

Abstract
Stabilization of the lipid bilayer membrane in red blood cells by its association with an underlying membrane-associated cytoskeleton has long been recognized as critical for proper red blood cell function. One of the principal connections between skeleton and bilayer is via linkages between band 3, the integral membrane protein that transports anions across the cell surface, and membrane skeletal elements including ankyrin, adducin, spectrin, and the junctional complex of the skeleton. Here, we use membrane tether formation coupled with fluorescent labeling of membrane components to examine the importance of band 3 in stabilizing the bilayer-skeletal association. In membranes from a patient deficient in band 3, the energy associated with the bilayer skeleton is approximately zero, whereas when band 3 is immobilized by ligation with the monoclonal antibody R10, the energy of association approximately doubles. Fluorescence images of tethers reveal that ∼40% of the band 3 on the normal cell surface can be pulled into the tether, confirming a lateral segregation of membrane components during tether formation. These results validate a critical role for band 3 in stabilizing the bilayer-skeletal association in red cells.
INTRODUCTION
The mechanical stability of the cell membrane is critically important for survival and proper function of red blood cells. Observations of the effects of alteration of cell surface area on the ability of cells to survive have shown that the amount of excess surface area a cell has is a critical predictor for the ability of the cell to function in the vasculature (1,2). The underlying cause of this is the constraint that the fixed area of the membrane bilayer and the fixed volume of the cell place on the ability of the cell to deform. If there is insufficient membrane area to enclose the cell volume within the confined spaces of the microvasculature, the cell must either reduce its volume or be eliminated from circulation (1). From our early understanding of the fluid character of the membrane bilayer (3,4), it has been clear that stabilization must be provided by associations between the fluid bilayer and the underlying membrane-associated cytoskeleton (5). Thus, a critical function of the membrane-associated cytoskeleton in red blood cells is to prevent the loss of surface area by fragmentation of the bilayer.
Additional evidence for this viewpoint comes from the identification of an underlying defect in one or more of the membrane skeletal proteins as a fundamental cause of many forms of inherited hemolytic anemia (6–8). Identified abnormalities include defects in proteins known to mediate lateral associations within the membrane skeleton itself, including spectrin and protein 4.1, as well as molecules that form linkages between the membrane skeleton and the membrane bilayer, including ankyrin and band 3 (the anion transporter AE-1) (7,9). Many inherited molecular abnormalities have also been shown to be associated with abnormal mechanical behavior of the cell membrane. For example, deficiency in the major skeletal protein 4.1 is associated with an increase in the rate of cell fragmentation in shear flow (10). In a study of a collection of families exhibiting various forms of hemolytic anemia, the degree of deficiency in the cytoskeletal protein spectrin was found to correlate directly with a decrease in the elastic resistance of the membrane to extension (surface shear deformation) (11). Spectrin deficiency also correlates with the susceptibility of the cells to fragmentation in shear (12) and to the clinical severity of the disorder (13).
In this study, we examine the importance of integral membrane proteins in maintaining membrane stability by measuring the effects of two structural modifications on membrane stability in human red blood cells: immobilization of integral membrane proteins by antibodies and band 3 deficiency due to a genetic abnormality. Chasis and co-workers have shown that the binding of a monoclonal antibody (R10) targeted to glycophorin A results in the immobilization of glycophorin A and band 3 on the surface of the membrane (14). This immobilization is accompanied by a marked increase in the mechanical rigidity of the cell membrane (15). In addition, we have tested cells from a patient showing a rare form of hemolytic anemia in which the cell membranes have a severe deficiency (>80%) in the integral protein band 3. Membrane stability is tested by the mechanical formation of thin bilayer cylinders (tethers) from the cell surface. Tether formation has emerged as an important biophysical method for assessing membrane properties and can be used to obtain a direct assessment of the strength of the bilayer-skeletal association (16–18).
MATERIALS AND METHODS
Cell preparation
Whole blood was collected from human donors by venipuncture into acid-citrate dextrose tubes, following informed consent and procedures approved by the human subjects review board at the Lawrence Berkeley Laboratories. Cells from a patient with band 3 deficiency (19) were generously supplied by Dr. Perrotta Silverio (Seconda Università degli Studi di Napoli, Naples, Italy) along with a normal travel control sample. These samples were obtained by the same procedure and shipped on ice via overnight carrier. For shear modulus measurements, cells were suspended at very low concentration in isotonic phosphate-buffered saline (PBS) (130 mM NaCl, 25 mM Na2HPO4, 6.2 mM NaH2PO4, pH 7.4, 290 mOsm) containing 4% fetal calf serum (FCS). For tether formation studies, membrane proteins on the red cell surface were biotinylated with sulfo-N-hydroxysuccinimido-biotin (s-NHS-biotin; Pierce Chemical, Rockford, IL) to provide attachment sites for adhesion to streptavidin-coated paramagnetic beads. Whole blood was incubated at room temperature for 5 min with s-NHS-biotin (0.2 mg/mL). The red blood cells were then suspended and washed three times in PBS plus 4% FCS.
Fluorescent labeling of membrane components
To label band 3, 5 μL of packed red blood cells were added to 500 μL of 100 μM eosin-5-maleimide (EMA; Molecular Probes, Eugene, OR) and incubated for 1 h at room temperature or 30 min at 37°C. After incubation, the cells were washed three times and resuspended in PBS/FCS. In control experiments, 4,4′-diisothiocyanodihydro-stilbene-2,2′-disufonic acid (DIDS) at ∼100 μM was added to cells for 30 min at room temperature, followed by three washes before eosin maleimide labeling. For tether fluorescence experiments, cells were treated with CO for 20 min before eosin labeling to displace O2 from cell hemoglobin and reduce photobleaching. To label cells with the lipid marker Texas Red-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (Texas Red-PE, Molecular Probes), 10 μg dissolved in chloroform was dried onto a glass vial under nitrogen, then resuspended into 400 μL of 50% hematocrit of red cells, incubated for 30 min, and then washed three times and resuspended for measurement in PBS/FCS. To label cells with the lipid marker 1,1′dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI, Molecular Probes), 100 ng DiI in 10 μL of methanol was premixed with PBS then added to a cell suspension to a final hematocrit of 10% in 1.0 mL. Cells were incubated for 30 min then washed extensively to remove excess label before resuspension in PBS/FCS for measurement.
