

Abstract
Introduction
About 30% of rheumatoid arthritis patients fail to respond adequately to TNFα-blocking therapy. There is a medical and socioeconomic need to identify molecular markers for an early prediction of responders and nonresponders.
Methods
RNA was extracted from peripheral blood mononuclear cells of 19 rheumatoid arthritis patients before the first application of the TNFα blocker etanercept as well as after 72 hours. Clinical response was assessed over 3 months using the 28-joint-count Disease Activity Score and X-ray scans. Supervised learning methods were applied to Affymetrix Human Genome U133 microarray data analysis to determine highly selective discriminatory gene pairs or triplets with prognostic relevance for the clinical outcome evinced by a decline of the 28-joint-count Disease Activity Score by 1.2.
Results
Early downregulation of expression levels secondary to TNFα neutralization was associated with good clinical responses, as shown by a decline in overall disease activity 3 months after the start of treatment. Informative gene sets include genes (for example, NFKBIA, CCL4, IL8, IL1B, TNFAIP3, PDE4B, PPP1R15A and ADM) involved in different pathways and cellular processes such as TNFα signalling via NFκB, NFκB-independent signalling via cAMP, and the regulation of cellular and oxidative stress response. Pairs and triplets within these genes were found to have a high prognostic value, reflected by prediction accuracies of over 89% for seven selected gene pairs and of 95% for 10 specific gene triplets.
Conclusion
Our data underline that early gene expression profiling is instrumental in identifying candidate biomarkers to predict therapeutic outcomes of anti-TNFα treatment regimes.
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease of unknown aetiology that is characterized by recruitment and activation of inflammatory cells, synovial hyperplasia, and destruction of cartilage and bone. The proinflammatory cytokine TNFα is a key mediator in the pathogenesis of RA [1]. Etanercept (Enbrel®; Wyeth, Cambridge, MA, USA), a soluble TNFα receptor immunoglobulin fusion protein, has been recognized as a potent biological that neutralizes TNFα [2-4]. Clinical studies on the efficacy of TNFα-blocking agents clearly show that about 30% of patients receiving this expensive therapy are nonresponders [3,5]. Although many efforts have been made to identify biomarkers for therapy response [6], no clinical or single laboratory marker exists today that allows a prediction of TNFα therapy efficacy in the individual patient. This lack of biomarker includes the newly identified specific serological marker for RA – antibodies to cyclic citrullinated peptides [7,8] – as well as genetic markers [9-12].
A number of studies have shown that the expression of individual proteins – particularly cytokines such as TNFα, IL-1β, IL-6 and IFNγ [13,14], chemokines like IL-8 and MCP1, as well as matrix metalloproteinases such as MMP1 and MMP3 [15,16] – changes during etanercept therapy. These studies were limited to a small number of genes and their corresponding proteins, and were not able to identify new markers for characterizing disease activity or to determine discriminatory markers for the prediction of therapy outcome. Van der Pouw and coworkers [17] used gene expression profiling of synovial tissue to identify subsets of RA based on molecular criteria; see also Glocker and colleagues [18].
Lequerre and colleagues described changes in gene expression signatures of mononuclear cells in RA patients 3 months after the start of treatment that were correlated with the treatment response to another TNFα inhibitor, infliximab, in combination with methotrexate [19]. They reported a significant decrease of transcript levels of eight genes regulated by TNFα-dependent pathways in nonresponders, whereas transcript levels in responders did not change significantly but were slightly increased. The effects of infliximab treatment on the long-term changes of gene expression pattern of synovial tissue and their potential to predict the outcome of infliximab-treated RA patients was investigated by Lindberg and coworkers [20]. Differentially expressed genes were involved in processes such as chemotaxis, immune function, signal transduction and inflammatory responses. The value of tissue biopsies is still under debate, and biopsies repeated in quick succession are not feasible.
The present study uses global transcriptome analysis to determine RNA expression signatures in peripheral blood cells that specify the response to anti-TNFα therapy within the first days of treatment. The objective of our approach is to discover predictive markers by analysing gene sets that are distinctly regulated in the first 3 days after anti-TNF (etanercept) administration. This short time interval was chosen to identify initially perturbed gene expression not influenced by possible changes in comedication and environmental factors occurring during longer follow-up.
We report the application of established DNA array technology (Affymetrix®; St. Clara, CA, USA) to monitor changes in the expression levels of mononuclear cells from peripheral blood during etanercept treatment. Among about 14,500 genes, 42 candidate genes were found suitable for use as prognostic markers for the therapeutic outcome. Using supervised learning methods, pairs and triplets derived from these genes were found to have a high prognostic value – reflected by prediction accuracies of over 89% for seven gene pairs and of 95% for 10 specific gene triplets.
Patients and methods
Patients
Nineteen patients (15 females, four males; mean age, 50.8 ± 11.0 years; mean duration of disease, 15.8 ± 9.4 years; all Caucasian) who met the American College of Rheumatology criteria for RA [21] were studied; for details, refer to Table 1. More than three different disease-modifying antirheumatic drugs had failed to control disease activity before etanercept was administered. The study was approved by the ethics committee of the University of Magdeburg (71/99) and all patients were asked for written consent.
Table 1.
