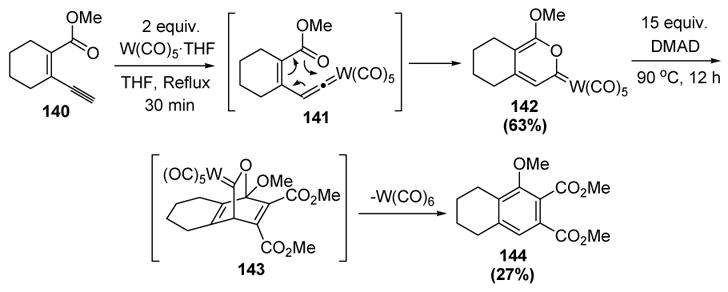
Uemura’s Synthesis and Diels-Alder Reaction of Pyranylidenes. DMAD= dimethylacetylene dicarboxylate, THF=tetrahydrofuran.
Iwasawa and colleagues contemporaneously reported a similar set of reactions. Thus o-ethynyl arylketones, such as 145, are converted into benzopyranylidenes, such as 147, via [3,3]-sigmatropic rearrangement of vinylidene 146 (Scheme 46).[66] Diels-Alder reaction with n-butyl vinyl ether, followed by retrocycloaddition, provides 149, whereby elimination of n-butanol provides naphthalene 150.
Scheme 46.
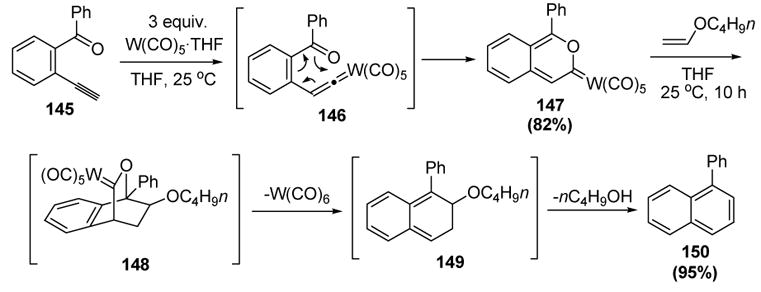
Iwasawa’s Synthesis and Diels-Alder Reaction of Benzopyranylidenes. THF=tetrahydrofuran.
Whereas these cases involved stoichiometric metal vinylidenes, Uemura and coworkers reported the first catalytic interception of metal vinylidene complexes by carbonyl groups. Treatment of cis-1-acyl-2-ethynylcyclopropanes, such as 151, with catalytic amounts of either tungsten or chromium pentacarbonyl complexes led to 2-substituted phenols (152) in excellent yield (Scheme 47).[66,68]
Scheme 47.
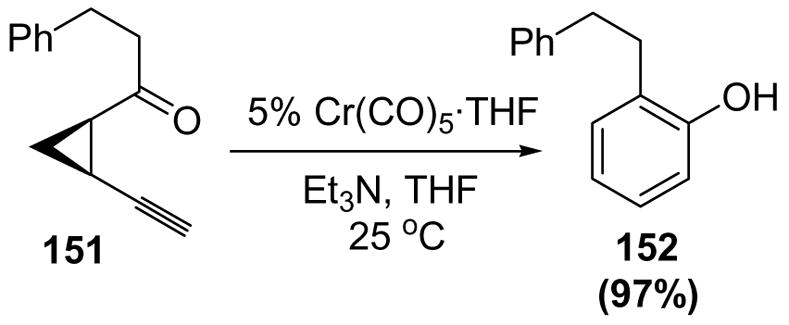
Uemura’s Catalytic Synthesis of Phenols from cis-1-Acyl-2-ethynylcyclopropanes. THF=tetrahydrofuran.
The proposed catalytic cycle is depicted in Scheme 48. Vinylidene formation between catalyst I and substrate 151, followed by [3,3]-sigmatropic rearrangement, affords III. A [1,5]-hydrogen shift, and reductive elimination, then liberates the catalyst and arene oxide V. A second [3,3]-sigmatropic rearrangement then gives VI, and subsequent rearrangement then yields product 152.
Scheme 48.
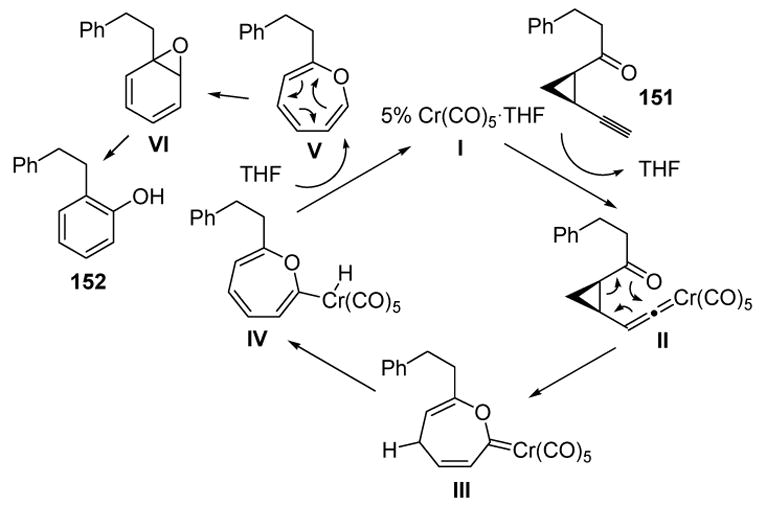
Mechanism of Phenol Formation from cis-1-Acyl-2-ethynylcyclopropanes. THF=tetrahydrofuran.
2.7 Thiol Nucleophiles
Despite the success of homopropargyl alcohol cycloisomerization, there has only been one report of homopropargyl thiols undergoing cyclization to the corresponding dihydrothiophenes.[69] In this case, a stoichiometric amount of chromium hexacarbonyl was required to give the product in good yield (Scheme 49). The lack of catalytic processes is presumably due to the strong coordination of thiols to metals, thus leading to catalyst deactivation.
Scheme 49.
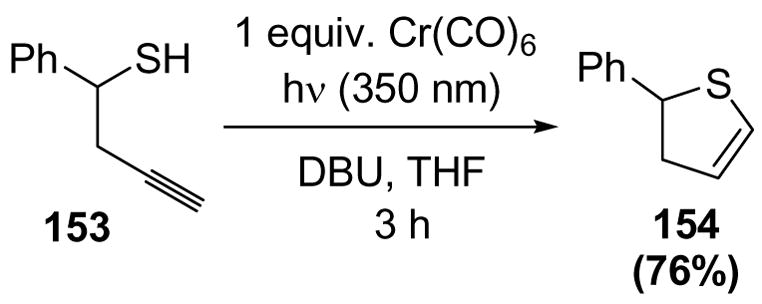
Cycloisomerization of Homopropargyl Thiols. DBU=1,8-diazabicyclo[5.4.0]undec-7-ene, THF=tetrahydrofuran.
2.8 Amine Nucleophiles
2.8.1 Intermolecular Reactions
There have been no reports of simple primary or secondary amines adding to metal vinylidene intermediates in a catalytic reaction, again possibly due to catalyst deactivation by the amine substrates. However, Watanabe and coworkers have reported the ruthenium-catalyzed addition of an amide (acetanilide, 156) to 1-octyne (155) to give the enamide product 157 (Scheme 50).[70] There were only a limited number of examples reported, and the yields were moderate, but the regio- and stereoselectivity of the process were excellent.
Scheme 50.
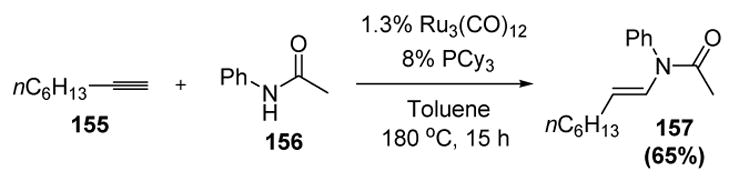
Watanabe’s Synthesis of an Enamide from Acetanilide and 1-Octyne
Several years later, Gooβen and colleagues reported that a number of amides could be added to terminal alkynes bearing a wide array of functional groups using a different ruthenium catalyst (Scheme 51).[71] For example, 2-pyrrolidinone (158) reacts with n-hexyne (12) in excellent yield, with complete regioselectivity and good E/Z selectivity (equation 1). Product 160 can be prepared from enyne 24 and amide 58 (equation 2), thus constituting an atom economical synthesis of activated dienes for Diels-Alder reactions.
Scheme 51.
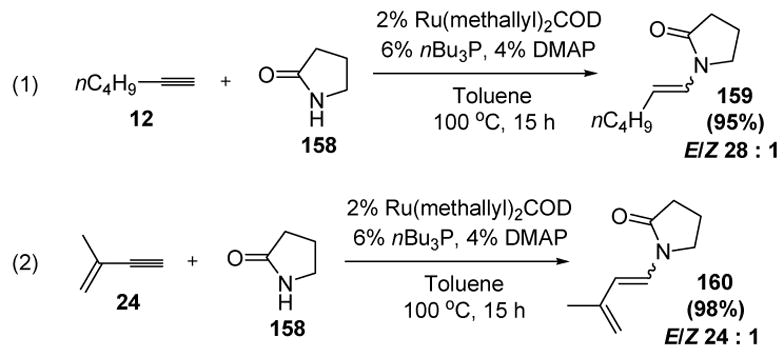
Gooβen’s Synthesis of Enamides from Amides and Alkynes. COD=1,5-cyclooctadiene, DMAP=4-dimethylaminopyridine.
Fukumoto and coworkers recently reported the use of N,N-dialkylhydrazines as nucleophiles for vinylidene complexes.[72] A variety of aromatic and aliphatic alkynes were reported to combine with N,N-dimethylhydrazine to give nitrile products, an example of which is depicted in Scheme 52.
Scheme 52.

Fukumoto’s Nitrile Synthesis from N,N-Dimethylhydrazine and Alkynes. Tp=tris(pyrazolyl)borate.
The proposed mechanism of this process is outlined in Scheme 53, starting with loss of chloride ion from the ruthenium complex to generate cationic species I. Vinylidene formation with the alkyne substrate then gives II. Nucleophilic addition of N,N-dimethylhydrazine provides III, whereby tautomerization and proton transfer leads to V. Finally, fragmentation releases the product 163, N,N-dimethylamine (164), and regenerates the active catalyst. It is interesting that the N,N-dimethylamine byproduct does not shut the reaction down by coordination with the catalyst, a fact that bodes well for future examinations of catalytic amine additions to vinylidenes.
Scheme 53.
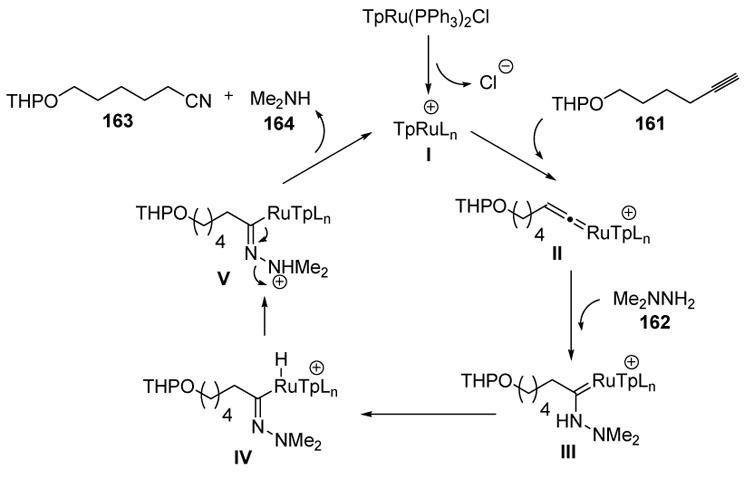
Proposed Mechanism of Fukumoto’s Nitrile Synthesis. Tp=tris(pyrazolyl)borate.
2.8.2 Intramolecular Reactions
McDonald was the first to report the cyclization of amines on to vinylidene intermediates. Thus N-Boc homopropargylamines (165) and bis-homopropargyl amines (167) undergo endo cyclization in the presence of stoichiometric amounts of molybdenum and tungsten complexes, respectively (equations 1 and 2, Scheme 54).[73] The related amide and sulfonamide substrates were inert to the reaction conditions. Although the corresponding free amine substrates led to decomposition, o-ethynylaniline (169) was found to cyclize to indole (170) in the presence of a catalytic amount of the molybdenum complex (equation 3).
Scheme 54.
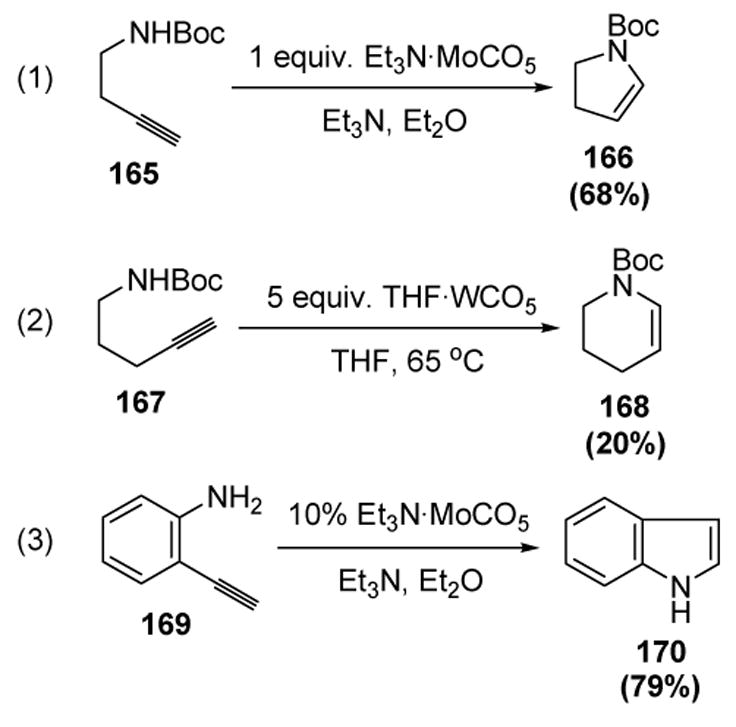
McDonald’s Cycloisomerization of Homo- and Bis-homopropargyl Amines. THF=tetrahydrofuran.
More recently, Trost has shown that a variety of o-ethynylanilines could cyclize to the corresponding indoles in the presence of a catalytic amount of a rhodium complex (equation 1, Scheme 55).[74] The exclusive participation of the terminal alkyne in equation 2 lends credence to the proposed vinylidene pathway. This process was further extended to tethered phenol and carboxylic acid nucleophiles.