Antibody liganding of glycophorin A
Monoclonal antibodies (R10), specific to the extracellular domain of glycophorin A (20), were a gift from Dr. David Anstee (International Blood Group Reference Laboratory, Bristol, England). Glycophorin A was liganded with R10 by incubating 15 μl of packed red cells in a total volume of 250 μl antibody solution (25 μg/mL in 290 mOsm PBS) for 30 min at room temperature. After incubation the cells were washed three times and resuspended in PBS/FCS.
Measurement procedures
Experiments were carried out on inverted light microscopes (Nikon Instruments, Melville, NY) equipped with custom-built digital imaging systems and, in one case, a magnetic force transducer (21,22), and in the other, a microcantilever device (17,23). Thin lipid cylinders (tethers) were pulled off the cell body by first attaching streptavidin-coated bead to the biotinylated cell surface and then applying force either via the cantilever or the “magnetic tweezers” (Fig. 1). The two different approaches are a consequence of the fact that some measurements were performed in Rochester, where cantilevers have become the method of choice, and some were performed at Berkeley, where the magnetic tweezers system was operational. The two approaches each entail some technical challenges but offer comparable precision in the measurement of force.
FIGURE 1.

A schematic overview of the experimental apparatus (adapted from Heinrich and Waugh (22)). A paramagnetic bead was attached to a red blood cell held in a micropipette. The current to an electromagnet was adjusted to control the force on the bead. The deformation of the cell and the position of the bead could be observed under a light microscope. All measurements were performed at a fixed distance between the bead and the magnet. A computerized feedback system was used to adjust the current and keep the bead at the desired position in the digitized image of the experiment. The magnitude of the current was monitored and saved to disk upon a keystroke.
Cells were placed into a thin chamber formed by sandwiching a plexiglass spacer between two cover glasses. The chambers were constructed to allow access for the pipette and to provide access for the cantilever or close apposition of the electromagnet. Images were formed in brightfield with monochromatic (436 nm) illumination and a 60× dry objective. The microscope stage was fitted with hydraulic micromanipulators and positioning devices to enable positioning of the magnet or cantilever and the micropipette. Adjustable height water reservoirs were used to control the aspiration pressure in the pipette, and the pressure was monitored using pressure transducers (Model DP 103, Validyne, Northridge, CA). Thus both the force on the tether and the membrane tension could be controlled and measured.
Magnetic force method
The magnetic force method required both high- and low-magnification views of the cell and the magnetic bead. Two charge-coupled device (CCD) video cameras were mounted to a beam splitter on the side port of the microscope, one providing the low-magnification image and the other showing a region of the field at higher magnification. The higher magnification camera could be translated laterally so that different ROIs could be selected for viewing. Both video signals were sent to a Pulsar channel-selectable digital imaging board (Matrox, Quebec, Canada) capable of real-time video display and image capture on a computer with a peripheral component interconnect (PCI) bus. In addition to the digital imaging system, a Super-VHS video recorder stored a multiplexed composite of the two video channels encoded with the time and the instantaneous current to the electromagnet. Automated control of the experimental system and data acquisition were accomplished using computer code (written in C++) running on a Windows-based workstation.
The electromagnet was designed and built at the University of Rochester. It consisted of a rod of HyMu paramagnetic alloy (Carpenter Technology, Rochester, NY) machined to a pointed tip and encased in a water-cooled wire coil wound onto a brass spindle. Current to the magnet was controlled using the HP 6655A power supply (Hewlett Packard, Palo Alto, CA). The power supply was interfaced to the computer via an HP-interface bus peripheral component interconnect expansion card. The set point for the electric current could be controlled by software on the computer or manually by the operator. The experimental current range was kept between 0.0 and 1.0 A with a resolution of 2.0 mA.
To form a tether with the magnet, biotinylated red blood cells were suspended in isotonic buffer with a low concentration of streptavidin-coated paramagnetic beads (diameter = 4.5μm, Dynal, Oslo, Norway) and placed in a “holding” chamber on the microscope stage. A large bore micropipette (d > 7 μm) was used to transfer a few cells and beads from the holding chamber to the center of the measurement chamber. Only a few cells and beads were transferred to avoid image degradation and the induction of spurious magnetic fields caused by excess paramagnetic material near the magnet tip. The measurement chamber was filled with either hypotonic or isotonic buffer depending on the type of cells being tested. After transferring cells, a smaller pipette (1.8–2.3 μm, inside diameter) was introduced and a single bead was attached to one of the biotinylated cells. The cell-bead pair was positioned close to the edge of the chamber in line with the electromagnet.
A reference location was chosen as the home position of the bead (Fig. 2 a). This location remained the same throughout the measurement series. The magnet was turned on and a feedback loop was engaged to maintain the magnetized bead at the desired location. The cell was slowly withdrawn from the home position, pulling a thin tether out of the membrane capsule. During tether growth, high-magnification images encoded with the time, pipette aspiration pressure, and magnet current (Fig. 2 b) were saved to disk for subsequent measurement of cell radius (Rc) and projection length (Lp). Meanwhile, the low-magnification video channel (Fig. 2 a) was used to track the bead position and monitor tether growth. A composite of the two video signals was also saved on videotape. Once the desired tether length (Lt) was achieved, the magnitude of the current was recorded at constant Lt for a series of increasing and decreasing aspiration pressures. After the tests on the cell, a force calibration was performed to convert the measured currents into forces.
FIGURE 2.

Images of a tether experiment in progress. A dichroic mirror on the side-port of the microscope passed 20% of the light collected from the objective to a CCD video camera to provide a low-magnification video signal (a), which was fed into one channel of the digitizer on the computer. The image from the other 80% of the light was diverted and magnified on the chip of a second translatable camera. The signal from this high-magnification camera was fed into another channel on the digitizer and saved to disk on request for cell geometry measurements (b). Micropipette aspiration pressure was also digitized and encoded onto the high-magnification image along with the date, time, videotape time stamp, and the instantaneous electric current to the electromagnet. (Scale bars = 10 μm.) Note that the tether shown in a is much longer than most of those formed during the study. Typical tether length was 20–30 μm (b).
The magnetic force was calibrated by measuring the velocity of the bead freely moving in the magnetic field. At the end of each experiment, the bead was separated from the cell and aspirated onto the tip of the pipette. The electromagnet was turned on at a fixed current, and the bead was videotaped as it was pulled through the solution. The velocity of the bead (νb) at the home position (where the bead was held for tether formation) was measured, and the corresponding force on the bead was found using the Stokes equation for the drag on a sphere moving in a fluid:
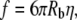 |
(1) |
where Rb is the radius of the bead and η is the viscosity of the fluid. A third order polynomial regression of the force as a function of the current fit the calibration data with an R2 > 0.95 (22) (Fig. 3).