Patient characteristics
| Patient number | Age (years) | Gender | RA duration (years) | Disease-modifying antirheumatic drugs | Steroids (mg/day) | CCP-Ab (U/ml) (t0) | DAS28 | X-ray progression | Response after 3 months | |
| Baseline | 3 months | |||||||||
| 1 | 77 | Male | 21 | None | 5.0 | 644 | 5.45 | 4.69 | No | Nonresponder |
| 2 | 64 | Male | 27 | Leflunomide | 10.0 | 610 | 5.18 | 4.61 | No | Nonresponder |
| 3 | 43 | Female | 33 | Methotrexate | 7.5 | 81 | 4.82 | 0.69 | No | Responder |
| 4 | 65 | Female | 45 | None | 15.0 | 187 | 6.00 | 6.44 | Yes | Nonresponder |
| 5 | 63 | Female | 8 | None | 15.0 | >1,600 | 5.83 | 8.37 | Yes | Nonresponder |
| 6 | 51 | Female | 17 | Methotrexate | 20.0 | Negative | 6.16 | 4.40 | Yes | Nonresponder |
| 7 | 34 | Female | 9 | None | 0.0 | 806 | 5.37 | 5.47 | Yes | Nonresponder |
| 8 | 44 | Male | 9 | None | 15.0 | Negative | 5.51 | 2.55 | No | Responder |
| 9 | 39 | Male | 1 | Methotrexate | 5.0 | Negative | 5.12 | 2.09 | No | Responder |
| 10 | 42 | Female | 29 | Methotrexate | 7.5 | Negative | 6.52 | 1.79 | No | Responder |
| 11 | 26 | Female | 2 | None | 0.0 | Negative | 4.47 | 1.50 | No | Responder |
| 12 | 48 | Female | 24 | Leflunomide | 8.0 | 429 | 5.57 | 2.73 | No | Responder |
| 13 | 47 | Female | 13 | Cyclosporin A | 10.0 | 96 | 7.11 | 5.29 | No | Responder |
| 14 | 53 | Female | 5 | Leflunomide | 8.0 | 1064 | 3.29 | 2.42 | No | Nonresponder |
| 15 | 62 | Female | 13 | Methotrexate | 0.0 | Neg. | 5.88 | 4.40 | No | Responder |
| 16 | 65 | Female | 2 | Sulfasalazine/hydroxychloroquin | 15.0 | >1,600 | 7.68 | 5.90 | No | Responder |
| 17 | 42 | Female | 14 | None | 5.0 | 61 | 5.6 | 3.36 | No | Responder |
| 18 | 52 | Female | 8 | Methotrexate | 0.0 | 436 | 5.59 | 2.38 | No | Responder |
| 19 | 70 | Female | 14 | Leflunomide | 7.5 | 855 | 5.08 | 2.55 | No | Responder |
Therapeutic response was defined clinically by changes of 28-joint-count Disease Activity Score (DAS28) determined at the beginning of the study (baseline) and 3 months after the start of etanercept treatment and additionally by X-ray analysis of hands and feet after 9 to 12 months. An improvement of the DAS28 by >1.2 was considered a good response (if no progression of joint destruction were observed by X-ray analysis), a DAS28 reduction by ≤ 1.2 was considered a nonresponse. Serum antibodies to cyclic citrullinated peptide (CCP-Ab) were analysed using the Immunoscan RA ELISA CCP2 test (Euro-Diagnostica, Malmö, Sweden) according to the manufacturer's instructions (cutoff point = 25 U/ml). RA, rheumatoid arthritis.
Each patient was given a standard dose of 2 × 25 mg etanercept per week subcutaneously. Disease-modifying antirheumatic drugs and steroids remained unchanged in all patients for the first week of TNF-blocking therapy. Blood samples were taken at 7:00 a.m. before treatment (time t0; baseline), and at 72 hours after the first application of etanercept (time t1). Comedication was given after blood was taken.
Patients were assessed for overall disease activity using the 28-joint-count Disease Activity Score (DAS28) as described elsewhere [22]. Patients were categorized according to the European Leage against Rheumatism (EULAR) recommendations 3 months after the start of treatment, considering an improvement of the DAS28 >1.2 a good response. X-ray scans were read by two independent experienced physicians, but the sequence of the X-ray scans was not blinded. After reviewing X-ray scans of hands and feet, the responder group was further characterized by the absence of new bone erosions after a time interval of at least 9 to 12 months of follow up.
Sample preparation
Peripheral blood mononuclear cells from 25 ml blood were separated on a Ficoll density gradient [23]. Using a FACSCalibur Flow Cytometer (Becton Dickinson, San Diego, CA, USA) the populations of CD3+, CD14+, CD19+ and CD56+ cells were determined to ensure comparability of peripheral blood mononuclear cell fractions of individual patients in the course of the study. Extraction of total RNA was performed using the Qiagen RNeasy kit (Qiagen, Hilden, Germany) including a DNA digest on-column according to the manufacturer's instructions.
Microarray analysis
Affymetrix® microarray technology (Human Genome U133A gene chip) was used to analyse the expression levels of about 18,400 transcripts interrogated by more than 22,000 probe sets. The Human Genome U95A gene chip was applied to verify array data with selected patients. Labelling and microarray processing was performed according to the manufacturer's protocol. The scanning was carried out with 3 μm resolution, 488 nm excitation and 570 nm emission wavelengths employing the GeneArray Scanner (Affymetrix, St. Clara, CA, USA). The microarray data were stored according to the MIAME standard and are available from ArrayExpress [24] (accession number E-MTAB-11).
Quantitative real-time RT-PCR
Expression levels of a subset of genes were measured by quantitative real-time RT-PCR performed with TaqMan assay reagents according to the manufacturer's instructions on a 7900 High Throughput Sequence Detection System (Applied Biosystems, Foster City, CA, USA) using predesigned primers and probes (GAPDH Hs99999905_m1, ICAM1 Hs00164932_m1, TNFAIP3 Hs00234713_m1, IL1β Hs00174097_m1, PDE4B Hs00277080_m1, PPP1R15A Hs00169585_m1, NFKBIA Hs00153283_m1, CCL4 Hs00237011_m1, IL-8 Hs00174103_m1, ADM Hs00181605_m1).
To calculate the gene expression change of selected genes, the ΔΔCT method was used. According to this method, the threshold cycle values (CT) for specific mRNA expression in each sample were normalized to the CT values of GAPDH mRNA in the same sample. This provides ΔCT values that were used to calculate the changes of gene expression levels. Thereby, for each gene, the gene expression change in the first 3 days (ΔΔCT) is defined by the difference of the ΔCT value at day 3 (t1) and the ΔCT value before treatment (t0).
Data processing and analysis
The microarray data were preprocessed using the Microarray Suite, version 5.0 (MAS5.0; Affymetrix, Santa Clara, CA, USA) in the default configuration, and were analysed by a set of algorithms.
First, an algorithm for calculation of a score J to rank differentially regulated genes. Basically, the J score introduced here is a t statistic, which compares the logarithm of the expression ratios t1/t0 (signal log ratios) between responders and nonresponders. Thereby, the confidence intervals of the signal log ratios provided by MAS5.0 are used. In this way, the J score considers interindividual differences as well as measurement errors. A higher J score represents a more significant differential regulation. J > 0 was used as the cutoff point to define genes as differentially regulated.
