

Abstract
The Golgi apparatus is comprised of stacked cisternal membranes forming subcompartments specialized for posttranslational processing of newly synthesized secretory cargo. Recent experimental evidence indicates that the Golgi apparatus can undergo de novo biogenesis from the endoplasmic reticulum, but the mechanism by which the membranes self assemble into compartmentalized structures remains unknown. We developed a discrete-event computer simulation model to test whether two fundamental mechanisms—vesicle-coat-mediated selective concentration of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins during vesicle formation, and SNARE-mediated selective fusion of vesicles—suffice to generate and maintain compartments. Simulations verified that this minimal model is adequate for homeostasis of preestablished compartments, even in response to small perturbations, and for de novo formation of stable compartments. The model led to a novel prediction that Golgi size is in part dependent on target SNARE expression level. This prediction was supported by a demonstration that exogenous expression of the Golgi target SNARE syntaxin-5 alters Golgi size in living cells.
INTRODUCTION
Subcellular compartments optimize reactions by creating specialized environments and increasing surface area, and they dramatically increase regulatory potential. The Golgi apparatus provides a striking example of the compartmentalization and its dynamic aspects. The Golgi apparatus forms an array of stacked subcompartments carrying out sequential posttranslational modifications of secretory proteins and lipids (1). Golgi compartments maintain their identity despite a high degree of membrane and protein flux, and they can undergo sustained and, in some cases, dramatic changes in size due to altered differentiation states and cargo expression (2–5) . Further, the Golgi apparatus reversibly disassembles during cell division or in response to stress (6,7) and, significantly, it is capable of assembling de novo from the endoplasmic reticulum (ER), indicating that the mechanisms that establish and maintain the Golgi apparatus are inherently capable of self-assembly (8).
How is self-assembly, growth, and maintenance of Golgi compartments achieved? The core reactions are thought to be vesicle budding and fusion, which mediate exchange between biosynthetic secretory compartments (9). Vesicle budding is driven by soluble coat complexes that assemble at discrete locations on a donor compartment, selectively capture cargo-containing export signals, and deform the membrane into a curved structure to promote scission of the cargo-laden vesicle from the donor compartment (10). Cognate vesicle (v) and target (t) soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) then target the vesicles and promote fusion to a specific acceptor compartment, thereby completing cargo transfer (11).
It is possible that different coats have different affinities for various SNAREs and that this difference in SNARE-coat affinity could result in SNAREs being selectively targeted to different compartments, in turn resulting in gradients of SNARE concentrations along the secretory pathway (12,13). Essentially, the exit rate of a SNARE with a higher affinity for a particular vesicle coat will exceed that of a SNARE with a lower affinity, and this effect, acting over several compartments, would establish the differential SNARE gradient. It is, however, not obvious from first principles whether selective differences in SNARE-coat affinities alone would be sufficient to establish and maintain discrete compartment identities. In particular, de novo biogenesis of the Golgi apparatus requires a starting point at which all relevant SNAREs are inter-mixed in the ER membrane and selective SNARE targeting by itself offers no mechanistic explanation for the emergence of distinct identities from a single initial compartment. As SNAREs emerge from the ER in budded vesicles, new compartments must be established with their steady-state SNARE gradients despite the fact that, at least initially, cognate SNARE pairing will drive inappropriate fusion with the ER due to its contamination with Golgi tSNAREs. Even after compartments are established, a fraction of all SNAREs are mislocalized—due to imperfect sorting, transient presence of vSNAREs in target compartments after vesicle fusion, and SNARE replenishment beginning at the ER—thus creating the possibility of unwanted fusion that might degrade compartmentalization.
Computer simulations, which have proven to be a powerful tool for understanding many complex self-organizing systems in biology, have begun to show promise for distinguishing among theories of Golgi operation (14–16). However, to our knowledge, there is not yet a mechanistic model that can simultaneously explain de novo biogenesis of the Golgi and its steady-state maintenance. Although there have been great strides in improving models of cellular biochemistry in recent years, these models nonetheless fall short of what is needed for capturing realistic de novo Golgi behavior. The most widely accepted computational models for cellular simulation are those based on continuum mass action models, which represent simulation progress as the evolution of a system of coupled differential equations. Such a model was applied to great effect in simulating the restoration and maintenance of compartment identities in the Golgi (16) by abstracting the exchange of material through vesicle-mediated transport as a continuous flux of membrane and proteins between compartments. However, a continuum model does not provide any obvious way to incorporate initiation of new compartments, which is inherently a discrete, stochastic event. Stochastic simulation (Gillespie) models (17) provide a means of incorporating discretization and reaction noise into a reaction system by representing reaction progress as a series of discrete reaction steps occurring at random time intervals and affecting discrete, finite sets of reactants. These capabilities have recently led to the wide embrace of stochastic simulation methods for general simulation of cellular biochemistry (18–21). A true Gillespie model, however, would also be poorly suited to describing the exchange of material through vesicle formation and fusion. A fully discrete model of the process would require simulating the exchange of individual lipid and protein molecules with forming vesicles, making it intractable for the timescale of de novo formation of a full Golgi apparatus.
In this article, we develop a hybrid model of Golgi formation and maintenance designed to test whether a minimal model of vesicle trafficking involving selective concentration of protein components into vesicles and selective fusion of these vesicles to target compartments is sufficient to explain de novo biogenesis of Golgi-like compartments. The simulator uses a coarse-grained variant of the Gillespie model in which vesicle budding and fusion are treated as single reaction events. However, the exchange of membrane and protein content between vesicles and compartments before budding and after fusion are treated as deterministic, continuum processes. This approach allows us to examine discontinuous compartment formation events that are not accessible to a pure continuum model but are essential to our focus on Golgi biogenesis without sacrificing the tractability of the continuum approach. It further implicitly captures stochastic behaviors that may be significant to the robustness and parameter sensitivity of the system. The simulation experiments herein show that compartment biogenesis and homeostasis can be explained by the core membrane trafficking reactions. They further lead to testable predictions regarding parameters that determine trafficking kinetics and compartment size, which are consistent with the hypothesis that interaction affinity between vesicle coat complexes and SNARE molecules establishes and controls the size of Golgi compartments (5). We validate the model by demonstrating that Golgi size can be controlled by SNARE syntaxin 5 expression levels, as the model predicts.
METHODS
Model
Our computer model keeps track of a discrete set of membrane structures occupying one of three classes: vesicle, vesicular tubular cluster (VTC), and compartment. Each structure has a complement of three kinds of proteins—guanine-nucleotide exchange factors (GEFs), which initiate vesicle budding; vSNAREs, which mark vesicles and control their target preference; and tSNAREs, which mark compartments and act with vSNAREs to determine the relative probability of a particular vesicle binding to a particular target. Each of these proteins has one of a small number of “identities” that correspond to the compartment with which a given protein is preferentially associated. Membrane and protein are exchanged among structures through budding and fusion of vesicles, with vesicle budding rates determined by the budding structure's GEF and contaminant protein concentrations, and fusion times and probabilities dependent on vesicle and target SNARE marker concentrations. Each vesicle also has an identity that is determined by the GEF initiating its budding and that determines which vSNARE labels it, as well as which marker proteins it selectively concentrates. Each vesicle contains proteins of all identities, but preferentially concentrates a subset of the proteins based on its own identity. Successive fusions of vesicles will produce VTCs, which can in turn be upgraded to compartments upon accumulating sufficient membrane mass, making possible the emergence of new compartments de novo.