FIGURE 3.

Tether force calibration curve. After the experiment, a force calibration was obtained by measuring the velocity of a freely moving bead while electric currents of known magnitude were applied to the magnet. A separate image analysis program was designed to calculate the pixel distance traveled by the bead between successive video fields. The corresponding velocity was used in Stokes equation for the drag on a sphere moving in a fluid to find the force on the bead at each current. The force-current data were fit to a third-order polynomial that generally regressed to a fit with an R2 > 0.95.
The cantilever method
Shorter tethers were formed using microcantilevers from cells labeled with fluorescent markers to determine the distribution of band 3 between the cell body and the tether. The mechanical procedure for forming tethers is essentially identical to one published previously (17). Cantilevers were prepared by extruding thin glass fibers then coating them with biotinylated gelatin. These were mounted on a small manipulator and positioned in the chamber perpendicular to the axis of the pipette. Cells and beads were placed together in the measurement chamber, and beads were manipulated into adhesive contact with the cantilever. Force calibration was accomplished using a large-bore pipette (∼4.0 μm diameter) and using a cell attached to the cantilever as a piston to transmit the suction force in the pipette and deflect the cantilever. To form a tether, a smaller pipette (1.8–2.3 μm) was used to hold the cell, attach it to the cantilever via an adherent bead, and pull the cell away, forming a tether between the cell and the bead. The deflection of the cantilever was used to obtain the force, and the dimensions of the cell and the tether length were obtained from video recordings of the experiment.
Analysis
Analysis of mechanical equilibrium for tethered red blood cells leads to a prediction that the square of tethering force ft should be proportional to the tension τo (isotropic force resultant) in the membrane bilayer over the surface of the cell (17,22,24):
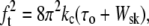 |
(2) |
where kc is the membrane bending stiffness and Wsk is the work required to separate the membrane bilayer from the underlying membrane skeleton. A subtle but important point is that the tension τo is not the total membrane tension, which includes contributions from both the bilayer and the underlying skeleton, but only the tension in the bilayer itself. The total membrane tension is related to the aspiration pressure applied to the cell by a simple force balance:
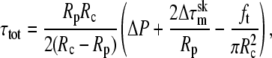 |
(3) |
where Rp is the pipette radius, Rc is the maximum radius of the portion of the cell outside the pipette (measured perpendicular to the axis of symmetry), ΔP is the pressure difference between the inside of the pipette and the suspending fluid, and 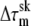 is the difference between the skeletal tension at the tip of the pipette and the skeletal tension at the cell rim. When the skeletal tensions are small in comparison with the total tension, the bilayer tension can be approximated by
is the difference between the skeletal tension at the tip of the pipette and the skeletal tension at the cell rim. When the skeletal tensions are small in comparison with the total tension, the bilayer tension can be approximated by
 |
(4) |
where we have also neglected the contribution from the tether force, which is also small compared to ΔP. Unfortunately, the approximation given in Eq. 4 is not accurate over the range of membrane tensions used in this study, and a separate calculation of the skeletal tension was required. These calculations were based on elastic constitutive relationships of the red cell membrane skeleton. Details are given in a separate manuscript.
Fluorescence distribution measurements
The distribution of red blood cell membrane components in normal and experimental cell types was measured using fluorescence-imaged microdeformation (25). Two types of fluorescence measurements were performed, one in which cells were aspirated into pipettes and the distribution of fluorescence along the projection was measured, and one in which a tether was formed from the cell and the fluorescence intensity of the tether was compared with the intensity of the cell body. For the pipette projection measurements, a red blood cell coated with a fluorescent label to a specific membrane component was aspirated at high pressure into a micropipette. For optimal resolution, a 100× oil immersion objective lens was used for these measurements. (During manipulation, brightfield illumination in the emission wavelength of the fluorophore (546 nm) was used to avoid photobleaching.) For fluorescence distribution measurements, a shuttering system was used to switch off brightfield illumination and switch on epiillumination at the appropriate wavelength for the fluorophore.
The fluorescent image was captured digitally using a liquid-cooled CCD camera (Photometrics, Tucson, AZ). Cells were typically exposed for 200–1000 ms using image acquisition and analysis software to control the exposure time and display the images. The distribution of the fluorescently labeled membrane elements was obtained by measuring the distribution of grayscale intensity over the cell surface using Scion Image (Scion, Fredrick, MD). The ratio of the fluorescence intensity at the cap of the membrane projection ρc to the fluorescence intensity at the entrance of the pipette ρe was obtained from a linear fit to the gradient of the intensity along the projection. This ratio is characteristic of the extent to which the labeled component is associated with the membrane skeleton. (For example, lipid labels show no gradient in intensity at all, but a label on actin gives the steepest gradient and the largest ratio of ρe/ρc (25).)
For measurements of tether fluorescence, cells were labeled with a lipid dye (DiI or Texas Red-PE) or with eosin maleimide, a covalent label that preferentially labels band 3. Tethers were formed using the microcantilever method, and a series of fluorescent images was captured over a period of ∼30 min. An automated system was used to control illumination and the sensitivity of the camera. A shuttering system switched illumination from transillumination to epiillumination, and the camera sensitivity was adjusted via a computer interface. The camera was a silicon-intensified target device coupled to a CCD (DAGE-MTI, DSP 2000, Michigan City, IN, or Solamere, Palo Alto, CA), and both the sensitivity and exposure time could be adjusted. Calibration determined that the gray scale of the video image increased linearly with the time of exposure within the dynamic range of the cameras. Thus, short exposure times could be used to measure the (relatively) high fluorescence intensity on the body of the cell, and longer exposures could be used to detect faint fluorescence signals from tethers. Measurements were made over a period of 20–30 min. A measurement of cell body intensity was taken at the beginning and at the end of the sequence so that the intensities could be corrected for bleaching. Typically, <15% of the signal was lost over the course of measurement. For tether intensity, an estimate of the tether radius was made from measured values of the force at each time point. (Force decreased gradually with time after tether formation.) The radius Rt was assumed to follow an inverse relationship with force f (26)
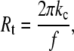 |
(5) |
where the bending stiffness kc was taken to be 2 × 10−19 J. To determine the fluorescence intensity per unit area of membrane, a region of interest (ROI) was placed around the cell body or the tether, and the mean fluorescence intensity was measured for the ROI. The mean background fluorescence was also measured and subtracted from mean for the cell body or the tether to obtain the mean fluorescence above background (Fnet). The fluorescence per unit area of membrane per frame was then calculated as
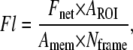 |
(6) |
where AROI is the area of the ROI, Nframe is the number of frames integrated, and Amem is the area of the cell body or the tether in the ROI. For the cell body,
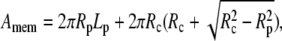 |
(7) |
and for the tether,
 |
(8) |
where LROI is the length of the tether in the ROI, and Eq. 5 has been used to substitute for the tether radius Rt. The ratio of tether fluorescence at a given time to cell body fluorescence is then calculated using a value for the fluorescence of the cell body corrected for bleaching.
