

Abstract
The association of receptor tyrosine kinases is a key step in the initiation of growth factor-mediated signaling. Although the ligand-induced dimerization of inactive, monomeric receptors was the central dogma of receptor tyrosine kinase activation for decades, the existence of larger oligomers is now accepted. Both homoassociations and heteroassociations are of extreme importance in the epidermal growth factor (EGF) receptor family, leading to diverse and robust signaling. We present a statistically reliable, flow-cytometric homo-fluorescence resonance energy transfer method for the quantitative characterization of large-scale receptor clusters. We assumed that a fraction of a certain protein species is monomeric, whereas the rest are present in homoclusters of N-mers. We measured fluorescence anisotropy as a function of the saturation of fluorescent antibody binding, and fitted the model to the anisotropy data yielding the fraction of monomers and the cluster size. We found that ErbB2 formed larger homoclusters than ErbB1. Stimulation with EGF and heregulin led to a decrease in ErbB2 homocluster size, whereas ErbB1 homoclusters became larger after EGF stimulation. The activation level of ErbB2 was inversely proportional to its homocluster size. We conclude that homoclusters of ErbB1 and ErbB2 behave in a fundamentally different way. Whereas huge ErbB2 clusters serve as a reservoir of inactive coreceptors and dissociate upon stimulation, small ErbB1 homoclusters form higher-order oligomers after ligand binding.
INTRODUCTION
The dimerization of membrane proteins is believed to play a fundamental role in the activation of receptor tyrosine kinases (RTKs). The groundbreaking discovery of the importance of receptor oligomerization in the 1970s (1) was supported by the crystal structure of the epidermal growth factor (EGF) receptor (2). The ligand-induced dimerization of RTKs became textbook dogma, and because of the limited potential of classical molecular biological methods to detect clusters composed of more than two receptors, larger oligomers were neglected. Promiscuous homoassociation and heteroassociation are prominent features of the EGF receptor family (also known as the ErbB family) of RTKs consisting of four members, i.e., ErbB1 (EGF receptor) and ErbB2–4 (3–5). The prototypical RTK, ErbB1, is believed to undergo ligand-induced homodimerization and activation (2). The ligandless coreceptor, ErbB2, is activated either by heterodimerization with other ErbB proteins or by overexpression-driven constitutive homoassociation (6,7). The kinase-deficient ErbB3 receptor can only transmit signals by heterodimerization, primarily with ErbB2 (8). The simplistic view of inactive monomers and active dimers was complicated by discovering preformed clusters of inactive ErbB receptors (9–12). A further twist in the story involved the repeated identification of large receptor clusters in both quiescent and stimulated cells. Fluorescence correlation microscopy revealed clusters containing 10 to 30 EGF receptors (13). Webb et al. also reported on 4 to 5 EGF receptors/cluster, based on the number of photobleaching steps in single-molecule fluorescence time traces (14). A combined fluorescence resonance energy transfer (FRET)-correlation microscopic study confirmed the existence of ErbB1 tetramers and large homoclusters containing 15 to 30 receptors (15). The EGF receptor was shown to undergo activation-induced dimer-tetramer transition (16), whereas ErbB3 was shown to behave differently. It forms homoclusters whose upper size limit is a dodecamer. Heregulin, the ligand of ErbB3, reverses the homoassociation of ErbB3 (17,18). The ErbB3 molecules that break free from the homoclusters most probably heteroassociate with ErbB2, because kinase-dead ErbB3 can only transmit signals by heteroassociating with other ErbB proteins. Scanning near-field optical microscopy revealed clusters containing ∼1000 ErbB2 molecules in microdomains with a diameter of ∼500 nm, which increased upon stimulation (19). These reports present convincing evidence for the existence and importance of large-scale protein associations.
Apart from biochemical methods, which are usually semiquantitative and are not appropriate for the investigation of living cells, several approaches are available for the quantitative characterization of large-scale receptor associations in intact cells. Fluorescence correlation spectroscopy (13,15), single-molecule imaging (14,20), and measurement of the rotational diffusion rate (21) were applied for this purpose. Probably the most widely used quantitative technique for the analysis of protein-clustering is FRET, in which energy is transferred in a nonradiative fashion from an excited donor molecule to a nearby acceptor. The rate of FRET varies inversely with the sixth power of the donor-acceptor separation, turning it into a useful distance-measuring tool (22). When the process takes place between a donor and a spectroscopically different acceptor, it is termed hetero-FRET. Although a careful analysis of hetero-FRET signals can reveal the stoichiometry of association, the physical interaction in hetero-FRET takes place between a single pair of donor and acceptor, limiting the potential of hetero-FRET measurements to analyze large protein clusters (16,23,24). In homo-FRET, the interaction takes place between a donor and a spectroscopically identical “acceptor” which can serve as the donor in the next homo-FRET step. This sequential aspect of the interaction is reflected in the term energy migration FRET (emFRET), used as a synonym for homo-FRET (10). Because energy is distributed among the interacting molecules, homo-FRET can be used for the quantitative analysis of large protein clusters. Because the only manifestation of homo-FRET is decreased fluorescence anisotropy, Runnels and Scarlata derived a formula for the steady-state anisotropy of clusters of N molecules (25). Steady-state anisotropy is ambiguously related to homo-FRET, because factors other than energy transfer can also change its value. Therefore, anisotropy is either measured in the time domain (23), or its dependence on fluorophore density is analyzed to enumerate the number of interacting fluorophores. The latter strategy was applied by measuring the increase in anisotropy as a function of fluorophore photobleaching (10,26,27). Alternatively, the loss of homo-FRET occurring at red-edge excitation was used to isolate the contribution of homo-FRET to the measured anisotropy in microscopy (28).
Microscopy suffers from low statistical accuracy because of the limited number of cells measured. Flow cytometry offers superior statistics, without the capability to quantitate photobleaching. Here we present a flow-cytometric method for the measurement of homo-FRET, based on varying the amount of saturation of antibody binding to cell-surface receptors. We derived a formula for the dependence of fluorescence anisotropy on the fraction of monomers and on the number of proteins in a single cluster, and fitted the model to anisotropy data measured on cells labeled with anti-ErbB1 or anti-ErbB2 antibodies. The data presented here provide significant new insights into the large-scale association properties of ErbB receptors.
MATERIALS AND METHODS
Cell lines
The human breast-cancer cell line SKBR-3 and the human epithelial carcinoma cell line A431 were obtained from the American Type Culture Collection (Rockville, MD), and were grown according to their specifications. For flow cytometry, cells were harvested by trypsinization.