The model is illustrated in Fig. 1. Fig. 1 A illustrates the budding reaction, which is initiated by a GEF protein that determines the budded vesicle's vSNARE marker and identifies the subset of compartment markers that the vesicle selectively concentrates during its budding. Fig. 1 B shows the fusion reaction, which is presumed to be driven by pairing of cognate vesicle vSNAREs with target compartment tSNAREs. The model assumes that the choice of target will therefore depend on the relative tSNARE complements of the different compartments, VTCs, or other vesicles. Fig. 1 C gives a high-level picture of the desired steady-state behavior of the model. Each compartment, vesicle, or VTC in the system contains some concentration of every marker present, but is identified by relatively higher concentration of markers of a particular identity. Precise details of this model are explained in the Methods section below.
FIGURE 1.

(A) Schematic diagram of the simplified Golgi model assumed in our simulator. The model uses three compartment identities in this work, corresponding to ER and cis and trans Golgi compartments. Coloring represents a sample distribution of the GEFs (G1–G3) and tSNAREs (T1–T3) corresponding to each compartment. Vesicles are marked by their respective vSNAREs (V1–V3). VTCs can act as an intermediate form between vesicles and full compartments in the generation of new compartments. Note that the example shown illustrates a vesicle emerging from the ER enriched in both cis and trans components. Homotypic fusion of such vesicles will produce VTCs with similar mixed identities. (B) Coat (C1) recruitment is determined by compartment GEF concentration. Coats form vesicles containing components based on sorting affinities and component abundance. (C) Vesicles bearing particular vSNAREs fuse with other vesicles, VTCs, or compartments in proportion to the abundance of cognate tSNAREs.
For these experiments, we ran the model with three protein identities and thus potentially three distinct compartments. We refer to these three compartments in our discussion below as the ER, cis-Golgi, and trans-Golgi. However, it is important to note that the model itself makes no inherent distinction between the compartments. The compartment labeled ER is distinguished from the other two by the initial conditions of the simulations, either by having a larger initial size or by being the sole compartment in experiments on de novo Golgi formation. The cis-Golgi and trans-Golgi distinctions are arbitrary in our model and are based only on which of two equivalent but distinct markers has the higher concentration. We further note that the model itself can handle arbitrary numbers of identities and has verifiably generated stable compartments for up to five distinct identities (data not shown), although we limited ourselves to three here because a three-identity model is the simplest variant capable of modeling the separation of multiple distinct Golgi identities from a starting ER identity.
Computer simulation methods
Our simulations combine a stochastic discrete-event model at the scale of vesicle budding and fusion with a deterministic continuum model of exchange of proteins and membrane during the budding and fusion steps. In a discrete-event model, a simulation can be described by specifying the quantities that define the status of the system (called the state) and the operations that can change the state (called events). The state of our system encodes a finite set of membrane structures of three kinds:
Compartments, which represent the ER and subcompartments of a Golgi stack
Vesicles, which represent COPI- or COPII-derived vesicles in the process of transport
VTCs, an intermediate state formed by vesicle-vesicle fusion that has not yet reached sufficient size to become a compartment
Each such structure has a current size, encoded internally as a radius but representing the total volume it encloses. It further contains concentrations of three kinds of proteins:
GEFs, which are presumed to initiate budding of vesicles and thus influence the rate of budding
vSNAREs, which mark vesicle identities and, with tSNAREs, determine the rate and selectivity of vesicle fusion events
tSNAREs, which mark compartment identities and, with vSNAREs, determine the rate and selectivity of vesicle fusion events.
Each kind of protein comes in one of a fixed number of “identities” specified at the start of a simulation, which control how the proteins are sorted during vesicle formation and thus, in principle, should determine which proteins sort to common compartments. In this way, for example, a simulation will have a GEF1, which initiates the formation of vSNARE1-marked vesicles, which in turn target tSNARE1-marked compartments. It will likewise have a GEF2 with a similar relationship to vSNARE2 and tSNARE2, and so on. The complete state of a simulation is then described by the full complement of membrane structures present in the simulation, the sizes of each of those structures, and the concentration of each type of protein in each structure. Fig. 2 provides an example of a hypothetical state that might be encountered in a simulation with three protein identities.
FIGURE 2.

Illustration of a hypothetical simulation state. The state is defined by a set of compartments, VTCs, and vesicles. Each of these objects has a defined size and a defined concentration of each of three types of the three protein classes (vSNARE, tSNARE, and GEF).
The simulation state evolves through two kinds of events: vesicle budding and vesicle fusion. Each of these is treated as an instantaneous event but with an exponentially distributed waiting time, as in a standard stochastic simulation algorithm model (17). Exchange of individual proteins during budding and fusion is, however, assumed to proceed deterministically to equilibrium, as described below. The result is therefore a coarse-grained hybrid model that allows us to look at millions of vesicle budding and fusion events by neglecting the details of molecular interactions during the budding and fusion process.
The discrete budding reaction represents the overall process of formation of a vesicle from a budding compartment and exchange of proteins between the compartment and vesicle. Vesicles may bud from any compartment or VTC with a radius >60 nm. The model implicitly assumes that during the vesicle budding process, each protein type rapidly exchanges between the vesicle and compartment. If we denote a given protein by P before vesicle formation, P′ when found in the compartment during vesicle formation, and Pv when found in the vesicle during vesicle formation, then we can describe the exchange process in terms of a reversible first-order reaction:
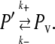 |
We then have keq = k+/k−, which describes the equilibrium ratio of protein concentration in vesicle versus compartment, keq = [Pv]eq/[P′]eq. The value of keq, and thus of [Pv]eq/[P′]eq, depends on the specific protein and vesicle type, as described below. Our model assumes that this exchange reaction goes to completion before the vesicle completes budding. We further assume for these experiments that vesicles always have a fixed radius of 60 nm and, thus, a surface area of Av = 14,400π nm2. Additional experiments beyond those presented here (data not shown) verified that the results of these tests do not vary substantially with changes in the presumed vesicle radius. After budding, the volume of the compartment is set such that the total volume of the vesicle and postbudding compartment are equal to the volume of the prebudding compartment. The postbudding surface area, A′, is then estimated from the volume by assuming spherical compartments. Given the preceding assumptions, we can compute [P′]eq and [Pv]eq analytically as follows:
 |
(1) |
 |
(2) |
Because our model assumes that the exchange reactions go to equilibrium for each protein P, we can thus fix [P′] = [P′]eq and [Pv] = [Pv]eq postbudding. The keq value that models the sorting affinity of a given vesicle type for a given protein is a parameter of the model. We assume that there is a single user-supplied keq > 1 that is used for all proteins that are selectively concentrated by a given vesicle type, and that keq = 1 for all other proteins. Note that the proteins selectively concentrated for a vesicle type are those proteins that are targeted for export from the compartment with the corresponding identity. Thus, keq > 1 ensures that the budded vesicle will have a higher concentration of those proteins than the compartment from which it buds. Any protein not targeted for export, because it has keq = 1, will have the same concentration in the budding vesicle as in the compartment from which it buds. Thus, every vesicle, regardless of its identity, will contain every protein in varying concentrations, but will preferentially concentrate a subset of proteins targeted for export from the compartment. The assumption that keq is never <1 is equivalent to assuming that there is no mechanism for active retention to prevent compartment markers from being exported during budding. Collectively, these equations describe one key assumption of the model, that vesicles selectively concentrate a subset of cargoes with some sorting affinity, keq, and enforce conservation of protein content given that assumption.