Scheme 55.
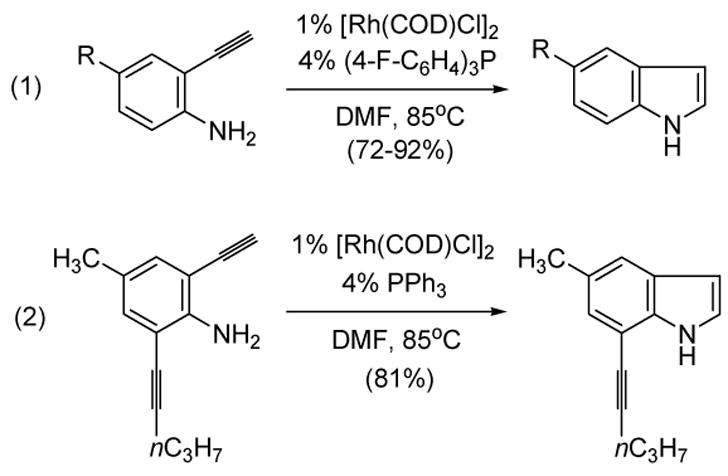
Trost’s Cycloisomerization of o-Ethynylanilines to Indoles
In an interesting transformation, Jun and coworkers have recently reported that terminal alkynes may be dimerized in the presence of water, Wilkinson’s catalyst, and 2-aminopicoline (171), the latter reagent acting as a cofactor for the reaction (Scheme 56).[75]
Scheme 56.
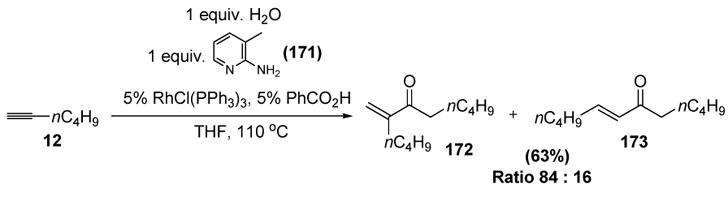
Jun’s Chelation-Assisted Hydrative Dimerization of Terminal Alkynes. THF=tetrahydrofuran.
The mechanism of this transformation is thought to involve vinylidene formation (I), followed by coordination of the 2-amino-3-picoline with the metal (II, Scheme 57). This serves to template the amino group for an intramolecular addition to the vinylidene. Following attack and tautomerization, one obtains IV, a metalacycle that may insert a molecule of alkyne. Depending on the bulk of the alkyne, this insertion will favor either V or VI. The former case is favored for small substituents (Ex. nC4H9), and after the subsequent ring contraction one obtains VII. Reductive elimination and hydrolysis then gives product 172 and the active catalyst. The minor product 173 is obtained from VI in a series of similar steps.
Scheme 57.
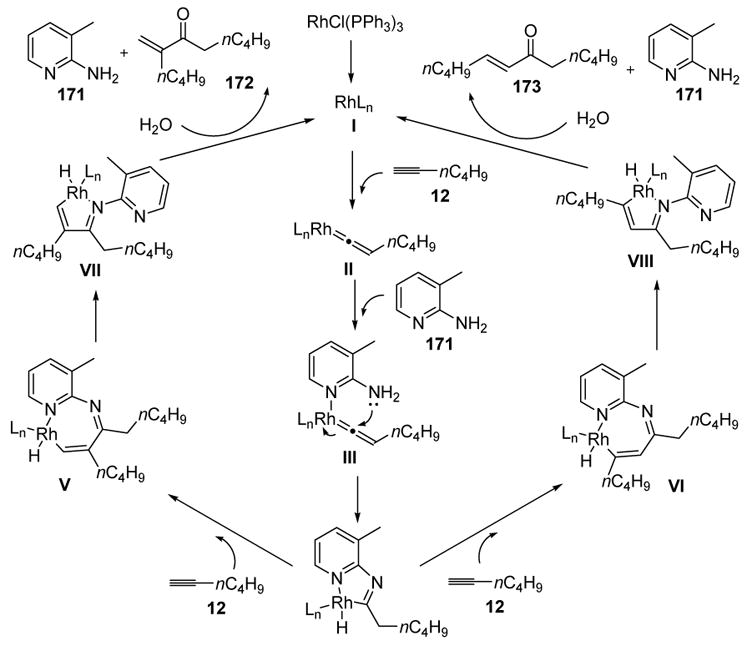
Mechanism of Jun’s Chelation-Assisted Hydrative Dimerization of Terminal Alkynes
2.9 Phosphine Nucleophiles
There has only been one example of anti-Markovnikov hydrophosphination of alkynes, although the utility of such products has not been demonstrated. Dixneuf and coworkers have found that diphenylphosphine (175) adds to propargyl alcohols in good yield and Z/E selectivity (Scheme 58).[76]
Scheme 58.
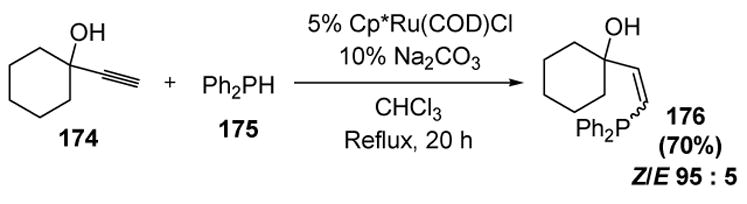
Hydrophosphination of Propargyl Alcohols. COD=1,5-cyclooctadiene, Cp=η5-cyclopentadienyl.
3. Carbon Nucleophile Reactions with Vinylidenes
McDonald and coworkers reported the first trapping of vinylidene intermediates by pendant carbon nucleophiles. Homopropargyl malonates, β-ketoesters, and β-diketones (i.e. stabilized nucleophiles) undergo cyloisomerization (5-endo attack) in the presence of approximately stoichiometric amounts of a molybdenum pentacarbonyl complex (equation 1, Scheme 59).[77] The use of a bis-homopropargyl β-dicarbonyl substrate, however, only resulted in 5-exo attack (equation 2).
Scheme 59.
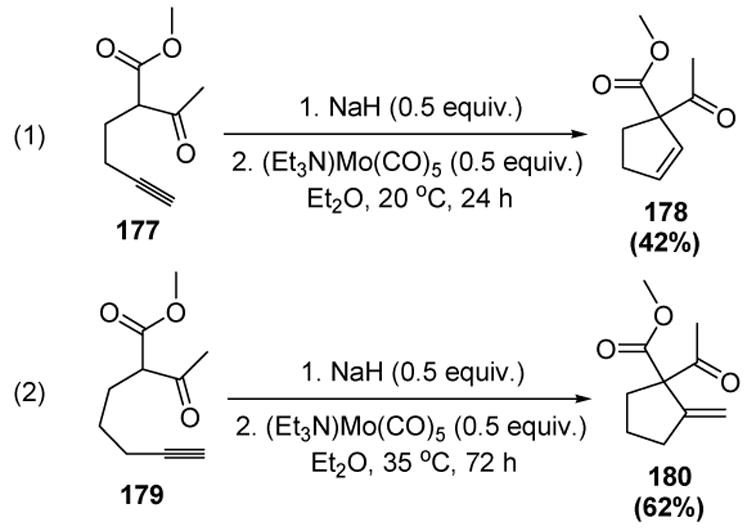
Cyclization of Homo- and Bis-homopropargyl β-Dicarbonyl Compounds
A possible mechanism involves deprotonation of the β-dicarbonyl substrate, followed by vinylidene formation, to give III (Scheme 60). Nucleophilic trapping then leads to IV, whereby protonation at the metal by the substrate, followed by reductive elimination, gives the product and the metal complex I.
Scheme 60.
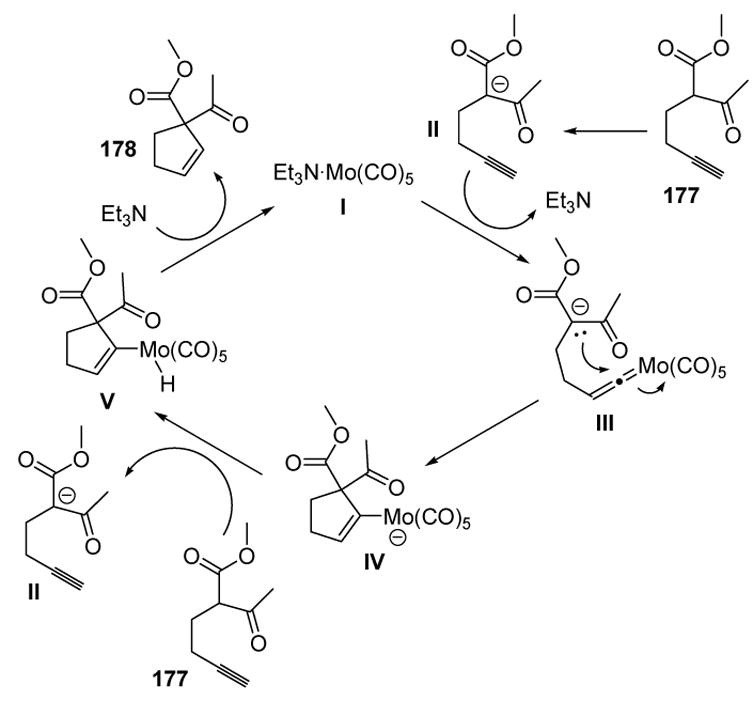
Mechanism of McDonald’s Homopropargyl β-Dicarbonyl Cycloisomerization
Iwasawa and coworkers have further investigated this reaction. Less stabilized nucleophiles (silyl enol ethers) were found to function in the presence of lower loadings of a tungsten catalyst. Five-membered rings (equation 1) as well as six-membered rings (equations 2 and 3) have been reported to form in good to excellent yield (Scheme 61).[78]
Scheme 61.
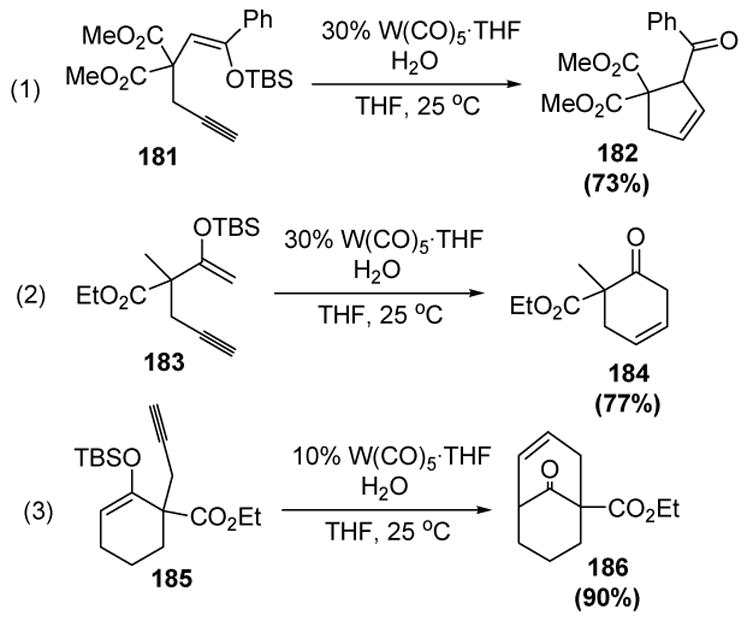
Iwasawa’s Cycloisomerization of Homo- and Bis-homopropargyl Silyl Enol Ethers. THF=tetrahydrofuran.
Iwasawa subsequently extended this reaction into a general method for cyclopentene annulation (Scheme 62).[79,80] Indium-mediated propargylation of α,β-unsaturated ketones (ex. (R)-carvone, 187), in the presence of dimethyl sulfide and t-butyldimethylsilyl triflate, provides the requisite substrates (189). Cycloisomerization in the presence of catalytic amounts of a tungsten carbonyl complex then give the unsubstituted cyclopentene products, such as 190.
Scheme 62.
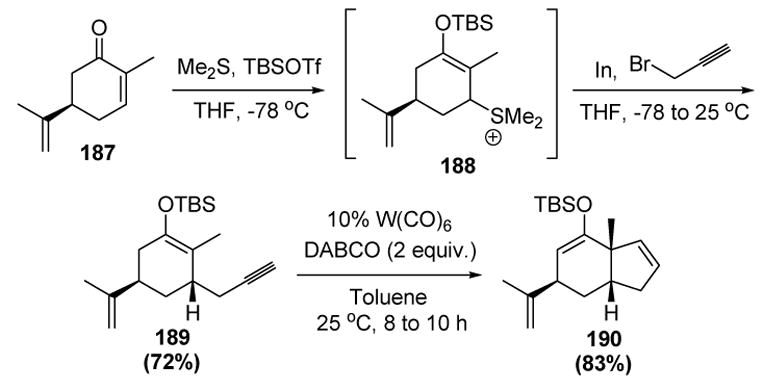
Iwasawa’s Cyclopentene Annulation Method. DABCO=1,4-diazabicyclo[2.2.2]octane, TBSOTf= tert-butyldimethylsilyl trifluoromethanesulfonate, THF=tetrahydrofuran.
The use of iodoalkynes, such as 191, led to the iodocyclopentene products such as 192 via migration of the iodine atom during vinylidene formation, thus extending the synthetic utility of the method (Scheme 63).
Scheme 63.
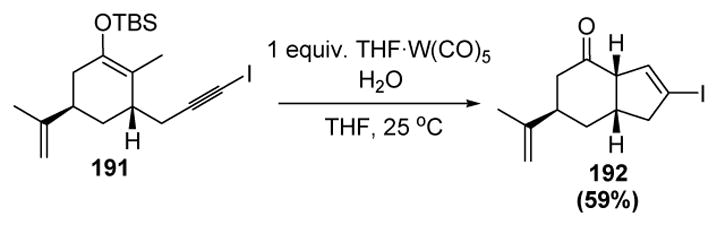
Iwasawa’s Cyclopentene Annulation Method: Iodine Migration. THF=tetrahydrofuran.
More recently, Lee and colleagues have reported that N-propargyl enamines undergo cycloisomerization in the presence of a rhodium catalyst developed by Trost for the cycloisomerization of homo- and bis-homopropargyl alcohols (vide supra).[51] Thus N-tosyl propargyl enamine 193 underwent cyclization to provide 1,3-diene product 194 (equation 1), while N-benzoyl propargyl enamine 195 cyclized to form 1,4-diene 196 in excellent yield (equation 2), both products containing the indolizidine skeleton (Scheme 64).[81]
Scheme 64.