Shear modulus
The elastic resistance of the red cell to deformations in the plane of the membrane was measured using micropipettes (27). The resistance to deformation was measured in different cell types, and this information was used in the analysis of the tether experiments to calculate the tension in the membrane. The flat region of the disk was aspirated at low pressures into a small micropipette (inside diameter ≈ 1 μm), and the projection length was measured as a function of the pressure. A 100× oil immersion objective and monochromatic (436 nm) illumination were used for optimal optical resolution. These data were compared to predictions of the projection length as a function of aspiration pressure.
RESULTS
HS cell characterization
Protein expression
A sample of blood was obtained from a patient with hereditary spherocytosis (HS) identified to have levels of erythrocyte band 3 that were were 12 ± 4% that of normal red blood cells (19). Western blot analysis was used to confirm the previously reported (19) reduced levels of band 3 as well as glycophorin A and normal levels of protein 4.1 and glycophorin C in HS red cell membranes (data not shown).
Cell morphology
The HS cells' morphology is shown at low and high magnification in Fig. 4. The HS cells were smaller and more spherical than those in the travel control sample, and cell fragments were observed within the circulating population. The variety of shapes, varying from discocyte to stomatocyte, and the presence of a few noticeably spherical cells are typical of cell morphology in HS (28). Furthermore, in the band 3 deficient blood, early stage reticulocytes were seen in most of the microscope fields sampled (Fig. 4 c), consistent with reticulocytosis seen in many HS patients and the expectation of accelerated erythropoeisis in anemic patients (29).
FIGURE 4.

Morphological evaluation of HS cells. Wide-field video micrographs of normal (a) and band 3 deficient (c) cells were recorded with the low-magnification video channel while high-magnification images of the same video fields were also taken for the normal (b) and HS (d) cells. The white arrows in tile (d) identify late-stage reticulocytes that were evident in many of HS sample fields. These cells were avoided when selecting candidates for experiments. (Scale bar = 10 μm on left, 5 μm on right.)
R10 antibody modified cells
Antibodies to glycophorin A have been shown to increase the stiffness of the erythrocyte membrane up to 10-fold (14,30). This rigidification is thought to be a result of a cooperative interaction between glycophorin A and band 3 that induces a higher degree of association between the cytoplasmic tail of band 3 and the peripheral network (15). At an incubation concentration of 25 μg/mL of R10, the average shear modulus (μ) of the antibody-treated cells was found to be 33 ± 13 μN/m. This value corresponds to a fourfold increase over the average value obtained for unmodified cells (8.8 ± 0.7 μN/m), which is in agreement with previously published estimates of the shear elasticity (6–9 μN/m) (27).
Fluorescence-imaged microdeformation
The distribution of fluorescence along the length of aspirated membrane projections was measured for normal and band 3 deficient cells labeled with EMA. The density ratios (ρe/ρc) were compared to theoretical predictions for the skeletal density based on constitutive models of the membrane skeleton that account for local changes in skeletal density in response to deformation. (These predictions have been verified in other studies.) The ratio ρe/ρc in the control cells matched previous measurements of band 3 distribution collected in our laboratory (25,31) (N. Mohandas, unpublished data), and fell well below the predicted values for actin. This result is consistent with expectations based on measurements of band 3 mobility by fluorescence recovery after photobleach, which show that ∼60% of the band 3 in the membrane is mobile and only 40% connected to the skeleton (32).
When compared with the travel controls, EMA-labeled HS cells exhibited steeper gradients in intensity along the membrane projection, and this is reflected in higher ratios of ρe/ρc for these cells (Fig. 5). Surprisingly, the ρe/ρc ratios in the band 3 deficient cells surpassed even the theoretical predictions for actin in the peripheral skeleton of normal cells. This could be because either all of the residual band 3 in these cells is linked to membrane skeleton or the ratio KN/μN in the HS cells may be slightly lower than the accepted value for normal cells (KN/μN = 1.5–2) and could reflect alterations in network organization in these cells.
FIGURE 5.

Band 3 density gradients along the pipette projection. The ratio of entrance density to cap density is an indication of the steepness of the molecular distribution in the pipette. Measured values of ρe/ρc for EMA-labeled band 3 deficient (squares) cells and travel controls (diamonds) are shown as a function of pipette projection length. Open circles and triangles represent the theoretical predictions for ρe/ρc of actin and band 3, respectively, in normal cells with 
Tether experiments
Tether experiments were performed on three types of cells: normal cells, band 3 deficient cells, and normal cells labeled with R10 monoclonal antibody. For each of the three cell types tested, the measured value of the force squared was examined as a function of the membrane tension. The dimensions of each cell at each equilibrium point were used as a basis for calculating the skeletal contribution to the total membrane tension on the cell body, so that the bilayer tension could be determined. A linear regression to the force squared as a function of the bilayer tension was used to calculate the separation work Wsk and the apparent bending stiffness kc (Fig. 6). The values presented here are based on the constitutive model derived from the molecular simulations of Discher and co-workers (33). Similar results were obtained when other models were used (J. Butler, M. Wegman, and R. E. Waugh, unpublished). For normal human red blood cells, the mean bending stiffness was 2.0 ± 0.2 × 10−19 J, and the work of separation Wsk was 0.078 ± 0.018 mJ/m2 for 10 cells tested.
FIGURE 6.

Tether force squared as a function of bilayer tension for control, antiglycophorin A liganded, and band 3 deficient human erythrocytes. Data were obtained using the magnetic tweezers approach. The bilayer tension was calculated using the Mohandas and Evans compressible skeleton model. The bending stiffness (kc) and the bilayer-skeletal separation energy (Wsk) were found from the slope and the  -intercept of the least squares linear fit to each data series.