Although the exchange of proteins during budding is modeled as a deterministic process, the time of budding is handled stochastically. Times between events are distributed according to exponential random variables, sampled as in the stochastic simulation algorithm first-reaction method (17). Budding-event times are chosen based on protein complement of the budding compartment. Vesicle budding is presumed to be initiated by one of the GEF types and thus to have first-order dependence on the GEF concentration. It is further assumed that the budding rate depends on the presence of the identifying vSNARE of the vesicle that is determined by the GEF initiating its budding, and which must be present to decorate the vesicle surface. The overall rate at which vesicles of a given type, i, bud from a compartment is then given by
 |
(3) |
where r is the compartment radius and c is a scaling constant set to 10−6 to approximately balance overall budding and fusion rates in the experiments described here. When a given GEF_i buds a vesicle of type i, that vesicle selectively concentrates all proteins not native to compartment i. The assertion that budding rate is modulated by a contaminating vSNARE protein concentration derives from a prior observation that the model can produce stable steady-state behavior but not de novo biogenesis in the absence of some such mechanism for cargo-modulated exit-rate control. This observation is backed up by experimental observation of a cargo-based exit rate control mechanism in yeast (5). The specific choice of vSNAREs, as opposed to tSNAREs, GEFs, or some other factor omitted from our model, as the control mechanism is arbitrary; the various markers sort together and we believe any one would be an effective proxy for the others.
Fusion of membranes is simulated by an analogous coarse-grained model, with stochastic waiting time between fusion events but instantaneous fusion once the waiting time is over. A fusion event may involve two vesicles or VTCs, or a compartment and one vesicle or VTC. Vesicle fusion is presumed to be cooperative, with rate equal to
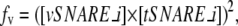 |
(4) |
where [vSNARE_i] is the vesicle's marker vSNARE concentration and [tSNARE_i] is the target compartment's corresponding marker tSNARE concentration. Vesicles are thus presumed to be decorated with only a single active vSNARE and have fusion rates determined only by interactions of that vSNARE with its cognate tSNARE on the target vesicle. This identifying vSNARE is determined by the identity of the GEF that initiated the vesicle's budding, with each GEF presumed by the model to select a single vSNARE for activation. VTCs and compartments, by contrast, are presumed to be able to fuse via any vSNAREs and tSNAREs they contain, and thus have fusion rates equal to the sum over all v-t-SNARE types of the square product of vSNARE concentration in one structure and tSNARE concentration in the other:
 |
(5) |
VTCs and compartments thus do not have a definite identity except that encoded in the relative concentrations of their markers. For purposes of plotting simulation results, we label each compartment according to the identity of its tSNARE of highest concentration. The cooperativity of the fusion reaction is based in part on prior observations using the model (not shown) that it does not produce successful de novo biogenesis without a cooperative fusion. In the absence of cooperativity, vesicles budding from an ER contaminated with Golgi markers preferentially back-fuse to the ER rather than to one another. The cooperativity of fusion is experimentally supported by results from Stewart et al. (22). The specific use of a second-order reaction is meant only to model cooperativity qualitatively, and we have no empirical basis for asserting that the reaction is second-order versus any other order. In a fusion event, two structures merge, pooling their membrane and protein complements.
Structures can be “promoted” from vesicle to VTC or VTC to compartment based on increases in mass. If a fusion of vesicles reaches a size of at least five vesicles, then it is reclassified as a VTC. If a fusion of vesicles and/or VTCs reaches a size of at least 10 vesicles, then it is reclassified as a compartment. The primary distinction between vesicle and VTC is that a VTC can bud new vesicles, whereas a vesicle cannot. The distinction between VTCs and compartments is that VTCs can fuse with vesicles, other VTCs, or compartments, whereas compartments can fuse only with vesicles and VTCs, not with other compartments. We have no mechanistic hypothesis for how this prohibition on compartment/compartment fusion would be enforced, but some such prohibition appears to be necessary to prevent back-fusion of nascent compartments to the ER in the model. Furthermore, as noted in the preceding paragraph, vesicles have only a single active vSNARE, which defines a discrete vesicle identity, whereas VTCs and compartments can use all of their vSNARE and tSNARE complement for fusion and have no discrete identities except those implied by their relative marker concentrations.
The overall operation of the simulator is described in pseudocode in Fig. 3. The pseudocode presents a high-level description of the central event loop of the simulator. Each pass through the loop results in one simulation event, either budding a new vesicle or fusing two structures (vesicle, VTC, or compartment). The simulator first generates the set of possible candidate budding events (lines 1 and 2) and fusing events (lines 3–5). It then chooses the event with the shortest waiting time (line 6). It then updates the simulation state to reflect that next event (lines 7–10) and updates the system timer (line 11). Finally, it returns to the start of the loop to sample new possible events using the update state (line 12).
FIGURE 3.

Pseudocode describing the central event loop of the simulator. The simulator considers all possible budding and fusion events, sampling a waiting time for each from an exponential distribution whose rate depends on protein concentrations in the participating vesicles, VTCs, or compartments. The simulator then selects the event with the smallest time for execution before returning to the start of the loop to identify the next event.
We can illustrate the operation of the model with a simple example. Suppose we start a simulation with a single compartment with concentration 1.0 of all proteins and surface area 28,800π nm2 (equivalent to the surface area of two vesicles of radius 60 nm). We further assume a sorting affinity of 10. The compartment has three possible budding events. The first possibility is budding through GEF1 with rate  The other possibilities, budding through GEF2 or GEF3, have the same rate by symmetry. The simulator will sample three exponentially distributed waiting times for these events with the same mean rate and choose the event with nearest time. Let us suppose that the simulation buds through GEF1, the ER-identity GEF, resulting in a vesicle targeted to the cis-Golgi, with vSNARE2 active. The vesicle will have surface area 14,400π nm2, leaving the compartment with surface area 14,400π nm2 as well. ER markers GEF1, vSNARE1, and tSNARE1 will not be selectively concentrated during budding and will therefore have concentration 1.0 in both the vesicle and the remaining compartment. The remaining proteins will be concentrated 10-fold in the vesicle relative to the compartment, yielding concentrations of 20/11 in the vesicle and 2/11 in the compartment. At this point, the compartment will be too small to bud again, so the only possible next event is for the vesicle to back-fuse to the compartment. Because the vesicle has active vSNARE2, the fusion rate will be given by the square product of the vesicle's vSNARE2 concentration and the compartment's tSNARE2 concentration:
The other possibilities, budding through GEF2 or GEF3, have the same rate by symmetry. The simulator will sample three exponentially distributed waiting times for these events with the same mean rate and choose the event with nearest time. Let us suppose that the simulation buds through GEF1, the ER-identity GEF, resulting in a vesicle targeted to the cis-Golgi, with vSNARE2 active. The vesicle will have surface area 14,400π nm2, leaving the compartment with surface area 14,400π nm2 as well. ER markers GEF1, vSNARE1, and tSNARE1 will not be selectively concentrated during budding and will therefore have concentration 1.0 in both the vesicle and the remaining compartment. The remaining proteins will be concentrated 10-fold in the vesicle relative to the compartment, yielding concentrations of 20/11 in the vesicle and 2/11 in the compartment. At this point, the compartment will be too small to bud again, so the only possible next event is for the vesicle to back-fuse to the compartment. Because the vesicle has active vSNARE2, the fusion rate will be given by the square product of the vesicle's vSNARE2 concentration and the compartment's tSNARE2 concentration: 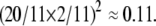 This fusion event will pool protein and membrane between the vesicle and compartment, restoring the compartment to surface area 28,800π nm2 and concentration 1.0 of all proteins. At this point, the model will have returned to its initial state and the simulator will again consider the three possible budding events from the compartment.
