

Abstract
Retrotransposons are currently active in the human and mouse genomes contributing to novel disease mutations and genomic variation via de novo insertions. However, little is known about the interactions of non-Long Terminal Repeat (non-LTR) retrotransposons with the host DNA repair machinery. Based on the model of retrotransposition for the human and mouse LINE-1 element, one likely intermediate is an extension of cDNA that is heterologous to the genomic target, a flap intermediate. To determine whether a human flap endonuclease could recognize and process this potential intermediate, the genetic requirement for the ERCC1/XPF heterodimer during LINE-1 retrotransposition was characterized. Reduction of XPF in human cells increased retrotransposition whereas complementation of ERCC1-deficiency in hamster cells reduced retrotransposition. These results demonstrate for the first time that DNA repair enzymes act to limit non-LTR retrotransposition and may provide insight into the genetic instability phenotypes of ercc1 and xpf individuals.
Keywords: LINE, genetic instability, L1, transpose, DNA damage, nucleotide excision repair, target primed reverse transcription
1. Introduction
The Long Interspersed Element-1 (LINE-1 or L1) is an active retrotransposon in primate and mouse genomes [1;2]. L1 insertion has resulted in at least 15 disease causing insertions in recent human generations [3;4]. Other retroelements, such as Alu and SVA which utilize the L1 protein machinery for insertion [5–7], have also caused at least 33 disease insertions in humans in recent generations [3;4]. While insertional mutagenesis is an easily recognizable form of genetic instability caused by these elements, the processing of insertion intermediates has been shown to have several alternate mutagenic outcomes. Utilizing a molecular assay several labs have shown that about 5–10% of insertions cause genomic deletions either by L1-L1 recombination or via processing of the target genomic sequence [8–10]. Some insertions are accompanied by complex chromosomal rearrangements [9]. These types of events have been observed in the human and chimpanzee genomes for both L1 and Alu [11;12]. L1 expression has also been shown to induce the formation of γ-H2AX foci (a marker of double-strand breaks) in an L1-encoded endonuclease-dependent manner [13]. This latter experiment directly demonstrates that host DNA repair proteins recognize and process L1-induced lesions in DNA. Consistent with a host DNA repair response to L1 integration, the double-strand break repair protein ATM is required for L1 integration [13]. The specific role of ATM in L1 integration is unknown. However, given the complexity of the L1 integration event, it seems likely that several DNA repair proteins are able to recognize and process L1 integration intermediates.
L1 is a non-LTR retroelement that amplifies through an RNA intermediate in a process termed retrotransposition [1]. The proposed mechanism of L1 insertion is Target Primed Reverse Transcription (TPRT, see Figure 1). TPRT was originally proposed for the Bombyx mori R2 retrotransposons based on the observation that the endonucleolytic action of R2 is coupled with the initiation of reverse transcription [14;15]. In the case of L1, cleavage of the target site adjacent to a run of thymidines allows basepairing with the polyadenosine tail of L1 mRNA to prime first strand cDNA synthesis [6;16]. This heterologous extension of cDNA is thought to result in a "flap" intermediate (step 3 of Figure 1A). A second nick in the other genomic DNA strand is inferred from the observation that target site duplications frequently flank L1 insertions [1;2]. This second genomic nick is likely required for the final integration of the second strand of DNA as a priming site for second DNA strand synthesis. Frequent microhomologies present at genomic-L1 junctions suggest the role of this double-nicked (essentially a DSB) intermediate creating a primer for DNA synthesis [17]. The timing of the DSB relative to cDNA synthesis is unknown for L1. However, recent data from biochemical analyses of the Bombyx mori non-LTR retrotransposon R2 suggests that cDNA synthesis may be required for the second genomic nick [18]. This would imply that the “flap” intermediate could represent a relatively long-lived or stable intermediate for L1. If so, then DNA repair proteins that recognize this type of 3' flap intermediate may be able to process this L1 integration intermediate.
Figure 1.
