

Abstract
For proteins to enter the secretory pathway, the membrane attachment site (M-site) on ribosomes must bind cotranslationally to the Sec61 complex present in the endoplasmic reticulum membrane. The signal recognition particle (SRP) and its receptor (SR) are required for targeting, and the nascent polypeptide associated complex (NAC) prevents inappropriate targeting of nonsecretory nascent chains. In the absence of NAC, any ribosome, regardless of the polypeptide being synthesized, binds to the endoplasmic reticulum membrane, and even nonsecretory proteins are translocated across the endoplasmic reticulum membrane. By occupying the M-site, NAC prevents all ribosome binding unless a signal peptide and SRP are present. The mechanism by which SRP overcomes the NAC block is unknown. We show that signal peptide-bound SRP occupies the M-site and therefore keeps it free of NAC. To expose the M-site and permit ribosome binding, SR can pull SRP away from the M-site without prior release of SRP from the signal peptide.
Proteins destined for subcellular localization to the secretory pathway are generally synthesized on membrane bound ribosomes (1). It has long been known that ribosomes have affinity for binding sites at the endoplasmic reticulum (ER) membrane (2), and it has been shown that ribosome binding alone can target nascent polypeptide chains to translocation sites at the ER membrane (3). In the absence of cytosolic components, this targeting and the translocation that subsequently occurs is nonspecific—that is, even signal-less polypeptides were targeted efficiently to translocons in the ER membrane. Although lacking fidelity, this result revealed that targeting actually can occur via the direct interaction of a ribosomal membrane attachment site (M-site) with translocon components. The ability of the translocon to discriminate ribosomes synthesizing signal peptide-containing as opposed to signal-less nascent chains could be reconstituted from just two purified cytosolic components, namely signal recognition particle (SRP) and nascent polypeptide associated complex (NAC) (3). SRP, a ribonucleoprotein complex, was found to be required for cotranslationally directing secretory nascent polypeptides to the ER membrane. On interacting with its membrane bound receptor (SR), SRP releases the signal peptide in a GTP-regulated manner (4). NAC is an abundant heterodimeric protein that interacts exclusively with nascent (but not terminated and released) chains as they emerge from the ribosome and is likely to be one of the first cytosolic factors that contacts nascent polypeptides (5, 6). NAC is conserved phylogenetically and is essential for viability in both drosophila and mice (7, 8). Because they interact at the interface of the ribosome and the cytosol, factors such as NAC and SRP are in a unique position to influence events such as ribosome binding. Purified NAC prevents the membrane binding of ribosome nascent chain complexes (RNCs) whether or not the nascent chains harbor a signal peptide. Contrary to our published data, two reports claim that NAC is not involved in regulating ribosome binding (9, 10). We have shown recently (I.M., B.B., G.K., Hideaki Sakai, B.L., and M.W., unpublished data), however, that the inability to detect NAC’s activity resulted from use of a modified version of the standard in vitro system in which the NAC concentration was subphysiologic and in which even SRP is not required for cotranslational targeting. In this system, readdition of NAC to near its physiologic concentration restored both specificity in targeting and SRP dependence. Concomitant addition of SRP rescued ribosome binding only for signal peptide containing RNCs (3). Although it seemed probable that SRP’s ability to rescue binding after engaging an emerged signal peptide was a result of preventing NAC from occupying the M-site (11), the mechanism by which this occurred remained obscure. Below, we present evidence that SRP and NAC compete with one another for occupancy of the M-site and that the SRP receptor facilitates ribosome binding by disengaging SRP from the M-site, which in turn allows for Sec61 complex, the central component of the translocon, to engage the M-site.
MATERIALS AND METHODS
In Vitro Transcription and Translation and Isolation of Nascent Chain Complexes.