This fusion event will pool protein and membrane between the vesicle and compartment, restoring the compartment to surface area 28,800π nm2 and concentration 1.0 of all proteins. At this point, the model will have returned to its initial state and the simulator will again consider the three possible budding events from the compartment.
Simulation experiments
The simulation experiments fall into three groups. The first, which we call the steady-state model, describes a system initiated from an approximately steady state with three discrete compartments. Compartment 1 (representing the ER) has initial radius 1000 nm and compartments 2 and 3 have initial radii of 600 nm. All three have a 10-fold excess of native over nonnative marker types. Detailed parameters for this baseline simulation are provided in Table 1. The simulation was run for 1,000,000 steps, which was determined empirically to be sufficient for the system to reach a steady state under all conditions we subsequently examined. This baseline system was then perturbed for a series of simulations testing the effects of parameter variations on the model. Simulations were run for variations of sorting affinity for nonnative markers of 1, 2, 5, and 10–100 in increments of 10. Effects of changing GEF concentrations were assessed by varying the initial concentration of GEF2 in ER from 0.5 to 1 and then to 10 in increments of 1. Initial tSNARE2 concentration was varied in cis from 0.1 to 1.0 in increments of 0.1. Initial vSNARE2 concentration was varied in cis from 1 to 10 in increments of 1.0. Each parameter variation experiment was conducted 10 times, and mean values and error bars corresponding to the standard deviations over the experiments were plotted (see Fig. 7).
TABLE 1.
Steady-state model input parameters
| Initial compartment radius (nm)
| |||
|---|---|---|---|
| ER | cis | trans | |
| 1000.0
|
600.0
|
600.0
|
|
| Initial protein concentrations | |||
| Protein | ER | cis | trans |
| GEF1 | 1.0 | 0.1 | 0.1 |
| GEF2 | 0.1 | 1.0 | 0.1 |
| GEF3 | 0.1 | 0.1 | 1.0 |
| tSNARE1 | 1.0 | 0.1 | 0.1 |
| tSNARE2 | 0.1 | 1.0 | 0.1 |
| tSNARE3 | 0.1 | 0.1 | 1.0 |
| vSNARE1 | 1.0 | 0.1 | 0.1 |
| vSNARE2 | 0.1 | 1.0 | 0.1 |
| vSNARE3 | 0.1 | 0.1 | 1.0 |
| Affinity constants | |||
| Native proteins | Affinity | Nonnative proteins | Affinity |
| GEF (marker) | 1.0 | GEF (nonmarker) | 10.0 |
| tSNARE (marker) | 1.0 | tSNARE (nonmarker) | 10.0 |
| vSNARE (native) | 1.0 | vSNARE (nonmarker) | 10.0 |
FIGURE 7.

Sensitivity of the model to changes in starting protein concentrations. The figure shows steady-state radii for ER (solid line), and cis (asterisks) and trans (triangles) Golgi resulting from varying the amounts of starting GEF2, tSNARE2, and vSNARE2 initially in ER. (A–C) Varying GEF2, tSNARE2, and vSNARE2 concentrations, respectively, for the steady-state model. (D–F) Varying GEF2, tSNARE2, and vSNARE2 concentration, respectively, in the templated model. (G–I) Varying GEF2, tSNARE2, and vSNARE2 concentrations, respectively, in the de novo model.
The second group of simulations, which we call the templated model, tests the ability of the simulator to restore itself to a steady-state condition from an initial state in which the Golgi compartments have been reduced to vesicle-sized remnants. Cis and trans Golgi each have size 60 nm and ER a size of 1000 nm. Because this model is meant to represent a state in which most of the Golgi mass has collapsed into the ER, it is further assumed that most Golgi markers have returned to the ER, contaminating its identity. Table 2 provides detailed parameter values for this baseline system. Note that the total protein concentrations across all compartments are different from those of the steady-state model. For example, we keep the initial tSNARE concentrations in all three compartments the same as in the steady-state model, and thus have smaller total amounts of Golgi marker tSNAREs, requiring that we compensate with relatively higher amounts of the other Golgi marker proteins. Parameter variation experiments around this template baseline were performed using the same parameter ranges and experimental protocol as with the steady-state system.
TABLE 2.
Template model input parameters
| Initial compartment radius (nm)
| |||
|---|---|---|---|
| ER | cis | trans | |
| 1000.0
|
60.0
|
60.0
|
|
| Initial protein concentrations | |||
| Protein | ER | cis | trans |
| GEF1 | 1.0 | 0.1 | 0.1 |
| GEF2 | 2.0 | 1.0 | 0.1 |
| GEF3 | 2.0 | 0.1 | 1.0 |
| tSNARE1 | 1.0 | 0.1 | 0.1 |
| tSNARE2 | 0.1 | 1.0 | 0.1 |
| tSNARE3 | 0.1 | 0.1 | 1.0 |
| vSNARE1 | 1.0 | 0.1 | 0.1 |
| vSNARE2 | 1.0 | 1.0 | 0.1 |
| vSNARE3 | 1.0 | 0.1 | 1.0 |
| Affinity constants | |||
| Native proteins | Affinity | Nonnative proteins | Affinity |
| GEF (marker) | 1.0 | GEF (nonmarker) | 10.0 |
| tSNARE (marker) | 1.0 | tSNARE (nonmarker) | 10.0 |
| vSNARE (native) | 1.0 | vSNARE (nonmarker) | 10.0 |
The third group of simulations, which we call the de novo model, test the ability of the simulator to generate new compartments de novo when initially all protein and membrane in the system is in the ER. The simulation initially has an ER with radius 1000 nm and no initial cis or trans. The ER has a 10-fold excess of tSNARE1 over tSNAREs 2 and 3 to establish its ER identity. It has equal quantities of all three vSNAREs and a twofold excess of GEF2 and GEF3 over GEF1. Table 3 provides detailed parameter values for this baseline system. Parameter variation experiments around this de novo baseline were performed using the same parameter ranges and experimental protocol as with the steady-state system.
TABLE 3.