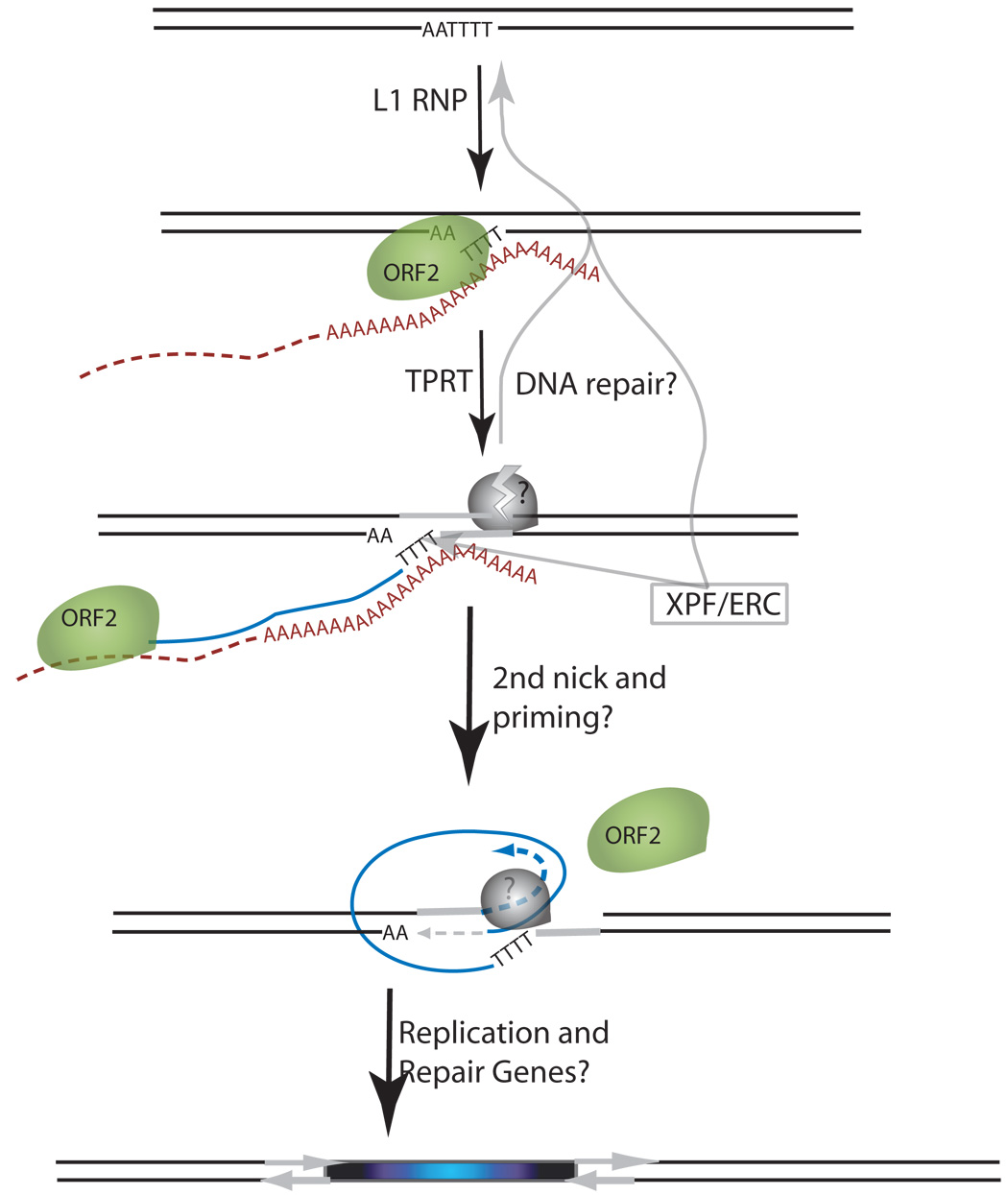
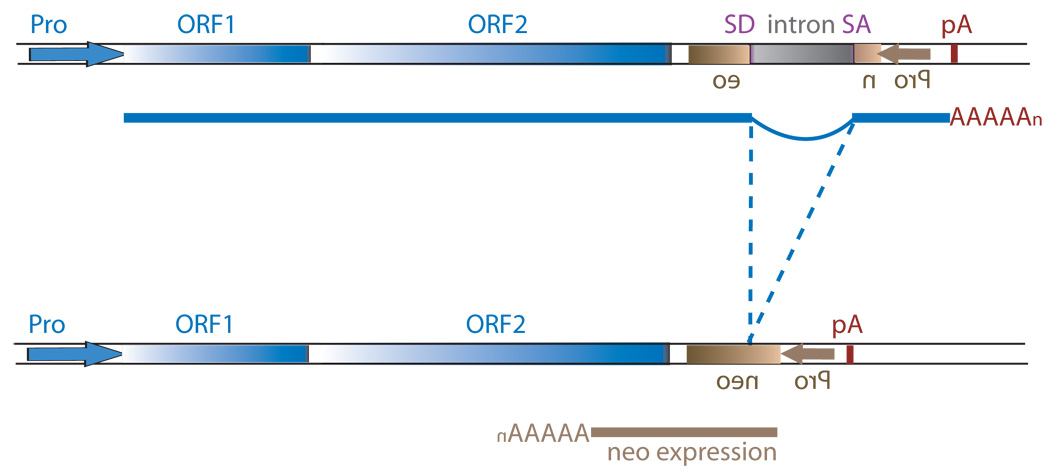
Schematic of the L1 TPRT reaction and retrotransposition assay. (A) A genomic site with a typical L1 endonuclease cleavage site (5’TTTTAA) (step 1) is exposed in the nucleus to the L1 RNP after the ORF2 cleaves the consensus site (step 2). The exposed T-rich region primes reverse transcription on the L1 mRNA polyA tail (step 3), and L1’s reverse transcriptase activity of ORF2 synthesizes cDNA (blue) forming a “flap intermediate” with a 3’ end. This intermediate is a known substrate for the ERCC1/XPF heterodimer. Processing by ERCC1/XPF is predicted to result in restoration of the original target DNA sequence. A nick occurs on the second strand via an unknown mechanism. The segment between the two nicks is highlighted in gray to illustrate the eventual formation of flanking direct repeats by the duplication of these segments. Second-strand synthesis is primed by microhomology-mediated priming which results in synthesis of a second copy of the gray segment (dotted arrow, step 4). Replication from the gray arrow completes synthesis across the 2nd strand of the cDNA creating a new L1 insert and completing synthesis across the other side of the direct repeat (step 5). (B) Schematic of the L1 retrotransposition assay. The L1.3 ORF1 and ORF2 are expressed by the CMV promoter without the L1 5’ UTR. The neomycin resistance gene is under the control of the SV40 promoter in the reverse orientation. The neoR coding sequence is interrupted by the human gamma-globin or the TNF intron which is spliced in the forward orientation. Subsequent integration via retrotransposition creates a neoR transgene that allows for colony growth under selection.