In vitro transcription and translation of truncated mRNAs were as described (12). Truncated mRNAs were translated for 20 min at 26°C, a temperature that best preserves the ribosome/nascent chain complexes. After translation, 9 volumes of dilution buffer (40 mM Hepes/0.5 M KOAc/5 mM Mg(OAc)2/2 mM DTT/pH 7.5) was added, and the ribosome/nascent chain complexes were recovered by centrifugation (100,000 rpm, 40 min, 4°C; TLA 100.4 rotor, Beckman) through a 1.5-ml high salt-containing sucrose cushion [0.5 M sucrose in dilution buffer supplemented with protease inhibitors (13) and 0.8 units/μl RNasin (Promega)]. The complexes were resuspended in translation blank buffer as described (14). Recovery of the nascent chains was typically 40–75%. These complexes were free of NAC as assessed by Western blotting (not shown) or by a photocrosslinking approach (5). Photocrosslinking in which trifluoromethyldiazirinobenzoic acid-modified lys-tRNA was added to a reticulocyte lysate translation system (15) was according to Görlich et al. (16).
Nascent Chain Binding Assay.
This assay is as described (14). Basically, truncated ribosome/nascent chain complexes were incubated with the membrane preparations indicated in the figure legends. The reconstituted proteoliposomes were prepared as described (17), and the Sec61 complex proteoliposomes bind ribosomes with affinities similar to the published data (20). After incubation, 20-μl samples were mixed with 205 μl of 2.3 M sucrose in ribosome binding buffer (RBB; 50 mM Hepes/100 mM KOAc/5 mM Mg(OAc)2/2 mM DTT/0.8 units/μl RNasin/protease inhibitors) to give a final sucrose concentration of 2.1 M. Samples were transferred to 750-μl tubes and were overlaid with 360 μl of 1.9 M sucrose in RBB. Tubes were filled with RBB and then were centrifuged (45,000 rpm, 2 h, 4°C; SW 55 rotor, Beckman). Gradients then were frozen in liquid nitrogen and were cut into top and bottom fractions. The content of radiolabeled nascent chains in each fraction was analyzed by SDS/PAGE and fluorography or scintillation counting. Canine pancreas 80 S ribosomes were prepared and radiolabeled with a [35S]-Met labeling reagent (Amersham) as described (18).
RESULTS
NAC and SRP are the two cytosolic complexes that are necessary and sufficient for fidelity in cotranslational targeting in vitro. The membrane components that contribute to fidelity are as yet uncharacterized. Fidelity in cotranslational targeting could be reconstituted in vitro from purified components by using only SRP and NAC (3). Although the Sec61 complex, when reconstituted into proteoliposomes, supports basal translocation as well as the binding of nontranslating ribosomes (17, 18, 19), it may be that other translocon components are required to regulate ribosome binding such that only those ribosomes synthesizing proteins with signal peptides actually bind to the translocon.
We therefore asked whether Sec61 complex proteoliposomes could support the binding of RNCs and, as a measure of specificity, whether this binding could be prevented by NAC. A homogeneous population of targeting/translocation intermediates was generated by in vitro translation of truncated mRNAs lacking stop codons (12). Because termination of translation cannot occur, nascent chains remain stably associated with the ribosomes. Associated cytosolic factors such as NAC and elongation factors are removed from the RNCs by high salt stripping, and RNCs are collected by sedimentation (5). The salt stripping does not impair the ability of the ribosomes to continue translational elongation or cotranslationally translocate their nascent chains on readdition of fresh cytosol to the system (3).
A truncated mRNA encoding the amino terminal 77 amino acids of firefly luciferase was used to produce high salt-stripped 77-aa firefly luciferase (77aaffLuc) RNCs devoid of an ER signal sequence. The RNCs were incubated with microsomes containing the full complement of integral ER membrane proteins (Fig. 1, lanes 1–4) or with reconstituted proteoliposomes containing only the Sec61 complex (19) (Fig. 1, lanes 5–8) either in the presence or absence of 1.5 μM NAC as indicated in Fig. 1 before analysis in the RNC binding assay. Bound and free RNCs were separated by centrifugation in discontinuous sucrose density gradients. Membranes containing bound RNCs were recovered in the top fraction (T) of the gradients whereas unbound RNCs remained in the bottom fraction (B). Fig. 1 shows that Sec61 complex proteoliposomes bind the RNCs similarly to native microsomal membranes that had been stripped of their ribosomes by puromycin/high salt washing (PKRMs) and that this binding is blocked by NAC. This demonstrates that the interaction of the M-site with Sec61 complex is a NAC-sensitive interaction. Put another way, NAC blocks ribosome binding at least partly by preventing ribosome engagement with Sec61 complex.