De novo model input parameters
| Initial compartment 1 protein concentrations (Initial radius = 1000 nm)
| ||
|---|---|---|
| Proteins | Concentration | Affinity |
| GEF1 | 1.0 | 1.0 |
| GEF2 | 2.0 | 10.0 |
| GEF3 | 2.0 | 10.0 |
| tSNARE1 | 1.0 | 1.0 |
| tSNARE2 | 0.1 | 10.0 |
| tSNARE3 | 0.1 | 10.0 |
| vSNARE1 | 1.0 | 1.0 |
| vSNARE2 | 1.0 | 10.0 |
| vSNARE3 | 1.0 | 10.0 |
We conducted one additional set of simulation experiments to test the ability of the Golgi to disassemble in response to artificial blockage of the ER to Golgi transport. This simulation is meant to provide an alternative validation of the model by testing whether it can recapitulate the experimental disassembly of the Golgi in response to inhibition of the COPII vesicle coat. These simulations most directly model a series of experiments using a dominant-negative form of sar1, which is GDP-restricted due to a mutation that prevents nucleotide exchange, and which prevents ER exit and causes collapse of the Golgi and redistribution of Golgi enzymes into the ER (23–27). The ER GEF sec12 normally activates sar1 by generating sar1-GTP. Sar1-GTP then triggers COPII coat assembly. Thus, to simulate an inability to generate sar1-GTP, we simply set the ER GEF concentration to zero. Initial conditions are otherwise identical to those of the steady-state simulations. Elimination of the ER-identity GEF will block budding of any vesicles with the ER identity in the model. Note that this model may continue to allow some budding from the ER initiated by Golgi-identity GEFs contaminating the ER.
Golgi size determination
Hela cells were transfected with plasmids encoding C-terminally myc-tagged syntaxin-5 or N-terminally green fluorescent protein-tagged Sec61 using Transfectol (GeneChoice, Frederick, MD). After 24 h, the cells were paraformaldehyde-fixed and processed as previously described (8) using anti-myc and anti-giantin antibodies and rhodamine phalloidin (Molecular Probes, Eugene, OR). Microscopy was performed using a spinning disk confocal scan head equipped with three-line laser and independent excitation and emission filter wheels (PerkinElmer, Waltham, MA) and a 12-bit digital camera (Orca ER, Hamamatsu City, Japan) mounted on a microscope (Axiovert 200, Carl Zeiss MicroImaging, Inc.) with a 100x, 1.4 NA apochromat oil-immersion objective (Carl Zeiss MicroImaging, Oberkochen, Germany). Sections at 0.3 μm spacing were acquired using ImagingSuite software (PerkinElmer). Syntaxin-5 and Sec61 expression level was determined using the ImageJ “Measure” function by summing the total fluorescence per slice per cell across the entire image stack. For quantification of Golgi size in each cell, giantin staining was used to define pixels in each slice corresponding to the Golgi and their area was then summed for the entire stack using the ImageJ “Measure stack” function. Cell volume was estimated by summing the area in each slice outlined using the phalloidin staining pattern. Individual experiments were performed with identical laser output levels, exposure times, and scaling. To allow direct comparison of distinct experiments, given small changes in staining intensity, values were normalized by dividing by the mean values of the entire dataset for a given experiment.
RESULTS
Steady-state maintenance
We first examined the ability of the model to maintain discrete compartment identities at steady state when initiated on a system of three compartments with distinct identities and followed for 106 time steps. We can evaluate this steady-state behavior based on the ability to maintain stable compartment sizes and distinct compartment identities as assessed by concentrations of marker proteins. Fig. 4 A shows that all compartments remained close to their initial sizes—1000 nm for ER and 600 nm each for cis and trans Golgi—throughout the simulations. There are stochastic fluctuations over a range of ∼±50 nm around the initial values. Fig. 5 shows that tSNARE, vSNARE, and GEF marker concentrations were also stable after an initial expulsion of nonnative markers. Each compartment maintained a low and approximately constant concentration of its nonnative markers and a substantially higher level of its native markers. All three marker proteins partition similarly to one another, with the exception of a somewhat greater amount of stochastic fluctuation in tSNAREs (Fig. 5, A–C) versus vSNAREs (Fig. 5, D–F) and GEFs (Fig. 5, G–I). Fluctuations in all compartment marker concentrations are higher for the two Golgi compartments than for the ER. We attribute this observation to the significantly smaller surface area of two Golgi compartments, resulting in relatively larger changes in concentrations from individual budding and fusion events.
FIGURE 4.

Compartment radii versus simulation step for the three models. Each plot shows evolution over time of the compartment radii for ER (solid line), cis (short-dashed line), and trans (long-dashed line) of a single execution of the model. (A) Steady-state model. (B) Templated model. (C) De novo model.
FIGURE 5.

tSNARE, vSNARE, and GEF concentrations versus time for the baseline simulations in the steady-state model. Each plot shows concentrations of tSNAREs, vSNAREs, and GEFs of identities 1 (solid line), 2 (short-dashed line), and 3 (long-dashed line) for a single compartment. (A) ER, tSNARE. (B) cis Golgi, tSNARE. (C) trans Golgi, tSNARE. (D) ER, vSNARE. (E) cis Golgi, vSNARE. (F) trans Golgi, vSNARE. (G) ER, GEF. (H) cis Golgi, GEF. (I) trans Golgi, GEF.
Given that the model depends on several unknown parameters, we next sought to determine the degree to which successful sorting in the model depends on specific choices of the parameter values. The model first depends on the existence of a sorting reaction capable of separating native from nonnative cargos during vesicle formation. The strength of this reaction in the model is set by a user-specified affinity constant (keq) of the sorting reaction. We therefore examined the effects of the overall sorting of proteins into compartments as a function of this affinity constant. Integrity of the model was assessed by the mean fraction of contamination of the three compartments by nonnative tSNARE markers as a fraction of their total tSNARE complement. Fig. 6 A shows that the model is sensitive to affinities over a fairly narrow range (1–30), with contamination approaching 67% (complete loss of identity) for affinity 1 and dropping below 10% by affinity 30. We further assessed the effects of affinity variations on compartment sizes (Fig. 6 B). Compartment sizes appear unaffected by affinity constant except at extremely low affinities, where all of the membrane mass collapses into a single compartment. It thus appears that the model requires a minimum affinity on the order of 5–10 to be functional but is not otherwise highly sensitive to that parameter.
FIGURE 6.

Sensitivity of the models to changes in sorting affinity. (A, C, and E) Fractional contamination of compartments with nonnative tSNARE markers as a function of sorting affinity for the steady-state, templated, and de novo models, respectively. (B, D, and F) Average compartment radii as a function of sorting affinity for ER (solid line), cis (asterisks), and trans (triangles) for the steady-state, templated, and de novo models, respectively.
We then examined sensitivity of the model to variations in starting protein concentrations. Because the model is symmetric with respect to marker identities 2 and 3, we chose to examine variations in the three identity-2 proteins, implemented by varying the initial concentration of identity-2 proteins in the ER. Compartment size was found to be responsive to changes in initial concentrations of all three marker types. Increasing the amounts of GEF2 (Fig. 7 A), tSNARE2 (Fig. 7 B), or vSNARE2 (Fig. 7 C) initially found in the ER caused a reassortment of membrane from ER and trans Golgi to cis Golgi. The model therefore suggests that total concentrations of compartment markers in the system provide a mechanism for controlling relative compartment sizes. This finding is of particular interest because it provides a nonobvious testable prediction about Golgi behavior that can be used to validate the model. In the subsection Experimental validation, below, we validate one case of this prediction by examining compartment size changes in response to changing concentrations of the ER-Golgi tSNARE syntaxin-5.