One DNA repair protein complex known to recognize and process a 3' flap intermediate is the ERCC1/XPF heterodimer (XPF is also known as ERCC4) [19]. ERCC1 and XPF were initially recognized as components of Nucleotide Excision Repair (NER) in both human and hamster cells with defects in repair after UV irradiation [20–22]. The specific biochemical role of the ERCC1/XPF heterodimer in NER is to nick the unwound DNA 5’ of the lesion [23]. The ERCC1/XPF endonuclease was subsequently shown to act on minimal DNA substrates with 3’ “flaps” [24]. ERCC1/XPF has also been implicated in other DNA repair processes that are not repaired by NER nor require other components of NER. Cells completely lacking expression of ERCC1 or XPF are also sensitive to interstrand cross-linking agents [25–27]. These additional roles have been supported in rodent models of ERCC1/XPF deficiency [28–30]. Genetic instability and other hallmarks of unrepaired DNA damage are observed in tissues other than those exposed to ultraviolet light [28–34]. Specifically, genomic rearrangements and DNA strand breaks are observed in liver and male germ cells which are not canonical hallmarks of NER deficiency [32;35]. Altogether, ERCC1/XPF displays functions in processing DNA lesions independent of its specific role in processing bulky adducts observed in NER, and these additional functions appear to play a role in the fitness of mammalian cells. One potentially important substrate in mammalian cells may be active retrotransposon integration intermediates.
2. Materials and Methods
2.1 Cell lines and culture conditions
HeLa (ATCC Manassas, VA) were cultured in EMEM plus nonessential amino acids and sodium pyruvate supplemented with 10% FBS (Gibco/Invitrogen, Carlsbad, CA). CHO cells (ATCC Manassas, VA) were cultured in DMEM. G418 was used at 400 µg/mL (Invitrogen). Colony counts for experiments in T75s utilized the ColCount from Oxford Optronix.
2.2 Plasmids
Three similar vectors were utilized for the L1 assay in HeLa and CHO cells. SynL1_optORF1_neo is a modified L1 reporter plasmid whose construction is described previously [36] and was used for 2 of the 5 HeLa transfections. This vector was further modified with substitution of codon optimized G418 resistance sequences and substitution of the gamma-globin intron with the TNF intron and is termed SynL1_optORF1_optNeo. These sequences were synthesized by Blue Heron (Bothell, WA). Sequences are available upon request. This vector was used for the other 3 HeLa transfections. A modified SynL1_optORF1_neo vector in which part of the 3’UTR of L1 is deleted using flanking AleI sites (SynL1_optORF1_AleΔ_neo) was used for all of the CHO-UV20 experiments.
The NeoR expression plasmid used to control for transfection and toxicity is pIRES2-EGFP (BD Biosciences Clontech, Palo Alto, CA).
Hairpin expression constructs utilized a 7SL-Alu-Atail-hairpin expression cassette previously described, pSuper_AluA [36]. DNA oligoes for cloning into the plasmid were purchased from IDT (Coralville, IA). XPF-targeting DNA oligoes target nucleotides 263–285 of the XPF ORF (Accession number U64315) are sense-GATCCCCGTTTACACACAAGGTGGTGTTCAAGAGACACCACCTTGTGTGTAAACTTTTTGGAAA and antisense-AGCTTTTCCAAAAAGTTTACACACAAGGTGGTGTCTCTTGAACACCACCTTGTGTGTAAACGGG.
The human ERCC1 expression vector utilized a modified pCDNA3 vector in which the neoR portion was deleted using a PvuII digest and religation (pCDNA3_pvuΔ). An I.M.A.G.E. clone (#2824122, ATCC) containing the full-length ORF of human ERCC1 was digested with EcoRI and XhoI, and the fragment was cloned into pCDNA3_pvuΔ. digested with EcoRI and Xho.
2.3 Transfection
All colony plating assays utilized Lipofectamine with PLUS (InVitrogen) for transfection. PLUS was always used at a 1 µL : 1 µg DNA concentration. HeLa cells were plated at 100,000 cells per well then the next day 2.4 µg of pSuperAluA-term or -XPF was cotransfected with 1 µg of L1 plasmid or 0.3 µg of pIRES2-EGFP using 6 µL of Lipofectamine/PLUS per plate. G418 media was added 1 day post-transfection and maintained for approximately 14 days. To make protein extracts from HeLa or HCT-116 cells, a T75 was seeded and transfected equivalently as for the 6-well plates. For L1 retrotransposition assay in CHO cells, 250,000 cells were seeded in T75s and transfected the following day with 2 µg of the ERCC1 expression vector or empty vector control using 5 µL of Lipofectamine2000 (Invitrogen). The following day 1 µg of L1 plasmid or 0.3 µg of pIRES2-EGFP were transfected with 3 µL Lipofectamine/PLUS per T75. G418 media was added the following day and maintained for 14 days. For colony counting, cells were fixed and stained for 30 minutes with crystal violet (0.2% crystal violet in 5% acetic acid and 2.5% isopropanol).
