

Abstract
NMDA receptors play critical roles in synaptic modulation and neurological disorders. In this study, we investigated the developmental changes in NR2 cleavage by NMDA receptor-activated calpain in cultured cortical and hippocampal neurons. Calpain activity increased with development, associated with increased expression of NMDA receptors but not of calpain I. The activation of calpain in immature and mature cortical cultures was inhibited by antagonists of NR1/2B and NR1/2A/2B receptors, whereas the inhibition of NR1/2B receptors did not alter calpain activation in mature hippocampal cultures. The degradation of NR2 subunits by calpain differed with developmental age. NR2A was not a substrate of calpain in mature hippocampal cultures, but was cleaved in immature cortical and hippocampal cultures. NR2B degradation by calpain in cortical cultures decreased with development, but the level of degradation of NR2B in hippocampal cultures did not change. The kinetics of NMDA receptor-gated whole cell currents were also modulated by calpain activation in a manner that varied with developmental stage in vitro. In early (but not later) developmental stages, calpain activation altered the NMDA-evoked current rise time and time constants for both desensitization and deactivation. Our data suggest that the susceptibility of the NMDA receptor to cleavage by calpain varies with neuronal maturity in a manner that may alter its electrophysiological properties.
Keywords: excitotoxicity, glutamate, learning, NMDA receptor, protease
The NMDA receptor (NMDAR) is involved in many neuronal functions including synaptogenesis, learning, memory and excitotoxicity. NMDARs contain combinations of NR1 and NR2 subunits (NR2A–NR2D) (Hollmann and Heinemann 1994), both of which are usually required for a functional receptor. NR2 subunits have large cytoplasmic carboxyl termini containing phosphorylation sites for protein kinase C (PKC), protein tyrosine kinases and Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Omkumar et al. 1996; Liao et al. 2001; Grant et al. 2001; Yang and Leonard 2001), and a PSD-95, discs large, Z01 (PDZ) binding motif that facilitates interactions with post-synaptic density protein 95 (PSD-95) and other synaptic proteins (Kornau et al. 1995; Niethammer et al. 1996). The C-terminal region of NR2 subunits also contains internalization motifs that alter NMDAR turnover (Scott et al. 2004). Thus, C-terminal truncation of NR2 subunits could separate NMDARs from downstream signaling pathways and change the function, localization and turnover of NMDARs (Steigerwald et al. 2000; Kohr et al. 2003).
Calpain (EC3.4.22.52) is a neutral, calcium-dependent protease activated by calcium entry through NMDARs. This enzyme proteolyzes many intracellular neuronal substrates (Vanderklish et al. 1995; Johnson and Guttmann 1997). In vitro and in heterologous systems, calpain cleaves NR2A and NR2B in their C-terminal region and could be involved in the turnover of NMDARs (Guttmann et al. 2001, 2002). Following NMDAR activation in hippocampal cultures, calpain selectively cleaves NR2B (but not NR2A) in a manner in which the major fragment remains on the cell surface and is potentially active (Simpkins et al. 2003). This suggests that calpain-mediated cleavage of NR2B may serve a modulatory role rather than being entirely degradative.
The effect of calpain on NMDAR biochemistry and function differs among neuronal systems. In relatively mature [17 days in vitro (DIV17) or older] hippocampal neurons NR2A is associated with PSD-95 (Li et al. 1998; Sans et al. 2000), an association that hinders calpain-mediated cleavage (Dong et al. 2004). In other studies using less mature cortical cultures and acutely dissociated neurons, calpain cleaves both NR2A and NR2B and appears to decrease the number of functional NMDARs (Wu et al. 2005). In the present study, to provide a context for the biochemical and functional variability in the effect of calpain on NMDARs, we have extended our studies to both cortical and hippocampal cultures during development and examined the susceptibility of NR2A and NR2B to calpain-mediated cleavage.
Materials and methods
Materials
Glutamate, glycine and trypsin were purchsed from Sigma-Aldrich (St Louis, MO, USA); minimal essential medium, penicillin/streptomycin, glutamine and horse serum were from Invitrogen (Carlsbad, CA, USA). Fetal bovine serum was from Hyclone Laboratories (Logan, UT, USA); dizocilpine (MK801), ifenprodil and Ro25-6981 were from Research Biochemicals International (Natick, MA, USA). Z-Val-Phe-CHO (MDL 28170; calpain inhibitor III; CalI3), Ac-Val-Ala-Asp-CHO (caspase inhibitor II; CasI2), and Z-Phe-Gly-NHO-Bz (cathepsin inhibitor I; CatI1) were purchased from Calbiochem (San Diego, CA, USA). An N-terminal antibody to amino acids 25–130 of NR2C (A-6475) was from Molecular Probes (Eugene, OR, USA). This antibody recognizes NR2A and NR2B (both 170–175 kDa) as well as NR2C (135–140 kDa) (Guttmann et al. 2001). An anti-NR2B antibody (made to the N-terminal 251 amino acids) was from Zymed (San Francisco, CA, USA). An anti-NR2A antibody (made to the last 200 amino acids of the C-terminus) was from Upstate Biotechnology (Lake Placid, NY, USA). AB38, which recognizes calpain-cleaved spectrin, was produced as described previously (Roberts-Lewis et al. 1994). An anti-NR1 monoclonal antibody was from BD Transduction Laboratories (Lexington, KY, USA). An anti-actin antibody was from Sigma-Aldrich. An anti-calpain I antibody was from Chemicon (Temecula, CA, USA). Conantokin G (ConG) was a generous gift from Dr Frank Castellino and Dr Mary Prorok (University of Notre Dame).
Preparation of primary neuronal cultures
Primary rat cortical and hippocampal neurons were derived from embryonic day 17 Sprague–Dawley rat (Charles River Laboratories Inc., Wilmington, MA, USA) embryos as described previously using an Institutional Animal Care and Use Committee approved protocol (Estus et al. 1997). Cortical and hippocampal tissue were dissected, minced and trypsinized (0.027%, 37°C, 7% CO2 for 20 min), and then washed with 1X Hanks’ balanced salt solution. Cells were plated in Neurobasal medium supplemented with B27 and grown on poly-d-lysine coated wells at a density of 6 × 105 viable cells per 35-mm culture dish. Cultures were maintained at 37°C with 5% CO2. Non-neuronal cell growth was inhibited with cytosine arabinoside at 7–10 DIV. All individual experiments were performed in triplicate (using samples from three culture dishes) for each condition. All experiments are shown as replications (n) of 3–10 individual experiments.