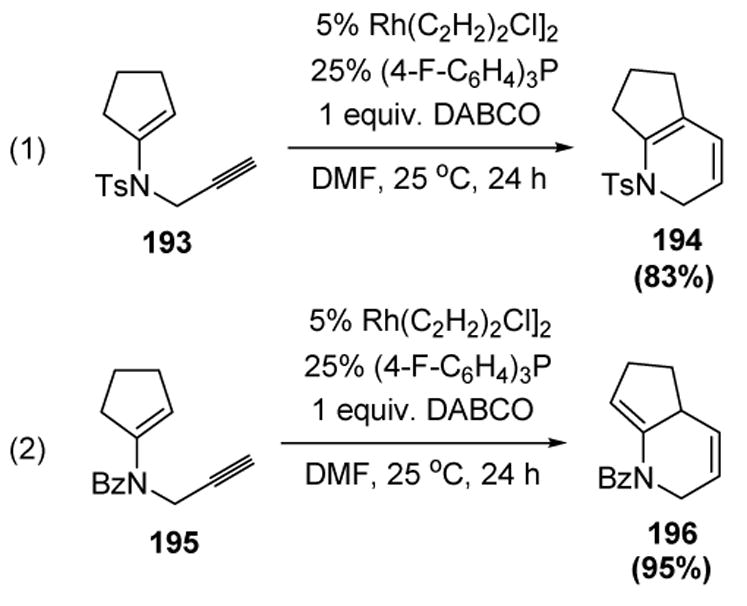
Lee’s N-Propargyl Enamine Cycloisomerization. DABCO=1,4-diazabicyclo[2.2.2]octane, DMF= N,N-dimethylformamide.
The different products obtained may be rationalized according to Scheme 65. Terminal alkyne substrates 193 or 195 form vinylidene intermediate II, which undergoes intramolecular attack by the pendant enamine to give zwitterionic iminium species III. In the case of the sulfonamide protecting group, the base (DABCO) deprotonates the iminium species to give IV. Protonation at the metal, followed by reductive elimination, then gives 1,3-diene product 194 and regenerates the active catalyst. On the other hand, when the protecting group is an amide, III may suffer a 1,5-H shift, followed by deprotonation to yield VII. Protonation of the metal and reductive elimination then gives 1,4-diene product 196 and the active catalyst.
Scheme 65.
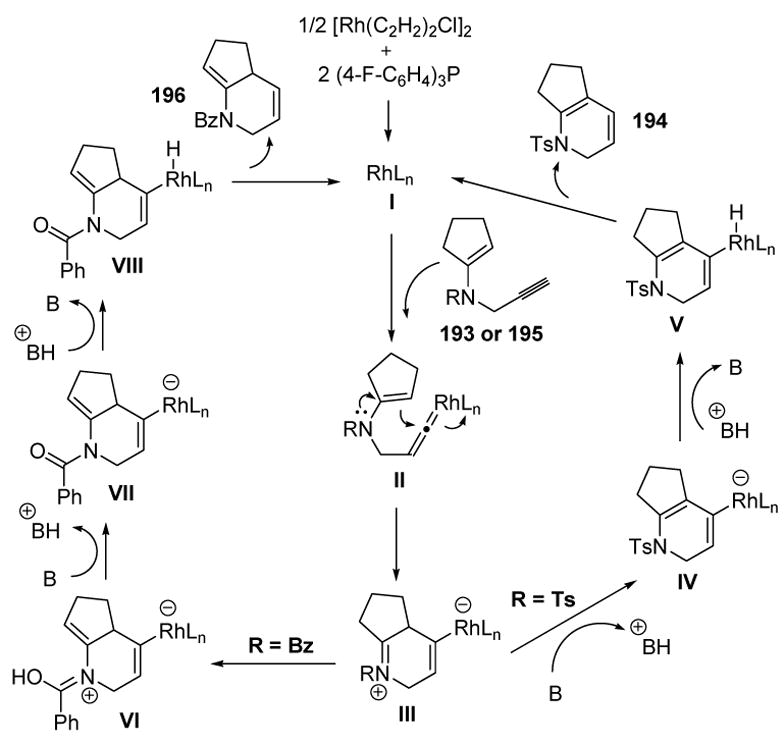
Proposed Mechanism of N-Propargyl Enamine Cycloisomerization
4. Pericyclic Reactions with Vinylidenes
4.1 Electrocyclization Reactions
The oxa 6π electrocyclization of carbonyl groups onto vinylidene species was seen in stoichiometric reactions earlier in this review (vide supra). The first example of a reaction involving an all-carbon 6π electrocyclization onto a catalytic vinylidene intermediate was reported by Merlic. In this reaction, dienylalkynes, such as 197, underwent conversion to benzene derivatives, such as 198 (Scheme 66).[82]
Scheme 66.
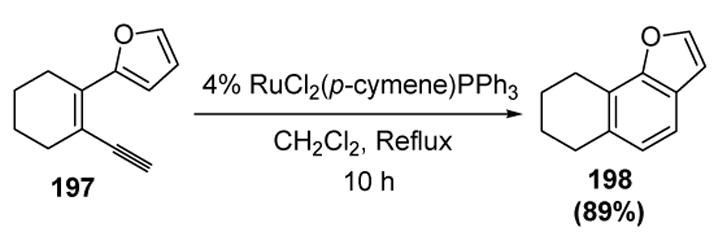
Merlic’s Dienyne Cycloisomerization
The catalytic cycle for this transformation is depicted in Scheme 67. Following vinylidene formation (II), the key 6π electrocyclic ring closure leads to III. The latter step may be either concerted or stepwise. Tautomerization, followed by reductive elimination, then gives the product and regenerates the active catalyst I.
Scheme 67.
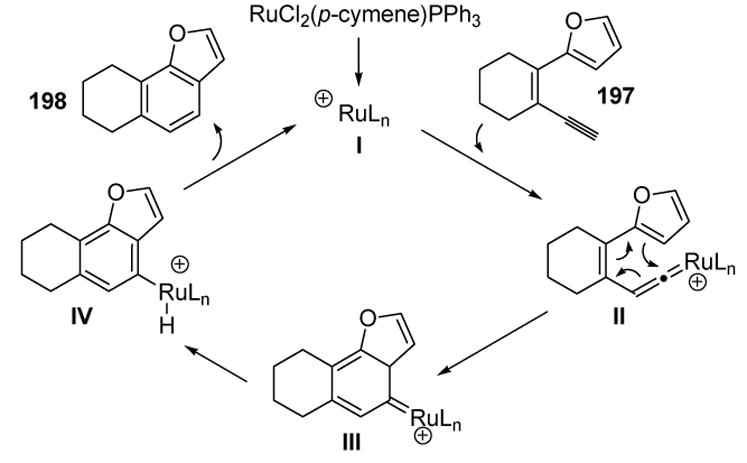
Plausible Mechanism for Dienyne Cycloisomerization
Scott has recently applied Merlic’s conditions in a naphthoannulation procedure (Scheme 68).[83] For example, anthraquinone (199) was olefinated to the bis-gem dibromide, and Sonogashira cross-coupling with trimethylsilylacetylene, followed by protodesilylation, provided substrate 200. Exposure to the ruthenium catalyst then led to a four-fold dienyne cycloisomerization, providing coronene (201). Although the yield was modest, the convergency of the approach and the amount of complexity generated in the reaction is impressive.
Scheme 68.
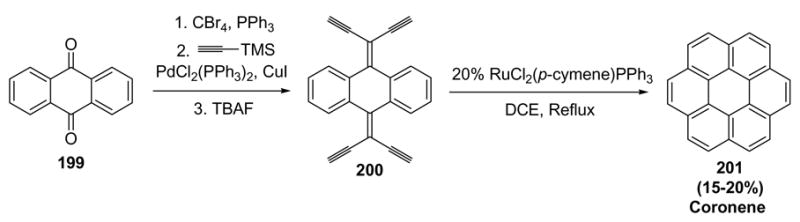
Scott’s Naphthonannulation Procedure. DCE=1,2-dichloroethane, TBAF=tetrabutylammonium fluoride.
Iwasawa has reported that a tungsten complex can also catalyze the 6π electrocyclization of dienynes (equation 1, Scheme 69).[84] Exclusive formation of iodonaphthalene 205 from iodoalkyne 204 increases the synthetic utility of the reaction (equation 2).[85]
Scheme 69.
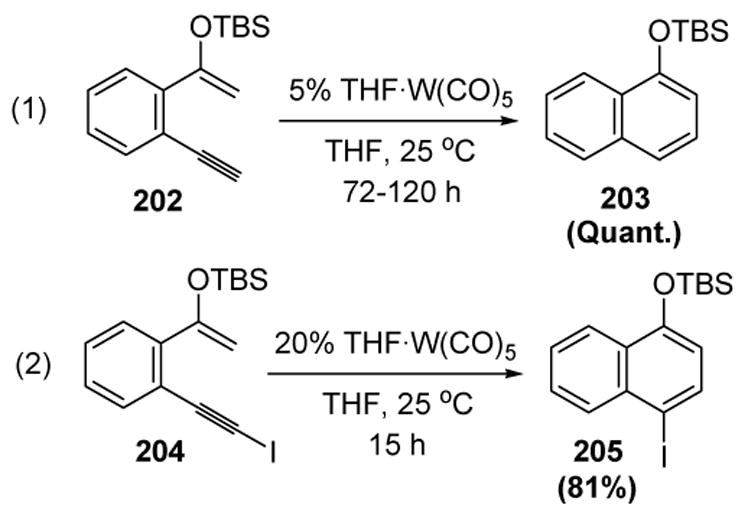
Iwasawa’s Dienyne Cycloisomerization. THF=tetrahydrofuran.
Akiyama and coworkers have expanded the substrate scope to include alkynyl imines (Scheme 70).[86] The yields were improved by treating the crude reaction mixture with N-methylmorpholine-N-oxide, presumably to decomplex the product from the tungsten catalyst.
Scheme 70.

Akiyama’s Cycloisomerization of Alkynyl Imines. NMO= N-methylmorpholine N-oxide, THF=tetrahydrofuran.
Liu and coworkers have recently reported a number of ruthenium-catalyzed electrocyclizations of dienynes, followed by a variety of rearrangements depending on the substitution pattern of the substrate. In one such process, iododienyne substrate 208 was converted to iodonaphthalene 209 in good yield (Scheme 71).[87]
Scheme 71.
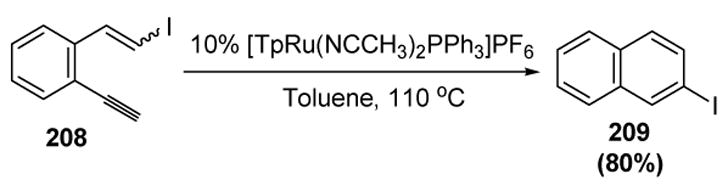
Liu’s Electrocyclization and Halide Migration. Tp=tris(pyrazolyl)borate.
To account for this migration of the iodine atom, the mechanism shown in Scheme 72 was proposed in which vinylidene II is proposed to undergo 6π electrocyclization. A 1,2-iodine migration leads to carbocation IV, which then rearranges to give the product.
Scheme 72.
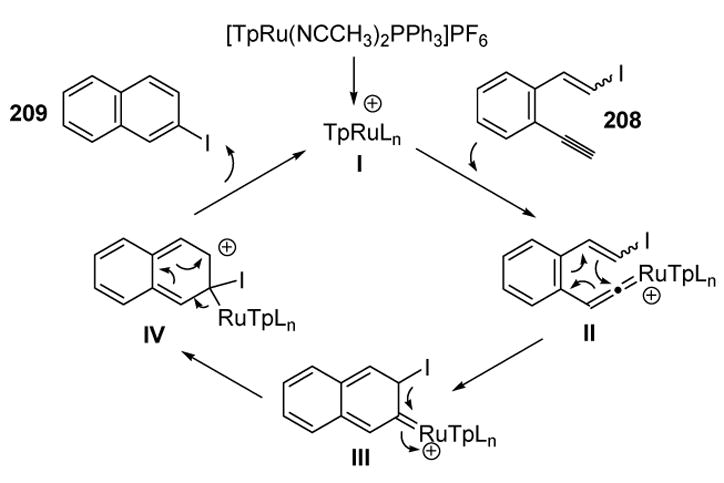
Mechanism of Liu’s Electrocyclization and Halide Migration. Tp=tris(pyrazolyl)borate.
An interesting divergence in mechanism is observed in the reaction of dienyne substrates 210 and 212 (Scheme 73). In the former case, a highly substituted benzene is formed (211, equation 1),[88] whereas in the latter case a 1,3-diene product is formed (213, equation 2).[89]
Scheme 73.
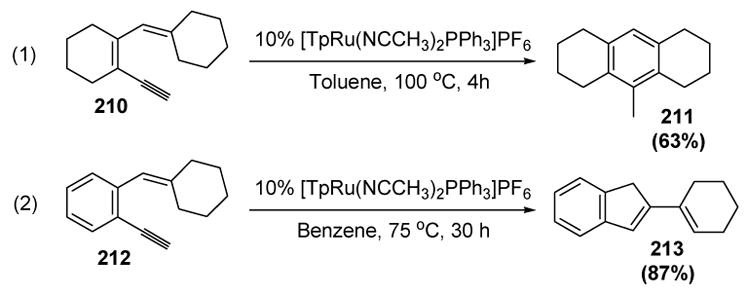
Liu’s Formation of Substituted Benzenes and 2-Vinyl 1H-Indenes. Tp=tris(pyrazolyl)borate.
The formation of product 211 can be explained by the proposed mechanism in Scheme 74. Following its formation, vinylidene II undergoes 6π electrocyclization to give III, whereby a 1,2-alkyl shift gives tertiary carbocation IV. A second 1,2-alkyl shift then gives cyclobutyl ruthenium species V. A 1,5-alkyl shift, followed by β-hydride elimination, leads to VII, and reductive elimination then yields the product and regenerates the active catalyst.
Scheme 74.
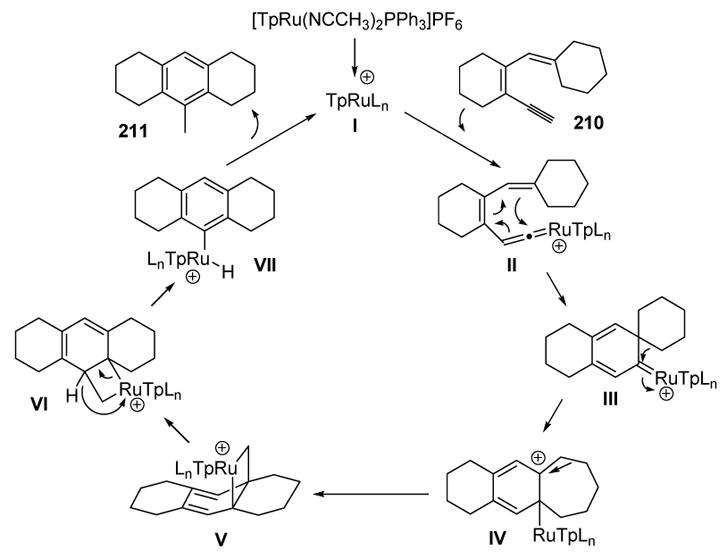
Mechanism of Liu’s Substituted Benzene Synthesis. Tp=tris(pyrazolyl)borate.