Second, an algorithm for learning of classifiers used for prediction of the therapy outcome on evaluation of the fold change of pairs and triplets of genes (Support-Vector Machine algorithm together with cross-validation by the leave-one-out method).
Finally, an algorithm for inference of hypothetic gene regulatory networks (modified LASSO algorithm).
These three algorithms are described in detail in Additional file 1.
Methods of multiple testing to control the type I error rates taking into account the large multiplicity (more than 22,000 probe sets) were not applied. This feature was circumvented by validating expression patterns of a selected set of genes (ICAM1, TNFAIP3, IL1B, PDE4B, PPP1R15A, NFKBIA, CCL4, IL8, ADM).
Results
Clinical evaluation
Before the start of treatment, all RA patients presented with a high disease activity reflected by a DAS28 (mean ± standard deviation) of 5.7 ± 0.7. Within 3 months of TNFα-blocking therapy, the disease activity decreased significantly looking at all patients as a group (DAS28 = 3.8 ± 2.1) (Table 1).
Twelve patients (patients 3, 8 to 13, and 15 to 19) were characterized by a good therapy response, as indicated by a significant reduction of the DAS28 >1.2 without progression of bone erosions as shown by X-ray scans of hands and feet. Three out of seven nonresponders (patients 4, 5 and 7) showed mild progression of bone erosion by X-ray reviewing. One patient (patient 6) was considered a nonresponder despite a good DAS28 response due to a progressive joint destruction as demonstrated by the X-ray scan. None of the clinical characteristics at baseline was significantly associated with the clinical outcome (Table 2)
Table 2.
Comparison of clinical characteristics at baseline
| Characteristic | Responder | Nonresponder | P value |
| Age (years) | 48.33 (± 12.29) | 58.14 (± 13.67) | 0.125a |
| Gender (male) | 2/12 | 2/7 | 0.603b |
| Rheumatoid arthritis duration (years) | 13.5 (± 10.46) | 18.86 (± 13.93) | 0.353a |
| Steroids (mg/d) | 6.71 (± 5.16) | 10.43 (± 6.80) | 0.195a |
| 28-joint-count Disease Activity Score baseline | 5.75 (± 0.94) | 5.33 (± 0.97) | 0.364a |
| Antibodies to cyclic citrullinated peptide-negative | 5/12 | 1/7 | 0.333b |
| Disease-modifying antirheumatic drugs | |||
| None | 3/12 | 4/7 | 0.326b |
| Leflunomide | 2/12 | 2/7 | 0.603b |
| Methotrexate | 5/12 | 1/7 | 0.333b |
| Cyclosporin A | 1/12 | 0/7 | 1.000b |
| Sulfasalazine/hydroxychloroquin | 1/12 | 0/7 | 1.000b |
Both patient groups show similar characteristics before therapy onset (mean ± standard deviation and number of patients, respectively). Statistical tests (atwo-sample t test, bexact Fisher test) were applied to check whether any of the parameters are associated with clinical outcome.
Gene expression profiling using the U133A array
Application of Affymetrix DNA-chip technology to monitor changes in the expression profile of about 14,500 known genes in peripheral blood mononuclear cells during anti-TNFα therapy reflected a differential response by our patients as evinced by changes in the DAS28 greater than 1.2. Forty-two genes represented by 46 probe sets (Table 3) were found to be differentially regulated in therapy responders and nonresponders. The majority (40 probe sets representing 36 genes) was stronger downregulated or lesser upregulated in responders compared with nonresponders.
Table 3.
Differentially regulated genes (probe sets) in responders and nonresponders
| Symbol | Accession number | Probe set | Function | J value | Directiona | Significanceb |
| Transcription/regulation of transcription | ||||||
| TNFAIP3 | AI738896 | 202643_s_at | TNFα-induced protein 3 | 1.1830 | - | + |
| TNFAIP3 | NM_006290 | 202644_s_at | TNFα-induced protein 3 | 0.9956 | - | + |
| NFKBIA | AI078167 | 201502_s_at | NFκB enhancer in B-cell inhibitor alpha | 0.4762 | - | + |
| RUNX1 | L21756 | 211620_x_at | Runt-related transcription factor 1 | 0.3940 | + | + |
| JUN | BG491844 | 201464_x_at | c-jun proto-oncogene | 0.1352 | - | - |
| ZFP36L2 | AI356398 | 201367_s_at | Zinc finger protein 36, C3H type-like 2 | 0.1308 | - | + |
| SRRM2 | AI655799 | 208610_s_at | Serine/arginine repetitive matrix 2 | 0.0081 | + | - |
| ASCL1 | AW950513 | 213768_s_at | Achaete-scute complex-like 1 | 0.0444 | - | - |
| FOXO3A | AF041336 | 210655_s_at | Forkhead box O3A | 0.0131 | - | - |
| Immune response | ||||||
| IL1B | NM_000576 | 205067_at | IL-1β | 0.9716 | - | + |
| IL1B | M15330 | 39402_ | IL-1β | 0.9523 | - | + |
| CCL4 | NM_002984 | 204103_at | Chemokine (C-C motif) ligand 4 | 0.8002 | - | + |
| CCL3 | NM_002983 | 205114_s_at | Chemokine (C-C motif) ligand 3 | 0.4621 | - | + |
| CXCR4 | AF348491 | 211919_s_at | Chemokine (C-X-C motif) receptor 4 | 0.2589 | - | - |
| CXCL2 | M57731 | 209774_x_at | Chemokine (C-X-C motif) ligand 2 | 0.2532 | - | + |
| LTF | NM_002343 | 202018_s_at | Lactotransferrin | 0.1884 | - | - |