-intercept of the least squares linear fit to each data series.
HS cells exhibited a similar value for the bending stiffness of the membrane (1.9 ± 0.4 × 10−19 J), but the separation work in these cells was not significantly different from zero (0.005 ± 0.017 × 10−19 mJ/m2). The rigidity of the R10 cells made it impossible to calculate the bilayer tension except at the highest values for the total membrane tension. (Numerical solutions for the bilayer tension could not be found for most cases because the skeletal rigidity accounted for essentially all the tension in the membrane.) Consequently, for these cells, there is little dependence of the equilibrium force on the membrane tension (Fig. 6) because even though the applied forces are causing an increase in the total membrane tension, the bilayer tension remains relatively low because of the large force resultants in the skeleton. Consequently, values for the bending stiffness of these membranes were not obtained. However, in the limit as the tension approaches zero, the extrapolated intercept provides a reliable estimate of the separation work, assuming that the bending stiffness of the membrane is approximately normal. For cells treated with mAb R10, the separation work was approximately twice that of normal cells. The values of the coefficients for each different cell type are summarized in Table 1.
TABLE 1.
Measurements of bending stiffness kc and skeletal separation energy Wsk
| kc (× 10−19 J) | Wsk (mJ/m2) | n | |
|---|---|---|---|
| Control | 2.0 ± 0.2 | 0.078 ± 0.018 | 10 |
| R10 bound | – | 0.138 ± 0.008 | 5 |
| Band 3 deficient | 1.9 ± 0.4 | 0.005 ± 0.017 | 14 |
Distribution of band 3 between cell body and tether
Tether formation is known to involve lateral segregation of lipid bilayer from the underlying membrane skeleton. Measurements of tether fluorescence were performed on five types of cells: normal cells with band 3 labeled with EMA, normal cells pretreated with DIDS and labeled with EMA, normal cells labeled with R10 monoclonal antibody and EMA, cells labeled with DiI, and cells labeled with Texas Red-PE. To the extent that band 3 is attached to or trapped by cytoskeletal associations, we expect there to be less band 3 appearing in the tether than would be expected for a molecule freely diffusing in the lipid membrane. Measurements shown in Fig. 7 confirm this expectation. The relative brightness of the tether in band 3 labeled cells is far less than the corresponding brightness in cells labeled with a lipid marker (DiI or Texas Red PE), indicating substantial lateral segregation of lipid from band 3 during tether formation. The specificity of the eosin label for band 3 was tested by first treating cells with the anion exchange inhibitor DIDS. This compound is known to block eosin maleimide labeling of band 3 (34). Comparison of the fluorescence intensity of cells with and without DIDS treatment (Fig. 7 b) indicates that 80% of the labeling was specific for band 3.
FIGURE 7.

Fluorescence images of tethered cells. (a) The first row shows short-exposure images used to obtain the fluorescence intensity on the cell body. Subsequent rows show the time course of fluorescence on the tether at 1, 10, 15, and 25 min, respectively. Cells in the left column were labeled with eosin maleimide; cells at the right were labeled with DiI. Fluorescence intensity was measured using Scion Image. Mean gray was determined for ROIs for the cell body or tether (see examples at the upper right). The mean background intensity was subtracted, and the total fluorescence obtained by multiplying by the area of the ROI. This value was divided by the area of the cell body or the tether to obtain the fluorescence intensity per unit membrane area on the cell body or the tether. The ratio of these two values provides a measure of the percentage of fluorophore concentration on the cell that is contained in the tether. These values are shown in Fig. 8. (b) When cells were pretreated with DIDS to block eosin labeling of band 3, fluorescence was reduced by 80%. Upper cell labeled with eosin without pretreatment, lower cell labeled with eosin after DIDS pretreatment. Exposure for the lower image was twice that of the upper image. (c) Treatment of cells with R10 antibody caused a significant reduction in tether fluorescence, indicating that little if any band 3 moved onto the tether for these cells. Upper image, cell treated with R10 mAb. Lower cell, no antibody treatment. Exposures in the two images are comparable. Scale bars, 5 μm.
The monoclonal antibody to glycophorin A (mAb R10) has been shown to reduce band 3 mobility and increase cell rigidity (14). When cells labeled with eosin maleimide and treated with mAbR10 were tested, little or no fluorescence was observed on the tether (Fig. 7 c). This indicates a more complete segregation of lipid from integral membrane protein in these cells and may explain the larger energies required to separate bilayer from skeleton in these cells (see Discussion).
The measurements depicted in Fig. 7 are quantitative in that they represent the ratio of fluorescence intensity per unit membrane area in the tether normalized by the intensity per unit membrane area on the cell. The time dependence of the ratio of fluorescence intensity in tethers to the fluorescence intensity of the cell body is summarized in Fig. 8. That this ratio for lipid labeled membranes is not statistically different from 1.0 and does not change over time matches expectations that lipid is more or less uniformly distributed between the membrane of the cell and the tether membrane. The mean ratio for the band 3 label eosin maleimide is 0.3–0.4, indicating that the majority of band 3 (60–70%) is confined to the cell body. The fluorescence intensity in the band 3 labeled tethers increases slightly with time, indicating that some band 3 initially trapped on the cell body may diffuse onto the tether over time. Unfortunately there is not sufficient resolution in these measurements to deduce a diffusion constant from this increase. Finally, the effect of mAb R10 treatment is clearly evident, as the normalized fluorescence intensity for these tethers approaches zero.
FIGURE 8.

Time course of tether fluorescence under different conditions. These are the averages and standard errors of the ratio of the fluorescence intensity per unit membrane area of the tether divided by the intensity on the cell body for 9 DiI-labeled tethers (open circles), 5 cells labeled with Texas Red PE (open squares), 3 cells treated with R10 antibody and labeled with eosin maleimide (solid triangles), and 11 labeled with eosin maleimide (solid circles), all shown as a function of time after tether formation. Note that both the lipid-labeled tethers (DiI and Texas Red PE) give a fluorescence ratio near 1.0 and that there is no significant variation in the mean values with time. This means that the fluorescence intensity per unit cell area is essentially the same for the cell body and the tether. For the eosin-labeled tethers, the fluorescence intensity per unit tether area is ∼40% of the intensity on the cell body, and there is a slight increase in tether intensity with time. Treatment of cells with mAb R10 reduced tether fluorescence to nearly zero. (Scale bars = 5 μm.)