To explain the formation of 1,3-diene product 213, the vinylidene species II is attacked by the pendant olefin to generate tertiary carbocation III, which may be represented by resonance structure IV (Scheme 75). Demetalation then gives methylene cyclopropane V, which is prone to the well-established ring opening to the diradical trimethylenemethane VI.[90] This structure can be represented as fulvene VII, and electrophilic attack by the ruthenium catalyst on this species gives carbocation VIII. E1 elimination then leads to 1,3-diene species IX, and protonation of the metal, followed by reductive elimination, completes the catalytic cycle.
Scheme 75.
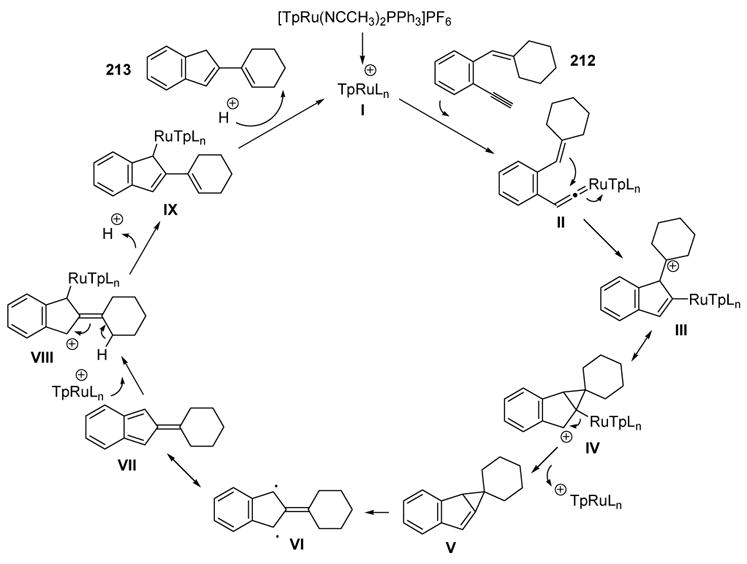
Mechanism of Liu’s 2-Vinyl 1H-Indene Synthesis. Tp=tris(pyrazolyl)borate.
4.2 [2+2] Cycloaddition Reactions
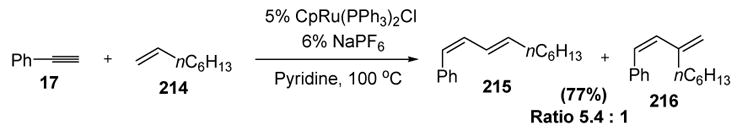
The first example of a [2+2] reaction on a catalytic vinylidene complex involved the intermolecular coupling of alkynes with alkenes to generate 1,3-diene products, as shown in Scheme 76.[91]
Scheme 76.
Murakami’s Intermolecular Alkene-Alkyne Coupling. Cp=η5-Cyclopentadienyl.
A proposed mechanism for the enyne coupling reaction is shown in Scheme 77. The formation of vinylidene intermediate II, followed by olefin coordination and oxidative coupling, leads to metalacyclobutane IV. The π-allyl structure V can then be formed by β-hydride elimination. Finally, reductive elimination releases the major product 215 and the catalyst. The minor isomeric 1,3-diene product 216 could arise from the alternative regiochemistry of metalacycle formation. Steric factors may then govern the product distribution, as the system would prefer to place the bulky olefin substituent closest to the developing (longer) carbon-ruthenium bond during metalacycle formation.
Scheme 77.
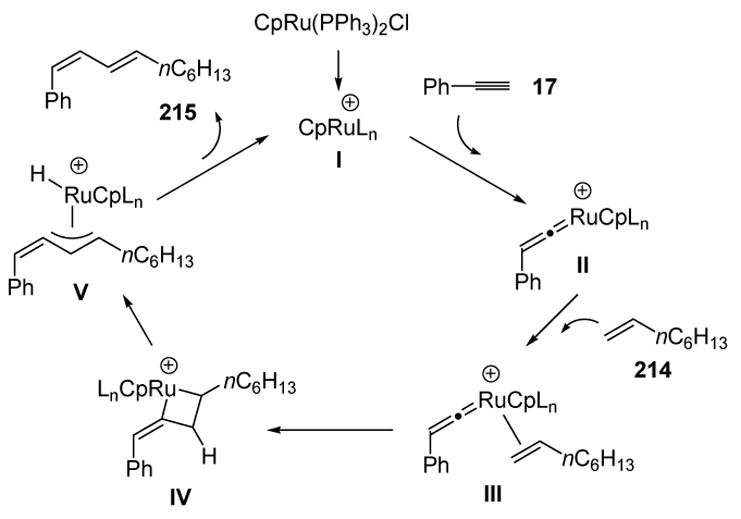
Mechanism of Intermolecular Enyne Coupling. Cp=η5-Cyclopentadienyl.
Murakami subsequently used the enyne coupling reaction in an interesting tandem transformation (Scheme 78). Treatment of enyne 217a with styrene, in the presence of a ruthenium catalyst, provided cyclohexadiene 220 in moderate yield.[92] In this transformation, Murakami takes advantage of the fact that a 1,3-diene is produced in the enyne coupling step, one of the olefins being Z. The presence of the third olefin (present in the substrate) sets up the conjugated triene intermediate 219 for a facile thermal disrotatory 6π electrocyclization to give the product. The use of the trimethylsilylalkyne (217b) provided the same product, but the yield was higher. This was explained as being due to the suppression of alkyne dimerization, a well-known reaction pathway for vinylidenes (vide infra). The silane presumably migrated during vinylidene formation, and was removed by protodesilylation at some point in the reaction.
Scheme 78.
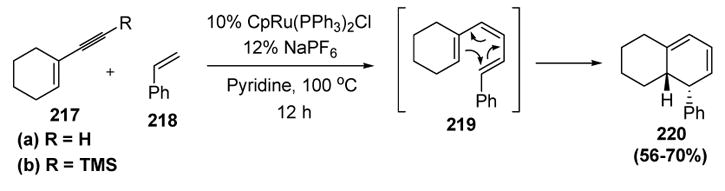
Murakami’s Tandem Enyne Coupling-Electrocyclization. Cp=η5-Cyclopentadienyl.
Murakami also discovered that pyridine is able to react with terminal alkynes through a vinylidene mechanism (Scheme 79).[93] Thus trimethylsilyl phenylacetylene reacts with pyridine to give the 2-alkenyl pyridine derivative 223 in good yield and with complete selectivity for the E olefin geometry (presumably through thermal or base-catalyzed isomerization). Although the catalyst loading and temperature are elevated, this nonetheless constitutes an intriguing method for regioselective pyridine functionalization; the selectivity results from templating the two reacting substrates by coordination of the pyridine to the metal of the ruthenium vinylidene intermediate.
Scheme 79.
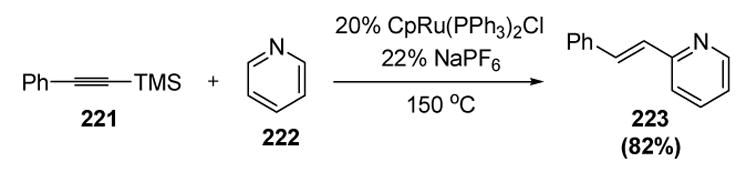
Murakami’s Alkenylation of Pyridine. Cp=η5-Cyclopentadienyl.
Tethering the two reacting partners can control the regioselectivity of enyne coupling. Grigg and colleagues reported some years ago that 1,6-enynes undergo cycloisomerization in the presence of catalytic Wilkinson’s catalyst to give the corresponding 1,3-dienes.[94] Recently, Lee and coworkers revisited this reaction and optimized the reaction conditions (equations 1 and 2, Scheme 80).
Scheme 80.
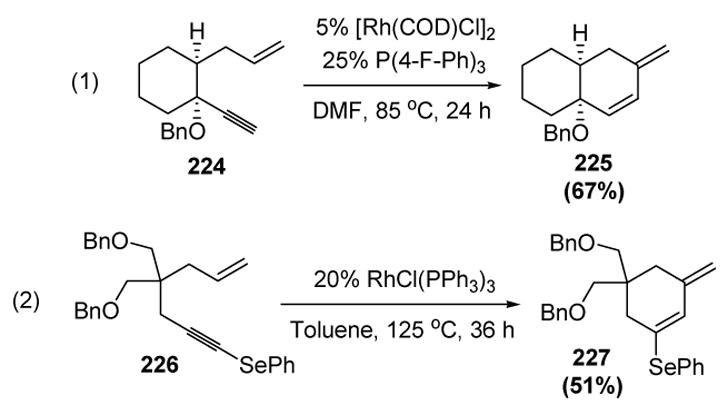
Rhodium-Catalyzed Enyne Cycloisomerization. COD=1,5-cyclooctadiene, DMF= N,N-dimethylformamide.
Lee proposed a mechanism for this reaction based on deuterium labeling studies, as well as the selective migration of the selenyl group in equation 2 above.[95] The process thus involves vinylidene formation (II), [2+2] cycloaddition to give III, β-hydride elimination to yield IV, and reductive elimination (Scheme 81).
Scheme 81.
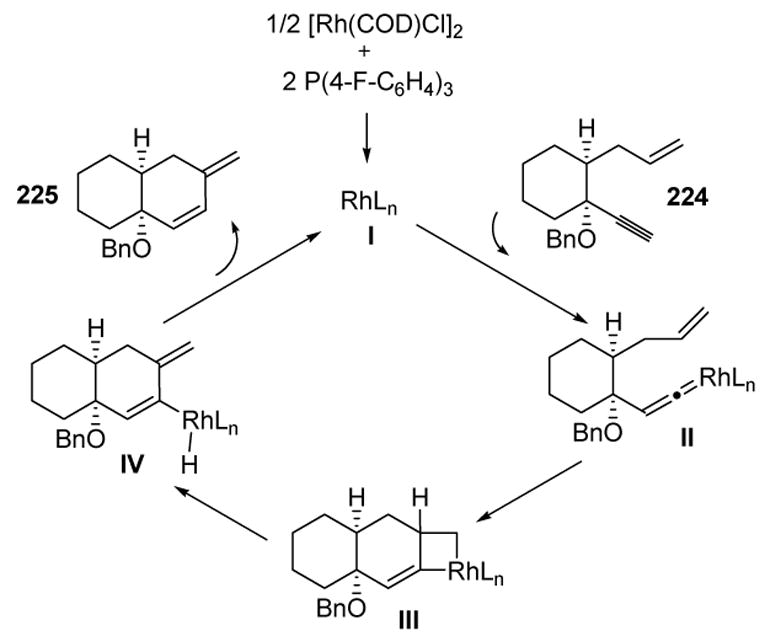
Mechanism of Rhodium-Catalyzed Enyne Cycloisomerization. COD=1,5-cyclooctadiene.
Lee has applied this transformation in a novel tandem process. Two rings are created when 1,6-enynes, bearing appropriately tethered alkyl halides, are treated with a catalytic rhodium species in the presence of triethylamine (Scheme 82).[96]
Scheme 82.
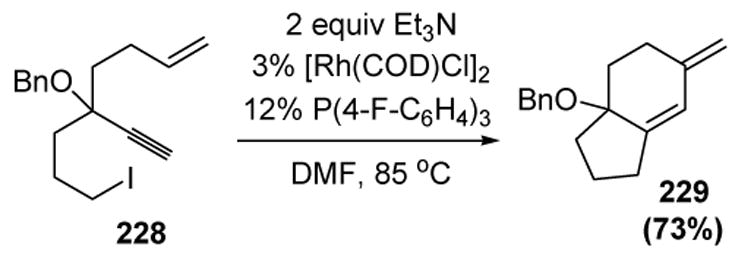
Lee’s Tandem Cyclization of 1,6-Enyne Systems. COD=1,5-cyclooctadiene, DMF= N,N-Dimethylformamide.
The proposed mechanism, depicted in Scheme 83, does not involve a metal vinylidene, but rather a metal alkenylidene (III). The first step involves insertion of the metal into the alkyne C-H bond to give II (which is presumably in equilibrium with the corresponding vinylidene species). This alkynyl metal species may be deprotonated by triethylamine, which induces displacement of the pendant iodide, leading to the alkenylidene species III. A [2+2] cycloaddition then gives IV, and β-hydride elimination, followed by reductive elimination, leads to product.
Scheme 83.
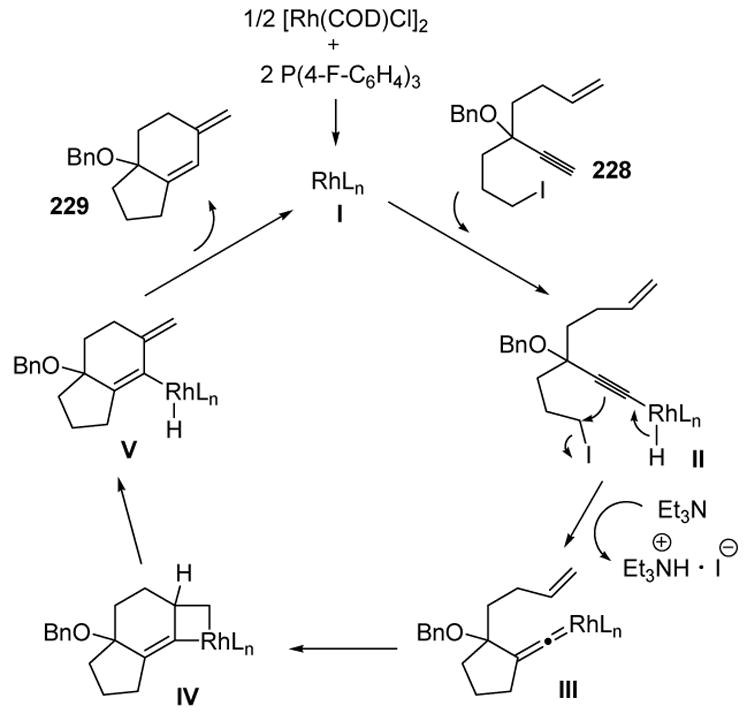
Mechanism of the Tandem 1,6-Enyne Cyclization. COD=1,5-cyclooctadiene.