Figure 1.

NAC prevents targeting by blocking the interaction of ribosomes with the Sec61 complex. High salt-stripped 77aaffLuc RNCs were prepared in a reticulocyte lysate translation system supplemented with [35]S-Met. RNCs at the final concentration of 3 OD260/ml were incubated with 1.5 μM NAC or NAC buffer for 2 min at 26°C and 5 min on ice before the addition of 1 equivalent (eq) of puromycin/KOAc-washed microsomes (PKRMs) (30) or reconstituted Sec61 complex containing proteoliposomes (17) as indicated. Total assay volumes were 20 μl. Binding was assessed with the flotation assay. Bound RNCs are recovered in top (T) fractions whereas free RNCs are in bottom (B) fractions. In vitro transcription and translation of truncated mRNAs were as described (12) for 20 min at 26°C. Salt stripping and sedimentation of RNCs was as described (5). The complexes were resuspended in blank buffer lacking nucleotides and energy-generating systems unless otherwise indicated. RNC binding was assayed as described (14). The content of radiolabeled nascent chains in each fraction was analyzed by SDS/PAGE and fluorography or scintillation counting.
SRP is required to prevent NAC from blocking the cotranslational targeting of secretory proteins (3).The mechanism by which SRP accomplishes this is unknown. We hypothesized that SRP may compete with NAC for occupancy of the M-site and that SR would remove SRP from the M-site so that ribosome binding could occur. A prediction of this model is that SRP will block the binding of signal peptide-containing RNCs to membranes lacking SR. Fig. 2 shows this to be the case. An mRNA encoding the amino-terminal 86 amino acids of the secretory protein preprolactin was used to produce high salt-stripped 86-aa preprolactin (86aapPL) RNCs, which were incubated with 20 nM SRP or buffer, as indicated in Fig. 2a. Reconstituted proteoliposomes containing only the Sec61 complex (Fig. 2a, lanes 1–4) or both the Sec61 complex and the SR (Fig. 2a, lanes 5–8) were added, and samples then were assayed for ribosome binding. SRP blocked ribosome binding only in the absence of the SRP receptor (Fig. 2a, compare lanes 3 and 4 with 7 and 8). Because some endogenous SRP from the reticulocyte lysate binds to the RNCs during translation and is not removed by high salt extraction (20), we wanted to confirm that binding still would occur in the absence of SRP. We therefore repeated the experiment by using wheat germ lysate 86aapPL RNCs in which one need not worry about contaminating SRP. As shown, the results are similar, indicating that there is not much, if any, residual SRP on our reticulocyte lysate 86aapPL RNCs.
Figure 2.

SRP blocks ribosome binding in the absence of the SR. (a) In the absence of SR, SRP blocks RNC binding to the Sec61 complex (lanes 1–4). The presence of the SR allows binding (lanes 5–8). Bound RNCs are recovered in top (T) fractions whereas free RNCs are in bottom (B) fractions. High salt-stripped 86aapPL RNCs were prepared in the reticulocyte or wheat germ lysate translation systems as described in the legend to Fig. 1. RNCs at the final concentration of 3 OD260/ml were incubated with 20 nM SRP or buffer as indicated in the figure for 5 min at 26°C and 5 min on ice before the addition of 1 eq reconstituted proteoliposomes containing either the Sec61 complex alone (lanes 1–4) or the Sec61 complex and the SR (lanes 5–8). Total assay volumes were 20 μl. After a second round of incubation as above, samples were fractionated with the RNC binding assay, and the fractions were analyzed by fluorography after SDS/PAGE. (b) Nontranslating 80 S ribosomes competitively inhibit the membrane association of 86aapPL RNCs that had been preincubated with SRP with Sec61 complex/SR proteoliposomes, indicating that RNC association occurs via interaction with the Sec61 complex rather than by the association of SRP with its receptor. Reticulocyte lysate-stripped 86aapPL RNCs were incubated with SRP as above and then were added to mixtures containing reticulocyte 80 S ribosomes at the indicated molar excesses over the RNCs and 1 eq of Sec61 complex/SR proteoliposomes in a 20-μl assay. After incubation for 3 min at 26°C and 5 min on ice, samples were analyzed for RNC binding. (c) 86aapPL RNCs containing photocrosslinker-modified lysines were prepared (16) and isolated under low salt conditions (5) before the addition of SRP to 20 nM. After 5 min at 26°C and 5 min on ice, the RNCs were irradiated, and an aliquot was analyzed (lane 1). PKRMs (1 eq) were incubated with the remainder of the RNCs for 5 min at 26°C and 5 min on ice (lane 2) in a 20-μl assay. The sample shown in lanes 3 and 4 was prepared like the one shown in lane 2 except that it was analyzed for RNC binding before SDS/PAGE and fluorography. Bound RNCs as well as the SRP 54-crosslinked nascent chains were recovered in the top fractions (lane 3). Because the RNCs containing irreversibly crosslinked SRP54 bound to the membranes, SR can free the M-site of SRP without prior release of SRP. The sample shown in lanes 5 and 6 was prepared the same as that in lanes 3 and 4, but ribosome binding was assessed under high salt conditions (0.5 M KOAc). (d) High salt-resistant binding of 15-aa-long nascent chains. The 15-aa stripped RNCs were produced by in vitro translation in a reticulocyte lysate of a truncated mRNA in which four codons encoding Met-Met-Met-Ile were engineered upstream of the sequence of the first 11 amino acids of firefly luciferase (15aaMMMI-ffLuc). 15aaMMMI-ffLuc or 86aapPL RNCs derived from 4 μl of translation reaction were incubated with 2 eq PKRMs, and, after incubation, samples were adjusted to either low (150 mM KOAc) or high salt (500 mM KOAc). Samples were fractionated in the flotation assay, in which gradients also were adjusted to 150 mM (LS) or 500 mM (HS) KOAc. Note that the short nascent chains bind as well as the longer chains (which are known to be membrane inserted) and that the fraction that binds in a salt-resistant manner is similar.
The simplest interpretation of the finding that SRP blocks binding in the absence of SR is that SRP occupies the ribosomal M-site, much as NAC does. Ribosome-bound NAC is in direct contact with the nascent chain (5), and NAC is capable of interacting with all domains of nascent chains tested, with the notable exception of signal peptides. We therefore imagine that, in the default mode, NAC is bound to the M-site. Because SRP has a very high affinity for signal peptide-containing RNCs (Kd < 10−9 M) and a low one for signal-less RNCs (Kd > 10−6 M) (21), it occupies the M-site only when a signal peptide emerges from the ribosome and comes to lie in the vicinity of the M-site.
SRP blocks ribosome binding in much the same manner as NAC does, and SR most likely abstracts SRP from the M-site to allow ribosome engagement with the translocon. It was necessary to demonstrate that ribosome–membrane interaction indeed had occurred and that RNC binding was not just mediated by the interaction of RNC-associated SRP with its receptor. Two lines of evidence presented below rule out this possibility. We reasoned that nontranslating 80 S ribosomes should act as competitive inhibitors of RNC binding to the Sec61 complex and SR containing proteoliposomes (14) only if SR does abstract SRP from the M-site but not if membrane association occurs via the interaction of SRP with SR. High salt-stripped 86aapPL RNCs were incubated with SRP and then were added to reaction mixtures containing 80 S ribosomes at the indicated molar excesses and Sec61 complex plus SR proteoliposomes (Fig. 2b). Samples were analyzed for ribosome binding. As membrane association of the RNCs was inhibited competitively by the 80 S ribosomes, SR apparently had freed the M-site of SRP. In cells, such competition probably is not observed because NAC prevents the 80 S ribosomes from interacting with the membrane (see Fig. 3).
Figure 3.

NAC prevents targeting of nontranslating ribosomes. NAC blocks the association of 80S ribosomes with ER membranes. Ribosomes prepared by puromycin and high salt treatment of dog pancreas microsomes were radiolabeled as described (18). Saturating concentrations of ribosomes (4 OD260/ml) were incubated with purified NAC at the concentrations indicated for 2 min at 26°C and 5 min on ice before the addition of 8 eq of EDTA/KOAc-washed microsomes (30, 31) in a 20-μl assay followed by another round of incubation. Binding was assessed by using the RNC binding assay described in Fig. 1. The ability of NAC to inhibit the binding of a similar amount of high salt-stripped 77aaffLuc RNCs was assayed in parallel.