Templated assembly
We next examined the ability of the model to reach a steady state from an initial configuration in which the ER contains most components and membrane, leaving only vesicle-sized cis and trans Golgi compartments with distinct identities. This condition is meant to model the assembly of a Golgi from Golgi remnants in the cytoplasm that have small sizes but proper marker identities, and is also similar to modeling the reassembly of Golgi from the ER after brefeldin A treatment. Brefeldin A, which is a fungal metabolite frequently used to inhibit activation of Arf1, causes redistribution of Golgi proteins into the ER and small structures termed Golgi remnants (28,29). Fig. 4 B shows changes in compartment sizes for a single baseline simulation, confirming a rapid transfer of membrane from compartment 1 to the two remnant compartments, followed by a stable partitioning of membrane mass similar to that of the steady-state model in Fig. 4 A. Compartment identities as assessed by marker tSNARE (Fig. 8, A–C), vSNARE (Fig. 8, D–F), and GEF (Fig. 8, G–I) concentrations all show the same rapid adjustment to a stable partitioning of compartments. Native marker concentrations are generally higher in the templated than in the steady-state model, largely because ER boosts its concentration through expulsion of membrane to cis and trans Golgi compartments, whereas the cis and trans Golgi never completely dilute the initially high marker concentrations of their remnants. Separation of native and nonnative concentrations is generally comparable between the two models, with the exception of the GEF concentrations in compartment 1 (Fig. 8 G), which show a comparatively higher degree of contamination in the templated model than the other markers or compartments. Fluctuations in marker protein concentrations are comparatively higher in the Golgi compartments than in the ER, as with the steady-state case, which we again attribute to the smaller Golgi compartment sizes.
FIGURE 8.

tSNARE, vSNARE, and GEF concentrations versus time for the baseline simulations in the templated model. Each plot shows concentration of tSNAREs, vSNAREs, and GEFs of identities 1 (solid line), 2 (short-dashed line), and 3 (long-dashed line) for a single compartment. (A) ER, tSNARE. (B) cis Golgi, tSNARE. (C) trans Golgi, tSNARE. (D) ER, vSNARE. (E) cis Golgi, vSNARE. (F) trans Golgi, vSNARE. (G) ER, GEF. (H) cis Golgi, GEF. (I) trans Golgi, GEF.
We next sought to determine whether the templated model exhibits parameter sensitivity similar to that of the steady-state model. Fig. 6, C and D shows a similar dependence of compartment integrity and size on sorting affinity. We attribute the quantitative difference between the peak contaminations of Fig. 6, A and C, to the fact that the templated model requires relatively higher amounts of ER markers to ensure that it has an ER identity at the beginning of the simulation, when it contains almost all membrane and markers that will eventually sort to the other compartments. Fig. 7, D–F, shows qualitatively similar effects of marker concentrations on compartment sizes, but with the templated model showing more pronounced effects at the extremes, especially for GEF concentrations. At very low GEF2 concentrations, the cis Golgi compartment cannot develop beyond its initial vesicle size, whereas at very high GEF2 concentrations, the trans Golgi compartment appears unable to form. We believe that excessively low GEF2 prevents nascent structures with cis-Golgi identity from expelling contaminants, causing them to lose their cis-Golgi identity and fuse with other compartments before they can reach compartment size. Excessively high GEF2, on the other hand, can be expected to contaminate nascent trans-Golgi compartments, causing them to expel their trans-Golgi markers and acquire cis-Golgi identity. This latter mechanism would similarly be expected to promote fusion with other compartments before trans-Golgi structures can reach compartment size.
De novo assembly
We then examined the ability of the model to generate distinct compartment identities de novo from a single compartment (the ER) without the need for any Golgi remnants to seed other compartment identities. Fig. 4 C confirms that two compartments form de novo from the ER and reach steady-states sizes similar to those of the template model by accumulating membrane from the ER. The two compartments emerge almost immediately, but their growth to full size is noticeably slower than with the template model. tSNARE marker concentrations (Fig. 9, A–C), vSNARE marker concentrations (Fig. 9, D–F), and GEF marker concentrations (Fig. 9, G–I) likewise evolve similarly to those of the templated model. There is, however, a short-lived transient state not observed with the templated model, in which cis and trans Golgi compartments each initially form from the ER with a hybrid 2/3 identity before differentiating from one another. Fig. 10 examines this transient state in more depth by showing the evolution of compartment sizes and marker concentrations over the first 10,000 simulation steps of the de novo model. The images show that cis and trans Golgi compartments are each initially established with approximately equal concentrations of identity-2 and identity-3 markers before rapidly expelling one or the other in approximately the first 500 steps of the simulation. Fluctuations in marker protein concentrations are again comparatively higher in the Golgi compartments than in the ER, presumably also reflecting the differences in steady-state compartment sizes.
FIGURE 9.

tSNARE, vSNARE, and GEF concentrations versus time for the baseline simulations in the de novo model. Each plot shows concentrations of tSNAREs, vSNAREs, and GEFs of identities 1 (solid line), 2 (short-dashed line), and 3 (long-dashed line) for a single compartment. (A) ER, tSNARE. (B) cis Golgi, tSNARE. (C) trans Golgi, tSNARE. (D) ER, vSNARE. (E) cis Golgi, vSNARE. (F) trans Golgi, vSNARE. (G) ER, GEF. (H) cis Golgi, GEF. (I) trans Golgi, GEF.
FIGURE 10.

Time progress of a single run of the de novo baseline simulation over 10,000 time steps. (A) Compartment radii versus time for ER (solid line), cis (short-dashed line), and trans (long-dashed line). (B–D) Concentrations of tSNARE1 (solid line), tSNARE2 (short-dashed line), and tSNARE3 (long-dashed line) in ER, cis, and trans, respectively, versus time. (E–G) Concentrations of vSNARE1 (solid line), vSNARE2 (short-dashed line), and vSNARE3 (long-dashed line) in ER, cis, and trans, respectively, versus time.
We next examined whether the requirements of de novo biogenesis caused any additional parameter sensitivity beyond that required for the steady-state or templated models. Fig. 6, E–F, shows that the de novo and templated models have a similar sensitivity to sorting affinity. The de novo model does, however, require a slightly higher minimum affinity to establish the three compartments (Fig. 6 F) than does the templated model (Fig. 6 D). Sensitivity to tSNARE (Fig. 7 H) and vSNARE (Fig. 7 I) concentrations are essentially identical between the de novo and templated models. However, the de novo model appears to be more tolerant of extremes of GEF concentration (Fig. 7 G) than is the templated model.
Golgi disassembly
We further examined the ability of the model to recapitulate experimentally induced disassembly of the Golgi by eliminating the ER-specific GEF from a model otherwise initialized identically to the steady-state model. In Fig. 11, we plot the ER, cis Golgi, and trans Golgi compartment sizes versus time from the initiation of this model over 10,000 simulation steps. The model shows a rapid disassembly of both Golgi compartments. Membrane mass redistributes from both Golgi compartments to the ER over 9,500 simulation steps, until the compartments reach vesicle size (60 nm). The simulation does not allow further budding once a compartment drops to vesicle size. The model appears stable for subsequent times.
FIGURE 11.