2.4 Western blotting
Protein was extracted from trypsinized and PBS-washed cells using standard Tris-SDS-glycerol buffer and boiling for 15 minutes with agitation. Extracts were eletrophoresed on 4–12% NuPage Bis-Tris gels (Invitrogen). Proteins were transferred to 0.2 micron Nitrocellulose using a Genie Transfer system (Idea Scientific Company, Minneapolis, MN). Antibodies used were mouse anti-XPF (Trevigen, Gaithersburg, MD), rabbit anti-beta actin (Sigma-Aldrich, St. Louis, MO), rabbit anti-H2AX (Novus Biologicals, Littleton, CO). Secondary HRP conjugated antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Epitopes were detected using the SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL) and imaged on a Kodak Imager
2.5 Statistical Analyses
ANOVA was used to determine the statistical significance of treatment (hERCC1 expression or XPF knockdown) versus control plasmid plating efficiencies. Specifically, the “ANOVA: two-factor with replication” function was used in Microsoft Excel with individual well counts as individual replicates within each trial. n in the figures refers to the number of trials.
3. Results
To determine whether ERCC1/XPF is able to recognize and process L1 integration intermediates, we utilized a quantitative tissue culture assay system for L1 retrotransposition [37]. In this assay, expression of L1 with a resistance marker cassette (in this case G418) results in colonies that are specifically due to individual retrotransposition insertions (Figure 1B). The majority of L1 retrotransposition occurs in a 48-hour timeframe using our protocols (A. Engel personal communication), so cells can be treated with other factors that impact gene expression or protein activity over a reasonable time interval. Two approaches were used to evaluate the effect of ERCC1/XPF on the L1 retrotransposition rate: Complementation of an ERCC1-deficient rodent cell line and depletion of the complex via RNA interference (RNAi).
To establish a role of ERCC1/XPF in processing L1 integration intermediates, experiments were carried out in the CHO-UV20 cell line. CHO UV20s were derived from an EMS mutagenesis screen for UV-sensitive mutants and is a member of complementation group 1 for UV sensitivity [38]. The human gene demonstrating cross complementation in these hamster cell lines was termed ERCC1 [39]. The mutation in UV20 lines results in very low expression of ERCC1 and XPF proteins, and UV20 cells demonstrate the non-NER defects [40;41]. To transiently complement ERCC1-deficiency, a hERCC1 expression construct was derived from human ERCC1 cDNA and cloned into a modified pCDNA3 vector. Serial transfection of the L1 retrotransposition vector 24 hours after transfection with the hERCC1 expression vector into CHO-UV20 cells resulted in a 72% decrease in colonies relative to transfection with the empty vector control (Figure 2, P=9.3 × 10^−4). The slight decrease in random plasmid integration observed with hERCC1 transfection was not statistically different (P=0.44). L1 retrotransposition in a wild-type CHO cell line (CHO-K1) was unaffected by additional exogenous hERCC1 expression relative to empty vector (data not shown).
Figure 2.

ERCC1 complementation decreases L1 retrotransposition. The Chinese hamster ovary cell line (UV-20, ercc1) was characterized for it ability to support L1 retrotransposition. One day prior to transfection of the L1 construct, the cells were transfected with a hERCC1 expression construct or with the corresponding empty vector. (A) Sample plates of the L1 retrotransposition assay after serial transfection with control and hERCC1 expression vectors in ERCC1-deficient CHO-UV20 cells. (B) Quantitation of the relative effect of ERCC1 complementation on the L1 retrotransposition assay. (n = 4)
To reduce the amount of XPF expression in HeLa cells, we used a previously published vector-based RNA interference (RNAi) strategy that is amenable to colony-plating assays [36]. A hairpin targeting XPF (XPF263) demonstrates minimal target knockdown in HeLa cells using the same tranfection conditions as for retrotransposition (data not shown). However, using the more efficiently transfected HCT-116 cell line, this vector reduced XPF protein expression 60% relative to the empty vector control 24 hours after transfection (Figure 3A). Cotransfection of AluA-XPF263 with the L1 retrotransposition assay vector resulted in a 71% increase in colonies relative to the control vector, AluA-term (Figure 3B, P = 1.1 × 10^−14). Cell growth and capability of generating G418-resistant colonies were also evaluated by the transfection of a vector containing a G418-resistant gene. Effects of the expression of the hairpin causing toxicity or alterations in cell growth would be observed as an alteration in colonies obtained. The XPF hairpin had a slightly negative effect on random plasmid integration (decrease of 27%, P = 4.3 × 10^−5, Figure 3B, “neoR”). ERCC1/XPF may play a role in circular plasmid integration because ERCC1/XPF has been shown to be required to process heterologous ends during plasmid integration or recombination [42;43] and for homologous gene targeting in mouse ES cells [25]. Modification of the random plasmid integration assay by linearization of the plasmid demonstrated no effect on colony formation by XPF knockdown (data not shown).