The second piece of evidence that shows that we were observing ribosome-Sec61 complex interaction rather than SRP-SR interaction exploits the fact that the latter but not the former interaction is high salt sensitive (22). In Fig. 2c, lanes 5 and 6, it is shown that the RNCs already have bound in a high salt-resistant manner before SRP is released, indicating that association could not be merely the result of an interaction between SRP and SR.
We next addressed the question of whether the ability of SR to clear SRP from the M-site can occur before release of the signal peptide from SRP. If it can, we reasoned that SR would still rescue binding even if the 54-kDa component of SRP was covalently crosslinked to the signal peptide before the addition of membranes to the reaction. To this end, a photocrosslinking approach in which the ɛ-amino group of lys-tRNA is modified with trifluoromethyldiazirinobenzoic acid, a photoactivatable, irreversible crosslinking reagent, was used. When this reagent is included in in vitro translation/translocation systems, the lysine residues bearing photoactivatable crosslinkers are incorporated into nascent polypeptides as dictated by lysine codons in the mRNA. Photoadducts formed between the nascent chain and proteins are evident by observing decreased electrophoretic mobility of the radiolabeled nascent chain. This crosslinking technology was used previously with pPL mRNA to identify the signal peptide binding component as SRP 54 kDa (23, 24).
Reticulocyte lysate low salt-stripped 86aapPL RNCs were incubated with SRP and then were irradiated, and a fraction of the nascent chains became crosslinked to SRP 54 kDa (Fig. 2c, lane 1). When GTP and microsomes containing the SR are present, the crosslink persists because SR cannot release SRP from the signal peptide (Fig. 2c, lane 2) as expected because of the prior crosslinking (25). In Fig. 2c, lanes 3 and 4, a sample prepared as in Fig. 2c, lane 2 was assayed for the ability of the RNCs with the crosslinked SRP to bind to microsomes. As expected, a large fraction of the nascent chains was targeted. More importantly, virtually all of the RNCs whose signal peptides were crosslinked to SRP 54 kDa bound to the membranes, suggesting that SR had removed SRP from the M-site without prior release of the signal peptide. As expected for productive ribosome binding, the RNCs with SRP crosslinked to the signal peptide bound in a high salt-resistant manner (Fig. 2c, lanes 5 and 6). This result also raises the possibility that high salt-resistant RNC binding does not depend on nascent chain insertion into the membrane. The experiment shown in Fig. 2d further investigates this possibility.
Twenty-five years ago, investigators observed that removal of maximal numbers of ribosomes from rough microsomes required both elevated salt concentrations and use of puromycin to discharge nascent chains (26). These data have been interpreted widely to mean that two forces tether ribosomes to the membrane—an electrostatic interaction between the ribosome and translocon and the tethering force of the membrane-inserted nascent chain. However, nobody ever experimentally investigated the possibility that the mere presence of a nascent chain, whether membrane-inserted or not, allows ribosomes to engage the translocon in a high salt-resistant manner, perhaps by altering ribosomal conformation.
When examining membrane-bound ribosomes on microsomes, all nascent chains will be ≈60-aa or longer in length if they were targeted by SRP (27)—a length long enough to be inserted into Sec61 complex (28). It was therefore reasonable to equate membrane insertion with high salt resistance. It was only with the discovery of NAC and the ability to deplete it that we could ask directly whether nascent chain insertion is required for high salt-resistant binding. Previously, we observed that NAC-depleted 77aaffLuc RNCs, although only inefficiently translocated, bound efficiently and in a high salt-resistant manner (14). This indicated that a signal peptide is not required for high salt-resistant binding but left open the possibility that insertion of the nascent chain is important (3).
We therefore prepared high salt-stripped RNCs (15aaMMMI-ffLuc) containing short 15-aa nascent chains, which should be too short to insert into the translocon/Sec61 complex (28), much less tether the huge ribosome to the ER. As is shown in Fig. 2d, the high salt-resistant binding is comparable to that observed for the longer 86aapPL nascent chains, which become membrane-inserted under these conditions.