Antibodies and growth factors
The anti-ErbB1 monoclonal antibody EGFR455, which does not block the binding of EGF, was produced by hybridoma 455 obtained from the European Collection of Cell Cultures (Wiltshire, UK). Mab528 against ErbB1, competing with EGF binding, was prepared from the supernatant of the HB-8509 hybridoma cell line obtained from American Type Culture Collection. Antibodies were purified from hybridoma supernatants, using protein A affinity chromatography. The anti-ErbB2 antibody, trastuzumab (Herceptin), was purchased from Roche (Budapest, Hungary). The conjugation of primary antibodies with AlexaFluor (Molecular Probes, Eugene, OR) dyes was performed according to the manufacturer's specifications. The OP15 monoclonal antibody against an intracellular epitope of ErbB2 was obtained from Calbiochem-Merck Biosciences (Schwalbach, Germany). Ab-18, recognizing the activated, tyrosine-phosphorylated form of ErbB2, was purchased from Lab Vision (Fremont, CA). Pertuzumab (Omnitarg) was a gift from Genentech (South San Francisco, CA). EGF and heregulin-β1 were obtained from R&D Systems (Minneapolis, MN). The Alexa488-conjugated and Alexa546-conjugated F(ab′)2 fragments of goat anti-mouse immunoglobulin G (IgG) and the Cy3-labeled F(ab′)2 fragment of goat anti-human IgG were from Molecular Probes/Invitrogen (Eugene, OR).
Labeling of cells with antibodies
Freshly harvested cells were washed twice in ice-cold phosphate-buffered saline (PBS; pH 7.4), and the cell pellet containing ∼1 million cells was suspended in 100 μL of Hanks' buffer with 1 mg/mL bovine serum albumin (BSA). For homo-FRET measurements, ErbB1 or ErbB2 receptors were labeled with different ratios of unlabeled and Alexa488-labeled antibodies for 30 min on ice in the dark. The total concentration of unlabeled and labeled antibodies was 50 μg/mL, and the fraction of labeled antibody varied from 0% to 100%, with increments of 10%. Labeled cells were washed twice with cold PBS and fixed with 1% formaldehyde in PBS. All samples were kept at 4°C before performing measurements, to prevent the modulation of antibody-induced receptor clustering.
To determine the limiting anisotropy of Alexa488-conjugated antibodies, ErbB1 or ErbB2 receptors were incubated with 50 μg/mL donor-conjugated antibody (Alexa488-trastuzumab, Alexa488-Mab528, or Alexa488-EGFR455) for 30 min on ice in the dark. After washing, the samples were divided into five parts, which were labeled by different concentrations (0.5 to 9 μg/mL) of acceptor-conjugated secondary antibody (Cy3-labeled F(ab′)2 fragments of goat anti-human IgG in the case of Alexa488-trastuzumab, or Alexa546- conjugated F(ab′)2 fragments of goat anti-mouse IgG in the case of Alexa488-Mab528 and Alexa488-EGFR455) for 30 min on ice in the dark. Labeled cells were washed and fixed with 1% formaldehyde in PBS.
For the measurement of ErbB2 phosphorylation, quiescent and stimulated cells were fixed with 3.7% formaldehyde in PBS for 30 min on ice, and washed twice with cold Tris-buffer (100 mM Tris + 100 mM NaCl; pH 7.4) to quench unreacted formaldehyde. Each sample was divided into two parts, which were separately labeled with OP15 (10 μg/mL) and Ab-18 (10 μg/mL) against ErbB2 and phosphorylated ErbB2 (p-ErbB2), respectively. Labeling was performed in PBS-BSA-TX permeabilization buffer (0.1% (v/v) Triton X-100, 1 mg/mL BSA in PBS, pH 7.4) for 30 min on ice. After washing with cold PBS-BSA-TX, cells were labeled with Alexa488-conjugated F(ab′)2 fragments of goat anti-mouse IgG for 30 min on ice in the dark. Labeled cells were washed twice, resuspended in 1% formaldehyde, and analyzed by flow cytometry.
Treatment of cells with EGF, heregulin, and pertuzumab
Cells were starved for 24 h before the experiment in medium containing 0.1% fetal calf serum (FCS). Freshly harvested cells were washed twice in cold PBS, and resuspended in 100 μL Hanks' buffer supplemented with 1 mg/mL BSA. Pretreatment with 20 μg/mL pertuzumab was performed for 15 min at 37°C. Control and pertuzumab-pretreated cells were stimulated with 100 ng/mL EGF or 100 ng/mL heregulin for 15 min at 37°C, and subsequently used for homo-FRET measurements, as described above.
Flow cytometry and data analysis
Cell-by-cell fluorescence anisotropy measurements were performed in the L-format arrangement on a FACSVantage SE instrument with a DiVa option (Becton Dickinson, Franklin Lakes, NJ). Alexa488 was excited by a vertically polarized 488-nm beam, which was produced by letting the 488-nm line of an Ar ion laser pass through a half-wave plate retarder (model 481AS, Newport, Irvine, CA). The angle of the half-wave plate corresponding to vertical polarization was adjusted by light-scatter measurements. Emitted photons passed through a 530 ± 15 nm band-pass filter, followed by a broadband polarizing beam-splitter cube (model 10FC16PB.3, Newport) separating the vertically and horizontally polarized components. Two polarized fluorescence intensities, designated by Ivv and Ivh, were detected, where the first and second indices refer to the polarization directions of the exciting laser light and of fluorescence, respectively. For the determination of the G-factor, cells were excited by horizontally polarized light produced by rotating the half-wave plate by 45° relative to its vertical position. The two polarized fluorescence intensities recorded with horizontal excitation are labeled by Ihv and Ihh. After subtracting the intensity of unlabeled cells from each polarized intensity, the G-factor compensating for the different sensitivities of the detection system to vertically and horizontally polarized light, the total fluorescence intensity (Itot), and the fluorescence anisotropy (r) were calculated as follows:
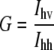 |
(1) |
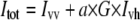 |
(2) |
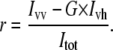 |
(3) |
The influence of high-aperture fluorescence collection on the determined anisotropy was compensated according to Jovin (constant a in Eq. 2) (29). Fifty thousand cells were recorded from each sample containing a certain fraction of labeled and unlabeled antibodies. Anisotropy was calculated on a cell-by-cell basis according to Eq. 3. Cells were gated on the forward scatter-side scatter dot plot and on the total fluorescence intensity-anisotropy dot plot, using ReFlex software (available at http://www.freewebs.com/cytoflex) (30); ∼10,000 to 20,000 cells were gated out, leaving 30,000 to 40,000 cells for calculating the average fluorescence intensity and anisotropy values. Anisotropy was plotted against saturation, and the graph from a single experiment was fitted by Eq. 7 (see Therory, below). Fitting and Monte Carlo simulation were performed using Mathematica (Wolfram Research, Champaign, IL). Cluster size and monomer percentage values reported in the text and in Table 1 were calculated by averaging the values obtained from the fitting of three separate experiments. The sequence of data collection and processing is summarized in Fig. 1.
TABLE 1.