Figure 3.

XPF deficiency increases L1 retrotransposition. To decrease XPF expression, hairpins targeting human XPF for RNAi-mediated knockdown were designed and cloned into the pSuper_AluA construct. (A) Transfection of HCT-116 cells with pSuper_AluA_XPF demonstrated knockdown relative to transfection with a no hairpin control vector. Cotransfection of the pSuper_AluA_XPF vector with an L1 retrotransposition construct was performed in HeLa cells. (B) Sample plates of the L1 retrotransposition assay and random plasmid integration after cotransfection with control and XPF-targeting RNAi vectors. (C) Quantitation of the relative effect of XPF knockdown versus RNAi control vector on the L1 retrotransposition assay and random plasmid integration. (n = 6)
4. Discussion
Our data indicate a role for ERCC1/XPF in limiting L1 insertion in a molecular retrotransposition assay. ERCC1/XPF‘s known biochemical properties are 5’ “bubble” and 3’ “flap” endonuclease activity. This latter activity can occur within flanking DNA duplex or at a DNA end [24]. The 5’ bubble endonuclease activity is important for processing NER intermediates, but its substrate would not be predicted to be a step of TPRT. In order for L1 to integrate into the genome, it must make at least one nick in DNA to create a primer site for reverse transcription in which the synthesis occurs 5’ to 3’ leading to a 3’ flap extension. This intermediate is a substrate for ERCC1/XPF activity, and failure to cleave it would result in an increase in the retrotransposition frequency. Therefore, the most straightforward interpretation of our results is that that cDNA synthesis step of TPRT is recognized and processed by ERCC1/XPF. However, a direct demonstration of this role awaits an in vitro or in vivo system capable of quantifying L1 cDNA intermediates.
These results further support that there is a complex interaction of host DNA repair genes with non-LTR retroelements. To date, the only demonstrated relationships of DNA repair genes with any endonuclease proficient non-LTR retrotransposon are H2AX and ATM in human cells [13]. H2AX’s phosphorylated form (γ-H2AX) localizes to chromatin in response to L1 expression dependent on L1’s endonuclease function which likely marks the DSB-like intermediate of TPRT. However, a direct role for γ-H2AX in facilitating or limiting TPRT is unknown. L1 retrotransposition shows a genetic dependency for ATM although the exact TPRT intermediates upon which ATM acts is speculative. ERCC1/XPF, in contrast, demonstrates a host defense role by limiting L1 retrotransposition. This is the first demonstration of a DNA repair protein limiting non-LTR retrotransposition in any organism. These results also extend the known substrates of the ERCC1/XPF protein complex to include TPRT intermediates in addition to interstrand crosslinks and NER intermediates.
A host defense to L1 retrotransposition at the level of DNA integration is another example of the multi-layered response of host cells to the damaging effects of retroelements at various stages of the L1 life cycle. L1 retrotransposition in molecular assays has been shown to be inhibited by the human APOBEC3 family of cytidine deaminases at a posttranscriptional stage [44–48]. In addition, RNA interference limits L1 mRNA expression in human and mouse cells [49–51]. Human L1 RNA is also subject to splicing and premature polyadenylation such that the retrotranspositionally competent full-length mRNA is limited [52;53]. The primary and first level of regulation of L1 activity is at the level of transcription, and methylation of L1 promoters is well documented as controlling L1 activity [49;54;55]. This work documents a host response to L1 at the terminal stage of its life cycle—the integration of the cDNA into the genome.
This work provides a possible explanation for the source of genetic instability in mouse models of ERCC1/XPF deficiency that has so far proved elusive. Endogenous L1 expression is seen in a variety of tissues in adult mice [56], during embryogenesis [57] [58;59], and in germline cells [49;54;58;59]. Some of these tissues demonstrate higher levels of ERCC1 or XPF expression [32;60]. How would increased L1 activity contribute to the phenotypes of the ercc1−/− and xpf−/− mice? An increase in L1 insertions would very likely disrupt genes. Accumulation over time could lead to disruption of cellular processes and result in a genetically unstable phenotype. However, what seems more likely is that the increase in the flap intermediate of L1 would lead to an increase in the genomic deletions or rearrangements that can occur concomitant with L1 insertions [8–10]. Stabilized L1 flap intermediates could also lead to an increase in DSBs and subsequent aberrant processing could “decap” chromosomes of their telomeres. This could induce chromosome fusion breakage cycles which are highly recombinogenic and would induce a persistent DSB repair response [61]. ercc1−/− and xpf−/− mice show phenotypes consistent with attempted repair of fusion breakage cycles like increased p53 and Rad51 expression [34]. The combination of persistent genomic instability from fusion breakage cycles and additional L1 insertion/rearrangements would then underlie the other phenotypes. Persistent genetic instability from endogenous sources is thought to contribute to the aging process as is typified by the progeria phenotypes of DNA repair deficiencies [62]. If L1-mediated genomic instability is a significant source for this damage, then treatment with reverse transcriptase inhibitors may alleviate the progeria phenotypes.