| PBEF1 | NM_005746 | 217739_s_at | Pre-B-cell colony-enhancing factor 1 | 0.0751 | - | - |
| IGHA1 | S55735 | 217022_s_at | Immunoglobulin heavy constant alpha 1 | 0.0475 | - | - |
| IER3 | NM_003897 | 201631_s_at | Immediate early response 3 | 0.0284 | - | - |
| Receptors, cell surface antigens, cell adhesion | ||||||
| ADAM12 | AU145357 | 215613_at | ADAM metallopeptidase domain 12 (meltrin alpha) | 0.5538 | - | + |
| ICAM1 | AI608725 | 202637_s_at | Intercellular adhesion molecule 1 (CD54) | 0.5399 | - | - |
| SCN2B | U87555 | 210364_at | Sodium channel, voltage-gated, type II, beta | 0.2294 | + | + |
| Signal transduction | ||||||
| PDE4B | L20966 | 211302_s_at | Phosphodiesterase 4B, cAMP-specific | 0.4374 | - | + |
| RAPGEF1 | NM_005312 | 204543_at | Rap guanine nucleotide-exchange factor 1 | 0.2890 | - | + |
| MYO10 | AI1561354 | 216222_s_at | Myosin X | 0.2066 | - | + |
| PTPRD | NM_002839 | 205712_at | Protein tyrosine phosphatase, receptor type, D | 0.1822 | + | + |
| SOCS1 | AI056051 | 209999_x_at | Suppressor of cytokine signaling 1 | 0.1239 | - | - |
| PDE4B | NM_002600 | 203708_at | Phosphodiesterase 4B, cAMP-specific | 0.0593 | - | + |
| Metabolism | ||||||
| LGALS13 | NM_013268 | 220440_at | Lectin, galactose-binding, soluble, 13 (galectin 13) | 0.5013 | + | + |
| SNCA | BG260394 | 204466_s_at | Synuclein, alpha | 0.0568 | - | - |
| CHST3 | AB017915 | 32094_at | Carbohydrate sulfotransferase 3 | 0.0366 | - | + |
| Cellular and oxidative stress response | ||||||
| CROP | AW089673 | 208835_s_at | Cisplatin resistance-associated overexpressed protein | 0.7500 | + | + |
| PPP1R15A | NM_014330 | 202014_at | Protein phosphatase 1, regulatory (inhibitor) subunit 15A | 0.6886 | - | + |
| PPP1R15A | U83981 | 37028_at | Protein phosphatase 1, regulatory (inhibitor) subunit 15A | 0.5939 | - | - |
| DDIT4 | M_019058 | 202887_s_at | DNA-damage-inducible transcript 4 | 0.2366 | - | - |
| SOD2 | W46388 | 215223_s_at | Superoxide dismutase 2, mitochondrial | 0.0724 | - | - |
| ADM | NM_001124 | 202912_at | Adrenomedullin | 0.0459 | - | + |
| Transport | ||||||
| ATP2A3 | AF068220 | 207521_s_at | ATPase, Ca2+ transporting, ubiquitous | 0.227 | - | - |
| CHRND | NM_000751 | 207024_at | Cholinergic receptor, nicotinic, delta | 0.1977 | - | + |
| Protein binding | ||||||
| PIGO | AC004472 | 214990_at | Phosphatidylinositol glycan, class O | 0.5216 | - | + |
| IBRDC3 | W27419 | 36564_at | IBR domain containing 3 | 0.1194 | - | + |
| EBP49 | NM_001978 | 204505_s_at | Erythrocyte membrane protein band 4.9 (dematin) | 0.0804 | - | - |
| FBX07 | NM_012179 | 201178_at | F-box protein 7 | 0.0080 | - | - |
| Unknown | ||||||
| FSD1 | NM_024333 | 219170_at | Fibronectin type III and SPRY domain containing 1 | 0.2935 | - | + |
| HCG4P6 | AF036973 | 215974_at | HLA complex group 4 pseudogene 6 | 0.1518 | - | + |
| C20orf103 | NM_013361 | 219463_at | Chromosome 20 open reading frame 103 | 0.0022 | - | - |
Genes were identified as differentially regulated using a modified t-statistic score, J (see Additional file 1), calculated using signal log ratios at t1 versus t0 considering 12 responders and seven nonresponders to etanercept therapy. aDirection denotes genes as stronger downregulated or lesser upregulated in responders compared with nonresponders (-), and vice versa (+). b+, significance approved by the resampling method with the modified t statistic on the significance level α = 0.05 (see Data processing and analysis section).
The mean of expression signals at t0 averaged over the responders (n = 12) and over the nonresponders (n = 7) did not differ significantly in these genes, with the exception of SCN2B with P < 0.05 (Additional file 1, Table S3a). A subset of 23 genes (represented by 27 probe sets) were approved to be differentially expressed according to the permutation test, with a significance level α = 0.05.
All 1,035 gene pairs resulting from the 46 preselected probe sets of differentially expressed genes were examined according to their ability to clearly discriminate responders and nonresponders. For each gene pair, a set of classifiers was constructed and evaluated by cross-validation using the leave-one-out method. Seven gene pairs (Table 4) produced a prediction accuracy Q > 89%. Baseline levels of the selected gene pairs were not reliable in predicting the outcome as reflected by Qt0log values between 42.1% and 79.0% (Additional file 1, Table S4a). The classification performance was also insufficient when using expression levels at t1 (Qt1log). Figure 1 shows a representative example of a discriminating gene pair (Q = 90.5%). Only one of the 19 patients (patient 16 – preclassified to be a clinical responder) matches with the pool of nonresponders. Owing to a DAS28 score that remained reasonably high, patient 16 eventually resembles a nonresponder according to EULAR criteria.
Table 4.