Cluster sizes and monomer percentages of ErbB1 and ErbB2
| ErbB1
|
ErbB2
|
||||
|---|---|---|---|---|---|
| Cluster size | Monomer % | Cluster size | Monomer % | ||
| Starved | Control | 4 ± 1 | 88 ± 4 | 111 ± 12 | 60 ± 5 |
| EGF | 11 ± 2 | 71 ± 3 | 71 ± 6 | 61 ± 4 | |
| Heregulin | – | – | 32 ± 4 | 59 ± 5 | |
| Pertuzumab | – | – | 84 ± 7 | 70 ± 4 | |
| Pertuzumab + heregulin | – | – | 73 ± 6 | 58 ± 3 | |
| Nonstarved | Control | 7 ± 1 | 75 ± 4 | 9 ± 2 | 53 ± 3 |
| Pertuzumab | – | – | 30 ± 4 | 55 ± 3 | |
ErbB1 and ErbB2 were labeled by Alexa488-EGFR455 and Alexa488-trastuzumab, respectively. The cluster size and monomer percentage are shown on cells pretreated and stimulated in different ways. Means (± SE) were calculated from the fitted cluster size and monomer percentage values obtained from three independent experiments. Cluster size is the average number of ErbB1 or ErbB2 proteins in the cluster. Monomer percentage is the fraction of ErbB1 or ErbB2 present outside homoclusters (either as monomers or heterodimers with other proteins).
FIGURE 1.

Data collection, gating, and processing strategy to calculate cluster size and monomer percentage from flow-cytometric anisotropy measurements; 50,000 cells, labeled by a mixture of labeled and unlabeled antibodies, were measured. Cells were first gated on the forward scatter-side scatter (FSC-SSC) dot plot (A), followed by gating on the anisotropy versus total fluorescence intensity dot plot (B). Data were recorded with 18-bit resolution, but were rescaled for the dot plots. The thick dashed line in B indicates the fluorescence intensity of unlabeled cells. Gates are represented by shaded polygons. The position of the gates was identical for every sample with a different concentration of the labeled antibody measured on the same day. The lower threshold of total fluorescence intensity was visually adjusted to gate out dim cells whose anisotropy had a large variation, and the threshold was kept constant for all samples. The average anisotropy was calculated, yielding a total intensity-anisotropy data point on the graph showing the dependence of anisotropy on the concentration of labeled antibody (saturation, in C). Equation 7 was fitted to the anisotropy-saturation plot, yielding the estimated cluster size and monomer percentage. The results of three independent experiments were averaged. Anisotropy was assumed to be normally distributed, and its error was estimated. The mean and error values were used to generate 250 random data sets, similar to the one shown in C. Every data set was fitted, yielding 250 pairs of cluster sizes and monomer percentages from which frequency histograms were generated.
Hetero-FRET efficiency was determined using a FACSVantage SE flow cytometer with a DiVa option, with dual-laser excitation at 488 and 532 nm. The donor (Alexa488) and FRET signals were excited at 488 nm and detected at 530 ± 15 nm and 585 ± 21 nm, respectively. Direct acceptor emission (Alexa546 or Cy3) was detected at 585 ± 21 nm upon 532-nm excitation with a diode-pumped, solid-state laser. The FRET efficiency was determined on a cell-by-cell basis, using ReFlex software as described previously (30,31).
Theory
Homo-FRET efficiency changes as a function of fluorophore density. Previously, photobleaching was used to create a range of fluorophore densities (10,27), but the same approach is not applicable in flow cytometry. Instead, we labeled cell-surface proteins with a mixture of fluorophore-labeled and unlabeled antibodies against the same epitope, so that the total antibody concentration saturated the available binding sites. In this way, the fraction of epitopes to which fluorophore-tagged antibody bound (saturation designated by s in the equations) was approximately equal to the fraction of labeled antibody in the mixture. The amount of saturation (s) was estimated by dividing the fluorescence intensities of each sample with that of the brightest one. We assumed that a fraction (mon) of the proteins is monomeric, and that the rest (1-mon) are present in homoclusters of N-mers. Essentially, any protein unable to undergo homo-FRET is regarded as monomeric in the calculations, e.g., a labeled protein in a heterodimeric complex, because it does not interact with another protein of the same kind. The probability that k out of N proteins is labeled by a fluorescent antibody in the N-mer when the fraction of labeled proteins is s is described by the binomial distribution:
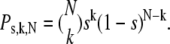 |
(4) |
The anisotropy of such a homocluster according to Runnels and Scarlata (25) is
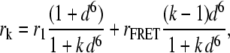 |
(5) |
where r1 and rFRET are the anisotropies of an initially excited molecule and that of a molecule excited by homo-FRET, respectively, and d is the distance between the fluorophores in the clusters normalized to R0. The determination of r1 will be explained in Results. We assumed a complete depolarization of fluorescence emitted by every fluorophore except for the initially excited one, and therefore, rFRET = 0 (25). Although membrane proteins are ordered in the plane of the membrane and their rotation is slow on the timescale of fluorescence, bending of the antibody around the epitope, segmental motions of the Fab arms around the Fc portion, and rapid rotation of the antibody-bound fluorophore make the system practically randomly oriented. If the assumption of rFRET = 0 is incorrect, it leads to an underestimation of cluster size or an overestimation of monomer percentage. The resultant anisotropy of the mixture of monomers and N-mers is the intensity-weighted average of the individual anisotropies (32):
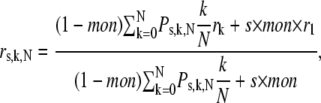 |
(6) |
where k/N is a correction factor normalizing the sum of the fluorescence intensity of N-mers and monomers to unity. Multiplication by k is necessary to weigh the anisotropy values according to the fluorescence intensity. Without dividing by N, the fluorescence intensity of clusters would be overweighted by a factor of N. By combining Eqs. 4–6, the total anisotropy is given by the following formula:
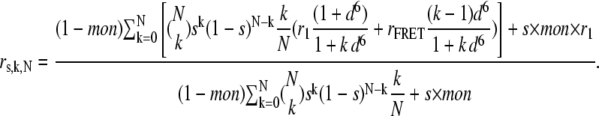 |
(7) |
Equation 7 predicts that the total anisotropy is constant, if only monomers are present, and that the anisotropy is a linear function of saturation if homoclusters are dimers. The higher the number of proteins in a homocluster, the steeper the anisotropy declines at low values of saturation (Fig. 2). Equation 7 was fitted to the measured anisotropy-saturation plot, with mon and N as the fitting parameters (Fig. 3). We then calculated the standard deviation of the anisotropy values and generated 250 random data sets, with mean and standard deviation values equal to the measured data. These random data sets were also fitted by Eq. 7, yielding 250 values for cluster size (N) and monomer percentage (mon). The distribution of both parameters was calculated, and the graphs for ErbB1 and ErbB2 under different conditions are given in Figs. 5 and 6.
FIGURE 2.

Theoretical curves show the dependence of fluorescence anisotropy on the saturation of binding sites as a function of cluster size and monomer percentage. The anisotropy of fluorescence antibodies as a function of the saturation of binding sites was determined according to Eq. 7, assuming a limiting anisotropy of 0.27. The calculation was performed for several cluster sizes, with no monomers present (numbers indicate number of proteins in the homocluster: •, 1; ○, 2; ▪, 3; □, 4; ▴, 5; Δ, 10; ♦, 20), and for the case when half of the proteins are monomers (⋄, 10; ★, 20).