Acknowledgements
We would like to thank Melanie Palmisano for technical assistance with DNA preparation and tissue culture. We also thank Deininger lab members for suggestions on the manuscript and with experiments. The P.L.D. lab is supported by grants from the USPHS grant R01GM45668, NIH P20 RR020152, National Science Foundation EPS-0346411 and the State of Louisiana Board of Regents Support Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Babushok DV, Kazazian HH., Jr Progress in understanding the biology of the human mutagen LINE-1. Hum.Mutat. 2007;28:527–539. doi: 10.1002/humu.20486. [DOI] [PubMed] [Google Scholar]
- 2.Ostertag EM, Kazazian HH., Jr Biology of mammalian L1 retrotransposons. Annu.Rev.Genet. 2001;35:501–538. doi: 10.1146/annurev.genet.35.102401.091032. [DOI] [PubMed] [Google Scholar]
- 3.Chen JM, Ferec C, Cooper DN. LINE-1 Endonuclease-Dependent Retrotranspositional Events Causing Human Genetic Disease: Mutation Detection Bias and Multiple Mechanisms of Target Gene Disruption. J.Biomed.Biotechnol. 2006;2006:56182. doi: 10.1155/JBB/2006/56182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen JM, Stenson PD, Cooper DN, Ferec C. A systematic analysis of LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease. Hum.Genet. 2005;117:411–427. doi: 10.1007/s00439-005-1321-0. [DOI] [PubMed] [Google Scholar]
- 5.Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nat.Genet. 2003;35:41–48. doi: 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- 6.Jurka J. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc.Natl.Acad.Sci.U.S.A. 1997;94:1872–1877. doi: 10.1073/pnas.94.5.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ostertag EM, Goodier JL, Zhang Y, Kazazian HH., Jr SVA elements are nonautonomous retrotransposons that cause disease in humans. Am.J.Hum.Genet. 2003;73:1444–1451. doi: 10.1086/380207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilbert N, Lutz-Prigge S, Moran JV. Genomic deletions created upon LINE-1 retrotransposition. Cell. 2002;110:315–325. doi: 10.1016/s0092-8674(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 9.Gilbert N, Lutz S, Morrish TA, Moran JV. Multiple fates of L1 retrotransposition intermediates in cultured human cells. Mol.Cell Biol. 2005;25:7780–7795. doi: 10.1128/MCB.25.17.7780-7795.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Symer DE, Connelly C, Szak ST, Caputo EM, Cost GJ, Parmigiani G, Boeke JD. Human l1 retrotransposition is associated with genetic instability in vivo. Cell. 2002;110:327–338. doi: 10.1016/s0092-8674(02)00839-5. [DOI] [PubMed] [Google Scholar]
- 11.Han K, Sen SK, Wang J, Callinan PA, Lee J, Cordaux R, Liang P, Batzer MA. Genomic rearrangements by LINE-1 insertion-mediated deletion in the human and chimpanzee lineages. Nucleic Acids Res. 2005;33:4040–4052. doi: 10.1093/nar/gki718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Callinan PA, Wang J, Herke SW, Garber RK, Liang P, Batzer MA. Alu retrotransposition-mediated deletion. J.Mol.Biol. 2005;348:791–800. doi: 10.1016/j.jmb.2005.02.043. [DOI] [PubMed] [Google Scholar]
- 13.Gasior SL, Wakeman TP, Xu B, Deininger PL. The human LINE-1 retrotransposon creates DNA double-strand breaks. J.Mol.Biol. 2006;357:1383–1393. doi: 10.1016/j.jmb.2006.01.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- 15.Luan DD, Eickbush TH. RNA template requirements for target DNA-primed reverse transcription by the R2 retrotransposable element. Mol.Cell Biol. 1995;15:3882–3891. doi: 10.1128/mcb.15.7.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morrish TA, Gilbert N, Myers JS, Vincent BJ, Stamato TD, Taccioli GE, Batzer MA, Moran JV. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat.Genet. 2002;31:159–165. doi: 10.1038/ng898. [DOI] [PubMed] [Google Scholar]
- 17.Zingler N, Willhoeft U, Brose HP, Schoder V, Jahns T, Hanschmann KM, Morrish TA, Lower J, Schumann GG. Analysis of 5′ junctions of human LINE-1 and Alu retrotransposons suggests an alternative model for 5′-end attachment requiring microhomology-mediated end-joining. Genome Res. 2005;15:780–789. doi: 10.1101/gr.3421505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christensen SM, Ye J, Eickbush TH. RNA from the 5′ end of the R2 retrotransposon controls R2 protein binding to and cleavage of its DNA target site. Proc.Natl.Acad.Sci.U.S.A. 2006;103:17602–17607. doi: 10.1073/pnas.0605476103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gillet LC, Scharer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem.Rev. 2006;106:253–276. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- 20.Busch D, Greiner C, Lewis K, Ford R, Adair G, Thompson L. Summary of complementation groups of UV-sensitive CHO cell mutants isolated by large-scale screening. Mutagenesis. 1989;4:349–354. doi: 10.1093/mutage/4.5.349. [DOI] [PubMed] [Google Scholar]