Time progress of compartment sizes in response to inactivation of the ER GEF. The model shows sizes of ER (solid line), cis-Golgi (short-dashed line), and trans-Golgi (long-dashed line) compartments from a simulation with an initial steady-state distribution of markers from which the ER GEF has been removed.
Experimental validation
Our next goal was to determine whether our computational model provides a realistic qualitative description of compartment-level sorting in the Golgi. There is currently no direct way to verify experimentally that the basic operations of sorting and fusion function as the model predicts, nor is it possible to experimentally prove that these operations alone are sufficient for establishing compartment identities in vivo. We therefore sought to verify the model indirectly by validating one nonobvious emergent feature of the model observed in the simulation experiments: the ability of varying marker concentrations to produce selective reassortment of membrane from the ER to the Golgi compartments. The simulation experiments reported in Fig. 7 showed that Golgi compartment sizes relative to one another and to the ER can be controlled by the initial concentrations of marker proteins present in the system. This finding leads to the testable prediction that experimental interventions that change the concentration of a single compartment marker in isolation of the analogous markers in other compartments should cause a redistribution of membrane away from compartments whose relative marker concentrations have decreased and toward compartments whose relative marker concentrations have increased. Specifically, we chose to test the model prediction that changes in initial tSNARE concentrations lead to changes in the relative sizes of Golgi compartments (Fig. 7, B, E, and H). We can explain the simulation observation by noting that greater availability of a Golgi marker tSNARE would be expected to increase the net flux of membrane into the compartment type that recruits that tSNARE. This increased influx, in the absence of some corresponding increased efflux, would be expected to lead to improved competition for membrane between a given Golgi compartment and the ER. Determining whether Golgi size does in fact respond similarly to changes in a single Golgi marker tSNARE thus provides a concrete test of whether sorting of membrane into compartments is in fact controlled by marker protein concentrations, as our model predicts.
To test this prediction, the size of the Golgi apparatus was quantified in HeLa cells expressing various levels of the ER-Golgi tSNARE syntaxin-5. To estimate Golgi size, 3D confocal image sets were used to determine the volume of the staining corresponding to the Golgi marker giantin, as opposed to its staining intensity, and this value was normalized using total cell volume. Volume normalization reduced the variation due to normal Golgi growth and did not alter the experimental outcome. Syntaxin-5 expression was varied using transient transfection of a myc-tagged version and, on a cell-by-cell basis, the relative expression level was determined by immunostaining (Fig. 12, A–C). As a control, Golgi size was also determined for cells expressing the ER-localized protein Sec61-GFP (Fig. 12, D–F). Strikingly, syntaxin-5 over-expressors yielded an average normalized Golgi size that was 1.3-fold higher than the Sec61 over-expressing control cells (Fig. 12 G). These findings, which report normalized averages derived from multiple experiments, were further supported when the data was analyzed on a cell-by-cell basis using a correlation matrix. As shown, when syntaxin-5 and Sec61 expression levels were plotted against the ratio of Golgi/cell size, it was clear that increased syntaxin-5, but not increased Sec61, was associated with increased Golgi size (Fig. 12 H). Thus, these findings support a significant prediction that arose from our model simulating Golgi biogenesis and point to a role for tSNARE expression level as a control point in the homeostasis of secretory compartments.
FIGURE 12.

ER-Golgi tSNARE expression level influences Golgi size in vivo. (A–F) HeLa cells expressing myc-syn5 (A–C) or Sec61-GFP (D–F) were paraformaldehyde-fixed and analyzed to reveal myc staining (A), GFP fluorescence (D) and actin staining (B and E), each shown as z-axis projections of optical sections, as well as giantin staining (C and F) shown after 3D rendering. Bars, 10 μm. (G) Relative expression level, Golgi size, and cell volume were quantified (see Methods). Values are normalized averages (±SE, n = 3, >25 cells each). (H) Relative expression level and the Golgi/cell size ratio were also compared on a cell-by-cell basis. The correlation plot yielded a slope of 0.0002 for syn5 versus −0.00002 for Sec61.
DISCUSSION
We have established a minimal model of the formation and maintenance of membrane compartments of distinct identity as a means of understanding biogenesis of the Golgi apparatus. The model simulates two basic mechanisms: the formation of vesicles with selective concentration of marker proteins, arising from SNARE-coat interactions during vesicle budding, and the ability of these vesicles to target specific compartments, based on cognate SNARE-SNARE interactions during fusion. Simulations show that these two mechanisms alone are sufficient to explain the formation, restoration, and robust maintenance of Golgi-like compartments with distinct identities. We have found the model to be robust over a wide range of parameter variations, providing confidence that the ability of the model to mimic features of true assembly and biogenesis is not a chance product of a specific set of parameter choices. Furthermore, the model is consistent with at least two distinct hypotheses for Golgi biogenesis—formation from remnant template vesicles and true de novo formation from the ER—and predicts observable differences in transitory marker distributions between these models.
The use of a minimal model necessarily makes many simplifying assumptions. Most significantly, the model treats the selective budding and fusion mechanisms as solely responsible for the formation of compartment identities. This is a necessary model assumption that allows us to test the sufficiency of those two specific mechanisms for producing the observed Golgi behaviors, but we do not expect that it is literally true. Rather, there are likely many other factors that assist in, but may not be essential to, the formation and maintenance of compartment identities. As a result, although our model allows us to test the sufficiency of the two mechanisms, we would not expect it to provide a close quantitative match to the real Golgi in relative compartment sizes or their responses to specific interventions. The model also eliminates many other cellular mechanisms that are known to interact with the Golgi, such as the cytoskeleton. The model can again suggest that such mechanisms are not necessary to producing compartment identities, but cannot suggest that they have no quantitative effects on Golgi size or marker distributions. Likewise, the model at present entirely neglects spatial arrangement of Golgi compartments relative to one another and to the ER and VTCs. Spatial proximity might be expected to significantly bias preferred targets for vesicle binding. The model also does not currently monitor compartment maturation, a process believed to be at least partly responsible for intra-Golgi traffic (9). Maturation is, however, an obvious future extension for which the current model should provide a strong foundation. Maturation is conceptually related to de novo biogenesis and is also presumed to be driven by the key reactions we have modeled: sorting during vesicle formation and targeted vesicle fusion. All of these approximations raise challenges for validation, as the model can make only qualitative, and not yet quantitative, predictions of Golgi function. Such problems may be addressed in future work by explicitly adding into the model some of these features that we can reasonably anticipate would affect quantitative Golgi function. An alternative approach would be to use data-fitting methods to build mathematical models of the influence of these unknown factors without explicitly building models of any particular physical processes.
The model may have several implications for productive avenues for laboratory experimentation. The development of the model itself has led to two nontrivial predictions about Golgi function. An earlier, unsuccessful attempt to develop de novo biogenesis in this framework suggested the need for exit rate control from compartments based on contamination with nonnative markers. Such a mechanism has since been experimentally verified (5). It also proved necessary to hypothesize a cooperative vesicle fusion reaction to prevent back-fusion from blocking de novo biogenesis in the model. Prior evidence does indeed suggest that vesicle fusion is a cooperative reaction, as the model requires (22). The model further suggests several specific parameters that influence compartment sizes in ways that should be experimentally detectable. One component of this prediction, the influence of tSNARE concentration on compartment size, has now been experimentally verified.