Drug treatments
For studies of cleavage by calpain, cells were rinsed twice with 1X phosphate-buffered saline and pre-incubated for 30 min in Hanks’ balanced salt solution with either inhibitors or vehicle. Glutamate and glycine were then added (100 μm each for routine experiments) for 30 min. MK801 was dissolved in water and used at a final concentration of 100 μm (Guttmann et al. 2001; Simpkins et al. 2003). CalI3, CasI2 and CatI1 were dissolved in dimethyl sulfoxide and used at 10 μm, 3 μm and 10 μm, respectively. The final dimethyl sulfoxide concentration was 0.1% or less. Ifenprodil (10 μm) and Ro25–6981 (10 μm) were dissolved in water at concentrations inhibiting >95% of NR1/2B receptors (Lynch et al. 2001). ConG (6 μm) was dissolved in water (Waxman and Lynch 2005). Cells were immediately scraped into 1X Laemmli stop buffer without bromophenol blue, EGTA or dithiothreitol (Guttmann et al. 2001). Samples were boiled for 5 min and protein concentrations were determined using the bicinchoninic assay (Pierce Chemical Co., Rockford, IL, USA). Bromophenol blue and dithiothreitol were then added, and the samples were then stored at −20°C until they were used.
Western blotting
Total protein (30 μg) was loaded on an 8% polyacrylamide gel. Following sodium dodecyl sulfate (SDS) gel electrophoresis, proteins were transferred to nitrocellulose, blocked with 3% dry milk, and incubated with primary antibody. Blots were then incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies and developed with enhanced chemiluminescence (Pierce Chemical Co.). Each blot was scanned using an Epson (Long Beach, CA) scanner in conjunction with Adobe Photoshop (San Jose, CA) (Guttmann et al. 2001). Data was quantitated using imaging densitometry. Briefly, blots were scanned and imported into Adobe Photoshop. The NIH Image program http://rsb.info.nih.gov/nih-image/ was used to identify an integrated signal from areas of interest. The gel subroutine was not used. Exposures were used that kept the maximum and minimum signals within the relatively linear range of the film response. However, data are presented as mean optical density units, recognizing the potential nonlinearity of the autoradiographic signal.
Biotinylation of surface NMDA receptors
Labeling of surface receptors was performed as described previously (Simpkins et al. 2003).
Electrophysiology
Whole-cell voltage clamp techniques were used to record cultured neurons. NMDA-elicited currents were recorded at room temperature (25°C) in tetrodotoxin-added (500 nm) HEPES solution. Neurons were voltage-clamped at −50 mV using an intrapipette solution containing (in mm) Trizma phosphate (dibasic) 100, Trizma base 28, EGTA 11, MgCl2 2, CaCl2 0.5, Mg-ATP 4, pH 7.35 and 290 mOsm/kg H2O. NMDAR-mediated currents were recorded in Mg2+-free solution. Recording signals were amplified using an Axopatch-1D amplifier (Axon Instruments/Molecular Devices Corporation Sunnyvale, CA), filtered at 5 kHz, and then saved using pclamp 8.01 software (Axon Instruments Inc., Foster City, CA, USA) for off-line analysis. NMDA application (2 s in duration) was accomplished using a computer-triggered step-perfusion device (Warner Instruments, Hamden, CT). Current density (defined as current amplitude divided by cell capacitance) and current kinetics (fitted with a single exponential component function; Levenberg–Marquardt non-linear least-squares algorithm) were calculated using Clampfit software (pclamp 8.01; Axon Instruments Inc.).
Data analysis
All data were analyzed using Instat software (GraphPad Software, San Diego, CA). Statistical significance of differences between two groups was evaluated using either the unpaired Student’s t-test or the Mann–Whitney Rank Sum test for groups with unequal variance. One-way anova and Tukey’s test for post-hoc comparison was used for comparisons of multiple groups. Significance was set at p < 0.05 and data were expressed as means ± SEM. For most experiments, in order to assess NMDAR-activated calpain activity, samples were compared with results from samples in the same experiment in which NMDARs were blocked with MK801. The variability of these triplicate samples within an experiment was routinely less than 10%. When comparisons were made with the MK801-treated samples, experimental conditions were compared using a one sample t-test with the null hypothesis being no change in levels of cleavage of the sample being assessed.
Results
Developmental changes in NMDAR-generated calpain activity
We first investigated the pharmacology of calpain activation by NMDAR stimulation in cortical cultures (as we previously had in hippocampal cultures; Simpkins et al. 2003), and its developmental course. Cortical neuronal cultures at DIV14 were incubated in glutamate and glycine and assessed for levels of a calpain-generated spectrin breakdown product (SBP) (Roberts-Lewis et al. 1994). Levels of the SBP markedly increased after 30 min of treatment with agonist. This increase was inhibited by the inclusion of either the NMDAR antagonist MK801 (100 μm) (Fig. 1a) or CalI3 (Fig. 1b). Inclusion of either CasI2 or CatI1 had no effect (Fig. 1b). These results confirmed that NMDAR activation leads to calpain activation in cortical neurons. Over the course of development, the level of the NMDAR-mediated calpain activation changed. In immature cortical cultures (DIV7) there was no significant change in the levels of SBP following NMDAR activation (Fig. 1c; 17% increase from 0 min). As neurons matured, NMDAR activation resulted in a 110% increase in SBP in DIV14 cortical cultures and a 156% increase in DIV21 cortical cultures compared with untreated (0 min) cultures. Compared with cortical neurons, hippocampal neurons developed NMDAR-activated calpain activity at earlier times in vitro, as agonist treatment significantly elevated the SBP in DIV7 hippocampal cultures (63% increase from 0 min). Calpain activity reached a plateau at DIV14 (244% increase from 0 min). No further increase in calpain activity was observed at DIV21 hippocampal cultures (245% increase from 0 min).
Fig. 1.

Cortical cultures at 14 days in vitro (DIV14) were treated with 100 μm glutamate and 100 μm glycine for 30 min and then probed for the calpain-produced spectrin breakdown product (SBP) with AB38. (a) A representative western blot and bar graph demonstrate elevated levels of SBP after stimulation of NMDA receptors (NMDARs) and inhibition by 100 μm dizocilpine (MK801) (p = 0.0017 for 0 min vs. 30 min; p = 0.0025 for MK801 vs. 30 min; n = 13). (b) The production of the SBP was also inhibited by calpain inhibitor III (CalI), but neither by caspase inhibitor II (CasI) nor cathepsin inhibitor I (CatI) (p = 0.001 by ANOVA; p = 0.0057 for CalI3 vs. 30 min; p > 0.05 for both CasI and CatI; n = 8). (c) The level of calpain activation (as measured by SBP levels) after NMDAR stimulation increased progressively in both cortical cultures and hippocampal cultures with development; no calpain activation was found in cortical neurons at DIV7 but was found in hippocampal neurons. Error bars indicate SEM. Data for (a) and (b) are shown as the percentage of the 30-min agonist treatment, whereas the data in (c) are shown as the increase of the 30-min time point over the 0-min time point.