- 21.Cleaver JE. Defective repair replication of DNA in xeroderma pigmentosum. Nature. 1968;218:652–656. doi: 10.1038/218652a0. [DOI] [PubMed] [Google Scholar]
- 22.Weerd-Kastelein EA, Keijzer W, Bootsma D. Genetic heterogeneity of xeroderma pigmentosum demonstrated by somatic cell hybridization. Nat.New Biol. 1972;238:80–83. doi: 10.1038/newbio238080a0. [DOI] [PubMed] [Google Scholar]
- 23.Sijbers AM, de Laat WL, Ariza RR, Biggerstaff M, Wei YF, Moggs JG, Carter KC, Shell BK, Evans E, de Jong MC, Rademakers S, de Rooij J, Jaspers NG, Hoeijmakers JH, Wood RD. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell. 1996;86:811–822. doi: 10.1016/s0092-8674(00)80155-5. [DOI] [PubMed] [Google Scholar]
- 24.de Laat WL, Appeldoorn E, Jaspers NG, Hoeijmakers JH. DNA structural elements required for ERCC1-XPF endonuclease activity. J.Biol.Chem. 1998;273:7835–7842. doi: 10.1074/jbc.273.14.7835. [DOI] [PubMed] [Google Scholar]
- 25.Niedernhofer LJ, Essers J, Weeda G, Beverloo B, de Wit J, Muijtjens M, Odijk H, Hoeijmakers JH, Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required for targeted gene replacement in embryonic stem cells. EMBO J. 2001;20:6540–6549. doi: 10.1093/emboj/20.22.6540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol.Cell Biol. 2000;20:7980–7990. doi: 10.1128/mcb.20.21.7980-7990.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoy CA, Thompson LH, Mooney CL, Salazar EP. Defective DNA cross-link removal in Chinese hamster cell mutants hypersensitive to bifunctional alkylating agents. Cancer Res. 1985;45:1737–1743. [PubMed] [Google Scholar]
- 28.Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ, Nigg A, van Steeg H, Bootsma D, Hoeijmakers JH. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr.Biol. 1997;7:427–439. doi: 10.1016/s0960-9822(06)00190-4. [DOI] [PubMed] [Google Scholar]
- 29.Tian M, Shinkura R, Shinkura N, Alt FW. Growth retardation, early death, and DNA repair defects in mice deficient for the nucleotide excision repair enzyme XPF. Mol.Cell Biol. 2004;24:1200–1205. doi: 10.1128/MCB.24.3.1200-1205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McWhir J, Selfridge J, Harrison DJ, Squires S, Melton DW. Mice with DNA repair gene (ERCC-1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nat.Genet. 1993;5:217–224. doi: 10.1038/ng1193-217. [DOI] [PubMed] [Google Scholar]
- 31.Kirschner K, Singh R, Prost S, Melton DW. Characterisation of Ercc1 deficiency in the liver and in conditional Ercc1-deficient primary hepatocytes in vitro. DNA Repair (Amst) 2007;6:304–316. doi: 10.1016/j.dnarep.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 32.Hsia KT, Millar MR, King S, Selfridge J, Redhead NJ, Melton DW, Saunders PT. DNA repair gene Ercc1 is essential for normal spermatogenesis and oogenesis and for functional integrity of germ cell DNA in the mouse. Development. 2003;130:369–378. doi: 10.1242/dev.00221. [DOI] [PubMed] [Google Scholar]
- 33.Melton DW, Ketchen AM, Nunez F, Bonatti-Abbondandolo S, Abbondandolo A, Squires S, Johnson RT. Cells from ERCC1-deficient mice show increased genome instability and a reduced frequency of S-phase-dependent illegitimate chromosome exchange but a normal frequency of homologous recombination. J.Cell Sci. 1998;111(Pt 3):395–404. doi: 10.1242/jcs.111.3.395. [DOI] [PubMed] [Google Scholar]
- 34.Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GT, Meinecke P, Kleijer WJ, Vijg J, Jaspers NG, Hoeijmakers JH. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–1043. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- 35.Dolle ME, Busuttil RA, Garcia AM, Wijnhoven S, van Drunen E, Niedernhofer LJ, van der HG, Hoeijmakers JH, van Steeg H, Vijg J. Increased genomic instability is not a prerequisite for shortened lifespan in DNA repair deficient mice. Mutat.Res. 2006;596:22–35. doi: 10.1016/j.mrfmmm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 36.Gasior SL, Palmisano M, Deininger PL. Alu-linked hairpins efficiently mediate RNA interference with less toxicity than do H1-expressed short hairpin RNAs. Anal.Biochem. 2006;349:41–48. doi: 10.1016/j.ab.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 37.Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH., Jr High frequency retrotransposition in cultured mammalian cells. Cell. 1996;87:917–927. doi: 10.1016/s0092-8674(00)81998-4. [DOI] [PubMed] [Google Scholar]
- 38.Thompson LH, Mooney CL, Burkhart-Schultz K, Carrano AV, Siciliano MJ. Correction of a nucleotide-excision-repair mutation by human chromosome 19 in hamster-human hybrid cells. Somat.Cell Mol.Genet. 1985;11:87–92. doi: 10.1007/BF01534738. [DOI] [PubMed] [Google Scholar]
- 39.Westerveld A, Hoeijmakers JH, van Duin M, de Wit J, Odijk H, Pastink A, Wood RD, Bootsma D. Molecular cloning of a human DNA repair gene. Nature. 1984;310:425–429. doi: 10.1038/310425a0. [DOI] [PubMed] [Google Scholar]