Combinations of genes predictive for the clinical outcome: gene pairs and gene triplets
| Combination | Gene 1 | Gene 2 | Gene 3 | Q (%) | |||
| Gene pair | |||||||
| 1 | TNFAIP3 | 202643_s_at | RAPGEF1 | 204543_at | 90.5 | ||
| 2 | TNFAIP3 | 202643_s_at | PTPRD | 205712_at | 90.5 | ||
| 3 | TNFAIP3 | 202644_s_at | PTPRD | 205712_at | 90.5 | ||
| 4 | IL1B | 205067_at | LGALS13 | 220440_at | 90.5 | ||
| 5 | CCL4 | 204103_at | ADAM12 | 215613_at | 89.5 | ||
| 6 | ADAM12 | 215613_at | CCL3 | 205114_s_at | 89.5 | ||
| 7 | FSD1 | 219170_at | HCG4P6 | 215974_at | 89.5 | ||
| Gene triplet | |||||||
| 1 | CCL4 | 204103_at | PDE4B | 211302_s_at | RAPGEF1 | 204543_at | 99.0 |
| 2 | PDE4B | 211302_s_at | RAPGEF1 | 204543_at | CXCR4 | 211919_s_at | 98.0 |
| 3 | CCL4 | 204103_at | PIGO | 214990_at | RAPGEF1 | 204543_at | 96.8 |
| 4 | CCL4 | 204103_at | FSD1 | 219170_at | RAPGEF1 | 204543_at | 96.8 |
| 5 | CCL4 | 204103_at | CCL3 | 205114_s_at | RAPGEF1 | 204543_at | 96.8 |
| 6 | PDE4B | 211302_s_at | RUNX1 | 211620_x_at | RAPGEF1 | 204543_at | 96.8 |
| 7 | CCL4 | 204103_at | LGALS13 | 220440_at | RAPGEF1 | 204543_at | 95.8 |
| 8 | TNFAIP3 | 202643_s_at | CCL4 | 204103_at | RAPGEF1 | 204543_at | 95.8 |
| 9 | TNFAIP3 | 202643_s_at | PDE4B | 211302_s_at | RAPGEF1 | 204543_at | 95.8 |
| 10 | TNFAIP3 | 202644_s_at | PDE4B | 211302_s_at | RAPGEF1 | 204543_at | 95.8 |
Gene pairs and triplets of genes with prognostic relevance for etanercept therapy in rheumatoid arthritis determined using support vector machines based on 46 selected probe sets of differentially regulated genes. Gene pairs with prediction accuracy Q > 89% and triplets of genes with prediction accuracy Q > 95% are shown. For gene function refer to Table 3.
Figure 1.
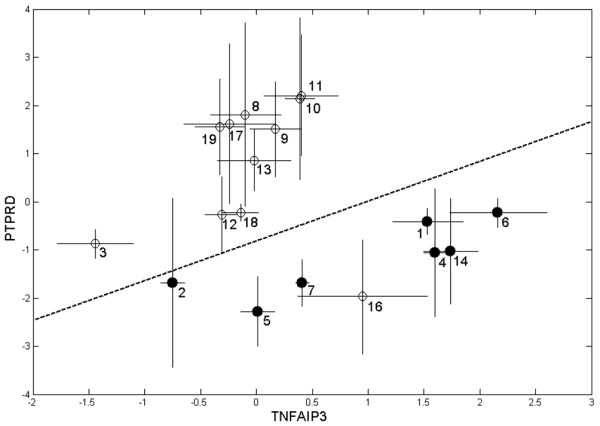
Gene expression changes of a representative predictive gene pair. Shown is the pair PTPRD [205712_at], TNFAIP3 [202643_s_at]. The pair is presented in Table 4 with a prediction accuracy of 90.5% determined using the support vector machine algorithm (signal log ratios for t1 versus t0: (○) 12 responders and (●) seven nonresponders, defined due to clinical response; bars denote the confidence intervals of the signal log ratios). Patient 16 was classified as a nonresponder based on gene expression data, but as a responder from clinical status.
Finally, the separation strength of classification could be further improved by taking triplets of differentially regulated genes. Thereto, 15,180 triplets as combinations of the 46 selected probe sets were computed. Ten triplets were identified to express a prediction accuracy >95%. Figure 2 shows a three-dimensional plot of one representative triplet gene set as presented in Table 4.
Figure 2.
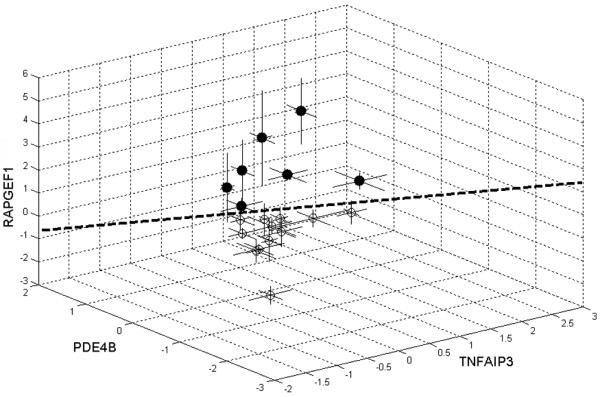
Gene expression changes of a representative predictive gene triplet. The triplet of genes TNFAIP3, PDE4B, RAPGEF1 is shown. The triplet is presented in Table 4 with a prediction accuracy of 95.8% determined using support vector machines (signal log ratios for t1 versus t0: (○) 12 responders and (●) seven nonresponders).
Validation of GeneChip U133A microarray data
Expression levels of a subset of genes were measured by quantitative real-time PCR for each patient and were compared with Human Genome arrays U133A and U95A (patients 1 to 11). As shown in Table 5, high correlations between the datasets obtained by three different methods of gene expression analysis were found.
Table 5.
Validation of array data by real-time quantitative RT-PCR
| Gene | Probe set | Correlation coefficient | |||
| U133A | U95A | U133A versus RT-PCR (n = 19) | U133A versus U95A (n = 11) | U95A versus RT-PCR (n = 11) | |
| ICAM1 | 202637_s_at | 32640_at | 0.9329 | 0.8916 | 0.8560 |
| TNFAIP3 | 202643_s_at | 595_at | 0.9437 | 0.9537 | 0.9792 |
| IL1B | 39402_at | 39402_at | 0.9443 | 0.9623 | 0.9667 |
| PDE4B | 211302_s_at | 33705_at | 0.8880 | 0.9583 | 0.6307 |
| PPP1R15A | 37028_at | 37028_at | 0.9519 | 0.9869 | 0.7649 |
Pearson correlation coefficients between real-time quantitative RT-PCR data (-ΔΔCT t1 versus t0) and the microarray data from the GeneChip U133A and U95A for five selected genes found to be differentially regulated in responders and nonresponders are presented.