To understand whether the developmental increase of NMDAR-stimulated calpain activity was caused either by the developmental increase of NMDARs or by increases in calpain, we measured the expression level of NMDARs and calpain I (the most abundant catalytic subunit in neurons) at different ages of cultured neurons. No change was found in the expression level of calpain I with development (Figs 2a–c). NR1, NR2A and NR2B all increased with development in cortical neurons (Figs 2a and b), matching previous results reported by other groups (Li et al. 1998). In hippocampal neurons, NR1 and NR2A increased with development, whereas in these high-density cultures, NR2B declined at later stages. Thus, NMDAR-controlled calpain activity developmentally increased in cultured neurons, which cannot be attributed to the increased expression of calpain I, but is instead attributed to developmental changes in the levels of NMDARs and synaptic development.
Fig. 2.
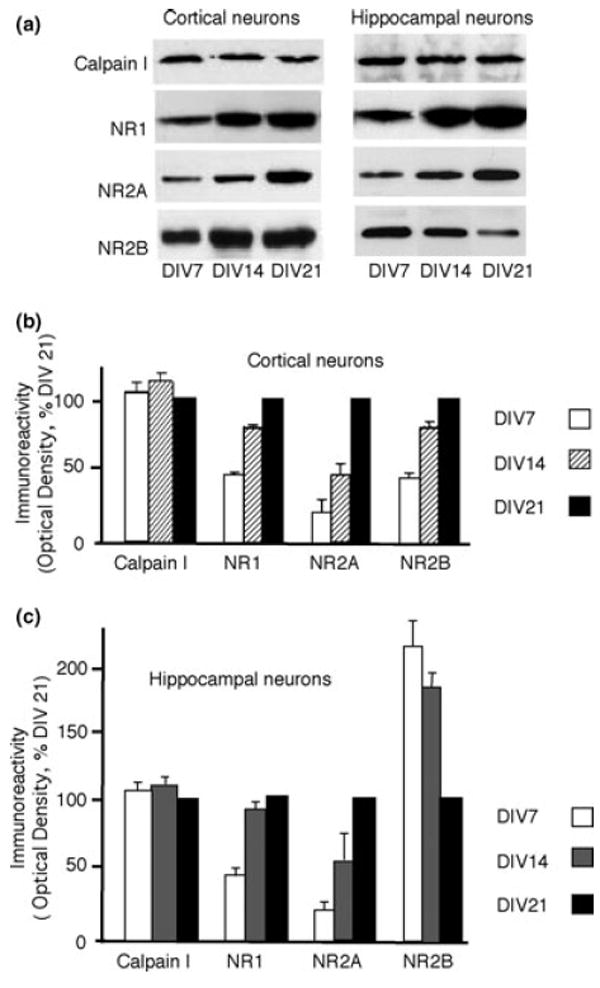
Cultured cortical and hippocampal neurons at different ages were collected and analyzed by western blots. A representative western blot (a) and bar graph (b) demonstrate increasing levels of NR1, NR2A and NR2B with age in cortical neurons. No change was found in the level of calpain I with age. In contrast, in hippocampal cultures, levels of NR1 and NR2A increased with development, but levels of NR2B decreased (a and c). No change was found in the levels of calpain I. Error bars indicate SEM, and all levels were normalized to 21 days in vitro (DIV21).
To identify the subtypes of NMDARs involved in calpain activation at different developmental stages we used NMDAR subtype-selective antagonists. Ifenprodil, 10 μm, an antagonist of NR1/2B receptors, inhibited calpain activation by 37% compared with the agonist application alone (Fig. 3a) in DIV14 cortical cultures. Similar results were seen with another NR1/2B receptor antagonist, Ro25-6981 (10 μm) (33% decrease compared with agonist application alone; Fig. 3a). ConG (6 μm), an antagonist of NR1/2B and NR1/2A/2B receptors (Klein et al. 2001), decreased calpain activation by 56% compared with agonist application alone (Fig. 3a). The extent of inhibition was substantially more than that caused by either ifenprodil or Ro25–6981, suggesting that both NR1/2B and NR1/2A/2B receptors contribute to calpain activation in DIV14 cortical cultures. When cortical neurons became more mature (DIV21), ifenprodil, Ro25–6981 and ConG still significantly inhibited calpain activity (23% decrease for ifenprodil; 21% decrease for Ro25–6981; 37% decrease for ConG; Fig. 3a). These results demonstrate that NR1/2B and NR1/2A/2B receptors are both necessary for the complete activation of calpain in immature and mature cortical neuronal cultures.
Fig. 3.

(a)Cortical and (b) hippocampal cultures at different ages were either treated with NR1/2B receptor antagonists (either 10 μm ifenprodil or 10 μm Ro25-6981) or with an antagonist of NR1/2B and NR1/2A/2B receptors (6 μm conantokin G, ConG) for 30 min, followed by glutamate/glycine exposure over a 30-min time period. Although ifenprodil, Ro25–6981 and ConG all significantly decreased the production of spectrin breakdown product (SBP) (compared with 30-min vehicle control) in cortical cultures (14 and 21 days in vitro; DIV14 and DIV21), with a greater role in less mature cortical cultures (DIV14), the effect of ConG was greater at all ages. This suggests a role for both NR1/2B and NR1/2A/2B receptors in activating calpain in both immature and mature cortical cultures. In contrast, treatment with 10 μm ifenprodil, 10 μm Ro25-6981 or 6 μm ConG significantly decreased the production of SBP in immature hippocampal cultures (≤ DIV14) (b), but only ConG significantly decreased the production of SBP in more mature hippocampal cultures. This suggests a role for both NR1/2B and NR1/2A/2B receptors in activating calpain in immature hippocampal cultures, but an absence of a necessity for NR1/2B activation in more mature cultures. Error bars indicate SEM; *p < 0.02, **p < 0.05, ***p < 0.01, ****p < 0.005, +p > 0.05. Data are shown as the percentage of the 30-min agonist treatment.
We also sought to identify the subtypes of NMDARs required for calpain activation in hippocampal neurons. Ifenprodil, Ro25–6981 and ConG inhibited calpain activity similarly in DIV7 neurons, suggesting that NR1/2B receptors mediate calpain activation in this population (50% decrease for ifenprodil; 47% decrease for Ro25–6981; 57% decrease for ConG; Fig. 3b). By DIV14, the inhibition of calpain activation by ifenprodil and Ro25-6981 decreased to 29% and 26%, respectively (Fig. 3b), whereas 6 μm ConG decreased spectrin degradation by 45% (Fig. 3b), suggesting that some NR1/2A/2B receptors rather than just NR1/2B receptors mediate calpain activation. In DIV21 hippocampal cultures, ifenprodil and Ro25-6981 had no effect on calpain activation, but ConG still significantly inhibited the calpain activation (38% decrease compared with the control group; Fig. 3b). These results demonstrated a developmental change in the NMDAR subtype associated with calpain activation in hippocampal neuronal cultures, with NR1/2B receptors participating in calpain activation in the least mature neurons (DIV7), and with NR1/2 A/2B and potentially NR1/2A receptors serving this function in more mature neurons.
