

Abstract
Mitochondrial dysfunction plays a central role in the selective vulnerability of dopaminergic neurons in Parkinson’s disease (PD) and is influenced by both environmental and genetic factors. Expression of the PD protein α-synuclein or its familial mutants often sensitizes neurons to oxidative stress and to damage by mitochondrial toxins. This effect is thought to be indirect, since little evidence physically linking α-synuclein to mitochondria has been reported. Here, we show that the distribution of α-synuclein within neuronal and non-neuronal cells is dependent on intracellular pH. Cytosolic acidification induces translocation of α-synuclein from the cytosol onto the surface of mitochondria. Translocation occurs rapidly under artificially-induced low pH conditions and as a result of pH changes during oxidative or metabolic stress. Binding is likely facilitated by low pH-induced exposure of the mitochondria-specific lipid cardiolipin. These results imply a direct role for α-synuclein in mitochondrial physiology, especially under pathological conditions, and in principle, link α-synuclein to other PD genes in regulating mitochondrial homeostasis.
Keywords: α-Synuclein, mitochondria, pH, Parkinson’s disease, oxidative stress, metabolic dysfunction
Introduction
Oxidative stress and metabolic dysfunction appear to be important factors in a number of neurodegenerative diseases, including Parkinson’s disease (PD), although the cellular pathways through which the various stresses converge are only slowly being uncovered [1, 2]. Many of the pathological features of PD, including the selective loss of dopaminergic neurons and α-synuclein aggregation can be reproduced by environmental toxins that principally target mitochondria [3, 4]. Impairments in the electron transport chain are associated with increased levels of reactive oxygen species and decreased energy production [4, 5], and can be amplified by proteasome inhibition [6, 7]. In addition to environmental factors, multiple genes mediating familial forms of PD or affecting PD risk have been identified, several of which encode proteins that localize to mitochondria and/or are associated with mitochondrial homeostasis. These include PINK1 [8], DJ-1 [9, 10], parkin [11, 12], and Omi/HtrA2 [13]. Since mitochondrial complex I deficiencies have been reported in patients with sporadic PD [14, 15], a common cellular network likely links familial and sporadic forms of the disease.
There is increasing evidence that the first familial PD gene to be identified, α-synuclein, also influences cellular responses to mitochondrial stress. Wild-type α-synuclein protects or sensitizes cells to apoptotic stimuli, depending on the cell type and insult examined [16, 17], whereas mutant α-synucleins (A30P or A53T) generally increase neuronal vulnerability to mitochondria-associated toxicity [16, 18, 19]. Conversely, α-synuclein knockout mice show marked resistance to several mitochondrial toxins [20]. Mitochondrial morphology or physiology may also be affected by α-synuclein overexpression [21, 22], and mitochondrial abnormalities are observed in α-synuclein-expressing mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [23]. There is, however, little evidence that α-synuclein exerts these effects by a direct physical association with mitochondria.
We now report that, in response to reduced intracellular pH, α-synuclein translocates from the cytosol onto the surface of mitochondria. This occurs under a number of oxidative and/or metabolic stress conditions, and is likely mediated by pH-dependent exposure of mitochondria-specific lipids. Thus, α-synuclein may play a direct role in mitochondrial physiology, ostensibly establishing a link between mitochondrial dysfunction and α-synuclein-associated toxicity in PD pathogenesis.
Materials and Methods
Cell lines, plasmids, and antibodies
All cell lines were from the ATCC. Stable cells were selected in G418 (500 µg/ml for HEK293) and 200 µg/ml (for SK-N-SH). Primary rat hippocampal neurons were provided by C. Winters (NINDS). Plasmids for expression of various synucleins in eukaryotic and prokaryotic cells were as previously described [24, 25]. Antibodies to synucleins included 202 (1/100, for immunofluorescence and immunoelectron microscopy, from V. Lee, Univ. of Pennsylvania; this antibody recognizes α- and β-synuclein) and anti-α-synuclein (1/2000 for immunoblotting, BD Biosciences). Anti-BclXL was from Cell Signaling Technology (54H6; 1:250); anti-VDAC from Alexis Biochemicals (210–785-C100; 1:2000); anti-Tom20 (FL-145; 1: 2500) from Santa Cruz, and anti cytochrome c (556432; 1:500) was from Pharmingen. MitoTracker (Red and Deep Red), and species-specific Alexa 488, 546, and 594-conjugated secondary antibodies were from Molecular Probes (Invitrogen). Anti-ADRP (a lipid droplet binding protein) and Bodipy 493/503 (to label lipid droplets) were described previously [24]. Additional anti-synuclein antibodies: anti- α-synuclein (recognizes amino acids 2–25; 1/1000 for immunofluorescence using Syn 1–102, Synaptic Systems); and anti γ-synuclein (1/500 for immunofluorescence; Abcam Limited).
Transfections and Immunohistochemistry
All cell lines were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen) in an incubator containing 95% air/5% CO2. Cells were transiently transfected with Lipofectamine 2000 (Invitrogen) or calcium phosphate (HEK 293), and incubated for 16–30 hours at 37°C. Cells were fixed, where indicated, in 3.7% formaldehyde for 20–30 minutes at RT in: phosphate buffered saline (PBS; 1.06 mM KH2PO4; 2.97 mM Na2HPO4-7H2O; 155 mM NaCl, pH 7.4; Invitrogen); 0.1 M sodium phosphate, pH 7.2, 150 mM NaCl; or 0.1 M sodium acetate buffer, 150 mM NaCl, pH 5.0. Stock 37% formaldehyde solutions (Sigma; Cat #252549) were deionized using mixed bed resin (Sigma, Cat # M8032) until no color change occurred. Immunofluorescence staining was performed as described [24]. Preembedding immunoelectron microscopy was performed, according to [26]. EM images were obtained on a JEOL-100 transmission electron microscope. Images were calibrated and gold particles counted using NIH Image.