DISCUSSION
Importance of bilayer-skeletal connectivity
From the earliest characterization of the structure of bilayer membranes as fluid mosaics, it was apparent that bilayer alone, as a two-dimensional fluid, could not account for the mechanical stability of the cell surface. The presence of an elastic network of membrane proteins at the intracellular surface of the bilayer was first indicated by the discovery of spectrin (35) and its associated proteins in the early 1970s. The important mechanical contribution that this network must make was also recognized and characterized at that time (3,36). Early observations of tether formation from cell surfaces were originally interpreted as a mechanical failure of the membrane skeleton itself (37), although subsequently it has been shown that this process involves the separation of membrane bilayer from the skeleton (16,38). The ability to measure the energy of separation by tether formation has created the opportunity to evaluate how specific molecular alterations affect the strength of the interaction.
The molecular connections between bilayer and skeleton primarily involve the protein ankyrin, which binds to both spectrin and the integral protein band 3 (39). Additional connections may exist in humans via the protein 4.1, which binds to glycophorin C and to the junctional complex formed around actin protofilaments. It is of interest that the gels from the patient show normal levels of these latter proteins, but the results here indicate almost complete loss of membrane stability. This argues for a dominant role for band 3 in maintaining the associations between the membrane bilayer and the membrane-associated cytoskeleton.
Importance of membrane stability for proper cell function
Relation between membrane stability and lifespan
Studies of red cell aging in mice have revealed a critical need for cells to maintain a proper surface area/volume (S/V) ratio (1). In acute studies, it was found that red blood cells with surface area removed responded by decreasing their volume to restore the S/V ratio and consequently regain levels of cellular deformability (2). It was also found that the extent of surface loss correlated with the long-term survival of the cells (1). It appears likely that accelerated surface loss as a result of molecular abnormalities is the underlying mechanism in many inherited anemias.
One of the more common inherited red blood cell disorders, HS (40,41), is characterized by abnormal osmotic fragility of mature red cells (42). This increased fragility reflects a decrease in the S/V ratio of the cell. Several molecular abnormalities have been associated with these disorders, many of which result in a reduced concentration of spectrin on the cell membrane. Spectrin deficiency has been shown to be associated with a weakness in the elastic strength of the membrane (11) and could also lead to a lessening of the force required to separate the lipid bilayer from the membrane skeleton (12). Bilayer-skeletal separation seems to be the most likely mechanism for the loss of red cell surface area with respect to the volume that presumably leads to increased fragility of the cells (9). Furthermore, the ability of erythrocytes to deform into small vessels is inhibited by the reduction in excess membrane area, which also has the effect of premature red cell removal from the circulation when the cells are unable to pass through splenic-sinus gaps. This latter effect is obvious from the improved condition of anemic HS patients after splenectomy (8,13,42).
Although anemia is not a ubiquitous symptom of HS, the band 3 deficient human blood studied in this work had a low hematocrit indicative of chronic anemia. In such cases the increased rate of erythropoeisis, typically seen in HS (43) and evidenced by the high reticulocyte levels in these samples, was not sufficient to compensate for the shortened cell lifetimes. In this study, we have shown for the first time to our knowledge that red blood cells from band 3 deficient membranes required lower forces to pull membrane material off the cells. This is the first documentation to our knowledge of reduction in the strength of bilayer-skeletal adhesion in red cells with integral membrane protein deficiencies.
The flip side: increased stability when band 3 interactions are increased
The importance of band 3 in determining the stability of the bilayer-skeletal interactions is underlined by the results in this study showing a marked increase in the energy of bilayer-skeletal association when interactions between band 3 and the underlying skeleton are enhanced. Prior studies have demonstrated that the monoclonal antibodies used in the experiments here markedly reduced the lateral mobility of band 3 on the membrane and (as confirmed here) increased the elastic rigidity of the membrane substantially (14,15). That this also increases bilayer-skeletal association has potential implications for the physical mechanism by which cells “hang onto” their membranes. As we (16) and others (44) proposed previously, lateral separation of lipid membrane components from integral proteins during tether formation results in a difference in lipid chemical potential analogous to the difference in chemical potential of water across a semipermeable membrane.
This “osmotic membrane tension” would tend to pull lipid from the tether back to the cell body and could account for much of the energy cost of tether formation from skeletally linked bilayers (16). As shown in this study, immobilization of band 3 by ligation of the mAb R10 results in greater retention of band 3 on the cell surface, thus increasing the concentration difference between the cell body and the tether and increasing the energy cost of tether formation. Our presumption is that increasing the number of species retained on the cell body will not increase the number of other mobile species that may still be transported onto the tether but that these other components will continue to seek their own chemical equilibrium. This rationale is consistent with our observations that the bilayer-skeletal separation energy is approximately double in cells labeled with R10.
Dynamic resistance to tether formation
Although it was not a goal of this study to examine the dynamic resistance of the membrane to tether formation, measurements of the magnitude of the force during tether extraction and the magnitude of force relaxation after tether extraction suggest that band 3 contributes substantially to this dynamic resistance. For the mechanical measurements of tether formation, tethers were extracted manually, i.e., by displacing the pipette with a manipulator by hand. Consequently, the tether formation rate was not steady. Furthermore, the operator adjusted the rate during pulling to maintain a relatively low force on the tether to avoid breaking it. Nevertheless, we were able to determine average pulling rates for the three different types of cells tested: 0.06–0.09 μm/s for band 3 deficient cells, 0.035–0.05 μm/s for normal cells, and 0.025–0.035 μm/s for R10-labeled cells. We were also able to estimate the percentage by which the force decreased when pulling was stopped and the system was allowed to come to steady state. This indirectly reflects the magnitude of the dynamic contribution to the force during pulling. The force for band 3 deficient cells dropped by ∼25%, whereas the force for the R10 cells decreased by ∼50%. This is the opposite of what one would expect based on the faster pulling rates for the band 3 deficient cells. It is also important to note that the force magnitudes were substantially higher for the R10 cells than for the band 3 deficient cells. Taken together, these observations support theoretical predictions (45–47) that increasing the concentration of integral proteins adds substantially to the dynamic resistance of the membrane to tether formation.