Acknowledgments
We are grateful to Kuok-Chiang Kim and Jonathan Lustgarten for their efforts in coding earlier prototypes of the simulation approach used in this work.
This work was supported by grants from the U.S. National Science Foundation (0346981, to R.S.), U.S. National Institutes of Health (R01GM56779, to A.D.L.), the American Cancer Society (RSG0314801CSM, to A.D.L.), and the Eberly Family Foundation (to R.S.).
Editor: Klaus Schulten.
References
- 1.Farquhar, M. G., and G. E. Palade. 1998. The Golgi apparatus: 100 years of progress and controversy. Trends Cell Biol. 8:2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu, W., L. E. O'Brien, F. Wang, H. Bourne, K. E. Mostov, and M. M. Zegers. 2003. Hepatocyte growth factor switches orientation of polarity and mode of movement during morphogenesis of multicellular epithelial structures. Mol. Biol. Cell. 14:748–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu, Z., D. Joseph, E. Bugnard, K. J. Zaal, and E. Ralston. 2001. Golgi complex reorganization during muscle differentiation: visualization in living cells and mechanism. Mol. Biol. Cell. 12:795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knaapen, M. W., B. C. Vrolijk, and A. C. Wenink. 1997. Ultrastructural changes of the myocardium in the embryonic rat heart. Anat. Rec. 248:233–241. [DOI] [PubMed] [Google Scholar]
- 5.Guo, Y., and A. D. Linstedt. 2006. COPII-Golgi protein interactions regulate COPII coat assembly and Golgi size. J. Cell Biol. 174:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colanzi, A., C. Suetterlin, and V. Malhotra. 2003. Cell-cycle-specific Golgi fragmentation: how and why? Curr. Opin. Cell Biol. 15:462–467. [DOI] [PubMed] [Google Scholar]
- 7.Lee, T. H., and A. D. Linstedt. 1999. Osmotically induced cell volume changes alter anterograde and retrograde transport, Golgi structure, and COPI dissociation. Mol. Biol. Cell. 10:1445–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puri, S., and A. D. Linstedt. 2003. Capacity of the Golgi apparatus for biogenesis from the endoplasmic reticulum. Mol. Biol. Cell. 14:5011–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonifacino, J. S., and B. S. Glick. 2004. The mechanisms of vesicle budding and fusion. Cell. 116:153–166. [DOI] [PubMed] [Google Scholar]
- 10.Springer, S., A. Spang, and R. Schekman. 1999. A primer on vesicle budding. Cell. 97:145–148. [DOI] [PubMed] [Google Scholar]
- 11.Rothman, J. E. 1994. Mechanisms of intracellular protein transport. Nature. 372:55–63. [DOI] [PubMed] [Google Scholar]
- 12.Volchuk, A., M. Ravazzola, A. Perrelet, W. S. Eng, M. Di Liberto, O. Varlamov, M. Fukasawa, T. Engel, T. H. Sollner, J. E. Rothman, and L. Orci. 2004. Countercurrent distribution of two distinct SNARE complexes mediating transport within the Golgi stack. Mol. Biol. Cell. 15:1506–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Puthenveedu, M. A., and A. D. Linstedt. 2005. Subcompartmentalizing the Golgi apparatus. Curr. Opin. Cell Biol. 17:369–375. [DOI] [PubMed] [Google Scholar]
- 14.Glick, B. S., T. Elston, and G. Oster. 1997. A cisternal maturation mechanism can explain the asymmetry of the Golgi stack. FEBS Lett. 414:177–181. [DOI] [PubMed] [Google Scholar]
- 15.Weiss, M., and T. Nilsson. 2000. Protein sorting in the Golgi apparatus: a consequence of maturation and triggered sorting. FEBS Lett. 486:2–9. [DOI] [PubMed] [Google Scholar]
- 16.Heinrich, R., and T. A. Rapoport. 2005. Generation of nonidentical compartments in vesicular transport systems. J. Cell Biol. 168:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gillespie, D. T. 1976. Exact stochastic simulation of coupled reactions. J. Phys. Chem. 81:2340–2361. [Google Scholar]
- 18.Le Novere, N., and T. S. Shimizu. 2001. STOCHSIM: modelling of stochastic biomolecular processes. Bioinformatics. 17:575–576. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi, K., K. Kaizu, B. Hu, and M. Tomita. 2004. A multi-algorithm, multi-timescale method for cell simulation. Bioinformatics. 20:538–546. [DOI] [PubMed] [Google Scholar]
- 20.Ander, M., P. Beltrao, B. Di Ventura, J. Ferkinghoff-Borg, M. Foglierini, A. Kaplan, C. Lemerle, I. Tomas-Oliveira, and L. Serrano. 2004. SmartCell, a framework to simulate cellular processes that combines stochastic approximation with diffusion and localisation: analysis of simple networks. Syst. Biol. 1:129–138. [DOI] [PubMed] [Google Scholar]
- 21.Hoops, S., S. Sahle, R. Gauges, C. Lee, J. Pahle, N. Simus, M. Singhal, L. Xu, P. Mendes, and U. Kummer. 2006. COPASI: a COmplex PAthway SImulator. Bioinformatics. 22:3067–3074. [DOI] [PubMed] [Google Scholar]
- 22.Stewart, B. A., M. Mohtashami, W. S. Trimble, and G. L. Boulianne. 2000. SNARE proteins contribute to calcium cooperativity of synaptic transmission. Proc. Natl. Acad. Sci. USA. 97:13955–13960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barlowe, C., L. Orci, T. Yeung, M. Hosobuchi, S. Hamamoto, N. Salama, M. F. Rexach, M. Ravazzola, M. Amherdt, and R. Schekman. 1994. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 77:895–907. [DOI] [PubMed] [Google Scholar]
- 24.Storrie, B., J. White, S. Rottger, E. H. Stelzer, T. Suganuma, and T. Nilsson. 1998. Recycling of Golgi-resident glycosyltransferases through the ER reveals a novel pathway and provides an explanation for nocodazole-induced Golgi scattering. J. Cell Biol. 143:1505–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaal, K. J., C. L Smith, R. S. Polishchuk, N. Altan, N. B. Cole, J. Ellenberg, K. Hirschberg, J. F. Presley, T. H. Roberts, E. Siggia, R. D. Phair, and J. Lippincott-Schwartz. 1999. Golgi membranes are absorbed into and reemerge from the ER during mitosis. Cell. 99:589–601. [DOI] [PubMed] [Google Scholar]
- 26.Miles, S., H. McManus, K. E. Forsten, and B. Storrie. 2001. Evidence that the entire Golgi apparatus cycles in interphase HeLa cells: sensitivity of Golgi matrix proteins to an ER exit block. J. Cell Biol. 155:543–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ward, T. H., R. S. Polishchuk, S. Caplan, K. Hirschberg, and J. Lippincott-Schwartz. 2001. Maintenance of Golgi structure and function depends on the integrity of ER export. J. Cell Biol. 155:557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujiwara, T., K. Oda, S. Yokota, A. Takatsuki, and Y. Ikehara. 1988. Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. J. Biol. Chem. 263:18545–18552. [PubMed] [Google Scholar]
- 29.Lippincott-Schwartz, J., L. C. Yuan, J. S. Bonifacino, and R. D. Klausner. 1989. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell. 56:801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]