In eight out of 20 genes selected for real-time quantitative RT-PCR (NFKBIA, CCL4, IL8, IL1B, PDE4B, TNFAIP3, PPP1R15A and ADM), the means of the gene expression change differed significantly for responders and nonresponders at significance level α < 0.05, as shown in Table 6. For all these genes, the means of the gene expression changes measured by quantitative real-time RT-PCR averaged over the seven nonresponders are positive, whereas those averaged over the 12 responders are negative or less positive than for the nonresponders.
Table 6.
Gene expression analysis by real-time quantitative RT-PCR
| Gene | Responder | Nonresponder | P value | ||
| NFKBIA | -0.227 | (± 0.749) | 1.053 | (± 1.128) | 0.008 |
| CCL4 | -0.142 | (± 1.184) | 1.144 | (± 0.924) | 0.025 |
| IL8 | -0.025 | (± 1.871) | 2.429 | (± 2.489) | 0.028 |
| IL1B | -0.595 | (± 1.680) | 1.487 | (± 2.191) | 0.032 |
| TNFAIP3 | 0.002 | (± 0.895) | 1.266 | (± 1.510) | 0.034 |
| PDE4B | -0.276 | (± 0.846) | 0.534 | (± 0.544) | 0.037 |
| PPP1R15A | -0.280 | (± 0.935) | 0.825 | (± 1.225) | 0.040 |
| ADM | -0.931 | (± 1.289) | 0.279 | (± 1.016) | 0.049 |
Data shown are the changes of gene expression (-ΔΔCT t1 versus t0; mean ± standard deviation) of eight selected genes averaged over the 12 responders and seven nonresponders, and the corresponding P values determined by two-sample t test comparing the means of responders and nonresponders.
Genetic network modelling
A hypothetic dynamic network was calculated (Figure 3) to reveal the underlying regulatory network that characterizes responders to the TNFα inhibitor therapy. This responder model accentuates IL-6 functions through the highest number of edges (vertex degree of 22) (see Additional file 1).
Figure 3.
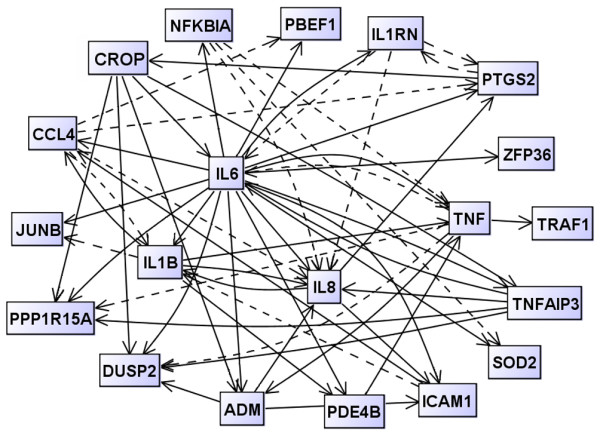
Visualization of the inferred dynamic gene regulatory network for the responder group. Each gene is represented by a node, and gene regulatory interactions are shown by directed edges. Solid lines, activating effects; dashed lines, inhibitory effects. The hypothesized network was reconstructed from quantitative real-time RT-PCR data by the modified LASSO method.
Discussion
The goal of the present study was to identify reliable biomarkers for predicting therapy outcomes in RA patients treated with the TNFα-blocking agent etanercept. Changes of the pre-existing gene activities were monitored following the neutralization of TNFα. The Affymetrix microarray technique produced reliable semiquantitative results confirmed by comparing real-time RT-PCR results of selected genes with Affymetrix microarray results.
By applying a newly implemented criterion that takes into account the confidence intervals of the signal log ratios of gene expression [25] (see Additional file 1), 42 candidate genes (46 probe sets) were found to be differentially regulated following a single application of etanercept (Table 2). The early downregulation of expression levels secondary to TNFα neutralization includes genes involved in different pathways and cellular processes such as TNFα signalling via NFκB (TNFAIP3, NFKBIA), NFκB-independent signalling via cAMP (PDE4B), and in the regulation of cellular and oxidative stress response (PPP1R15A, DDIT4, CROP, adrenomedullin, MnSOD). The differential expression of this gene set was associated with distinct clinical responses as evinced by changes in overall disease activities 3 months after the start of treatment. The majority of the identified genes (40 probe sets) were found to be downregulated in responders compared with nonresponders. The differential expression of 27 probe sets was confirmed to be significant using a resampling method. Most importantly, changes in the expression profiles of these selected genes, particularly of pairs or triplets of genes detected 3 days after the start of treatment, were identified as being closely associated with the outcome of therapy (Additional file 1, Tables S3a, S3b). Flow cytometry analysis ruled out that changes of the expression pattern within the first 3 days of treatment were due to an altered cellular distribution of peripheral blood cells.
Two patients (patients 2 and 16) who were not predicted properly were classified as outliers by correlating clinical data and gene expression changes. Patient 2 presents a highly destructive RA, making it difficult to distinguish joint destructions in RA from destructions due to secondary osteoarthritis. Patient 16 displays the highest DAS28 score of the cohort, making it difficult to classify the patient as responder when reaching a DAS28 of 5.9, which is exceptionally high. The stratification of these two cases is hampered in their overall assessment by the limitation of tools such as the DAS28.
In contrast to changes in gene expression pattern in the first days of treatment, gene expression signatures at a single time point, here at baseline, were not reliable in predicting the clinical outcome. Diversities between RA patients on the genetic, molecular and clinical levels [17] evinced by the presence of autoantibodies (rheumatoid factor, anti-cyclic citrullinated peptide antibodies) [26] probably underline the difficulty to predict therapy outcome solely based on pretreatment expression profiles. Eventually, the differences seen in transcriptional responses to etanercept administration might either reflect the state or type of the RA disease or describe epigenomic/genomic variabilities within the patient cohort.
The reconstructed dynamic network representing responders (Figure 3) indicates that not only TNFα may play a significant role in the response to TNFα inhibitors such as etanercept. IL-6-related functionalities seem to play a key role in the responder model, while TNFα-related mechanisms are underscored in nonresponders. The functional dynamics of TNFα and IL-6 might be crucial for the outcome of an etanercept therapy. In biological terms, functionalities of anti-TNFα responses observed in nonresponding patients in comparison with responding patients might emerge due to a differential dynamic regulation of TNFα and of TNFα-dependent target gene expression, possibly also flanked by TNFα-independent mechanisms.