- 40.Rolig RL, Layher SK, Santi B, Adair GM, Gu F, Rainbow AJ, Nairn RS. Survival, mutagenesis, and host cell reactivation in a Chinese hamster ovary cell ERCC1 knock-out mutant. Mutagenesis. 1997;12:277–283. doi: 10.1093/mutage/12.4.277. [DOI] [PubMed] [Google Scholar]
- 41.Rolig RL, Lowery MP, Adair GM, Nairn RS. Characterization and analysis of Chinese hamster ovary cell ERCC1 mutant alleles. Mutagenesis. 1998;13:357–365. doi: 10.1093/mutage/13.4.357. [DOI] [PubMed] [Google Scholar]
- 42.Adair GM, Rolig RL, Moore-Faver D, Zabelshansky M, Wilson JH, Nairn RS. Role of ERCC1 in removal of long non-homologous tails during targeted homologous recombination. EMBO J. 2000;19:5552–5561. doi: 10.1093/emboj/19.20.5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sargent RG, Meservy JL, Perkins BD, Kilburn AE, Intody Z, Adair GM, Nairn RS, Wilson JH. Role of the nucleotide excision repair gene ERCC1 in formation of recombination-dependent rearrangements in mammalian cells. Nucleic Acids Res. 2000;28:3771–3778. doi: 10.1093/nar/28.19.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O'Shea KS, Moran JV, Cullen BR. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc.Natl.Acad.Sci.U.S.A. 2006;103:8780–8785. doi: 10.1073/pnas.0603313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bogerd HP, Wiegand HL, Doehle BP, Lueders KK, Cullen BR. APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 2006;34:89–95. doi: 10.1093/nar/gkj416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kinomoto M, Kanno T, Shimura M, Ishizaka Y, Kojima A, Kurata T, Sata T, Tokunaga K. All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res. 2007 doi: 10.1093/nar/gkm181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muckenfuss H, Hamdorf M, Held U, Perkovic M, Lower J, Cichutek K, Flory E, Schumann GG, Munk C. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J.Biol.Chem. 2006;281:22161–22172. doi: 10.1074/jbc.M601716200. [DOI] [PubMed] [Google Scholar]
- 48.Stenglein MD, Harris RS. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J.Biol.Chem. 2006;281:16837–16841. doi: 10.1074/jbc.M602367200. [DOI] [PubMed] [Google Scholar]
- 49.Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, Hannon GJ. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science. 2007;316:744–747. doi: 10.1126/science.1142612. [DOI] [PubMed] [Google Scholar]
- 50.Yang N, Kazazian HH., Jr L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat.Struct.Mol.Biol. 2006;13:763–771. doi: 10.1038/nsmb1141. [DOI] [PubMed] [Google Scholar]
- 51.Soifer HS, Zaragoza A, Peyvan M, Behlke MA, Rossi JJ. A potential role for RNA interference in controlling the activity of the human LINE-1 retrotransposon. Nucleic Acids Res. 2005;33:846–856. doi: 10.1093/nar/gki223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Belancio VP, Hedges DJ, Deininger P. LINE-1 RNA splicing and influences on mammalian gene expression. Nucleic Acids Res. 2006;34:1512–1521. doi: 10.1093/nar/gkl027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perepelitsa-Belancio V, Deininger P. RNA truncation by premature polyadenylation attenuates human mobile element activity. Nat.Genet. 2003;35:363–366. doi: 10.1038/ng1269. [DOI] [PubMed] [Google Scholar]
- 54.Bourc'his D, Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431:96–99. doi: 10.1038/nature02886. [DOI] [PubMed] [Google Scholar]
- 55.Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13:335–340. doi: 10.1016/s0168-9525(97)01181-5. [DOI] [PubMed] [Google Scholar]
- 56.Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435:903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- 57.Packer AI, Manova K, Bachvarova RF. A discrete LINE-1 transcript in mouse blastocysts. Dev.Biol. 1993;157:281–283. doi: 10.1006/dbio.1993.1133. [DOI] [PubMed] [Google Scholar]
- 58.Branciforte D, Martin SL. Developmental and cell type specificity of LINE-1 expression in mouse testis: implications for transposition. Mol.Cell Biol. 1994;14:2584–2592. doi: 10.1128/mcb.14.4.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trelogan SA, Martin SL. Tightly regulated, developmentally specific expression of the first open reading frame from LINE-1 during mouse embryogenesis. Proc.Natl.Acad.Sci.U.S.A. 1995;92:1520–1524. doi: 10.1073/pnas.92.5.1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shannon M, Lamerdin JE, Richardson L, McCutchen-Maloney SL, Hwang MH, Handel MA, Stubbs L, Thelen MP. Characterization of the mouse Xpf DNA repair gene and differential expression during spermatogenesis. Genomics. 1999;62:427–435. doi: 10.1006/geno.1999.6016. [DOI] [PubMed] [Google Scholar]
- 61.Bailey SM, Murnane JP. Telomeres, chromosome instability and cancer. Nucleic Acids Res. 2006;34:2408–2417. doi: 10.1093/nar/gkl303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kirkwood TB. Understanding the odd science of aging. Cell. 2005:437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]