FIGURE 3.

Fitting of the anisotropy model to ErbB1 and ErbB2 anisotropy data. Starved (•) and heregulin-stimulated (▪) SKRB-3 cells were labeled with a mixture of unlabeled and Alexa488-labeled trastuzumab. Starved (◊) and EGF-stimulated (▿) A431 cells were labeled with a mixture of unlabeled and Alexa488-labeled EGFR455. The anisotropy of samples was determined and plotted as a function of saturation of binding sites. The lines show the results of fitting according to Eq. 7.
FIGURE 5.

Monomer percentage and cluster size of ErbB2 in quiescent and stimulated SKBR-3 cells (A and B) SKBR-3 cells starved in the presence of 0.1% FCS for 24 h (solid line) were stimulated with heregulin (dashed line) or EGF (dotted line). The distributions of cluster size (A) and monomer percentage (B) were determined by Monte Carlo simulation, based on the anisotropy data. (C and D) Starved SKBR-3 cells were pretreated with pertuzumab (solid line) and stimulated with heregulin (dotted line). (E, F) SKBR-3 cells cultured under normal serum conditions (10% FCS, solid line) were treated with pertuzumab (dotted line) and labeled with a mixture of Alexa488-labeled and unlabeled trastuzumab IgG. SKBR-3 cells cultured in the presence of 10% FCS were also labeled with a mixture of Alexa488-labeled and unlabeled trastuzumab Fab (dashed line).
FIGURE 6.

Monomer percentage and cluster size of ErbB1 in quiescent and stimulated A431 cells. A431 cells were either cultured under normal serum conditions (10% FCS, dotted line) or starved in the presence of 0.1% FCS for 24 h (solid line). Starved cells were stimulated with EGF (dashed line). The distributions of cluster size (A) and monomer percentage (B) were determined by Monte Carlo simulation.
RESULTS
Determination of the limiting anisotropy of antibody-bound fluorophores
We approximated the anisotropy of the initially excited fluorophore (r1) by the limiting anisotropy of the antibody-bound fluorophore (r0). The emission anisotropy of a fluorophore (r) changes as a function of its rotational correlation time and fluorescence lifetime, according to the Perrin equation:
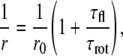 |
(8) |
where τfl is the fluorescence lifetime and τrot is the apparent rotational correlation time of the fluorophore, taking into account all depolarizing processes taking place during the excited-state lifetime (rotational diffusion of the antibody, and segmental motion of parts of the antibody and that of the fluorophore). The apparent limiting anisotropy of the fluorophore is given as r0. Because hetero-FRET decreases the fluorescence lifetime, a Perrin plot can be constructed by varying the hetero-FRET efficiency (33):
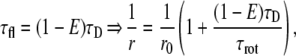 |
(9) |
where τD is the lifetime of the fluorophore (“donor”) in the absence of FRET. The y-intercept of a line fitted on the 1/r versus 1 − E plot is the reciprocal of the limiting anisotropy. To create a range of donor fluorescence lifetimes, i.e., FRET efficiencies, cells labeled with saturating concentrations of the donor-tagged antibody were labeled by different concentrations of an acceptor-tagged secondary antibody. Both the hetero-FRET efficiencies and the fluorescence anisotropies of the donor were measured and displayed on a Perrin plot (Fig. 4), and the limiting anisotropies were calculated for each of the IgGs and Fab fragments (r0,A488-trastuzumab = 0.268; r0,A488-trastuzumab Fab = 0.289; r0,A488-Mab528 = 0.226; and r0,A488-Mab EGFR455 = 0.170).
FIGURE 4.

Determination of limiting anisotropy. Cells were labeled with saturating concentration of the primary antibody (Alexa488-tagged trastuzumab, •; EGFR455, ▪; or Mab528, ▿) followed by secondary labeling by Cy3-anti-human F(ab′)2 or Alexa546-anti-mouse F(ab′)2. The fluorescence anisotropy and the hetero-FRET efficiency were calculated for each sample, and the y-intercept of the line fitted on the Perrin plot yielded the reciprocal of the limiting anisotropy.
Calculation of the separation distance between two antibodies bound to a homocluster
The distance between two antibodies bound to homoclusters has to be known in units of R0. We first determined R0 for the Alexa488 homo-FRET case, according to the following formula in units of nm:
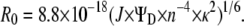 |
(10) |
The overlap integral (J) was calculated from spectra available from the Invitrogen web site, according to the following formula:
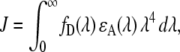 |
(11) |
where fD and ɛA are the normalized fluorescence spectrum and the molar absorption coefficient of Alexa488, respectively. The quantum yield (ΨD) of Alexa488 was previously reported to be 0.6 (34). We assumed a value of 1.4 and 2/3 for the index of refraction (n) and the orientation factor (κ2), respectively, yielding 4.8 nm for the R0 between two Alexa488 molecules undergoing homo-FRET. The hetero-FRET efficiency for ErbB2 homodimerization, measured by Cy3-trastuzumab and Cy5-trastuzumab, was found to be 20% (35). Using 5 nm as the R0 between Cy3 and Cy5 (35), the above FRET efficiency corresponds to an average separation distance of 6.29 nm, according to the formula:
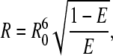 |
(12) |
where E is the FRET efficiency. Therefore, the average separation between two trastuzumab molecules (d in Eq. 7) in units of R0 is 6.29 nm/4.8 nm = 1.3 × R0. Using the same approach, the average distance between two EGFR455 antibodies and between two Mab528 antibodies was found to be 1.1 × Ro and 1.4 × Ro, respectively.
Stimulation of ErbB2 leads to a decrease in its cluster size
The homo-FRET model described above was used for evaluation of the homocluster formation of ErbB1 and ErbB2 in quiescent and stimulated cells. SKBR-3 cells expressing ∼8 × 105 ErbB2 proteins (36) were starved in low-serum medium (0.1% FCS) for 24 h, and the percentage of monomeric ErbB2 and the cluster size of homoassociated ErbB2 were determined; ∼60% of ErbB2 molecules were monomeric, but the rest formed huge clusters composed of ∼110 ErbB2 proteins (Fig. 5, A and B; Table 1). Stimulation of starved cells with EGF or heregulin did not change the percentage of monomers significantly, but led to a substantial decrease in the size of ErbB2 homoclusters. The effect of heregulin was more pronounced than that of EGF. Pretreatment of cells with pertuzumab led to a slight increase in the percentage of ErbB2 monomers and to a significant decrease in cluster size (Fig. 5, C and D; Table 1). Pertuzumab pretreatment practically abolished the effect of heregulin on ErbB2 cluster size, although the effect of heregulin on ErbB2 monomer percentage was unaffected. If cells were not starved before the experiment, the percentage of monomers was lower than in starved cells, and the size of ErbB2 homoclusters was even smaller than in heregulin-stimulated or EGF-stimulated cells. To examine the possible effect of labeling with bivalent antibodies on ErbB2 clustering, nonstarved SKBR3 cells (cultured in the presence of 10% FCS) were labeled with a mixture of Alexa488-labeled and unlabeled Fab fragments of trastuzumab. The observation that both the cluster size and monomer percentage were identical in cells labeled with trastuzumab IgG and Fab implies that labeling with bivalent IgG does not modify the clustering properties of ErbB2 (Fig. 5, E and F). Treatment of nonstarved cells with pertuzumab significantly increased the size of ErbB2 homoclusters, without an effect on monomer percentage (Fig. 5, E and F; Table 1). We concluded that ErbB2 is homoclustered to different degrees in quiescent and stimulated cells, and its cluster size changes as a function of its activation state.