Responders show complex network functions of cytokines including IL-6-mediated, IL-1-mediated, and IL-8-mediated activities. Once TNFα signals are therapeutically downregulated, cytokines such as IL-6 and IL-8 become visible, possibly modulating and eventually attenuating TNF-driven inflammatory processes. This observation is in line with reports on the pleiotropic/anti-inflammatory actions of IL-6 [27], which demonstrated the role of endogenous IL-6 in controlling the levels of proinflammatory cytokines in acute inflammatory responses. The particular role of IL-6 in inflammatory conditions such as RA is presently considered in therapeutic interventions that target IL-6 or its receptor [28].
Differential changes in the expression pattern following anti-TNFα treatment can most probably be attributed to the presence of genetic heterogeneities within the group of RA patients, suggesting the presence of polymorphisms (single nucleotide polymorphisms) and/or epigenetic differences (DNA methylation patterns) in the identified genes. These polymorphisms – found in regulatory gene elements of central cytokines or downstream cascades – or the combination of single nucleotide polymorphisms as well as other types of genetic variations within these differentially regulated or associated genes, such as copy number variations, might possibly turn out to be responsible for mediating therapeutic responses as observed. This hypothesis is supported by findings that some population differences in gene expressions are attributable to allele frequency differences, in particular at regulatory polymorphisms [29].
Conclusion
The present findings demonstrate that it is possible to predict the response of RA patients to anti-TNFα therapy at an early stage of treatment with likelihood >89% (95%) based on differentially expressed gene pairs or gene triplets. By knowing gene sets differentially regulated by TNFα-blocking therapy, additional epigenetic/genetic marker information might be obtained to circumvent the necessity of conducting cost-intensive expression studies. Along these lines, the real challenge of the listed predictory gene sets (pairs and triplets) is to validate in prospectively designed clinical trials the true accuracy and clinical value of this approach in selecting patients that profit most from a TNFα-blocking therapy.
Abbreviations
CT = treshold cycle; DAS = 28-joint-count Disease Activity Score; IFN = interferon; IL = interleukin; NF = nuclear factor; PCR = polymerase chain reaction; Q = prediction accuracy; RA = rheumatoid arthritis; RT = reverse transcription; TNF = tumour necrosis factor.
Competing interests
Based on these studies a patent has been applied for (PCT Patent PCT/EP03/05701, submitted 30 May 2003). The authors declare that they have no further competing interests.
Authors' contributions
H-JT initiated and coordinated the project. JK directed the study design and the patient recruitment and clinical assessment. SD and DK played a very substantial part in the experimental work, data collection and interpretation. RG was responsible for data entry and bioinformatic analysis, assisted by MH. AD was involved in the quantitative real-time RT-PCR analysis. All authors contributed to discussions and to several drafts of the paper. All authors have seen and approved the final version.
Supplementary Material
describing in detail the microarray hybridization as well as the data processing and analysis.
Acknowledgments
Acknowledgements
The present study was supported by grants from the German Federal Ministry of Education and Research (BMBF) (01GG0201), BioChancePlus/BMBF (0313692D), and BMBF-Leitprojekt Proteom-Analyse des Menschen (FKZ 01GG9831).
Contributor Information
Dirk Koczan, Email: dirk.koczan@med.uni-rostock.de.
Susanne Drynda, Email: susanne.drynda@med.ovgu.de.
Michael Hecker, Email: michael.hecker@hki-jena.de.
Andreas Drynda, Email: andreas.drynda@vodafone.de.
Reinhard Guthke, Email: reinhard.guthke@hki-jena.de.
Joern Kekow, Email: joern.kekow@med.ovgu.de.
Hans-Juergen Thiesen, Email: hans-juergen.thiesen@med.uni-rostock.de.
References
- Brennan FM, Maini RN, Feldmann M. Role of pro-inflammatory cytokines in rheumatoid arthritis. Springer Semin Immunopathol. 1998;20:133–147. doi: 10.1007/BF00832003. [DOI] [PubMed] [Google Scholar]
- Bathon JM, Genovese MC. The Early Rheumatoid Arthritis (ERA) trial comparing the efficacy and safety of etanercept and methotrexate. Clin Exp Rheumatol. 2003;21:S195–S197. [PubMed] [Google Scholar]
- Keystone EC, Schiff MH, Kremer JM, Kafka S, Lovy M, DeVries T, Burge DJ. Once-weekly administration of 50 mg etanercept in patients with active rheumatoid arthritis: results of a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2004;50:353–363. doi: 10.1002/art.20019. [DOI] [PubMed] [Google Scholar]
- Brennan A, Bansback N, Reynolds A, Conway P. Modelling the cost-effectiveness of etanercept in adults with rheumatoid arthritis in the UK. Rheumatology. 2004;43:62–72. doi: 10.1093/rheumatology/keg451. [DOI] [PubMed] [Google Scholar]
- Klareskog L, Heijde D van der, de Jager JP, Gough A, Kalden J, Malaise M, Martín Mola E, Pavelka K, Sany J, Settas L, Wajdula J, Pedersen R, Fatenejad S, Sanda M. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet. 2004;363:675–681. doi: 10.1016/S0140-6736(04)15640-7. [DOI] [PubMed] [Google Scholar]
- Hyrich KL, Watson KD, Silman AJ, Symmons DP. Predictors of response to anti-TNF-α therapy among patients with rheumatoid arthritis: results from the British Society for Rheumatology Biologics Register. Rheumatology. 2006;45:1558–1565. doi: 10.1093/rheumatology/kel149. [DOI] [PubMed] [Google Scholar]