Of interest, in performing controls for experiments addressing different questions than we have done in this paper, Neuhof et al. (9) found that SRP blocks targeting to Sec61 complex-only membranes but not to Sec61 complex/SR proteoliposomes by using protease protection as a measure of RNC binding. As we have shown by using a different assay, which more directly measures ribosome binding (Fig. 2), our interpretation of their data is that SRP could not be removed from the M-site by SR and therefore sterically blocked targeting. In other words, the nascent chains in their experiment were digested because the ribosomes synthesizing them probably never bound to the membrane.
Although NAC prevents the mistargeting of ribosomes synthesizing nonsecretory proteins, the fact that nontranslating 80 S ribosomes also bind to Sec61 complex reconstituted membranes (18, 19) and act as competitive inhibitors of RNC binding (ref. 14 and Fig. 2b) raises the following problem: How does the cell prevent inactive ribosomes from binding to translocation sites, thereby potentially blocking targeting and translocation? A potential mechanism would be for NAC to block the membrane association of nontranslating ribosomes with ER membranes.
To test this, saturating concentrations of [35S]-Met-labeled 80 S ribosomes, isolated from dog pancreas rough microsomes (18), were incubated with increasing amounts of NAC, as indicated in Fig. 3, before being assayed for binding to microsomes. Additionally, binding of 77aaffLuc RNCs, whose binding is known to be blocked by NAC, was assayed in parallel. NAC blocks the binding of nontranslating ribosomes.
DISCUSSION
It is well established that proteins destined for the secretory pathway can begin their synthesis on ribosomes that are free in the cytosol and that SRP is required for bringing these ribosomes to the ER membrane to allow for cotranslational translocation. The SRP receptor releases SRP from the polypeptide chain so translocation can occur (for review, see ref. 4). Because purified ribosomes bind to microsomes in vitro, it was unclear why SRP is required for targeting in systems containing cytosol (2). The paradox was resolved with the discovery that NAC is an abundant cytosolic protein complex that prevents ribosome binding. Indeed, in the absence of NAC, mistargeting and some mistranslocation of signal-less nascent chains occurs (14). The finding that SRP is required to overcome NAC’s inhibitory action explained why SRP is required for ribosome binding in complete systems but not in highly purified systems lacking NAC. This result also showed that fidelity in cotranslational targeting requires both NAC and SRP (3). We imagined that NAC and SRP may compete directly for occupancy of the M-site and provide more evidence for this model in the current work.
The crucial function in regulating ribosome binding is to determine who occupies the M-site. Indeed, in the absence of SRP, neither RNCs nor nontranslating ribosomescan bind to ER membranes, given the high amount of NAC present in the cytosol. Thus far, the three components (NAC, Sec61 complex, and SRP) have been described as interacting with the M-site, and all three are required to faithfully reconstitute RNC targeting in vitro. Consistent with the M-site model, Blobel and colleagues recently have provided electron microscopic evidence for direct binding of yeast ribosomes with Sec61 complex (29). SR, in addition to its previously characterized role in releasing SRP from the signal peptide, is essential for regulating exposure of the M-site by removing SRP. SRP effects a translational elongation arrest, which prevents secretory proteins from elongating and folding before insertion into the translocon (27). By both removing SRP from the signal peptide and from the M-site, SR temporally coordinates the resumption of translation with ribosome binding, perhaps ensuring that the exposed nascent chain has no place to go but into the translocon.
Although the Sec61 complex alone supports translocation (17), we have now defined the minimal requirements for fidelity and specificity in the targeting. From the perspective of the cytosol, both NAC and SRP are required for fidelity in targeting. Although the interaction of the M-site with the Sec61 complex is NAC-sensitive (see Fig. 1), a minimal system for faithful targeting also requires the SRP receptor because without its action, SRP acts as a binding prevention factor (see Fig. 4 for model). Identification of the specific components that comprise the ribosomal M-site should allow for one to examine the interaction of Sec61 complex, NAC, and SRP with these factors in greater detail to see whether they bind to the same molecules.
Figure 4.