Implications about the mobility of band 3
The proportion of band 3 found in the tethers in these experiments are consistent with other estimates of the proportion of mobile band 3 on the native red cell membrane (48). This suggests that the band 3 that does appear in the tether is a result of the native diffusibility of the molecules on the surface. A recent report suggests that tether formation from red cells may involve a rate-dependent tearing away of band 3 from its membrane-skeletal anchorage (45). In this study, tether formation rates were typically <0.1 μm/s. This is below the threshold estimated by those authors for separation of band 3 from the membrane skeleton by mechanical loading. Thus, our finding that tether formation appears not to augment the presence of “free” band 3 in the tether does not contradict their theory. A more critical test would be to form tethers at higher rates and determine if the surface concentration of band 3 increases with formation rate. Unfortunately, such data are not yet available.
This report documents a critical role for band 3 skeletal interactions in maintaining the mechanical stability of red cell membranes. Given the evidence that associations between glycophorin C and junctional complexes within the skeleton may also contribute to bilayer-skeletal association, this result may not have been expected. Waugh and Agre (11) found a correlation between the spectrin content and the shear modulus in spectrin-deficient cells, and it is interesting to speculate whether a similar correlation might exist between membrane skeletally linked integral proteins (e.g., band 3) and the separation work Wsk. Unfortunately the paucity of subjects with this abnormality makes it impossible to draw firm conclusions. Nevertheless, the association energy in membranes lacking band 3 approaches zero, indicating a critical role for band 3 in governing membrane stability.
Acknowledgments
The authors gratefully acknowledge the technical support of Richard Bauserman and Donna Brooks and thank Andrew Rape and Martin Wegman for assistance with data analysis.
This work was supported by the U.S. Public Health Service under National Institutes of Health grant Nos. HL 31524 (to R.E.W.) and DK26263, DK32094, and HL31579 (to N.M.).
James Butler's present address is Nikon Instruments, 1300 Walt Whitman Rd., Melville, NY 11747.
Editor: Thomas J. McIntosh.
References
- 1.Murdock, R. C., C. Reynolds, I. H. Sarelius, and R. E. Waugh. 2000. Adaptation and survival of surface-deprived red blood cells in mice. Am. J. Physiol. Cell Physiol. 279:C970–C980. [DOI] [PubMed] [Google Scholar]
- 2.Waugh, R. E., and I. H. Sarelius. 1996. Effects of lost surface area on red cells and red cell survival in mice. Am. J. Physiol. 271:C1847–C1852. [DOI] [PubMed] [Google Scholar]
- 3.Evans, E. A., and R. Skalak. 1979. Mechanics and thermodynamics of biomembranes. CRC Crit. Rev. Bioeng. 3:181–418. [PubMed] [Google Scholar]
- 4.Singer, S. J., and G. L. Nicolson. 1972. The fluid mosaic model of the structure of cell membranes. Science. 175:720–731. [DOI] [PubMed] [Google Scholar]
- 5.Evans, E. A., and R. M. Hochmuth. 1977. A solid-liquid composite model of the red cell membrane. J. Membr. Biol. 30:351–362. [DOI] [PubMed] [Google Scholar]
- 6.Palek, J., and S. E. Lux. 1983. Red cell membrane skeletal defects in hereditary and acquired hemolytic anemias. Semin. Hematol. 20:189–224. [PubMed] [Google Scholar]
- 7.Gallagher, P. G., and B. G. Forget. 1993. Spectrin genes in health and disease. Semin. Hematol. 30:4–21. [PubMed] [Google Scholar]
- 8.Walensky, L. N. Mohandas, and S. E. Lux. 2003. Disorders of the red blood cell membrane. In Principles and Practice of Hematology. R. I. Handin, S. E. Lux, and T. P. Stossel, editors. Lippincott, Williams & Wilkins, Philadelphia. 1709–1858.
- 9.Mohandas, N., and E. A. Evans. 1994. Mechanical properties of the red cell membrane in relation to molecular structure and genetic defects. Annu. Rev. Biophys. Biomol. Struct. 23:787–818. [DOI] [PubMed] [Google Scholar]
- 10.Takakuwa, Y., G. Tchernia, M. Rossi, M. Benabadji, and N. Mohandas. 1992. Restoration of normal membrane stability to unstable protein 4.1 deficient erythrocyte membranes by incorporation of purified protein 4.1. J. Clin. Invest. 78:80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waugh, R. E., and P. Agre. 1988. Reductions of erythrocyte membrane viscoelastic coefficients reflect spectrin deficiencies in hereditary spherocytosis. J. Clin. Invest. 81:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chasis, J. A., P. Agre, and N. Mohandas. 1988. Decreased mechanical stability and in vivo loss of surface area reflect spectrin deficiencies in hereditary spherocytosis. J. Clin. Invest. 82:617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agre, P., J. F. Casella, W. H. Zinkham, C. McMillan, and V. Bennett. 1985. Partial deficiency of spectrin in hereditary spherocytosis. Nature. 314:380–383. [DOI] [PubMed] [Google Scholar]
- 14.Chasis, J. A., N. Mohandas, and S. B. Shohet. 1985. Erythrocyte membrane rigidity induced by glycophorin A-ligand interaction. Evidence for a ligand-induced association between glycophorin A and skeletal proteins. J. Clin. Invest. 75:1919–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knowles, D. W., J. A. Chasis, E. A. Evans, and N. Mohandas. 1994. Cooperative action between band 3 and glycophorin A in human erythrocytes: Immobilization of band 3 induced by antibodies to glycophorin A. Biophys. J. 66:1726–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waugh, R. E., and R. G. Bauserman. 1995. Physical measurements of bilayer-skeletal separation forces. Ann. Biomed. Eng. 23:308–321. [DOI] [PubMed] [Google Scholar]
- 17.Waugh, R. E., A. Mantalaris, R. G. Bauserman, W. C. Hwang, and J. H. Wu. 2001. Membrane instability in late-stage erythropoiesis. Blood. 97:1869–1875. [DOI] [PubMed] [Google Scholar]
- 18.Hochmuth, R. M., and W. D. Marcus. 2002. Membrane tethers formed from blood cells with available area and determination of their adhesion energy. Biophys. J. 82:2964–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perrotta, S., A. Borriello, A. Scaloni, L. De Franceschi, A. M. Brunati, F. Turrini, V. Nigro, E. M. del Giudice, B. Nobili, M. L. Conte, F. Rossi, A. Iolascon, A. Donella-Deana, V. Zappia, V. Poggi, W. Anong, P. Low, N. Mohandas, and F. Della Ragione. 2005. The N-terminal 11 amino acids of human erythrocyte band 3 are critical for aldolase binding and protein phosphorylation: implications for band 3 function. Blood. 106:4359–4366. [DOI] [PubMed] [Google Scholar]