Developmental changes in NR2B degradation by calpain in neuronal cultures
As NR2B is readily cleaved by calpain following NMDAR activation in hippocampal cultures (Simpkins et al. 2003; Dong et al. 2004), we sought to compare such results directly with NR2B cleavage in cortical cultures. Cortical neurons cultured at DIV14 were incubated with glutamate and glycine and analyzed by western blots using an N-terminal antibody that recognizes NR2B (Dong et al. 2004). Agonist treatment decreased the full-length NR2B immunoreactivity, compared with the 0-min control, with a simultaneous increase in levels of a 115-kDa breakdown product (Figs 4a, b and d). We previously used an N-terminal antibody that recognizes both NR2A and NR2B subunits in hippocampal cultures (A-6475); this antibody recognized the 115-kDa breakdown product of NR2B, further confirming that the 115-kDa breakdown product resulted from the cleavage of NR2B by calpain (Simpkins et al. 2003). Inclusion of 100 μm MK801 prevented the decrease in full-length NR2B (Figs 4a and b) and the increase in the levels of the 115-kDa breakdown product (Figs 4a and d). Neither NR1 nor actin was degraded (data not shown). We confirmed directly that calpain was the protease involved in NR2B subunit degradation in cultured cortical neurons (DIV14). CalI3, but neither inhibitors of caspase nor cathepsin, prevented the loss of full-length NR2B immunoreactivity (Fig. 4f). The increase in the levels of the NR2B breakdown product was also inhibited by CalI3, but not by the caspase and cathepsin inhibitors (Fig. 4g). These data show that calpain degrades NR2B in cultured cortical neurons.
Fig. 4.

Cortical and hippocampal cultures at either 14 days in vitro (DIV14) or DIV21 were exposed to NMDAR agonist stimulation over a 0–30-min time period and subjected to western blot analysis with an antibody to the N-terminal of NR2B (170 kDa). In cortical neurons a representative western blot (a) and bar graph (b and d) demonstrate the decrease in full-length NR2B (170 kDa) [40% decrease compared with 30 min with agonist and dizocilpine (MK801); 55% decrease from 0 min, p =0.0185 for 30 vs. 0 min; n = 5] as well as the simultaneous increase in levels of a 115-kDa breakdown product (80% increase over agonists plus MK801; 131% increase from 0 min, p = 0.0097 for 30 vs. 0 min; n = 5). The decrease of full-length NR2B and the increase in breakdown product were inhibited by the inclusion of 100 μm MK801 in the treatment conditions (p = 0.0014 for full-length NR2B and p = 0.0035 for NR2B breakdown product; MK801 vs. 30 min; n = 7). As neurons matured (DIV21), there was a reduction in both the decrease in full-length NR2B (15% decrease compared with agonist and MK801; 26% decrease from 0 min, p = 0.0049 for 30 min vs. 0 min; n = 5) and the increase in the levels of NR2B breakdown product following agonist stimulation (50% increase over agonists and MK801; 87% increase from 0 min, p = 0.0133 for 30 min vs. 0 min; n = 5) in cortical cultures. NR2B immunoreactivity was quantitated as a percentage of the MK801 control condition. In (f) and (g), cortical cultures at DIV14 were treated with 100 μm glutamate and glycine for 30 min in the presence of calpain inhibitor III (CalI3), caspase inhibitor II (CasI2), or cathepsin inhibitor I (CatI1). The inhibition of calpain by CalI3 (p < 0.0001 by anova; p = 0.0085 for full-length NR2B; p = 0.0051 by anova; p =0.001 for NR2B breakdown product; CalI3 vs. 30 min; n = 5) but neither caspase nor cathepsin (p > 0.05; n = 5 for both) prevented the decrease of full-length NR2B and the appearance of the NR2B breakdown product. In hippocampal cultures of different ages a representative western blot (a) and bar graph (c and e) demonstrate the unchanged cleavage pattern of NR2B with development. Agonist exposure resulted in a 35% decrease in full-length NR2B (compared with agonist and MK801 control; p = 0.001 for MK801 vs. 30 min; 43% decrease from 0 min; p = 0.0088 for 30 min vs. 0 min; n = 6) (c) and a 70% increase of the NR2B breakdown product (compared with agonist and MK801 control; p = 0.0078 for MK801 vs. 30 min; 81% increase from 0 min; p = 0.0021 for 30 min vs. 0 min; n = 6) (e) in less mature cultures (DIV14). As cultures matured (DIV21), the level of NR2B cleavage was unchanged for full-length NR2B (40% decrease from 0 min, p = 0.0165 for 30 min vs. 0 min; n = 5) (c) and NR2B breakdown products (116 kDa) (90% increase from 0 min; p = 0.0055 for 30 min vs. 0 min; n = 5) (e). Error bars indicate SEM; *p < 0.02 vs. 30 min; **p < 0.01 vs. 30 min; ***p < 0.005 vs. MK801 (b–e) or 30 min (f–g); ****p < 0.01 vs. MK801 (b–e) or 30 min (f–g); *****p > 0.05.
We then examined the degradation pattern of NR2B by calpain in more mature cortical cultures (DIV21). Compared with immature cortical neurons, the level of NR2B degradation by calpain following agonist treatment was reduced from that noted in younger cultures, with only a 15% decrease in full-length NR2B immunoreactivity (n = 5) (Figs 4a and b) and a 45% increase in the levels of NR2B breakdown product (Figs 4a and d) observed at DIV21. These results demonstrate that the calpain-mediated degradation of NR2B decreases with development in cortical neurons.
In contrast, no change in NR2B degradation by calpain was evident between immature and mature hippocampal neurons. Agonist exposure decreased full-length NR2B immunoreactivity by 35% (Figs 4a and c) and increased the levels of the NR2B breakdown product by 70% (from the 0-min control) (Figs 4a and e) in immature hippocampal cultures (DIV14). Inclusion of 100 μm MK801 prevented the decrease in full-length NR2B (Figs 4a and c) and the increase in levels of the 115-kDa breakdown product (Figs 4a and e). As neurons matured (DIV21), the level of NR2B cleavage was unchanged as assessed by the loss of full-length NR2B (a 40% decrease from 0 min) (Figs 4a and c) and the appearance of the calpain-generated NR2B breakdown product (115 kDa; 99% increase from 0 min) (Figs 4a and e).