A recent paper from Buono and coworkers suggests that catalytic vinylidene reactions may not be the exclusive domain of ruthenium, rhodium, molybdenum, and tungsten. Palladium, when combined with the appropriate ligands, may in fact participate in vinylidene processes. Norbornadiene (or norbornene) and phenylacetylene has been reported to undergo a [2+1] cycloaddition reaction, catalyzed by palladium diacetate and phosphinous acid ligand 231 (Scheme 84).[97]
Scheme 84.
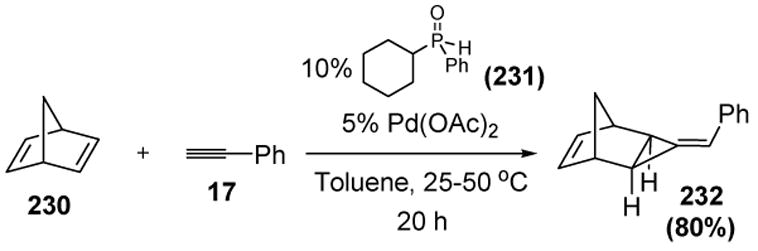
Buono’s Palladium-Catalyzed [2+1] Cycloaddition
The proposed mechanism, outlined in Scheme 85, suggests vinylidene formation (II, supported by labeling experiments), followed by coordination of norbornadiene to give III. A [2+2] cycloaddition, followed by reductive elimination, then gives the product and regenerates the catalyst.
Scheme 85.
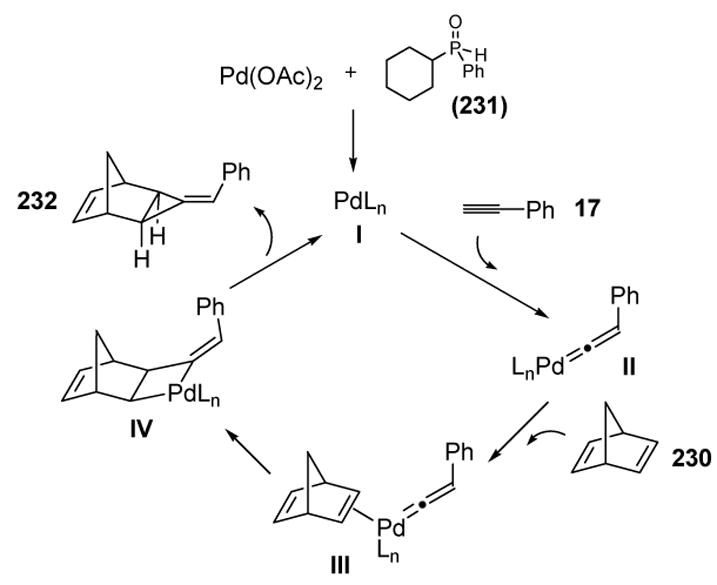
Mechanism of Buono’s Palladium-Catalyzed [2+1] Cycloaddition
4.3 [4+2] Cycloaddition Reactions
Tagliatesta and colleagues reported the only example of a Diels-Alder reaction involving a catalytic metal vinylidene complex.[98] In this transformation, rhodium and ruthenium porphyrin complexes were used to catalyze the conversion of arylacetylenes to 1-arylnaphthalenes (Scheme 86).
Scheme 86.
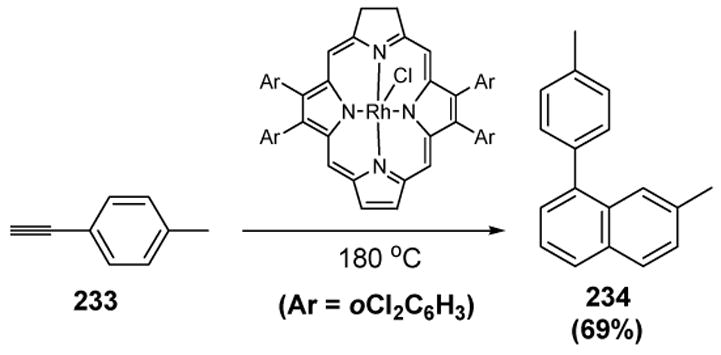
Tagliatesta’s Synthesis of 1-Arylnaphthalenes
The mechanism of the reaction can be rationalized according to Scheme 87. The rhodium porphyrin, represented by I, combines with the alkyne to form vinylidene II. An intermolecular Diels-Alder reaction then leads to III, and tautomerization, followed by reductive elimination, gives the product and releases the porphyrin catalyst. Although this reaction produced significant amounts of cyclotrimerization byproducts for most arylacetylenes, it does suggest the viability of a Diels-Alder manifold for vinylidene intermediates.
Scheme 87.
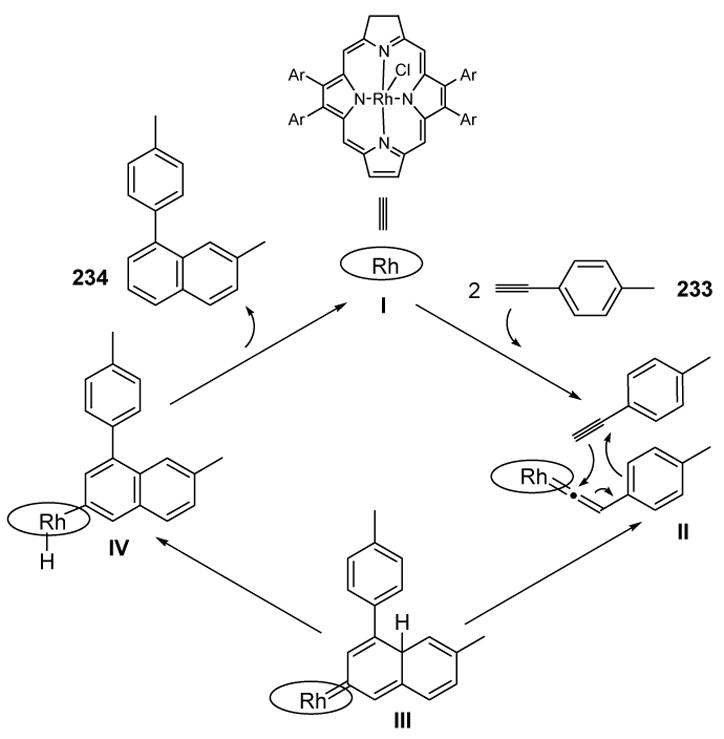
Mechanism of 1-Arylnaphthalene Formation from Arylacetylenes
4.4 [1,5]-Sigmatropic Rearrangement
Liu and coworkers recently reported another reaction pathway for metal vinylidenes. In a novel cyclopentadiene synthesis, the researchers made use of a [1,5]-hydrogen atom migration onto a vinylidene intermediate as a key step in the catalytic cycle.[99] In an example of this process, enyne 235 (or the corresponding propargyl alcohol) is converted to cyclopentadiene 236 in the presence of a ruthenium catalyst (Scheme 88).
Scheme 88.
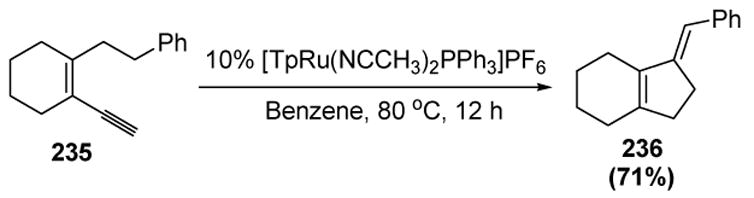
Liu’s Cyclopentadiene Synthesis. Tp= tris(pyrazolyl)borate.
The mechanism of this transformation, as outlined in Scheme 89, is thought to involve vinylidene formation (II), followed by a [1,5]-hydrogen atom migration onto the vinylidene moiety. This event is followed by a 6π electrocyclization, leading to IV, and then reductive elimination to give the active catalyst I and compound V. This species may then undergo two successive [1,5]-hydrogen migrations to give the observed product 236.
Scheme 89.
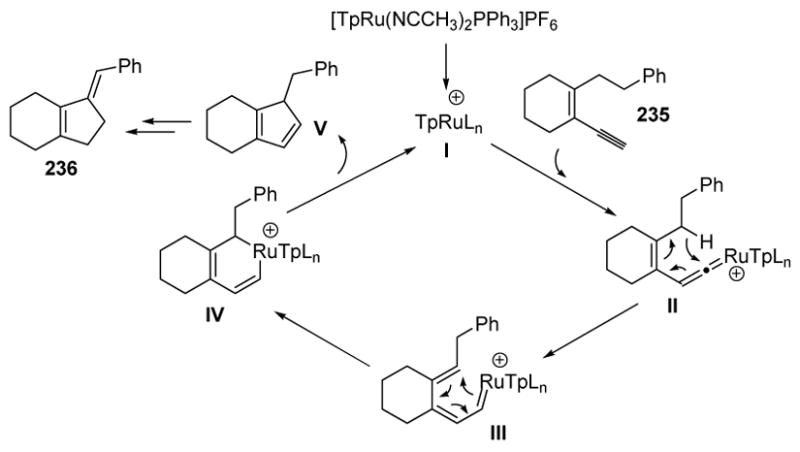
Mechanism of Cyclopentadiene Formation. Tp= tris(pyrazolyl)borate.
5. Cycloaromatization
Finn was the first to report a metal vinylidene mediated cycloaromatization reaction (Scheme 90).[100] This process begins with conversion of diyne 237 to the vinylidene complex 238 at room temperature. Subsequent treatment with cyclohexadiene in acetonitrile then induced Myers-Saito rearrangement (i.e. cycloaromatization, at a lower temperature than required for the all-carbon version) to give diradical 239. The carbon centered radical then induced a 5-exo-dig cyclization to give diradical 240. Hydrogen atom abstraction, followed by reductive elimination of the cationic metal fragment, then provided 242. The entire sequence (237 → 242) could conceivably be promoted by a catalytic amount of ruthenium, since the metal complex is released in the final step of the mechanism, but in practice a stoichiometric amount of metal was required.
Scheme 90.
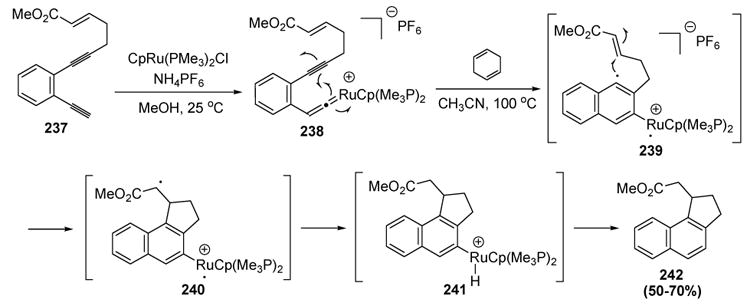
Finn’s Stoichiometric Metal-Mediated Cycloaromatization. Cp=η5-Cyclopentadienyl.
In switching to a rhodium catalyst, Uemura was able to render the cycloaromatization process catalytic. Thus treatment of enediyne 243 with a rhodium catalyst led to aromatic product 244 (Scheme 91).[101]
Scheme 91.
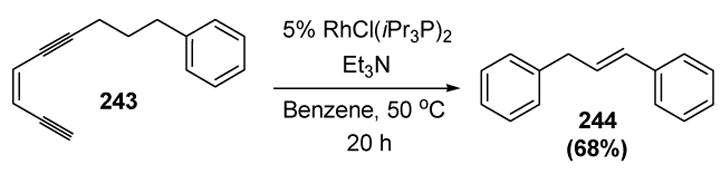
Uemura’s Rhodium-Catalyzed Cycloaromatization: 1,5-Hydrogen Migration
The proposed mechanism of this reaction involves vinylidene formation, followed by cycloaromatization, to give intermediate diradical III (Scheme 92). Intramolecular 1,5-hydrogen migration then gives IV, and radical-radical coupling leads to metalacycle V. β-Hydride elimination, followed by reductive elimination, then regenerates the catalyst and provides product 244.
Scheme 92.
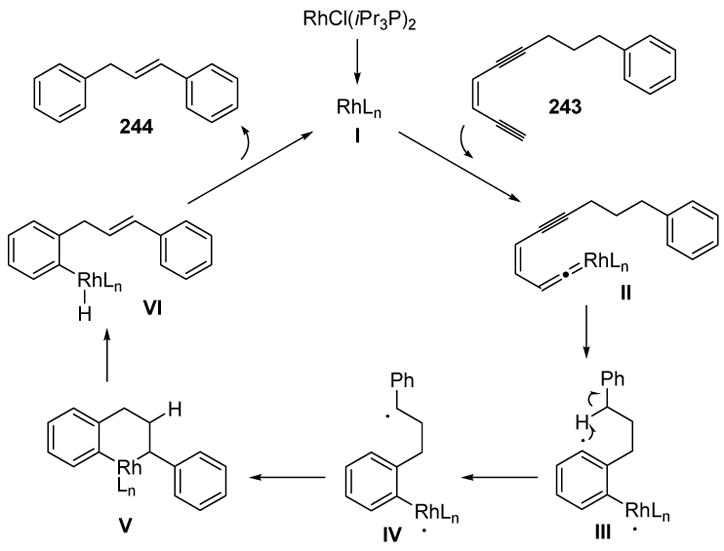
Mechanism of Rhodium-Catalyzed Cycloaromatization: 1,5-Hydrogen Migration
In the absence of an available hydrogen atom for the [1,5] shift, the reaction can follow a different course. This is seen in the following example, in which enediyne 245 was converted to silacycle 246 in moderate yield (Scheme 93).[102]
Scheme 93.
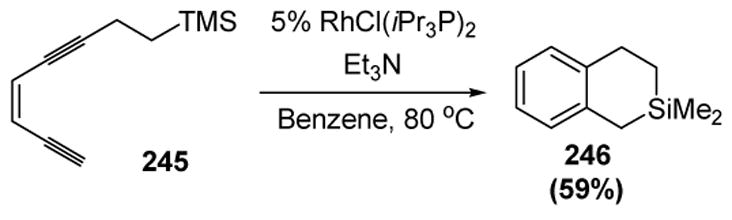
Uemura’s Rhodium-Catalyzed Cycloaromatization: 1,6-Hydrogen Migration
The formation of this product can be rationalized by assuming vinylidene formation, followed by cycloaromatization, to give diradical III (Scheme 94). A 1,6-hydrogen atom migration then leads to IV, and radical-radical coupling gives metalacycle V. Reductive elimination then provides the product.