Stimulation of ErbB1 induces an increase in its cluster size
Next, we compared the homoclustering properties of ErbB1 to those of ErbB2. The fluorescence anisotropy of Alexa488-EGFR455 antibody was evaluated in A431 cells starved in low-serum medium for 24 h. The A431 cell line expresses ∼2 million copies of ErbB1 per cell (37); ∼90% of ErbB1 was monomeric, and the remaining ∼10% formed homoclusters containing ∼4 ErbB1 molecules (Fig. 6; Table 1). Stimulation of cells with EGF reduced the monomer percentage of ErbB1 to ∼70%, and increased its homocluster size to ∼10 receptors/homocluster. In cells cultured under normal serum conditions (10% FCS), the homocluster size of ErbB1 was larger, and the monomer percentage was lower, than in starved cells. The experiments were repeated with A431 cells labeled with Mab528 against ErbB1. Because Mab528 sterically blocks the EGF binding site, the labeling of EGF-stimulated cells by Mab528 would have been unpredictable. Therefore, only cells cultured in the presence of 0.1% and 10% FCS were measured. Both the ErbB1 monomer percentages and the cluster sizes determined by Mab528 labeling were practically identical to those obtained by EGFR455 (cluster size, 4 and 10 in the presence of 0.1% and 10% FCS, respectively; monomer percentage, 90% and 78% in the presence of 0.1% and 10% FCS, respectively). We concluded that ErbB1 forms substantially smaller homoclusters than ErbB2, and that its cluster size increases in response to activation.
ErbB2 activation state and cluster size are inversely related to each other
Factors known to induce ErbB2 activation (EGF, heregulin, and serum) decreased its homocluster size (Fig. 5; Table 1). To analyze the correlation between ErbB2 cluster size and activation, the amount of phosphorylated and total ErbB2 were determined in parallel with its cluster size. We calculated the level of ErbB2 tyrosine phosphorylation by normalizing the intensity of the antibody against phosphorylated ErbB2 to the intensity characterizing the total amount of ErbB2. A plot of ErbB2 tyrosine phosphorylation against ErbB2 homocluster size shows an inverse correlation between the two parameters, both in starved and nonstarved SKBR-3 cells (Fig. 7 A).
FIGURE 7.

Relationship between the homoclustering of ErbB proteins and the tyrosine phosphorylation of ErbB2. (A) The normalized tyrosine phosphorylation level of ErbB2 in starved (•) and nonstarved (○) SKBR-3 cells was determined under different conditions, and plotted against the cluster size of ErbB2. (B) The clustering of ErbB1 (E1), ErbB2 (E2), and ErbB3 (E3) in quiescent (middle), EGF-stimulated (left), and heregulin (HRG)-stimulated (right) cells.
DISCUSSION
The potential of widely used approaches to analyze the composition of large protein aggregates is limited. Therefore, the unique capability of homo-FRET for measuring the size of homoclusters makes it a valuable tool in cell biological research (10,27,38). The fact that the relationship between anisotropy and homocluster size is ambiguous increases the complexity of the equations and necessitates the incorporation of simplifying assumptions into the model. Here, we present a flow-cytometric method for the quantitative analysis of protein homoclustering. The model relies on the correlation between fluorescence anisotropy and the local density of fluorophores, and in this sense, it is similar to the fluorescence microscopic approach of Yeow and Clayton (27). We assumed that a fraction of the investigated protein species is monomeric, whereas other proteins of the same species form homoclusters with a constant size. This is obviously not true, but given the low number of data points and the measurement error achievable by flow-cytometric anisotropy measurements, this simplification was required to make fitting of the model to measurement data possible. The theoretical curves derived from the model show that the shape of the anisotropy curves and the anisotropy values at low fluorophore density (low saturation) are substantially different between different cluster sizes (Fig. 2). Therefore, the distinct cluster sizes of ErbB1 and ErbB2, and their changes, were reliably determined.
Emitted fluorescence is split into horizontally and vertically polarized components in anisotropy measurements. In addition, the method described here involves the gradual increase in concentration of the unlabeled antibody, at the expense of the labeled one, leading to a decrease in fluorescence intensity. These circumstances set a relatively high fluorescence limit for the reliable detection of cluster size by our method. Based on extrapolation of the gradual increase of detection error at low fluorescence intensities, we estimate that the expression level of the investigated protein has to be ∼105/cell for accurate cluster-size determination. However, the shape of the anisotropy versus saturation curve strongly depends on the cluster size in the case of small clusters, and the curve is significantly different for large (N > 30) and small clusters. These observations allow us to conclude that a rough estimation of cluster size is possible even at lower protein expression levels. In addition, the accuracy of measurements at low fluorescence intensities can be increased by increasing the number of cells measured, which is easily achievable in flow cytometry. The fact that the total fluorescence intensity of a single cell is measured in flow cytometry, whereas it is distributed to several pixels in microscopy, increases the signal/noise ratio in flow cytometry compared with microscopy. This circumstance makes the measurement of dim samples more accurate in flow cytometry. However, one must not ignore the fact that subcellular resolution is not possible in flow-cytometric measurements.
Labeling of cells was usually performed using bivalent antibodies. To minimize the possibility that antibodies cross-link their target, labeling was performed on ice. Although antibodies are expected to bind monovalently in the saturation concentration range used throughout the study, we performed control experiments in which ErbB2 clusters in SKBR-3 cells labeled with trastuzumab IgG or Fab were compared. The curves obtained using the two labeling protocols showed remarkable overlap, indicating that labeling with bivalent IgG did not alter the clustering properties of membrane proteins under the labeling conditions used in the experiments.