Developmental decrease in NR2A degradation by calpain in hippocampal cultures
In previous studies of hippocampal cultures at either DIV18 or older, NR2A is resistant to calpain-mediated cleavage whereas NR2B is readily cleaved (Simpkins et al. 2003). To test whether NR2A is a substrate for calpain in younger hippocampal cultures, we treated DIV14 hippocampal cultures with glutamate and glycine and analyzed NR2A levels by western blots using a C-terminal antibody to NR2A. In DIV14 hippocampal cultures, a 30% decrease in full-length NR2A immunoreactivity was observed after agonist treatment (compared with the MK801-treated control), with only a 5% decrease in DIV21 hippocampal cultures (Figs 5a and b). We have previously noted similar results in cortical cultures (Dong et al. 2004), suggesting a developmental decrease in NR2A degradation by calpain in multiple cell types.
Fig. 5.

Representative bar graph and western blot (a and b) demonstrating the developmental decrease in NR2A cleavage by calpain in hippocampal cultures. NR2A was cleaved by calpain [30% decrease compared with agonist and dizocilpine (MK801) control; 50% decrease from 0 min, n = 5] at 14 days in vitro (DIV14) and became unable to be cleaved at DIV21 of development. The inclusion of MK801 prevented the loss of NR2A during agonist treatment at DIV14. Error bars indicate SEM; *p = 0.0089 vs. 0 min. Data are shown as the percentage of the 30-min agonist with MK801 treatment.
Physiological changes in calpain-mediated effects on NMDAR
We then sought to establish the physiological significance of calpain-mediated events in the regulation of NMDAR function. Using hippocampal cultures at DIV10 (in which NR2B is the major NR2 expressed) and DIV21 (in which only NR2B is cleaved, but both NR2A and NR2B are expressed), we assessed whether the activation of calpain altered the electrophysiological properties of NMDAR-generated currents. Exposure of cultures to agonists in a manner identical to the biochemical experiments described above decreased NMDAR current density in DIV21 cultures, but not in DIV10 cultures (Figs 6 and 7a). This change was not altered by calpain inhibition during the 30-min agonist-exposure period, suggesting that calpain-mediated cleavage of NMDARs in hippocampal neurons does not alter the overall density of NMDAR-mediated currents in this cell type at both levels of maturity. This is consistent with the possibility that although calpain cleaves NR2B subunits, these receptors remain on the cell surface in DIV18 hippocampal neurons (Simpkins et al. 2003) and remain potentially active. We confirmed this observation in the present work in DIV10 cultures by observing that although surface levels of full-length NR2B decrease after the activation of calpain, the truncated form of NR2B appeared almost exclusively in the cell-surface fraction, creating similar levels of potentially active cell-surface receptors (Fig. 8).
Fig. 6.

Differential NMDA receptor (NMDAR) functional changes between 10 days in vitro (DIV10) and DIV21 of hippocampal cultures after glutamate/glycine treatment. Hippocampal neuronal cultures were treated with buffer (control), buffer with agonists (Glu) 100 μm glutamate and 100 μm glycine, or buffer with agonists and 10 μm calpain inhibitor III (CalI3) (Glu + Cal-I). Cells were then prepared for electrophysiology to measure NMDA-generated responses. Representative traces are shown for both DIV10 and DIV21 neurons in response to a 2-s administration of 100 μm NMDA and 10 μm glycine.
Fig. 7.
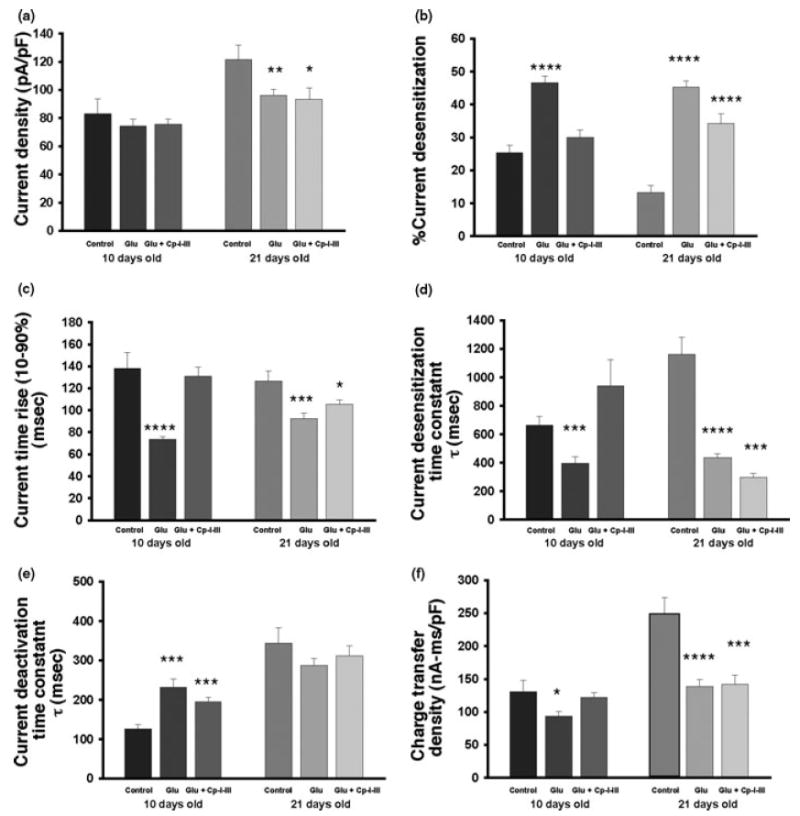
Functional changes in NMDA receptors (NMDAR) after calpain activation. Hippocampal neuronal cultures were treated with buffer (control), buffer with agonists (Glu) 100 μm glutamate and 100 μm glycine, or agonists and 10 μm CalI3 (Glu + Cp I–III). Cells were then prepared for electrophysiology to measure NMDA-evoked currents. (a) Patch-clamp techniques were used to record the NMDAR-gated current density (the current amplitude was normalized to cell capacitance) in hippocampal neurons after 10 days in vitro (DIV10) and DIV21. After exposure to glutamate/glycine for 30 min, NMDAR-evoked current density (compared with buffer-treated) was reduced in DIV21 but not in DIV10 cultures. (b)–(e) Possible changes in the kinetics of NMDAR-evoked currents were investigated under conditions in which calpain was activated. After treatment with glutamate/glycine for 30 min, the percentage of current desensitization was increased in both DIV10 and DIV21 neurons within the 2-s period of NMDA application. This alteration was prevented by the blockade of calpain activation in DIV10 hippocampal neurons, but only partially reversed in more mature neurons (b). A similar pattern of changes was observed in the rise time of the NMDA-evoked currents (c). In addition, agonist exposure increased the decay time constant of NMDAR-evoked current desensitization in both DIV10 and DIV21 cultures. However, calpain inhibitor treatment prevented the effects in DIV10 but not in DIV21 neurons (d). Furthermore, agonist treatment for 30 min prolonged the NMDAR-gated current deactivation time constant in DIV10 but not in DIV21 neurons. This effect was not prevented by the blockade of calpain (e). (f) All the kinetic parameters mentioned above contributed to the overall charge transfer during the current induced by NMDA application, which was calculated by integrating the area of the response (total charge transfer density). This measurement can be used as a measure of overall NMDAR function. After glutamate/glycine treatment, NMDAR-mediated function was reduced in both DIV10 and DIV21 neurons. The inhibition of calpain prevented the changes in DIV10, but not in DIV21, hippocampal neurons. Overall, calpain modulates NMDAR current kinetics in DIV10 neurons, but only had smaller physiological effects in more mature hippocampal neurons. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Fig. 8.