Scheme 94.
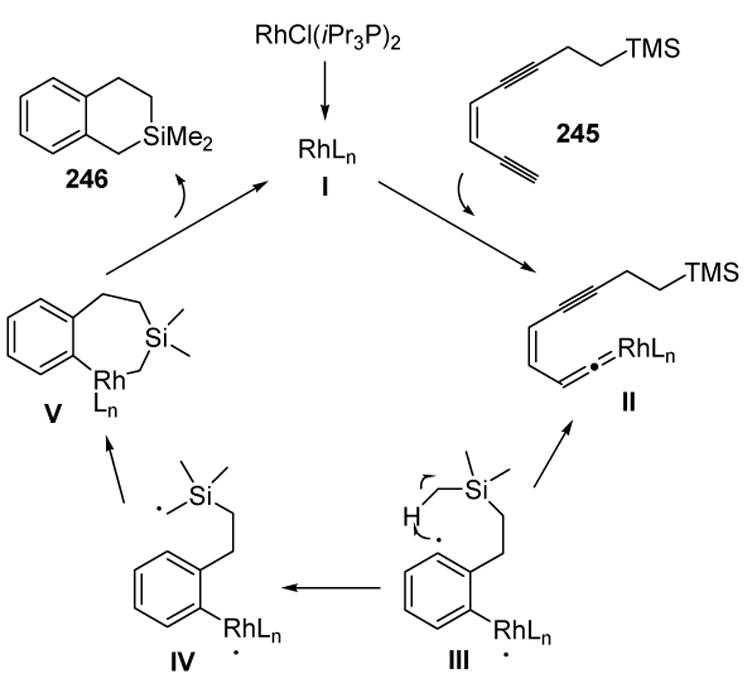
Mechanism of Rhodium-Catalyzed Cycloaromatization: 1,6-Hydrogen Migration
6. 1,2-Migration of Metal Ligands to Vinylidenes
6.1 Alkyne Dimerization
In principle, the metal-catalyzed head-to-head dimerization of terminal alkynes constitutes a direct method for the selective formation of conjugated enynes. In practice, there has been difficulty in controlling the selectivity of such processes. This is seen in the early work of Yamazaki, in which t-butylacetylene (247) was dimerized in the presence of a catalytic amount of a ruthenium complex to give a mixture of cumulenes 248 and 249, head-to-head enynes 250 and 251, and head-to-tail enyne 252 (Scheme 95).[103]
Scheme 95.

Yamazaki’s Dimerization of t-Butylacetylene
The preferential formation of 248 was later explained by Yamazaki and Wakatsuki (Scheme 96).[104] The formation of the active catalyst I was studied through identification of several intermediates leading to its formation. The catalytic cycle then commences with coordination of t-butylacetylene to give II. Vinylidene formation then leads to III, and intramolecular migration of an alkynyl ligand to the vinylidene leads to the Z σ-enynyl complex IV (the E σ-enynyl species would place a t-butyl group in close proximity with the ligand). This species leads to Z-enyne 251, but it is also in equilibrium with cumulene species V through a 1,3-shift. Although IV is more stable than V, coordination of the bulky t-butylacetylene to IV is disfavored due to steric effects, and thus the reaction preferentially continues on through V. Thus coordination of the alkyne leads to VI, and oxidative addition of another alkyne, followed by reductive elimination, then gives the cumulene product 248 and regenerates the active catalyst.
Scheme 96.
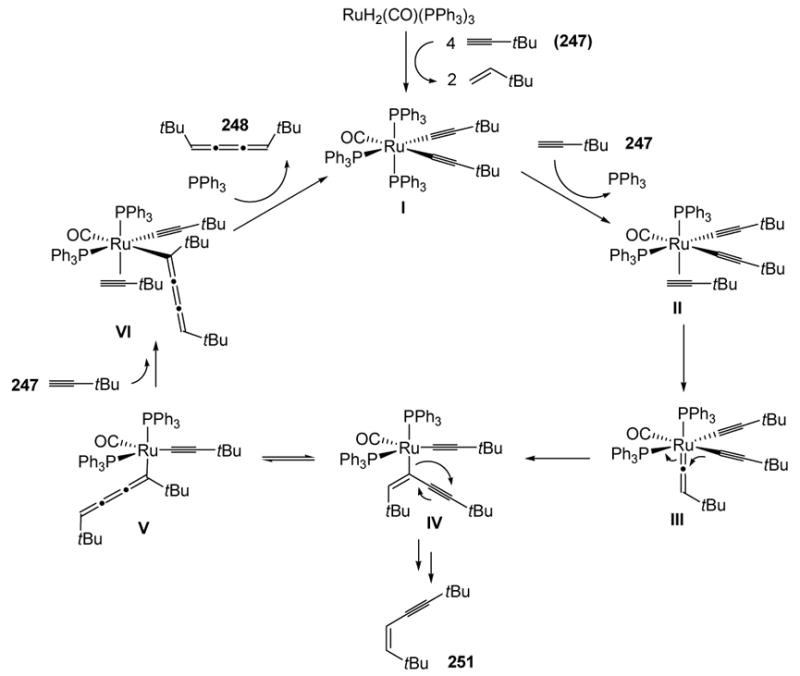
Catalytic Cycle for Yamazaki’s Dimerization of t-Butylacetylene
Bianchini subsequently reported that a σ-alkynyl ruthenium complex catalyzes the head-to-head dimerization of trimethylsilylacetylene and phenylacetylene in good yield and Z-selectivity (Scheme 97).[105] This mechanism proceeds through a series of steps similar to those leading to 232 above.[106]
Scheme 97.
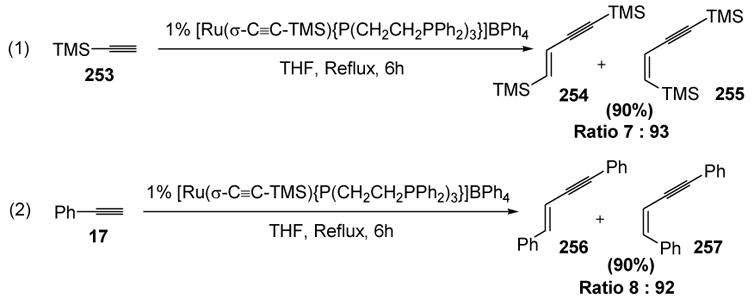
Bianchini’s Dimerization of Trimethylsilylacetylene and Phenylacetylene. THF=tetrahydrofuran.
Yi later reported an interesting ligand effect for the ruthenium-catalyzed dimerization of phenylacetylene. When Cp*Ru(L)H3 (L = PCy3) was used as the catalyst, phenylacetylene was dimerized in good yield to the Z-isomer, whereas with L = PMe3 the E-isomer was obtained as the major product (Scheme 98).[107]
Scheme 98.
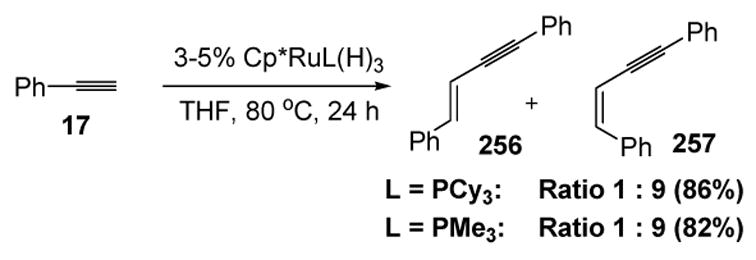
Yi’s Stereoselective Dimerization of Phenylacetylene: Ligand Effect. Cp*=1,2,3,4,5-η5-pentamethylcyclopentadienyl, THF=tetrahydrofuran.
A mechanism that accounts for this ligand effect was proposed, the features of which are summarized in Scheme 99. A series of steps leads to active catalyst I, which can be converted to the equilibrating vinylidene complexes II and III. When the ligand is large (L = PCy3), III is favored due to repulsion between L and the phenyl group on the vinylidene in II, thus reaction preferentially leads to Z-enyne 257. Conversely, when L = PMe3, the steric interactions dictate that reaction proceeds through II to give E-enyne 256.
Scheme 99.
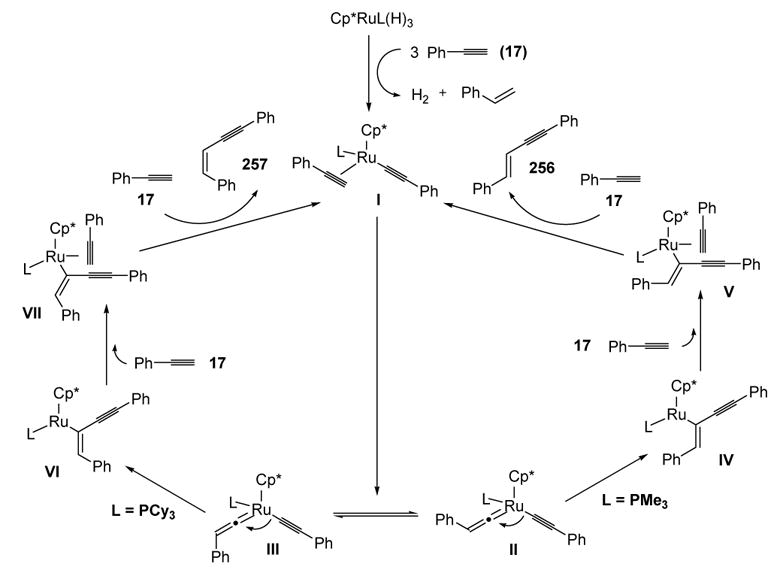
Alkyne Dimerization: Rationale for Product Formation. Cp*=1,2,3,4,5-η5-pentamethylcyclopentadienyl.
There have since been numerous reports of ruthenium-catalyzed head-to-head dimerization of phenylacetylene through a vinylidene mechanism, with various levels of Z/E selectivity. Some of the catalysts that have been employed include: TpRu(PPh3)2Cl,[108] TpRu(MeiPr2P)C(Ph)=C(Ph)C≡CPh,[109] Ru(ma)2(PPh3)2,[110] (η5-C9H7)Ru(PPh3)2C≡CPh,[111] and RuCl2(PCy3)2=CHPh.[112] The selective head-to-head dimerization of aliphatic alkynes through a vinylidene mechanism, however, was not successfully addressed for some time. Towards this goal, Hidai and coworkers reported that a dimeric ruthenium complex was able to catalyze the Z-selective dimerization of a variety of aliphatic terminal alkynes (Scheme 100).[113]
Scheme 100.
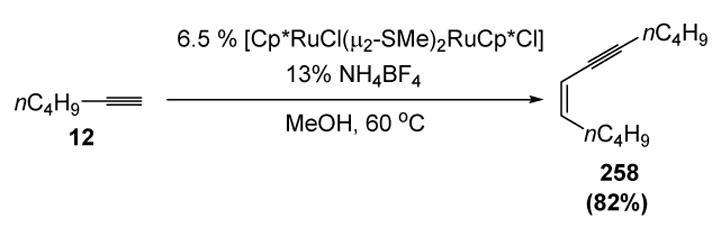
Hidai’s Head-to-Head Dimerization of Aliphatic Terminal Alkynes. Cp*=1,2,3,4,5-η5-pentamethylcyclopentadienyl.
Bianchini has reported the most general catalyst to date, capable of head-to-head dimerization of both aromatic and aliphatic terminal alkynes with good Z-selectivity. The catalyst loadings are quite low and the scope is broad, with even t-butylacetylene participating as a substrate (equations 1 → 3, Scheme 101).[114]
Scheme 101.
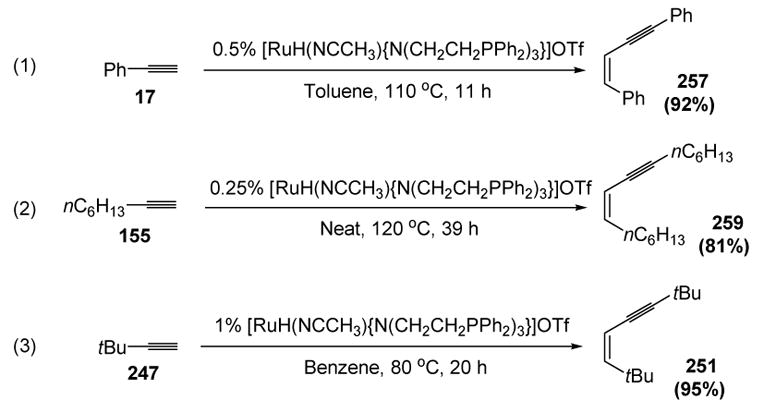
Bianchini’s Dimerization of Aromatic and Aliphatic Terminal Alkynes
The first Z-selective head-to-head cross-dimerization of arylacetylenes with silylacetylenes was recently reported, thus significantly enhancing the synthetic utility of this process (Scheme 102).[115] In this reaction, Katayama and Ozawa make use of the fact that arylacetylenes preferentially form metal vinylidene intermediates, and so by using an excess of trimethylsilylacetylene they were able to selectively capture these intermediates.
Scheme 102.

Katayama’s Cross-Dimerization of Arylacetylenes with Silylacetylenes
Recent reports suggest that alkynes may add to metal vinylidenes in a different manner than 1,2-migration from the metal center. For example, when 1,6-diyne substrates are exposed to a ruthenium catalyst system and one equivalent of a carboxylic acid, cyclic enol esters are obtained in moderate to good yields (Scheme 103).[116]
Scheme 103.
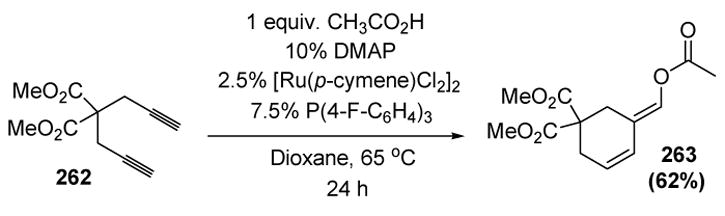
Lee’s Carboxylative Cyclization of 1,6-Diynes
The mechanism of this interesting process is depicted in Scheme 104. The key step of the catalytic cycle is thought to involve carboxylate anion-induced cyclization of an alkyne onto the electrophilic vinylidene intermediate (II → III), a process that would be consistent with the observed geometry of the enol ester. Finally, protonation and reductive elimination releases the product and the active catalyst.
Scheme 104.