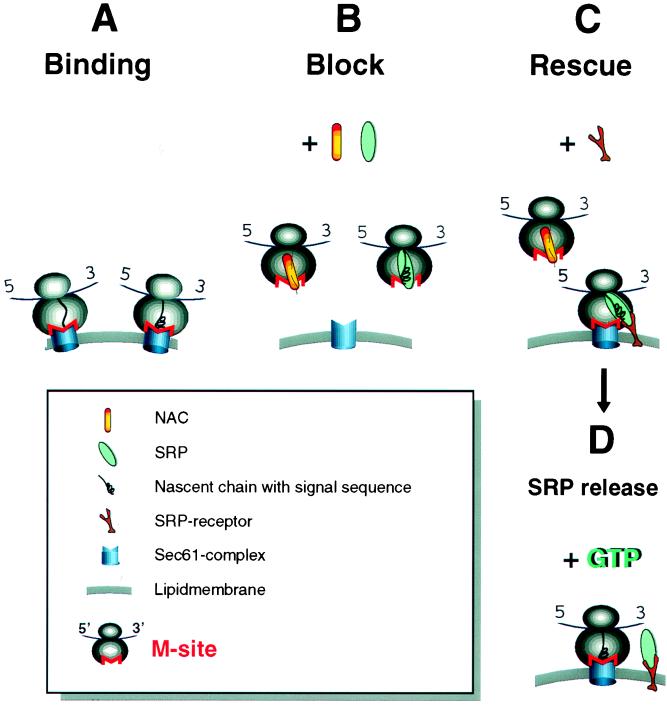
Model. (a) Ribosomes have an intrinsic ability to bind to the Sec61 complex via the M-site. In the absence of NAC and SRP, specificity is lost and any RNC can bind to the translocon. (b) NAC and SRP compete for binding to the M-site with the characteristics of the nascent chain in the vicinity of the M-site, determining which factor binds. NAC binds unless a signal peptide emerges to which SRP binds with high affinity. By experimentally “removing” SR, an intermediate where SRP blocks the M-site was detected. (c) If in the M-site, SRP first engages its receptor and then SR clears the M-site so that the ribosome can engage Sec61 complex. This can occur before the GTP-mediated release of SRP from the signal peptide (d).
Acknowledgments
We dedicate this paper to Waldo Schramm, who was actively involved in this work and who died in 1997. We thank members of the Wiedmann laboratory for critically commenting on the manuscript. This work was supported by a Fellowship from the Deutsche Forschungsgemeinschaft to B.B.; a grant from the Deutsche Forschungsgemeinschaft to R.Z.; the American Cancer Society (Grant CB 111A to G.K.), the Sloan-Kettering Institute (M.W.), and the National Institutes of Health (Grant GM50920–01 to M.W.)
ABBREVIATIONS
- ER
endoplasmic reticulum
- M-site
membrane attachment site
- SRP
signal recognition particle
- NAC
nascent polypeptide associated complex
- SR
SRP receptor
- RNC
ribosome nascent chain complex
- pPL
preprolactin
- eq
equivalent
- ffLuc
firefly luciferase
References
- 1.Blobel G, Dobberstein B. J Cell Biol. 1975;67:835–851. doi: 10.1083/jcb.67.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borgese N, Mok W, Kreibich G, Sabatini D D. J Mol Biol. 1974;88:559–580. doi: 10.1016/0022-2836(74)90408-2. [DOI] [PubMed] [Google Scholar]
- 3.Lauring B, Kreibich G, Wiedmann M. Proc Natl Acad Sci USA. 1995;92:9435–9439. doi: 10.1073/pnas.92.21.9435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walter P, Johnson A E. Annu Rev Cell Biol. 1994;10:87–119. doi: 10.1146/annurev.cb.10.110194.000511. [DOI] [PubMed] [Google Scholar]
- 5.Wiedmann B, Sakai H, Davis T A, Wiedmann M. Nature (London) 1994;370:434–440. doi: 10.1038/370434a0. [DOI] [PubMed] [Google Scholar]
- 6.Wang S, Sakai H, Wiedmann M. J Cell Biol. 1995;130:519–528. doi: 10.1083/jcb.130.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Markesich D, Beckingham K. 38th Annual Drosophila Research Conference. Bethesda, MD: Genetics Soc. Am.; 1997. p. 52C. [Google Scholar]
- 8.Deng J M, Behringer R R. Transgenic Res. 1995;4:264–269. doi: 10.1007/BF01969120. [DOI] [PubMed] [Google Scholar]
- 9.Neuhof A, Rolls M, M, Jungnickel B, Kalies K-U, Rapoport T A. Mol Biol Cell. 1998;9:103–115. doi: 10.1091/mbc.9.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raden D, Gilmore R. Mol Biol Cell. 1998;9:117–130. doi: 10.1091/mbc.9.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powers T, Walter P. Curr Biol. 1996;6:331–338. doi: 10.1016/s0960-9822(02)00484-0. [DOI] [PubMed] [Google Scholar]