Surface localization of cleaved NR2B subunits in hippocampal cultures at 10 days in vitro (DIV10). Hippocampal neuronal cultures were treated with agonists (100 μm glutamate and 100 μm glycine). Cell membranes were then labeled with N-Hydroxy-succinimidyl (NHS)-biotin, and separated into cell surface and intracellular fractions. NR2B was detected by western blotting (panel a, top blot). Although the levels of optical density representing full-length NR2B declined in both the intracellular (p = 0.0021) and cell surface (p = 0.011) fractions, the appearance of the 115-kDa breakdown product (arrow) was confined to the cell surface fraction (p = 0.018) (western blot in a, bar graphs in b and c). Cell surface actin levels were unchanged (panel a, bottom blot).
We also examined the kinetic changes of NMDAR-gated current under the conditions in which calpain was activated. After glutamate/glycine treatment for 30 min, NMDAR current desensitization increased in both DIV10 and DIV21 neurons; this change was prevented by the blockade of calpain activation during the 30-min agonist exposure in DIV10 hippocampal neurons, but only partially reversed in more mature neurons (Fig. 7b). Similar observations were noted for the rise time of the NMDA-evoked currents (Fig. 7c). In addition, glutamate/glycine treatment increased the time constant of the NMDAR current desensitization in both DIV10 and DIV21 hippocampal neurons. However, the effects observed in DIV10 (but not in DIV21) neurons were reversed by the blockade of calpain (Fig. 7d). Furthermore, agonist treatment prolonged the current deactivation time constant in DIV10 but not in DIV21 neurons; this effect was partially, but not totally, prevented by the blockade of calpain (Fig. 7e). In order to integrate the parameters mentioned above, the total charge transferred during an NMDAR current response was calculated and used to evaluate the alteration of NMDAR function after glutamate/glycine treatment. This measure demonstrated that NMDAR-mediated function was reduced after agonist treatment in both DIV10 and DIV21 neurons, but the blockade of calpain prevented the changes in DIV10 but not in DIV21 hippocampal neurons (Fig. 7f). Overall the data show that calpain modulates the kinetic features of NMDAR-mediated currents in DIV10 neurons, but has less effect in more mature neurons.
Discussion
In the present study, we have defined the relationship between calpain and NR2 subunits over neuronal maturation in culture. NMDAR-activated calpain activity increases over neuronal development, and the activation of calpain becomes more associated with NR1/2A/2B receptors over time after its initial association with NR1/2B receptors. Although NR2A and NR2B are both substrates for calpain in immature neurons, the cleavage of the subtypes varies as a course of neuronal development. The degradation of both NR2A and NR2B by calpain developmentally decreases in cultured cortical neurons, with NR2A showing more resistance to calpain-mediated cleavage. In hippocampal cultures, the degradation of NR2A by calpain decreases but the degradation of NR2B remains unchanged with development. These data suggest that the susceptibility of NMDAR degradation by calpain is developmentally modulated and cell-type selective.
Our data resolve some conflicts in the susceptibility of NR2 subunits to cleavage by calpain. NR2A has been reported as a substrate for calpain in neuronal culture, although our previous results did not identify a calpain-dependent degradation of NR2A (Simpkins et al. 2003; Wu et al. 2005). The present data suggest that the susceptibility of NR2A to cleavage varies with neuronal type and maturity. Anatomically, cortical cultures are derived from a more diverse group of precursor cells, suggesting that there is likely to be more variability between neurons cultured from cortical preparations (Dichter 1978). As synaptic development in both hippocampal and cortical neuronal cultures is used as a model of synaptogenesis in vivo, the exact role of calpain and its cleavage of NMDAR may vary between different neurons and at developmental stages in vivo.
The increased interaction of NR2A and NR2B with other postsynaptic proteins, such as PSD-95, may explain the decreased susceptibility of NR2 subunits to calpain with development. As neurons mature, NR2 subunits and PSD-95 expression concurrently increase, as does the interaction of NR2 subunits with PSD-95 (Sans et al. 2000). PSD-95 protects both NR2A and NR2B from calpain-mediated cleavage (Dong et al. 2004), and the disruption of this interaction produces NR2A with increased susceptibility to cleavage by calpain in mature cortical cultures (Dong et al. 2004). In parallel, synaptic development increases the ability of the C-terminal domain of NR2 subunits to be phosphorylated by PKC, protein tyrosine kinases and CaMKII (Omkumar et al. 1996; Grant et al. 2001; Liao et al. 2001; Yang and Leonard 2001). Phosphorylation of NR2 subunits by either PKC or Src protects them from cleavage by calpain in vitro and enhances NR2 binding to PSD-95 in vitro (Bi et al. 1998; Rong et al. 2001).
The developmental change in the subtype of NMDARs associated with calpain activation may also reflect the maturation of postsynaptic systems. In immature and mature cortical cultures, both NR1/2B and NR1/2A/2B receptor antagonists inhibit calpain activation, suggesting the involvement of these receptors in calpain activation. These results functionally support the existence of NR1/2A/2B receptors in cortical neurons and are consistent with the reports from other groups (Li et al. 1998; Gardoni et al. 2002; Zhou and Baudry 2006). In hippocampal cultures there was a greater developmental change in the subtype of NMDARs associated with calpain activation, with the inhibition of NR1/2B receptors blocking calpain activation in immature hippocampal cultures and the inhibition of NR1/2A/2B receptors being needed to decrease the calpain activation in mature hippocampal cultures. These results correspond with the developmental pattern of NR2 subunits in hippocampal neurons, where the expression of NR2B decreases and the expression of NR2A increases with development (Dichter 1978; Sans et al. 2000). Although we did not assess changes in the levels of either calpastatin or calpain II (the catalytic subunit of m-calpain), the levels of calpain I did not change over the course of development, suggesting that changes in NMDAR-activated calpain mainly reflect changes in NMDAR subtypes, rather than changes in the subtype of calpain. We cannot, however, rule out changes in other calpain subunits such as calpain II contributing to the effects noted here. In addition, intracellular sources of calcium (calcium-activated calcium release) and calcium buffering proteins may have different developmental profiles. Our data have not systematically investigated how the changing levels of these processes in developing synapses might alter the relationship of calpain and NMDARs. This remains an area for future investigation.