Mechanism of the 1,6-Diyne Carboxylative Cyclization
6.2 Arylative and Alkenylative Cyclization
Lee and coworkers have reported a new arylative (and alkenylative) cyclization reaction. Thus 1,5-enynes, where the alkene is conjugated to a carbonyl group, can undergo a tandem carbometalation-cyclization reaction to give cyclopentene products. An example of this transformation is depicted in Scheme 105.[117]
Scheme 105.
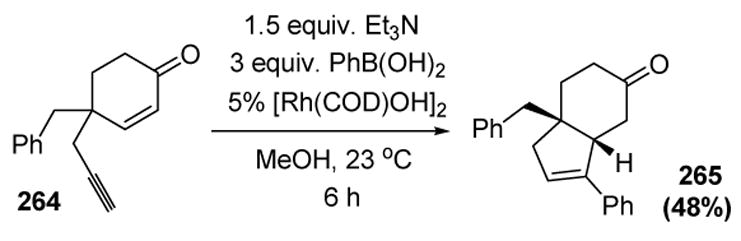
Lee’s Arylative Cyclization of 1,5-Enynes. COD=1,5-cyclooctadiene.
The product formation was rationalized according to Scheme 106. The active catalyst (I) may undergo ligand displacement with the solvent to give II. Transmetalation with the boronic acid then leads to III, which can form a vinylidene with the substrate. A 1,2-migration of the aryl group then leads to alkenyl rhodium species V, which can coordinate the proximal enone and undergo a 1,4-addition to give rhodium enolate VI. Finally, protonation and reductive elimination leads to product 265.
Scheme 106.
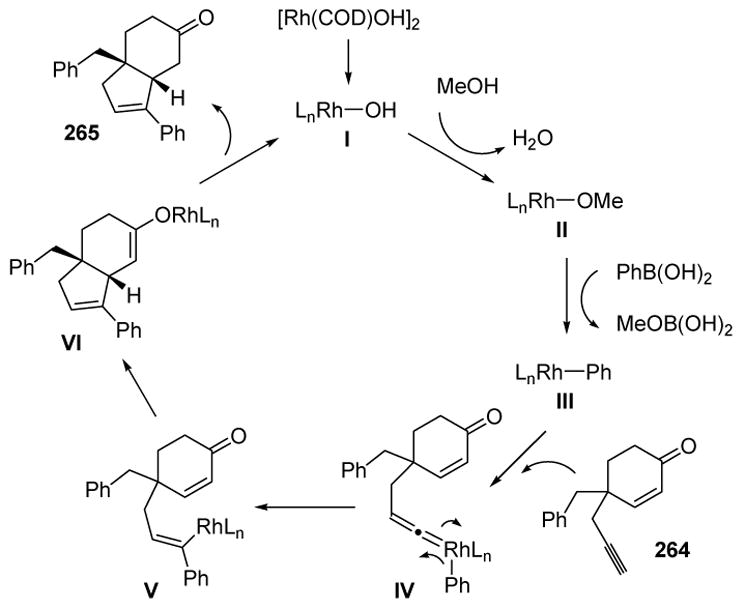
Mechanism of 1,5-Enyne Arylative Cyclization. COD=1,5-cyclooctadiene.
6.3 Hydroboration
Miyaura has reported the regio- and stereoselective hydroboration of terminal alkynes using either a rhodium or an iridium catalyst with electron-rich phosphines.[118] An example of this transformation is shown in Scheme 107.
Scheme 107.
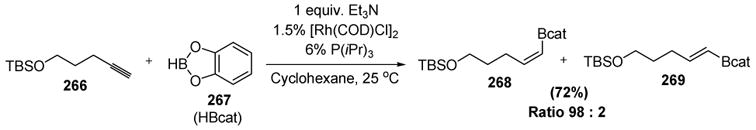
Miyaura’s Z-Selective Hydroboration of Terminal Alkynes. COD=1,5-cyclooctadiene.
This methodology has great synthetic promise as it offers Z-1-alkenylborane products, which can be valuable partners in Suzuki cross-coupling reactions. Traditionally one has only had direct access to E-1-alkenylboranes by rhodium-catalyzed hydroboration of terminal alkynes using Wilkinson’s catalyst[119] In order to explain the regio- and stereoselectivity, Miyaura proposed the mechanism shown in Scheme 108. The active catalyst (I) and the substrate form a vinylidene intermediate (II), a process that is consistent with deuterium labeling studies. Oxidative addition of catecholborane (267) then leads to III, whereby a 1,2-migration, followed by reductive elimination, gives the product 268. It was reasoned that the thermodynamic stability of the alkene geometry (E) of IV is reflected in the product distribution (Z product 268 preferentially formed).
Scheme 108.
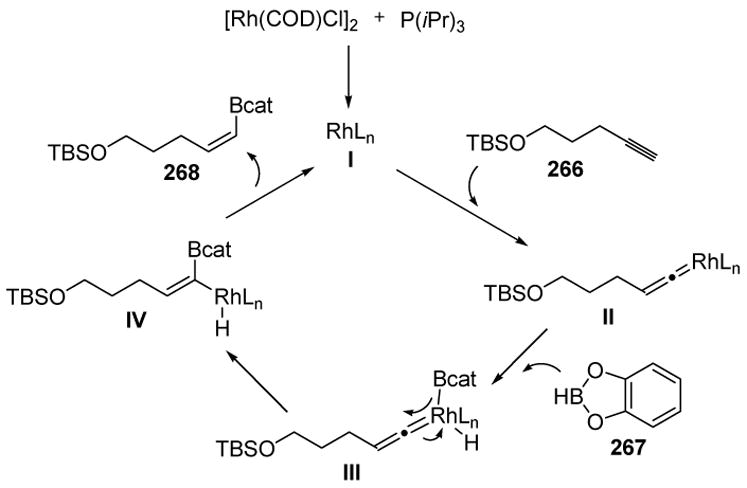
Z-Selective Hydroboration of Terminal Alkynes: Mechanistic Rationale. COD=1,5-cyclooctadiene.
7. Conclusion
The prime goal of organic synthesis should be to develop reactions that are efficient in terms of selectivity[120] and atom economy.[121] Selective addition and rearrangement reactions have the potential to fulfill these criteria perfectly. In this review it is evident that transition metals can catalyze these two types of reactions simply by taking advantage of the facile conversion of terminal alkynes to metal vinylidenes. The variety of products made in such reactions is impressive, and the list of such transformations will no doubt continue to grow, particularly as chemists apply these useful reactions in tandem bond-forming processes. Although most reactions involve ruthenium, rhodium, molybdenum, or tungsten complexes, other metals and ligands could potentially catalyze the same (or even new) reactions. The application of such reactions in target oriented synthesis remains limited, but this will change as chemists become aware of the advantages associated with converting readily available terminal alkynes into a wide variety of functionalized products.
Scheme 5.
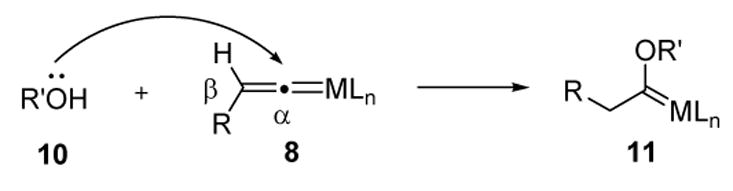
Nucleophilic Addition to Metal Vinylidenes: Fischer Carbene Formation
Biographies
Barry M. Trost was born in Philadelphia, PA in 1941 and studied at the University of Pennsylvania (BA, 1962). He obtained his Ph.D. in 1965 at MIT. He moved to the University of Wisconsin where he was made Professor in 1969 and subsequently Vilas Research Professor in 1982. He moved to Stanford University in 1987 and became Tamaki Professor of Humanities and Sciences in 1990. In addition to holding Visiting Professorships at several universities worldwide, he has been awarded numerous prizes. His interests span the entire field of organic synthesis, particularly in the development of novel methodology.

Andrew McClory was born in Montreal in 1976. He received his B.Sc. from Concordia University in 1999. He then carried out his graduate studies at Stanford University, where he worked on ruthenium and rhodium-catalyzed atom economical reactions under the guidance of Professor Barry M. Trost. He received his Ph.D. in 2007. He is currently a postdoctoral researcher in the laboratory of Professor Brian M. Stoltz at the California Institute of Technology.

References
- 1.(a) Huguet J, Karpf M, Dreiding AS. Tetrahedron Lett. 1983;24:4177–4180. [Google Scholar]; (b) Solaja B, Huguet J, Karpf M, Dreiding AS. Tetrahedron. 1987;43:4875–4886. [Google Scholar]
- 2.(a) Bruce MI, Swincer AG. Adv Organomet Chem. 1983;22:59. [Google Scholar]; (b) Werner H. Angew Chem Int Ed Engl. 1990;29:1077–1089. [Google Scholar]; (c) Bruce MI. Chem Rev. 1991;91:197–257. [Google Scholar]; (d) Bruce MI. Chem Rev. 1998;98:2797–2858. doi: 10.1021/cr970473x. [DOI] [PubMed] [Google Scholar]
- 3.Wolf J, Werner H, Serhadli O, Ziegler ML. Angew Chem Int Ed Engl. 1983;22:414–416. [Google Scholar]
- 4.Unimolecular pathway supported experimentally by double crossover studies: Grotjahn DB, Zeng X, Cooksy AL. J Am Chem Soc. 2006;128:2798–2799. doi: 10.1021/ja058736p.
- 5.Bimolecular pathway supported by computational studies: Wakatsuki Y, Koga N, Werner H, Morokuma K. J Am Chem Soc. 1997;119:360–366.
- 6.Review: Wakatsuki Y. J Organomet Chem. 2004;689:4092–4109.
- 7.Reviews: Bruneau C, Dixneuf PH. Acc Chem Res. 1999;32:311–323.Bruneau C, Dixneuf PH. Angew Chem Int Ed Engl. 2006;45:2176–2203. doi: 10.1002/anie.200501391.
- 8.Review: Varela JA, Saa C. Chem Eur J. 2006;12:6450–6456. doi: 10.1002/chem.200600388.
- 9.Sasaki Y, Dixneuf PH. J Chem Soc Chem Commun. 1986;790–791 [Google Scholar]
- 10.Bruneau C, Dixneuf PH. J Mol Cat. 1992;74:97–107. [Google Scholar]
- 11.Mahe R, Dixneuf PH, Lecolier S. Tetrahedron Lett. 1986;27:6333–6336. [Google Scholar]
- 12.(a) Sasaki Y, Dixneuf PH. J Org Chem. 1987;52:314–315. [Google Scholar]; (b) Mahe R, Sasaki Y, Bruneau C, Dixneuf PH. J Org Chem. 1989;54:1518–1523. [Google Scholar]
- 13.A comparison of ruthenium catalysts for this transformation: Bruneau C, Dixneuf PH, Lecolier S. J Mol Cat. 1988;44:175–178.
- 14.Hofer J, Doucet H, Bruneau C, Dixneuf PH. Tetrahedron Lett. 1991;32:7409–7410. [Google Scholar]
- 15.Ruppin C, Dixneuf PH. Tetrahedron Lett. 1986;27:6323–6324. [Google Scholar]
- 16.Review: Bruneau C, Dixneuf PH. Chem Commun. 1997:507–512.
- 17.Doucet H, Martin-Vaca B, Bruneau C, Dixneuf PH. J Org Chem. 1995;60:7247–7255. [Google Scholar]
- 18.Doucet H, Hofer J, Bruneau C, Dixneuf PH. J Chem Soc Chem Commun. 1993:850–851. [Google Scholar]
- 19.Doucet H, Hofer J, Derrien N, Bruneau C, Dixneuf PH. Bull Soc Chim Fr. 1996;133:939–944. [Google Scholar]
- 20.Ru(HB(pz)3)(COD)Cl: Gemel C, Trimmel G, Slugovc C, Kremel S, Mereiter K, Schmid R, Kirchner K. Organometallics. 1996;15:3998–4004. Ru(methallyl)2dppe: Doucet H, Derrien N, Kabouche Z, Bruneau C, Dixneuf PH. J Organomet Chem. 1997;551:151–157. RuClx(p-cymene)(triazol-5-ylidene): Melis K, Samulkiewicz P, Rynkowski J, Verpoort F. Tetrahedron Lett. 2002;43:2713–2716.
- 21.Review: Bruneau C, Dixneuf PH. Chem Commun. 1997:507–512.
- 22.Gooβen LJ, Paetzold J, Koley D. Chem Commun. 2003:706–707. [PubMed] [Google Scholar]
- 23.Picquet M, Bruneau C, Dixneuf PH. Chem Commun. 1997:1201–1202. [Google Scholar]
- 24.Picquet M, Fernandez A, Bruneau C, Dixneuf PH. Eur J Org Chem. 2000:2361–2366. [Google Scholar]
- 25.Swaminathan S, Narayanan KV. Chem Rev. 1971;71:429–438. [Google Scholar]
- 26.Jimenez-Tenorio M, Puerta MC, Valerga P, Moreno-Dorado FJ, Guerra FM, Massanet GM. Chem Commun. 2001:2324–2325. doi: 10.1039/b106647c. [DOI] [PubMed] [Google Scholar]
- 27.Melis K, Samulkiewicz P, Rynkowski J, Verpoort F. Tetrahedron Lett. 2002;43:2713–2716. [Google Scholar]
- 28.Fischer EO. Adv Organomet Chem. 1976;14:1–32. [Google Scholar]
- 29.Trost BM, Dyker G, Kulawiec RJ. J Am Chem Soc. 1990;112:7809–7811. [Google Scholar]
- 30.Trost BM, Kulawiec RJ. J Am Chem Soc. 1992;114:5579–5584. [Google Scholar]
- 31.Trost BM, Kulawiec RJ, Hammes A. Tetrahedron Lett. 1993;34:587–590. [Google Scholar]
- 32.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;13:3769–3772. [Google Scholar]
- 33.Trost BM, Flygare JA. J Org Chem. 1994;59:1078–1082. [Google Scholar]
- 34.Trost BM, Flygare JA. J Am Chem Soc. 1992;114:5476–5477. [Google Scholar]
- 35.Trost BM, Flygare JA. Tetrahedron Lett. 1994;35:4059–4062. [Google Scholar]
- 36.Reviews: Weyershausen B, Dotz KH. Eur J Inorg Chem. 1999:1057–1066.McDonald FE. Chem Eur J. 1999;5:3103–3106.McDonald FE, Reddy KS. J Organomet Chem. 2001;617–618:444–452.