The anisotropy of the initially excited fluorophore (r1) had to be determined for the model calculations. This anisotropy (r1) corresponds to the anisotropy of a single, isolated, fluorophore-tagged antibody, which is decreased by homo-FRET between antibodies bound to different proteins in the cluster. We assumed that the limiting anisotropy of the antibody-bound fluorophore determined by Perrin plots is a good approximation of r1 (Fig. 4). The data could be accurately fitted by a linear equation, indicating that the limiting anisotropies of the antibody-bound fluorophore could be reliably determined. The anisotropy of a single, isolated, fluorophore-tagged antibody is smaller than the limiting anisotropy of the fluorophore because of fast hindered rotations. These rotations are most likely missed by the Perrin plots, because they are expected to cause a curvature of the leftmost part of the graph, corresponding to very short lifetimes (FRET efficiencies close to 100%), which were not represented on the plots. Therefore, the determined r1 values are smaller than the canonical limiting anisotropy of the fluorophore. However, the anisotropy of a single fluorescent antibody (emitting fluorescence depolarized by these fast rotations), and not the canonical limiting anisotropy, is needed for Eq. 7; therefore, the fact that r1 most likely underestimates r0 is actually a benefit. We note that homo-FRET taking place between fluorophores bound to the same antibody can be neglected, because the number of fluorophore/antibody was ∼1. We conclude that the anisotropy of the initially excited fluorophore needed as an input parameter for Eq. 7 could be accurately measured.
We found in unstimulated cells that homoclusters of ErbB2 are larger than those of ErbB1, and a lower fraction of ErbB2 is monomeric than of ErbB1. These results imply that ErbB2 has a higher homoclustering tendency than ErbB1, in accordance with previous findings obtained by classical biochemical (7,39) and FRET methods (9,40). Two different anti-ErbB1 antibodies yielded identical results within experimental error, giving credence to our conclusion. Stimulation of A431 cells with EGF or serum induced an increase in ErbB1 homocluster size and a decrease in the percentage of monomers (Fig. 6). Although textbook dogma supported by crystallographic data suggests that ErbB1 undergoes a monomer-dimer transition upon ligand binding (2), several studies using FRET, correlation spectroscopic, and single molecule techniques reported higher-order ErbB1 oligomers in unstimulated cells, and a ligand-induced increase in ErbB1 cluster size (13–16). The cluster sizes of 4 and 11 that we found in quiescent and stimulated cells, respectively, are in agreement with what was reported previously.
Ligand-induced changes in ErbB1 cluster size always have to be interpreted in the context of the expression levels of all ErbB proteins. Because ErbB2 is the preferred heterodimerization partner of all other ErbB proteins (6), it competes with them for association partners. However, A431 cells express a huge excess of ErbB1 over other ErbBs (37) (also according to our unpublished observations). Therefore, the ligand-induced homodimerization and homoclustering of ErbB1 are not hindered by ErbB2.
In contrast to ErbB1, ErbB2 formed huge homoclusters containing ∼100 ErbB2 proteins in unstimulated cells, which became smaller upon stimulation with serum, EGF, or heregulin (Fig. 5).
Very large clusters of ErbB2 containing 50 to 80 receptors were observed by immunoelectron microscopy (41). Because serum, EGF, and heregulin stimulation leads to ErbB2 activation, the above results imply that activated ErbB2 forms smaller homoclusters. This assumption is corroborated by the negative correlation between the level of ErbB2 tyrosine phosphorylation and its cluster size (Fig. 7 A). Because ErbB2 is a ligandless coreceptor (6), its ligand-induced activation can only be achieved by heterodimerization with ErbB1 or ErbB3, whose ligands are EGF and heregulin, respectively, because ErbB4 is not expressed by SKBR-3 cells. The EGF-induced or heregulin-induced coclustering of ErbB2 with ErbB1 or ErbB3, respectively, were reported previously (41). We assume that ligand-binding to ErbB1 and ErbB3 recruits ErbB2 molecules from large ErbB2 homoclusters, leading to diminished ErbB2 homocluster size upon stimulation. The fact that neither EGF nor heregulin induced any change in the monomer percentage of ErbB2 needs explanation. The ErbB2 removed from large homoclusters can redistribute to smaller clusters in which several ErbB2 proteins heteroassociate with ErbB1 or ErbB3, i.e., ErbB2 will not become monomeric. Alternatively, if ErbB2 proteins expelled from large ErbB2 homoclusters form heterodimers with ErbB1 or ErbB3 (regarded as monomers by our method detecting only homoclusters), these heterocomplexes can be removed from the membrane by the low rate of endocytosis of ErbB2-containing heterodimers (42). If the rate of formation of ErbB2 heterodimers is comparable with their endocytosis rate, the fraction of ErbB2 monomers will remain constant.
Pertuzumab, an antibody sterically blocking the heterodimerization of ErbB2 (43), inhibited the heregulin-induced decrease in ErbB2 cluster size and the increase in tyrosine phosphorylation, and slightly decreased the effect of serum on tyrosine phosphorylation and cluster size of ErbB2 in SKBR-3 cells. These observations follow from the inability of ErbB2 to heterodimerize with ErbB1 or ErbB3. Consequently, ErbB2 remains in large homoclusters.
Pertuzumab slightly increased the tyrosine phosphorylation of ErbB2 in starved SKBR-3 cells, accompanied by a decrease in ErbB2 cluster size. The reciprocal change in tyrosine phosphorylation and cluster size of ErbB2 was expected, based on the negative correlation between them. However, the increased state of activation of ErbB2 was unexpected. Our previous unpublished results from hetero-FRET experiments showed that pertuzumab slightly blocks ErbB2 homoassociation. In starved cells, the majority of ErbB2 is inactive and present in large homoclusters, which are slightly disrupted by pertuzumab. Assuming that large ErbB2 homoclusters harbor inactive proteins, the ErbB2 expelled from these homoclusters will heterodimerize with other membrane proteins, somehow leading to its heteroactivation.
Although the higher-order oligomers of ErbB1 and ErbB2 described here were found in cells overexpressing these proteins, previous experimental evidence strongly indicates that the formation of such large-scale clusters (i.e., oligomers larger than dimers) is an inherent property of membrane proteins (15,19,41,44,45). We believe that proteins with both low and high expression levels form large-scale clusters, but the number of proteins/cluster and the cluster diameter are influenced by the expression level.
The model in Fig. 7 B integrates all these findings. The ErbB2 homoclusters are assumed to be depots of inactive proteins, which can be recruited by ligand-activated ErbB1 or ErbB3 to heterodimeric complexes, explaining the observed decrease in ErbB2 homocluster size upon activation. Because ErbB1 is a full-featured receptor tyrosine kinase with ligand-binding and tyrosine kinase activities, EGF not only induces ErbB1-ErbB2 heterodimers, but also ErbB1 homodimers, explaining the observed increase in ErbB1 homocluster size upon EGF stimulation. However, neither ErbB2 nor ErbB3 is a full-featured receptor tyrosine kinase, because the former has no ligand-binding activity, whereas the latter has no kinase activity. Therefore, they were likened to “the deaf and the dumb” (46). It was shown that unliganded ErbB3 forms homoclusters, which are disassembled by heregulin (18). Integrating this observation with ours, we can envisage that both ErbB2 and ErbB3 form large, inactive, and separate homoclusters in unstimulated cells. Heregulin stimulation removes ErbB3 molecules from ErbB3 homoclusters. Ligand-activated ErbB3 recruits ErbB2 proteins from inactive ErbB2 homoclusters to ErbB2-ErbB3 heterodimers. The fact that heregulin stimulation leads to the exclusive formation of ErbB2-ErbB3 heterodimers, whereas EGF induces both ErbB1 homodimers and ErbB1-ErbB2 heterodimers, explains why heregulin causes a much larger decrease in the size of ErbB2 homoclusters. Flow-cytometric homo-FRET measurements have the potential to shed light on the largely unrecognized role of higher-order oligomers in transmembrane signaling, which will significantly advance our understanding of the fine interactions governing the first steps of the activation cascade of receptor tyrosine kinases.