Some features of cleavage of NR2 subunits are consistent between neuronal populations. Compared with the N-terminal breakdown product (115 kDa) of NR2B, no breakdown product was identified for NR2A in either culture at any age. This most likely reflects the C-terminal location of the epitope of the NR2A-specific antibody, and is consistent with previous results from heterologous systems showing that calpain cleaves NR2A at amino acid 1279 and 1330 of the C-terminal region in vitro and in situ (Guttmann et al. 2001; Dong et al. 2004). However, using NR2 antibodies that detect all NR2 subunits, we previously did not identify any new cleavage products of NR2 subunits beyond the 115-kDa products derived from NR2B (Simpkins et al. 2003). This suggests that N-terminal breakdown products resulting from the calpain-mediated cleavage of NR2A are further degraded by either other proteases or rapid internalization. Endocytosis of NR2A is most notable in the early stages of development, consistent with our failure to identify truncated NR2A fragments (Roche et al. 2001; Lavezzari et al. 2004).
Interestingly, while the subtypes of NMDARs that are involved in the activation of calpain and their susceptibility to calpain changed with development, the electrophysiological properties of NMDAR also responded differently to calpain activation over time. The changes in electrophysiological response in association with calpain activation parallel the changes in the composition and localization of NMDAR with neuronal maturation in cultured hippocampal neurons. In DIV10 cultures, NR1/2B receptors are the major receptors cleaved by calpain, as NR2A levels are low at this point. Over time NR1/2B receptors become mainly extra-synaptic receptors, and NR2B becomes more associated with NR2A in synapses to form NR1/2A/2B receptors (Sans et al. 2000; Li et al. 1998; Li et al. 2003). Correlating these data with our findings in electrophysiological experiments suggests that calpain is a major modulator of NMDAR properties in developing synapses where its targets are likely to be the NR1/2B receptors. The cleaved NR2B subunit remains on the cell surface and is likely to remain active as the peak current density is unaffected by calpain activation in immature hippocampal cultures. In more mature cultures, calpain did not alter specific electrophysiological properties of the overall population NMDARs, suggesting that the effects of calpain on NMDAR, and specifically NR2B subunit function, are masked either by co-association with NR2A or by association with other elements of the PSD. Alternatively, the relative lack of calpain inhibition on electrophysiological properties may reflect the decrease in the abundance of the NR2B subunit in our cultures (Fig. 2). The calpain-independent effects on physiological properties of NMDARs may arise from several identified activity dependent modifications of either the NMDAR or the associated modulatory proteins including activity dependent internalization, use-dependent phosphatase activation or calcium-dependent proteosomal degradation of PSD-95 (Vissel et al. 2001; Colledge et al. 2003; Nong et al. 2003). In addition, it is possible that the changes in the ability of calpain to modulate NMDAR currents reflect effects of calpain on other aspects of NMDAR function, as many modulatory proteins of NMDARs are also substrates for calpain (Wu and Lynch 2006). Experiments in heterologous systems may be needed to further define these possibilities.
Taken together, our data suggest that calpain and NMDARs interact in a manner that is developmentally and cell-type selective. As calpain activity is ongoing during synaptic transmission, the results suggest that the role of calpain in the modulation of NMDARs at different synapses is likely to be complex (Vanderklish et al. 1995). Further examination of different physiological paradigms may more fully define the different roles of calpain-mediated modulation of NMDARs in different synaptic situations.
Acknowledgments
This work was supported by NIH grants NS45986, NS38572 and NS32403. We would like to thank Margaret Maronski for her preparation of neuronal cultures.
Abbreviations used
- CalI3
calpain inhibitor III
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- CasI2
caspase inhibitor II
- CatI1
cathepsin inhibitor I
- ConG
conantokin G
- DIV
days in vitro
- MK801
dizocilpine
- NMDAR
NMDA receptor
- PDZ
PSD-95, discs large, Z01
- PKC
protein kinase C
- PSD-95
post-synaptic density protein 95
- SBP
spectrin breakdown product
References
- Bi R, Bi X, Baudry M. Phosphorylation regulates calpain-mediated truncation of glutamate ionotropic receptors. Brain Res. 1998;797:154–158. doi: 10.1016/s0006-8993(98)00433-8. [DOI] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichter M. Rat cortical neurons in cell culture: culture methods, cell morphology, electrophysiology, and synapse formation. Brain Res. 1978;149:279–293. doi: 10.1016/0006-8993(78)90476-6. [DOI] [PubMed] [Google Scholar]
- Dong YN, Waxman EA, Lynch DR. Interactions of postsynaptic density-95 and the NMDA receptor 2 subunit control calpain-mediated cleavage of the NMDA receptor. J Neurosci. 2004;24:11035–11045. doi: 10.1523/JNEUROSCI.3722-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estus S, Tucker HM, van Rooyen C, Wright S, Brigham EF, Wogulis M, Rydel RE. Aggregated amyloid-beta protein induces cortical neuronal apoptosis and concomitant ‘apoptotic’ pattern of gene induction. J Neurosci. 1997;17:7736–7745. doi: 10.1523/JNEUROSCI.17-20-07736.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni F, Bellone C, Viviani B, Marinovich M, Meli E, Pellegrini-Giampietro DE, Cattabeni F, Di Luca M. Lack of PSD-95 drives hippocampal neuronal cell death through activation of an alpha CaMKII transduction pathway. Eur J Neurosci. 2002;16:777–786. doi: 10.1046/j.1460-9568.2002.02141.x. [DOI] [PubMed] [Google Scholar]
- Grant ER, Guttmann RP, Seifert KM, Lynch DR. A region of the rat N-methyl-D-aspartate receptor 2A subunit that is sufficient for potentiation by phorbol esters. Neurosci Lett. 2001;310:9–12. doi: 10.1016/s0304-3940(01)02085-7. [DOI] [PubMed] [Google Scholar]
- Guttmann RP, Baker DL, Seifert KM, Cohen AS, Coulter DA, Lynch DR. Specific proteolysis of the NR2 subunit at multiple sites by calpain. J Neurochem. 2001;78:1083–1093. doi: 10.1046/j.1471-4159.2001.00493.x. [DOI] [PubMed] [Google Scholar]