- 20.Anstee, D. J., and P. A. Edwards. 1982. Monoclonal antibodies to human erythrocytes. Eur. J. Immunol. 12:228–232. [DOI] [PubMed] [Google Scholar]
- 21.Guilford, W. H., and R. W. Gore. 1992. A novel remote-sensing isometric force transducer for micromechanics studies. Am. J. Physiol. 263:C700–C707. [DOI] [PubMed] [Google Scholar]
- 22.Heinrich, V., and R. E. Waugh. 1996. A piconewton force transducer and its application to measurement of the bending stiffness of phospholipid membranes. Ann. Biomed. Eng. 24:595–605. [DOI] [PubMed] [Google Scholar]
- 23.Tees, D. F., R. E. Waugh, and D. A. Hammer. 2001. A microcantilever device to assess the effect of force on the lifetime of selectin-carbohydrate bonds. Biophys. J. 80:668–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans, E., and A. Yeung. 1994. Hidden dynamics in rapid changes of bilayer shape. Chem. Phys. Lipids. 73:39–56. [Google Scholar]
- 25.Discher, D. E., N. Mohandas, and E. A. Evans. 1994. Molecular maps of red cell deformation: hidden elasticity and in situ connectivity. Science. 266:1032–1035. [DOI] [PubMed] [Google Scholar]
- 26.Waugh, R. E., and R. M. Hochmuth. 1987. Mechanical equilibrium of thick hollow liquid membrane cylinders. Biophys. J. 52:391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waugh, R., and E. A. Evans. 1979. Thermoelasticity of red blood cell membrane. Biophys. J. 26:115–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leblond, P. F., A. de Boisfleury, and M. Bessis. 1973. [Erythrocytes shape in hereditary spherocytosis. A scanning electron microscopic study and relationship to deformability (author's transl)]. Nouv. Rev. Fr. Hematol. 13:873–883. [PubMed] [Google Scholar]
- 29.Delhommeau, F., T. Cynober, P. O. Schischmanoff, P. Rohrlich, J. Delaunay, N. Mohandas, and G. Tchernia. 2000. Natural history of hereditary spherocytosis during the first year of life. Blood. 95:393–397. [PubMed] [Google Scholar]
- 30.Paulitschke, M., G. B. Nash, D. J. Anstee, M. J. Tanner, and W. B. Gratzer. 1995. Perturbation of red blood cell membrane rigidity by extracellular ligands. Blood. 86:342–348. [PubMed] [Google Scholar]
- 31.Discher, D. E., and N. Mohandas. 1996. Kinematics of red cell aspiration by fluorescence-imaged microdeformation. Biophys. J. 71:1680–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golan, D. E., J. D. Corbett, C. Korsgren, H. S. Thatte, S. Hayette, Y. Yawata, and C. M. Cohen. 1996. Control of band 3 lateral and rotational mobility by band 4.2 in intact erythrocytes: release of band 3 oligomers from low-affinity binding sites. Biophys. J. 70:1534–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Discher, D. E., D. H. Boal, and S. K. Boey. 1998. Simulations of the erythrocyte cytoskeleton at large deformation. II. Micropipette aspiration. Biophys. J. 75:1584–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cobb, C. E., and A. H. Beth. 1990. Identification of the eosinyl-5-maleimide reaction site on the human erythrocyte anion exchange protein: overlap with the reaction sites of other chemical probes. Biochemistry. 29:8283–8290. [DOI] [PubMed] [Google Scholar]
- 35.Tillack, T. W., S. L. Marchesi, V. T. Marchesi, and E. Steers Jr. 1970. A comparative study of spectrin: a protein isolated from red blood cell membranes. Biochim. Biophys. Acta. 200:125–131. [DOI] [PubMed] [Google Scholar]
- 36.Evans, E. A., and R. M. Hochmuth. 1978. Mechanochemical properties of membranes. In Current Topics in Membranes and Transport, Vol. 10. F. Bronner and A. Kleinzeller, editors, Academic Press, New York. 1–64.
- 37.Evans, E. A., and R. M. Hochmuth. 1976. Membrane viscoplastic flow. Biophys. J. 16:13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berk, D. A., and R. M. Hochmuth. 1992. Lateral mobility of integral proteins in red blood cell tethers. Biophys. J. 61:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bennett, V., and P. J. Stenbuck. 1979. The membrane attachment protein for spectrin is associated with band 3 in human erythrocyte membranes. Nature. 280:468–473. [DOI] [PubMed] [Google Scholar]
- 40.Becker, P. S., and S. E. Lux. 1985. Hereditary spherocytosis and related disorders. Clin. Haematol. 14:15–43. [PubMed] [Google Scholar]
- 41.Weed, R. I. 1975. Hereditary spherocytosis. A review. Arch. Intern. Med. 135:1316–1323. [DOI] [PubMed] [Google Scholar]
- 42.Cynober, T., N. Mohandas, and G. Tchernia. 1996. Red cell abnormalities in hereditary spherocytosis: relevance to diagnosis and understanding of the variable expression of clinical severity. J. Lab. Clin. Med. 128:259–269. [DOI] [PubMed] [Google Scholar]
- 43.Guarnone, R., E. Centenara, M. Zappa, A. Zanella, and G. Baroli. 1996. Erythropoietin production and erythropoiesis in compensated and anaemic states of hereditary spherocytosis. Br. J. Haematol. 92:150–154. [DOI] [PubMed] [Google Scholar]
- 44.Dai, J. W., and M. P. Sheetz. 1995. Mechanical properties of neuronal growth cone membranes studied by tether formation with laser optical tweezers. Biophys. J. 68:988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borghi, N., and F. Brochard-Wyart. 2007. Tether extrusion from red blood cells: integral proteins unbinding from cytoskeleton. Biophys. J. 93:1369–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bussell, S. J., D. L. Koch, and D. A. Hammer. 1995. Effect of hydrodynamic interactions on the diffusion of integral membrane proteins: diffusion in plasma membranes. Biophys. J. 68:1836–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dodd, T. L., D. A. Hammer, A. S. Sangani, and D. L. Koch. 1995. Numerical simulations on the effect of hydrodynamic interactions on diffusivities of integral membrane proteins. J. Fluid Mech. 293:147–180. [Google Scholar]
- 48.Golan, D. E. 1989. Red blood cell membrane protein and lipid diffusion. In Red Blood Cell Membranes: Structure, Function, Clinical Implications. P. Agre and J. C. Parker, editors. Marcel Dekker, New York. 367–400.