Acknowledgments
This work was supported by research grants from the Hungarian Scientific Research Fund (F49025, K72677, K62648, and K68763) and from the European Commission (LSHB-CT-2004-503467 and LSHC-CT-2005-018914).
Editor: Michael Edidin.
References
- 1.Schechter, Y., L. Hernaez, J. Schlessinger, and P. Cuatrecasas. 1979. Local aggregation of hormone-receptor complexes is required for activation by epidermal growth factor. Nature. 278:835–838. [DOI] [PubMed] [Google Scholar]
- 2.Ogiso, H., R. Ishitani, O. Nureki, S. Fukai, M. Yamanaka, J. H. Kim, K. Saito, A. Sakamoto, M. Inoue, M. Shirouzu, and S. Yokoyama. 2002. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 110:775–787. [DOI] [PubMed] [Google Scholar]
- 3.Citri, A., and Y. Yarden. 2006. EGF-ERBB signalling: towards the systems level. Nat. Rev. Mol. Cell Biol. 7:505–516. [DOI] [PubMed] [Google Scholar]
- 4.Nagy, P., A. Jenei, S. Damjanovich, T. M. Jovin, and J. Szöllősi. 1999. Complexity of signal transduction mediated by ErbB2: clues to the potential of receptor-targeted cancer therapy. Pathol. Oncol. Res. 5:255–271. [DOI] [PubMed] [Google Scholar]
- 5.Yarden, Y., and M. X. Sliwkowski. 2001. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2:127–137. [DOI] [PubMed] [Google Scholar]
- 6.Tzahar, E., H. Waterman, X. Chen, G. Levkowitz, D. Karunagaran, S. Lavi, B. J. Ratzkin, and Y. Yarden. 1996. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell. Biol. 16:5276–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Worthylake, R., L. K. Opresko, and H. S. Wiley. 1999. ErbB-2 amplification inhibits down-regulation and induces constitutive activation of both ErbB-2 and epidermal growth factor receptors. J. Biol. Chem. 274:8865–8874. [DOI] [PubMed] [Google Scholar]
- 8.Holbro, T., R. R. Beerli, F. Maurer, M. Koziczak, C. F. Barbas 3rd, and N. E. Hynes. 2003. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA. 100:8933–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gadella, T. W., Jr., and T. M. Jovin. 1995. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J. Cell Biol. 129:1543–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lidke, D. S., P. Nagy, B. G. Barisas, R. Heintzmann, J. N. Post, K. A. Lidke, A. H. Clayton, D. J. Arndt-Jovin, and T. M. Jovin. 2003. Imaging molecular interactions in cells by dynamic and static fluorescence anisotropy (rFLIM and emFRET). Biochem. Soc. Trans. 31:1020–1027. [DOI] [PubMed] [Google Scholar]
- 11.Liu, P., T. Sudhaharan, R. M. Koh, L. C. Hwang, S. Ahmed, I. N. Maruyama, and T. Wohland. 2007. Investigation of the dimerization of proteins from the epidermal growth factor receptor family by single wavelength fluorescence cross-correlation spectroscopy. Biophys. J. 93:684–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moriki, T., H. Maruyama, and I. N. Maruyama. 2001. Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J. Mol. Biol. 311:1011–1026. [DOI] [PubMed] [Google Scholar]
- 13.Keating, E., A. Nohe, and N. O. Petersen. 2007. Studies of distribution, location and dynamic properties of EGFR on the cell surface measured by image correlation spectroscopy. Eur. Biophys. J. 37:469–481. [DOI] [PubMed] [Google Scholar]
- 14.Webb, S. E., S. K. Roberts, S. R. Needham, C. J. Tynan, D. J. Rolfe, M. D. Winn, D. T. Clarke, R. Barraclough, and M. L. Martin-Fernandez. 2008. Single-molecule imaging and fluorescence lifetime imaging microscopy show different structures for high- and low-affinity epidermal growth factor receptors in A431 cells. Biophys. J. 94:803–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clayton, A. H., M. L. Tavarnesi, and T. G. Johns. 2007. Unligated epidermal growth factor receptor forms higher order oligomers within microclusters on A431 cells that are sensitive to tyrosine kinase inhibitor binding. Biochemistry. 46:4589–4597. [DOI] [PubMed] [Google Scholar]
- 16.Clayton, A. H., F. Walker, S. G. Orchard, C. Henderson, D. Fuchs, J. Rothacker, E. C. Nice, and A. W. Burgess. 2005. Ligand-induced dimer-tetramer transition during the activation of the cell surface epidermal growth factor receptor—a multidimensional microscopy analysis. J. Biol. Chem. 280:30392–30399. [DOI] [PubMed] [Google Scholar]
- 17.Kani, K., C. M. Warren, C. S. Kaddis, J. A. Loo, and R. Landgraf. 2005. Oligomers of ERBB3 have two distinct interfaces that differ in their sensitivity to disruption by heregulin. J. Biol. Chem. 280:8238–8247. [DOI] [PubMed] [Google Scholar]
- 18.Landgraf, R., and D. Eisenberg. 2000. Heregulin reverses the oligomerization of HER3. Biochemistry. 39:8503–8511. [DOI] [PubMed] [Google Scholar]
- 19.Nagy, P., A. Jenei, A. K. Kirsch, J. Szöllősi, S. Damjanovich, and T. M. Jovin. 1999. Activation-dependent clustering of the ErbB2 receptor tyrosine kinase detected by scanning near-field optical microscopy. J. Cell Sci. 112:1733–1741. [DOI] [PubMed] [Google Scholar]
- 20.Sako, Y., S. Minoghchi, and T. Yanagida. 2000. Single-molecule imaging of EGFR signalling on the surface of living cells. Nat. Cell Biol. 2:168–172. [DOI] [PubMed] [Google Scholar]
- 21.Jovin, T. M., and W. L. Vaz. 1989. Rotational and translational diffusion in membranes measured by fluorescence and phosphorescence methods. Methods Enzymol. 172:471–513. [DOI] [PubMed] [Google Scholar]
- 22.Jares-Erijman, E. A., and T. M. Jovin. 2003. FRET imaging. Nat. Biotechnol. 21:1387–1395. [DOI] [PubMed] [Google Scholar]
- 23.Sharma, P., R. Varma, R. C. Sarasij, I. K. Gousset, G. Krishnamoorthy, M. Rao, and S. Mayor. 2004. Nanoscale organization of multiple GPI-anchored proteins in living cell membranes. Cell. 116:577–589. [DOI] [PubMed] [Google Scholar]
- 24.Rao, M., and S. Mayor. 2005. Use of Förster's resonance energy transfer microscopy to study lipid rafts. Biochim. Biophys. Acta. 1746:221–233. [DOI] [PubMed] [Google Scholar]
- 25.Runnels, L. W., and S. F. Scarlata. 1995. Theory and application of fluorescence homotransfer to melittin oligomerization. Biophys. J. 69:1569–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varma, R., and S. Mayor. 1998. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature. 394:798–801. [DOI] [PubMed] [Google Scholar]
- 27.Yeow, E. K., and A. H. Clayton. 2007. Enumeration of oligomerization states of membrane proteins in living cells by homo-FRET spectroscopy and microscopy: theory and application. Biophys. J. 92:3098–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Squire, A., P. J. Verveer, O. Rocks, and P. I. Bastiaens. 2004. Red-edge anisotropy microscopy enables dynamic imaging of homo-FRET between green fluorescent proteins in cells. J. Struct. Biol. 147:62–69. [DOI] [PubMed] [Google Scholar]
- 29.Jovin, T. M. 1979. Fluorescence polarization and energy transfer: theory and application. In Flow Cytometry and Sorting. M. Melamed, P. Mullaney, and M. Mendelsohn, editors. John Wiley & Sons, New York. 137–165.