- Guttmann RP, Sokol S, Baker DL, Simpkins KL, Dong Y, Lynch DR. Proteolysis of the N-methyl-d-aspartate receptor by calpain in situ. J Pharmacol Exp Ther. 2002;302:1023–1030. doi: 10.1124/jpet.102.036962. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Johnson GV, Guttmann RP. Calpains: intact and active? Bioessays. 1997;19:1011–1018. doi: 10.1002/bies.950191111. [DOI] [PubMed] [Google Scholar]
- Klein RC, Prorok M, Galdzicki Z, Castellino FJ. The amino acid residue at sequence position 5 in the conantokin peptides partially governs subunit-selective antagonism of recombinant N-methyl-D-aspartate receptors. J Biol Chem. 2001;276:26860–26867. doi: 10.1074/jbc.M102428200. [DOI] [PubMed] [Google Scholar]
- Kohr G, Jensen V, Koester HJ, Mihaljevic AL, Utvik JK, Kvello A, Ottersen OP, Seeburg PH, Sprengel R, Hvalby O. Intracellular domains of NMDA receptor subtypes are determinants for long-term potentiation induction. J Neurosci. 2003;23:10791–10799. doi: 10.1523/JNEUROSCI.23-34-10791.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Lavezzari G, McCallum J, Dewey CM, Roche KW. Subunit-specific regulation of NMDA receptor endocytosis. J Neurosci. 2004;24:6383–6391. doi: 10.1523/JNEUROSCI.1890-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Otsu Y, Murphy TH, Raymond LA. Developmental decrease in NMDA receptor desensitization associated with shift to synapse and interaction with postsynaptic density-95. J Neurosci. 2003;23:11244–11254. doi: 10.1523/JNEUROSCI.23-35-11244.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JH, Wang YH, Wolfe BB, Krueger KE, Corsi L, Stocca G, Vicini S. Developmental changes in localization of NMDA receptor subunits in primary cultures of cortical neurons. Eur J Neurosci. 1998;10:1704–1715. doi: 10.1046/j.1460-9568.1998.00169.x. [DOI] [PubMed] [Google Scholar]
- Liao GY, Wagner DA, Hsu MH, Leonard JP. Evidence for direct protein kinase-C mediated modulation of N-methyl-D-aspartate receptor current. Mol Pharmacol. 2001;59:960–964. doi: 10.1124/mol.59.5.960. [DOI] [PubMed] [Google Scholar]
- Lynch DR, Shim SS, Seifert KM, Kurapathi S, Mutel V, Gallagher MJ, Guttmann RP. Pharmacological characterization of interactions of RO 25–6981 with the NR2B (epsilon2) subunit. Eur J Pharmacol. 2001;416:185–195. doi: 10.1016/s0014-2999(01)00868-8. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Kim E, Sheng M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci. 1996;16:2157–2163. doi: 10.1523/JNEUROSCI.16-07-02157.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nong Y, Huang YQ, Ju W, Kalia LV, Ahmadian G, Wang YT, Salter MW. Glycine binding primes NMDA receptor internalization. Nature. 2003;422:302–307. doi: 10.1038/nature01497. [DOI] [PubMed] [Google Scholar]
- Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT, Kennedy MB. Identification of a phosphorylation site for calcium/calmodulin dependent protein kinase II in the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 1996;271:31670–31678. doi: 10.1074/jbc.271.49.31670. [DOI] [PubMed] [Google Scholar]
- Roberts-Lewis JM, Savage MJ, Marcy VR, Pinsker LR, Siman R. Immunolocalization of calpain I-mediated spectrin degradation to vulnerable neurons in the ischemic gerbil brain. J Neurosci. 1994;14:3934–3944. doi: 10.1523/JNEUROSCI.14-06-03934.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- Rong Y, Lu X, Bernard A, Khrestchatisky M, Baudry M. Tyrosine phosphorylation of ionotropic glutamate receptors by Fyn or Src differentially modulates their susceptibility to calpain and enhances their binding to spectrin and PSD-95. J Neurochem. 2001;79:382–390. doi: 10.1046/j.1471-4159.2001.00565.x. [DOI] [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos J, Hell JW, 2nd, Wenthold RJ. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20:1260–1271. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DB, Michailidis I, Mu Y, Logothetis D, Ehlers MD. Endocytosis and degradative sorting of NMDA receptors by conserved membrane-proximal signals. J Neurosci. 2004;24:7096–7109. doi: 10.1523/JNEUROSCI.0780-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins KL, Guttmann RP, Dong Y, Chen Z, Sokol S, Neumar RW, Lynch DR. Selective activation induced cleavage of the NR2B subunit by calpain. J Neurosci. 2003;23:11322–11331. doi: 10.1523/JNEUROSCI.23-36-11322.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steigerwald F, Schulz TW, Schenker LT, Kennedy MB, Seeburg PH, Kohr G. C-Terminal truncation of NR2A subunits impairs synaptic but not extrasynaptic localization of NMDA receptors. J Neurosci. 2000;20:4573–4581. doi: 10.1523/JNEUROSCI.20-12-04573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderklish P, Saido TC, Gall C, Arai A, Lynch G. Proteolysis of spectrin by calpain accompanies theta-burst stimulation in cultured hippocampal slices. Brain Res Mol Brain Res. 1995;32:25–35. doi: 10.1016/0169-328x(95)00057-y. [DOI] [PubMed] [Google Scholar]
- Vissel B, Krupp JJ, Heinemann SF, Westbrook GL. A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat Neurosci. 2001;4:587–596. doi: 10.1038/88404. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Lynch DR. N-methyl-D-aspartate receptor subtype mediated bidirectional control of p38 mitogen-activated protein kinase. J Biol Chem. 2005;280:29322–29333. doi: 10.1074/jbc.M502080200. [DOI] [PubMed] [Google Scholar]
- Wu HY, Lynch DR. Calpain and synaptic function. Mol Neurobiol. 2006;33:215–336. doi: 10.1385/MN:33:3:215. [DOI] [PubMed] [Google Scholar]
- Wu HY, Yuen EY, Lu YF, Matsushita M, Matsui H, Yan Z, Tomizawa K. Regulation of N-methyl-D-aspartate receptors by calpain in cortical neurons. J Biol Chem. 2005;280:21588–21593. doi: 10.1074/jbc.M501603200. [DOI] [PubMed] [Google Scholar]
- Yang M, Leonard JP. Identification of mouse NMDA receptor subunit NR2A C-terminal tyrosine sites phosphorylated by co-expression with v-Src. J Neurochem. 2001;77:580–588. doi: 10.1046/j.1471-4159.2001.00255.x. [DOI] [PubMed] [Google Scholar]
- Zhou M, Baudry M. Developmental changes in NMDA neurotoxicity reflect developmental changes in subunit composition of NMDA receptors. J Neurosci. 2006;26:2956–2963. doi: 10.1523/JNEUROSCI.4299-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]