- 37.McDonald FE, Connolly CB, Gleason MM, Towne TB, Treiber KD. J Org Chem. 1993;58:6952–6953. [Google Scholar]
- 38.McDonald FE, Gleason MM. Angew Chem Int Ed Engl. 1995;34:350–352. [Google Scholar]
- 39.McDonald FE, Gleason MM. J Am Chem Soc. 1996;118:6648–6659. [Google Scholar]
- 40.McDonald FE, Reddy KS. Angew Chem Int Ed Engl. 2001;40:3653–3655. doi: 10.1002/1521-3773(20011001)40:19<3653::aid-anie3653>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 41.McDonald FE, Zhu HYH. J Am Chem Soc. 1998;120:4246–4247. [Google Scholar]
- 42.McDonald FE, Reddy KS, Diaz Y. J Am Chem Soc. 2000;122:4304–4309. [Google Scholar]
- 43.Wipf P, Graham TH. J Org Chem. 2003;68:8798–8807. doi: 10.1021/jo034813s. [DOI] [PubMed] [Google Scholar]
- 44.Cutchins WW, McDonald FE. Org Lett. 2002;4:749–752. doi: 10.1021/ol017195f. [DOI] [PubMed] [Google Scholar]
- 45.Davidson MH, McDonald FE. Org Lett. 2004;6:1601–1603. doi: 10.1021/ol049630m. [DOI] [PubMed] [Google Scholar]
- 46.McDonald FE, Schultz CC, Chatterjee AK. Organometallics. 1995;14:3628–3629. [Google Scholar]
- 47.McDonald FE, Bowman JL. Tetrahedron Lett. 1996;37:4675–4678. [Google Scholar]
- 48.Trost BM, Rhee YH. J Am Chem Soc. 1999;121:11680–11683. [Google Scholar]
- 49.Trost BM, Rhee YH. J Am Chem Soc. 2002;124:2528–2533. doi: 10.1021/ja011840w. [DOI] [PubMed] [Google Scholar]
- 50.Trost BM, Rhee YH. Org Lett. 2004;6:4311–4313. doi: 10.1021/ol048165q. [DOI] [PubMed] [Google Scholar]
- 51.Trost BM, Rhee YH. J Am Chem Soc. 2003;125:7482–7483. doi: 10.1021/ja0344258. [DOI] [PubMed] [Google Scholar]
- 52.Tokunaga M, Wakatsuki Y. Angew Chem Int Ed Engl. 1998;37:2867–2869. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2867::AID-ANIE2867>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 53.Suzuki T, Tokunaga M, Wakatsuki Y. Org Lett. 2001;3:735–737. doi: 10.1021/ol0003937. [DOI] [PubMed] [Google Scholar]
- 54.Alvarez P, Bassetti M, Gimeno J, Mancini G. Tetrahedron Lett. 2001;42:8467–8470. [Google Scholar]
- 55.(a) Grotjahn DB, Incarvito CD, Rheingold AL. Angew Chem Int Ed Engl. 2001;40:3884–3887. doi: 10.1002/1521-3773(20011015)40:20<3884::AID-ANIE3884>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]; (b) Grotjahn DB, Lev DA. J Am Chem Soc. 2004;126:12232–12233. doi: 10.1021/ja046360u. [DOI] [PubMed] [Google Scholar]; (c) Grotjahn DB. Chem Eur J. 2005:7146–7153. doi: 10.1002/chem.200500253. [DOI] [PubMed] [Google Scholar]
- 56.Chevallier F, Breit B. Angew Chem Int Ed Engl. 2006;45:1599–1602. doi: 10.1002/anie.200503826. [DOI] [PubMed] [Google Scholar]
- 57.Labonne A, Kribber T, Hintermann L. Org Lett. 2006;8:5853–5856. doi: 10.1021/ol062455k. [DOI] [PubMed] [Google Scholar]
- 58.Picquet M, Fernandez A, Bruneau C, Dixneuf PH. Eur J Org Chem. 2000:2361–2366. [Google Scholar]
- 59.Suzuki T, Tokunaga M, Wakatsuki Y. Tetrahedron Lett. 2002;43:7531–7533. [Google Scholar]
- 60.Chen Y, Ho DM, Lee C. J Am Chem Soc. 2005;127:12184–12185. doi: 10.1021/ja053462r. [DOI] [PubMed] [Google Scholar]
- 61.McDonald FE, Schultz CC. J Am Chem Soc. 1994;116:9363–9364. [Google Scholar]
- 62.Lo CY, Guo H, Lian JJ, Shen FM, Liu RS. J Org Chem. 2002;67:3930–3932. doi: 10.1021/jo020004h. [DOI] [PubMed] [Google Scholar]
- 63.Madhushaw RJ, Lin MY, Md S, Sohel A, Liu RS. J Am Chem Soc. 2004;126:6895–6899. doi: 10.1021/ja049943c. [DOI] [PubMed] [Google Scholar]
- 64.Lin MY, Maddirala SJ, Liu RS. Org Lett. 2005;7:1745–1748. doi: 10.1021/ol050317+. [DOI] [PubMed] [Google Scholar]
- 65.Ohe K, Miki K, Yokoi T, Nishino F, Uemura S. Organometallics. 2000;19:5525–5528. [Google Scholar]
- 66.Review: Miki K, Uemura S, Ohe K. Chem Lett. 2005;34:1068–1073.
- 67.Iwasawa N, Shido M, Maeyama K, Kusama H. J Am Chem Soc. 2000;122:10226–10227. [Google Scholar]
- 68.Ohe K, Yokoi T, Miki K, Nishino F, Uemura S. J Am Chem Soc. 2002;124:526–527. doi: 10.1021/ja017037j. [DOI] [PubMed] [Google Scholar]
- 69.McDonald FE, Burova SA, Huffman JG., Jr Synthesis. 2000:970–974. [Google Scholar]
- 70.Kondo T, Tanaka A, Kotachi S, Watanabe Y. J Chem Soc Chem Commun. 1995:413–414. [Google Scholar]
- 71.Gooβen LJ, Rauhaus JE, Deng G. Angew Chem Int Ed Engl. 2005;44:4042–4045. doi: 10.1002/anie.200462844. [DOI] [PubMed] [Google Scholar]
- 72.Fukumoto Y, Dohi T, Masaoka H, Chatani N, Murai S. Organometallics. 2002;21:3845–3847. [Google Scholar]
- 73.McDonald FE, Chatterjee AK. Tetrahedron Lett. 1997;38:7687–7690. [Google Scholar]
- 74.Trost BM, McClory A. Angew Chem Int Ed Engl. 2007;46:2074–2077. doi: 10.1002/anie.200604183. [DOI] [PubMed] [Google Scholar]
- 75.Park YJ, Kwon BI, Ahn JA, Lee H, Jun CH. J Am Chem Soc. 2004;126:13892–13893. doi: 10.1021/ja045789i. [DOI] [PubMed] [Google Scholar]
- 76.Jerome F, Monnier F, Lawicka H, Derien S, Dixneuf PH. Chem Commun. 2003:696–697. doi: 10.1039/b212408d. [DOI] [PubMed] [Google Scholar]
- 77.McDonald FE, Olson TC. Tetrahedron Lett. 1997;38:7691–7692. [Google Scholar]
- 78.Maeyama K, Iwasawa N. J Am Chem Soc. 1998;120:1928–1929. [Google Scholar]
- 79.Iwasawa N, Miura T, Kiyota K, Kusama H, Lee K, Lee PH. Org Lett. 2002;4:4463–4466. doi: 10.1021/ol026993i. [DOI] [PubMed] [Google Scholar]
- 80.For another method of cyclopentene annulation, see: Danheiser RL, Fink DM, Tsai Y-M. Org Synth. 1988;66:8–13.
- 81.Kim H, Lee C. J Am Chem Soc. 2006;128:6336–6337. doi: 10.1021/ja0619758. [DOI] [PubMed] [Google Scholar]
- 82.Merlic CA, Pauly ME. J Am Chem Soc. 1996;118:11319–11320. [Google Scholar]
- 83.Donovan PM, Scott LT. J Am Chem Soc. 2004;126:3108–3112. doi: 10.1021/ja038254i. [DOI] [PubMed] [Google Scholar]
- 84.Maeyama K, Iwasawa N. J Org Chem. 1999;64:1344–1346. [Google Scholar]
- 85.Miura T, Iwasawa N. J Am Chem Soc. 2002;124:518–519. doi: 10.1021/ja0113091. [DOI] [PubMed] [Google Scholar]
- 86.Sangu K, Fuchibe K, Akiyama T. Org Lett. 2004;6:353–355. doi: 10.1021/ol036190a. [DOI] [PubMed] [Google Scholar]
- 87.Shen HC, Pal S, Lian JJ, Liu RS. J Am Chem Soc. 2003;125:15762–15763. doi: 10.1021/ja0379159. [DOI] [PubMed] [Google Scholar]
- 88.Lian JJ, Odedra A, Wu CJ, Liu RS. J Am Chem Soc. 2005;127:4186–4187. doi: 10.1021/ja0504901. [DOI] [PubMed] [Google Scholar]
- 89.Madhushaw RJ, Lo CY, Hwang CW, Su MD, Shen HC, Pal S, Shaikh IR, Liu RS. J Am Chem Soc. 2004;126:15560–15565. doi: 10.1021/ja045516n. [DOI] [PubMed] [Google Scholar]
- 90.(a) Rule M, Salinaro RF, Pratt DR, Berson JA. J Am Chem Soc. 1982;104:2223–2228. [Google Scholar]; (b) Salinaro RF, Berson JA. J Am Chem Soc. 1982;104:2228–2232. [Google Scholar]
- 91.Murakami M, Ubukata M, Ito Y. Tetrahedron Lett. 1998;39:7361–7364. [Google Scholar]
- 92.Murakami M, Ubukata M, Ito Y. Chem Lett. 2002:294–295. [Google Scholar]
- 93.Murakami M, Hori S. J Am Chem Soc. 2003;125:4720–4721. doi: 10.1021/ja029829z. [DOI] [PubMed] [Google Scholar]
- 94.Grigg R, Stevenson P, Worakun T. Tetrahedron. 1988;44:4967–4972. [Google Scholar]
- 95.Kim H, Lee C. J Am Chem Soc. 2005;127:10180–10181. doi: 10.1021/ja052775j. [DOI] [PubMed] [Google Scholar]
- 96.Joo JM, Yuan Y, Lee C. J Am Chem Soc. 2006;128:14818–14819. doi: 10.1021/ja066374v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bigeault J, Giordano L, Buono G. Angew Chem Int Ed Engl. 2005;44:4753–4757. doi: 10.1002/anie.200500879. [DOI] [PubMed] [Google Scholar]
- 98.Elakkari E, Floris B, Galloni P, Tagliatesta P. Eur J Org Chem. 2005:889–894. [Google Scholar]
- 99.Datta S, Odedra A, Liu RS. J Am Chem Soc. 2005;127:11606–11607. doi: 10.1021/ja053674o. [DOI] [PubMed] [Google Scholar]
- 100.Wang Y, Finn MG. J Am Chem Soc. 1995;117:8045–8046. [Google Scholar]
- 101.Ohe K, Kojima M, Yonehara K, Uemura S. Angew Chem Int Ed Engl. 1996;35:1823–1825. [Google Scholar]
- 102.Manabe T, Yanagi S, Ohe K, Uemura S. Organometallics. 1998;17:2942–2944. [Google Scholar]
- 103.Yamazaki H. J Chem Soc Chem Commun. 1976:841–842. [Google Scholar]
- 104.Wakatsuki Y, Yamazaki H, Kumegawa N, Satoh T, Satoh JY. J Am Chem Soc. 1991;113:9604–9610. [Google Scholar]
- 105.Bianchini C, Peruzzini M, Zanobini F, Frediani P, Albinati A. J Am Chem Soc. 1991;113:5453–5454. [Google Scholar]
- 106.Bianchini C, Frediani P, Masi D, Peruzzini M, Zanobini F. Organometallics. 1994;13:4616–4632. [Google Scholar]
- 107.(a) Yi CS, Liu N. Organometallics. 1996;15:3968–3971. [Google Scholar]; (b) Yi CS, Liu N. Synlett. 1999:281–287. [Google Scholar]
- 108.(a) Slugovc C, Mereiter K, Zobetz E, Schmid R, Kirchner K. Organometallics. 1996;15:5275–5277. [Google Scholar]; (b) Slugovc C, Doberer D, Gemel C, Schmid R, Kirchner K, Winkler B, Stelzer F. Mon fur Chem. 1998;129:221–233. [Google Scholar]; (c) Pavlik S, Gemel C, Slugovc C, Mereiter K, Schmid R, Kirchner K. J Organomet Chem. 2001;617–618:301–310. [Google Scholar]
- 109.Tenorio MAJ, Tenorio MJ, Puerta MC, Valerga P. Organometallics. 2000;19:1333–1342. [Google Scholar]
- 110.Fryzuk MD, Jonker MJ, Rettig SJ. Chem Commun. 1997:377–378. [Google Scholar]
- 111.Bassetti M, Marini S, Tortorella F, Cadierno V, Diez J, Gamasa MP, Gimeno J. J Organomet Chem. 2000;593–594:292–298. [Google Scholar]
- 112.Melis K, De Vos D, Jacobs P, Verpoort F. J Organomet Chem. 2002;659:159–164. [Google Scholar]
- 113.Qu JP, Masui D, Ishii Y, Hidai M. Chem Lett. 1998:1003–1004. [Google Scholar]
- 114.Chen X, Xue P, Sung HHY, Williams ID, Peruzzini M, Bianchini C, Jia G. Organometallics. 2005;24:4330–4332. [Google Scholar]
- 115.Katayama H, Yari H, Tanaka M, Ozawa F. Chem Commun. 2005:4336–4338. doi: 10.1039/b504436g. [DOI] [PubMed] [Google Scholar]
- 116.Kim H, Goble SD, Lee C. J Am Chem Soc. 2007;129:1030–1031. doi: 10.1021/ja067336e. [DOI] [PubMed] [Google Scholar]
- 117.Chen Y, Lee C. J Am Chem Soc. 2006;128:15598–15599. doi: 10.1021/ja067125+. [DOI] [PubMed] [Google Scholar]
- 118.Ohmura T, Yamamoto Y, Miyaura N. J Am Chem Soc. 2000;122:4990–4991. [Google Scholar]
- 119.Manning D, Noth H. Angew Chem Int Ed Engl. 1985;24:878–879. [Google Scholar]
- 120.Trost BM. Science. 1983;219:245–250. doi: 10.1126/science.219.4582.245. [DOI] [PubMed] [Google Scholar]
- 121.(a) Trost BM. Science. 1991;254:1471–1477. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]; (b) Trost BM. Angew Chem Int Ed Engl. 1995;34:259–281. [Google Scholar]