- 12.Gilmore R, Collins P, Johnson J, Kellaris K, Rapiejko P. Methods Cell Biol. 1991;34:223–239. doi: 10.1016/s0091-679x(08)61683-0. [DOI] [PubMed] [Google Scholar]
- 13.Erickson A H, Blobel G. Methods Enzymol. 1983;96:38–50. doi: 10.1016/s0076-6879(83)96007-x. [DOI] [PubMed] [Google Scholar]
- 14.Lauring B, Sakai H, Kreibich G, Wiedmann M. Proc Natl Acad Sci USA. 1995;92:5411–5415. doi: 10.1073/pnas.92.12.5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson R J, Hunt T. Methods Enzymol. 1983;96:50–74. doi: 10.1016/s0076-6879(83)96008-1. [DOI] [PubMed] [Google Scholar]
- 16.Görlich D, Kurzchalia T V, Wiedmann M, Rapoport T A. Methods Cell Biol. 1991;34:241–262. doi: 10.1016/s0091-679x(08)61684-2. [DOI] [PubMed] [Google Scholar]
- 17.Görlich D, Rapoport T A. Cell. 1993;75:615–630. doi: 10.1016/0092-8674(93)90483-7. [DOI] [PubMed] [Google Scholar]
- 18.Kalies K U, Görlich D, Rapoport T A. J Cell Biol. 1994;126:925–934. doi: 10.1083/jcb.126.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Görlich D, Prehn S, Hartmann E, Kalies K U, Rapoport T A. Cell. 1992;71:489–503. doi: 10.1016/0092-8674(92)90517-g. [DOI] [PubMed] [Google Scholar]
- 20.High S, Flint N, Dobberstein B. J Cell Biol. 1991;113:25–34. doi: 10.1083/jcb.113.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walter P, Ibrahimi I, Blobel G. J Cell Biol. 1981;91:545–550. doi: 10.1083/jcb.91.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilmore R, Walter P, Blobel G. J Cell Biol. 1982;95:470–477. doi: 10.1083/jcb.95.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krieg U C, Walter P, Johnson A E. Proc Natl Acad Sci USA. 1986;83:8604–8608. doi: 10.1073/pnas.83.22.8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurzchalia T V, Wiedmann M, Girshovich A S, Bochkareva E S, Bielka H, Rapoport T A. Nature (London) 1986;320:634–636. doi: 10.1038/320634a0. [DOI] [PubMed] [Google Scholar]
- 25.Wiedmann M, Kurzchalia T V, Bielka H, Rapoport T A. J Cell Biol. 1987;104:201–208. doi: 10.1083/jcb.104.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adelman M R, Sabatini D D, Blobel G. J Cell Biol. 1973;56:206–229. doi: 10.1083/jcb.56.1.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walter P, Blobel G. J Cell Biol. 1981;91:557–561. doi: 10.1083/jcb.91.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mothes W, Prehn S, Rapoport T A. EMBO J. 1994;13:3973–3982. doi: 10.1002/j.1460-2075.1994.tb06713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beckmann R, Bubeck D, Grassucci R, Penczek P, Verschoor A, Blobel G, Frank J. Science. 1997;278:2123–2126. doi: 10.1126/science.278.5346.2123. [DOI] [PubMed] [Google Scholar]
- 30.Walter P, Blobel G. Methods Enzymol. 1983;96:682–691. doi: 10.1016/s0076-6879(83)96057-3. [DOI] [PubMed] [Google Scholar]
- 31.Walter P, Blobel G. Methods Enzymol. 1983;96:84–93. doi: 10.1016/s0076-6879(83)96010-x. [DOI] [PubMed] [Google Scholar]