- Bobbio-Pallavicini F, Alpini C, Caporali R, Avalle S, Bugatti S, Montecucco C. Autoantibody profile in rheumatoid arthritis during long-term infliximab treatment. Arthritis Res Ther. 2004;6:R264–R272. doi: 10.1186/ar1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kekow J, Wollenberg H, Kühne C, Drynda A, Drynda S. Clinical significance of anti-cyclic citrullinated peptide (CCP) antibodies in monitoring patients with rheumatoid arthritis (RA) treated with etanercept. Arthritis Rheum. 2005;52:s550. abstract. [Google Scholar]
- Ranganathan P. Pharmacogenomics of tumor necrosis factor antagonists in rheumatoid arthritis. Pharmacogenomics. 2005;6:481–490. doi: 10.2217/14622416.6.5.481. [DOI] [PubMed] [Google Scholar]
- Kang CP, Lee KW, Yoo DH, Kang C, Bae SC. The influence of a polymorphism at position -857 of the tumour necrosis factor alpha gene on clinical response to etanercept therapy in rheumatoid arthritis. Rheumatology. 2005;44:547–552. doi: 10.1093/rheumatology/keh550. [DOI] [PubMed] [Google Scholar]
- Lee YH, Rho YH, Choi SJ, Ji JD, Song GG. Association of TNF-alpha -308 G/A polymorphism with responsiveness to TNF-alpha-blockers in rheumatoid arthritis: a meta-analysis. Rheumatol Int. 2006;27:157–161. doi: 10.1007/s00296-006-0175-7. [DOI] [PubMed] [Google Scholar]
- Schotte H, Schluter B, Drynda S, Willeke P, Tidow N, Assmann G, Domschke W, Kekow J, Gaubitz M. Interleukin 10 promoter microsatellite polymorphisms are associated with response to long term treatment with etanercept in patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64:575–581. doi: 10.1136/ard.2004.027672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulfgren AK, Andersson U, Engstrom M, Klareskog L, Maini RN, Taylor PC. Systemic anti-tumor necrosis factor alpha therapy in rheumatoid arthritis down-regulates synovial tumor necrosis factor alpha synthesis. Arthritis Rheum. 2000;43:2391–2396. doi: 10.1002/1529-0131(200011)43:11<2391::AID-ANR3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Schotte H, Schluter B, Willeke P, Mickholz E, Schorat MA, Domschke W, Gaubitz M. Long-term treatment with etanercept significantly reduces the number of proinflammatory cytokine-secreting peripheral blood mononuclear cells in patients with rheumatoid arthritis. Rheumatology (Oxford) . 2004;43:960–964. doi: 10.1093/rheumatology/keh178. [DOI] [PubMed] [Google Scholar]
- Drynda S, Kuhne C, Kekow J. Soluble tumour necrosis factor receptor treatment does not affect raised transforming growth factor beta levels in rheumatoid arthritis. Ann Rheum Dis. 2002;61:254–256. doi: 10.1136/ard.61.3.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catrina AI, Lampa J, Ernestam S, af Klint E, Bratt J, Klareskog L, Ulfgren AK. Anti-tumour necrosis factor (TNF)-alpha therapy (etanercept) down-regulates serum matrix metalloproteinase (MMP)-3 and MMP-1 in rheumatoid arthritis. Rheumatology. 2002;41:484–489. doi: 10.1093/rheumatology/41.5.484. [DOI] [PubMed] [Google Scholar]
- Pouw Kraan TC van der, van Gaalen FA, Kasperkovitz PV, Verbeet NL, Smeets TJ, Kraan MC. Rheumatoid arthritis is a heterogeneous disease: evidence for differences in the activation of the STAT-1 pathway between rheumatoid tissues. Arthritis Rheum. 2003;48:2132–2145. doi: 10.1002/art.11096. [DOI] [PubMed] [Google Scholar]
- Glocker MO, Guthke R, Kekow J, Thiesen HJ. Rheumatoid arthritis, a complex multifactorial disease: on the way toward individualized medicine. Med Res Rev. 2006;26:63–87. doi: 10.1002/med.20045. [DOI] [PubMed] [Google Scholar]
- Lequerre T, Gauthier-Jauneau AC, Bansard C, Derambure C, Hiron M, Vittecoq O, Daveau M, Mejjad O, Daragon A, Tron F, Le Loët X, Salier JP. Gene profiling in white blood cells predicts infliximab responsiveness in rheumatoid arthritis. Arthritis Res Ther. 2006;8:R105. doi: 10.1186/ar1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg J, af Klint E, Catrina AI, Nilsson P, Klareskog L, Ulfgren AK, Lundeberg J. Effect of infliximab on mRNA expression profiles in synovial tissue of rheumatoid arthritis patients. Arthritis Res Ther. 2006;8:R179. doi: 10.1186/ar2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- Prevoo ML, van 't Hof MA, Kuper HH, van Leeuwen MA, Putte LB van de, van Riel PL. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995;38:44–48. doi: 10.1002/art.1780380107. [DOI] [PubMed] [Google Scholar]
- Boyum A. Isolation of lymphocytes, granulocytes and macrophages. Scand J Immunol. 1976;(Suppl 5):9–15. [PubMed] [Google Scholar]
- ArrayExpress http://www.ebi.ac.uk/arrayexpress-old/
- Thiesen HJ, Glocker MO, Guthke R, Kekow J. Patent PCT/EP03/05701. 30 May 2003.
- van Boekel MA, Vossenaar ER, Hoogen FH van den, van Venrooij WJ. Autoantibody systems in rheumatoid arthritis: specificity, sensitivity and diagnostic value. Arthritis Res. 2002;4:87–93. doi: 10.1186/ar395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto T. Interleukin-6: discovery of a pleiotropic cytokine. Arthritis Res Ther. 2006;8(Suppl 2):S2. doi: 10.1186/ar1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maini RN, Taylor PC, Szechinski J, Pavelka K, Broll J, Balint G, Emery P, Raemen F, Petersen J, Smolen J, Thomson D, Kishimoto T, CHARISMA Study Group Double-blind randomized controlled clinical trial of the interleukin-6 receptor antagonist, tocilizumab, in European patients with rheumatoid arthritis who had an incomplete response to methotrexate. Arthritis Rheum. 2006;54:2817–2829. doi: 10.1002/art.22033. [DOI] [PubMed] [Google Scholar]
- Spielman RS, Bastone LA, Burdick JT, Morley M, Ewens WJ, Cheung VG. Common genetic variants account for differences in gene expression among ethnic groups. Nat Genet. 2007;39:226–231. doi: 10.1038/ng1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
describing in detail the microarray hybridization as well as the data processing and analysis.