- 30.Szentesi, G., G. Horváth, I. Bori, G. Vámosi, J. Szöllősi, R. Gáspár, S. Damjanovich, A. Jenei, and L. Mátyus. 2004. Computer program for determining fluorescence resonance energy transfer efficiency from flow cytometric data on a cell-by-cell basis. Comput. Methods Programs Biomed. 75:201–211. [DOI] [PubMed] [Google Scholar]
- 31.Sebestyén, Z., P. Nagy, G. Horváth, G. Vámosi, R. Debets, J. W. Gratama, D. R. Alexander, and J. Szöllősi. 2002. Long wavelength fluorophores and cell-by-cell correction for autofluorescence significantly improves the accuracy of flow cytometric energy transfer measurements on a dual-laser benchtop flow cytometer. Cytometry. 48:124–135. [DOI] [PubMed] [Google Scholar]
- 32.Lakowicz, J. R. 1983. Fluorescence polarization. In Principles of Fluorescence Spectroscopy. Plenum Press, New York. 111–153.
- 33.Bene, L., M. J. Fulwyler, and S. Damjanovich. 2000. Detection of receptor clustering by flow cytometric fluorescence anisotropy measurements. Cytometry. 40:292–306. [PubMed] [Google Scholar]
- 34.Harikumar, K. G., and L. J. Miller. 2005. Fluorescence resonance energy transfer analysis of the antagonist- and partial agonist-occupied states of the cholecystokinin receptor. J. Biol. Chem. 280:18631–18635. [DOI] [PubMed] [Google Scholar]
- 35.Horváth, G., M. Petrás, G. Szentesi, A. Fábián, J. W. Park, G. Vereb, and J. Szöllősi. 2005. Selecting the right fluorophores and flow cytometer for fluorescence resonance energy transfer measurements. Cytometry A. 65:148–157. [DOI] [PubMed] [Google Scholar]
- 36.Mocanu, M. M., Z. Fazekas, M. Petrás, P. Nagy, Z. Sebestyén, J. Isola, J. Timar, J. W. Park, G. Vereb, and J. Szöllősi. 2005. Associations of ErbB2, beta1-integrin and lipid rafts on Herceptin (trastuzumab) resistant and sensitive tumor cell lines. Cancer Lett. 227:201–212. [DOI] [PubMed] [Google Scholar]
- 37.Nagy, P., D. J. Arndt-Jovin, and T. M. Jovin. 2003. Small interfering RNAs suppress the expression of endogenous and GFP-fused epidermal growth factor receptor (ErbB1) and induce apoptosis in ErbB1-overexpressing cells. Exp. Cell Res. 285:39–49. [DOI] [PubMed] [Google Scholar]
- 38.Bene, L., J. Szöllősi, G. Szentesi, L. Damjanovich, R. Gáspár, Jr., T. A. Waldmann, and S. Damjanovich. 2005. Detection of receptor trimers on the cell surface by flow cytometric fluorescence energy homotransfer measurements. Biochim. Biophys. Acta. 1744:176–198. [DOI] [PubMed] [Google Scholar]
- 39.Di Fiore, P. P., J. H. Pierce, M. H. Kraus, O. Segatto, C. R. King, and S. A. Aaronson. 1987. ErbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science. 237:178–182. [DOI] [PubMed] [Google Scholar]
- 40.Nagy, P., L. Bene, M. Balázs, W. C. Hyun, S. J. Lockett, N. Y. Chiang, F. Waldman, B. G. Feuerstein, S. Damjanovich, and J. Szöllősi. 1998. EGF-induced redistribution of ErbB2 on breast tumor cells: flow and image cytometric energy transfer measurements. Cytometry. 32:120–131. [DOI] [PubMed] [Google Scholar]
- 41.Yang, S., M. A. Raymond-Stintz, W. Ying, J. Zhang, D. S. Lidke, S. L. Steinberg, L. Williams, J. M. Oliver, and B. S. Wilson. 2007. Mapping ErbB receptors on breast cancer cell membranes during signal transduction. J. Cell Sci. 120:2763–2773. [DOI] [PubMed] [Google Scholar]
- 42.Lidke, D. S., P. Nagy, R. Heintzmann, D. J. Arndt-Jovin, J. N. Post, H. E. Grecco, E. A. Jares-Erijman, and T. M. Jovin. 2004. Quantum dot ligands provide new insights into ErbB/HER receptor-mediated signal transduction. Nat. Biotechnol. 22:198–203. [DOI] [PubMed] [Google Scholar]
- 43.Franklin, M. C., K. D. Carey, F. F. Vajdos, D. J. Leahy, A. M. de Vos, and M. X. Sliwkowski. 2004. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 5:317–328. [DOI] [PubMed] [Google Scholar]
- 44.Damjanovich, S., G. Vereb, A. Schaper, A. Jenei, J. Matkō, J. P. Starink, G. Q. Fox, D. J. Arndt-Jovin, and T. M. Jovin. 1995. Structural hierarchy in the clustering of HLA class I molecules in the plasma membrane of human lymphoblastoid cells. Proc. Natl. Acad. Sci. USA. 92:1122–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lillemeier, B. F., J. R. Pfeiffer, Z. Surviladze, B. S. Wilson, and M. M. Davis. 2006. Plasma membrane-associated proteins are clustered into islands attached to the cytoskeleton. Proc. Natl. Acad. Sci. USA. 103:18992–18997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Citri, A., K. B. Skaria, and Y. Yarden. 2003. The deaf and the dumb: the biology of ErbB-2 and ErbB-3. Exp. Cell Res. 284:54–65. [DOI] [PubMed] [Google Scholar]