Solutions, pH measurements, and microscopy
Nominally bicarbonate-free cell incubation buffer (PSS) contained: 120 mM NaCl; 5 mM KCl; 5 mM glucose; 1 mM CaCl2; 1 mM MgCl2; 1 mM Na2HPO4; and 20 mM Tris, pH 7.4. High K+ buffers were similar to PSS except for the substitution of 130 mM KCl and 20 mM NaCl. 20 mM MES buffer was used for pH calibrations between pH 4.5–6.4 and MOPS buffer from pH 6.8–8.0. For stress treatments, cells were rinsed once with PBS, and then incubated in PSS, as indicated. For intracellular pH measurements, cells were plated on 8-well Lab-Tek II chambered cover glass (Nalge Nunc International). For dye loading, cells were incubated in PSS with 2.5–5 µM SNARF-4F/AM (Molecular Probes) for 1 hour at 37°C. Cells were then incubated with the indicated treatments in PSS at 37°C before imaging. Cells were imaged at RT on a Zeiss Axiovert 200M with a 63X/1.4 NA Plan Apocromat oil/DIC objective, using an LSM 510 Meta scanning module. Measurements of pHi were performed using the dual-emission ratio method, with excitation at 514 nm (Argon laser) and emission collected at 576–608 nm and 630–673 nm. Mean fluorescence intensity ratio measurements were collected and analyzed using MIPAV (Medical Image Processing, Analysis, and Visualization; http://mipav.cit.nih.gov). A one-point calibration technique (at pH 6.4) was employed to convert background-subtracted ratio values to pHi [27]. For cells that failed to respond to the pH calibration buffer, fluorescence ratio measurements were fitted directly to the standard curve. Within each, ratios from ~30–60 individual cells were determined. Data are expressed as the mean ± s.e.m. from at least three independent experiments. Loss of SNARF 4F dye from cells occurred with extended DOG/Az and CCCP incubations; therefore, these measurements were generally performed after 30 minutes treatment. For the treatments and times indicated, cells remained trypan blue negative (data not shown). SNARF-4F/AM (Invitrogen) was stored as a 2.5 mM stock in DMSO. Nigericin was from Sigma and used at 10 µg/ml. The high K+ /nigericin solutions used for SNARF-F pH calibrations were the same used in pH clamping experiments, shown in Fig. 3. The pKa of the dye was determined to be ~6.4 for both in vitro spectrofluorometric and cell-based measurements, confirming the accuracy of the clamping procedure (data not shown).
Figure 3. Synuclein translocation to mitochondria is directly influenced by pHi.
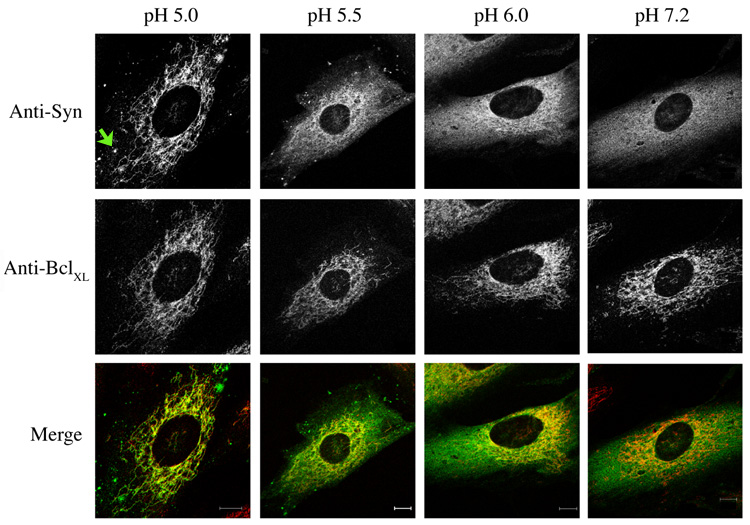
Immunofluorescence images of SK-N-SH cells expressing α-synuclein clamped to various intracellular pHs with high K+ buffers/nigericin. Cells were incubated for 15 minutes before fixation with 3.7% formaldehyde in 0.1 M PO4, pH 7.2. Cells were labeled with antibodies to synuclein and BclXL. Note the partial redistribution of synuclein at pHi 6.0 and below; complete redistribution occurs at pHi 5.0. The green arrow shows one of several synuclein clusters that form at low pH. Bar: 10 µm.
Biochemical Procedures
For western blotting of α–synuclein released into fixation buffer, 6-well dishes containing HEK293 cells stably expressing α-synuclein were washed twice with PBS, and then fixed as indicated in 2 ml fixative for 45 minutes at RT. The fixative was removed and neutralized with 107 µl 28% NH4OH for 10 minutes on ice. The pH was then adjusted to ~7 with HCl, before adding SDS-PAGE sample buffer. The sample was incubated at 37°C for 5 minutes before gel loading.
Mouse brain mitochondria were isolated [28] and stored at ~10 mg/ml in MB buffer (10 mM Tris-Cl, pH 7.4; 1 mM EDTA; 0.32 M sucrose) at −80°C. Recombinant synucleins were prepared, as described [25], and incubated (0.25 µg) with mitochondria (1 mg/ml; ~500 µM phospholipid) in assay buffer (60 mM KCl; 110 mM sucrose; 4 mM MgCl2; 0.5 mM EDTA; 20 mM MES, pH 6.0 or MOPS, pH 7.2) for 20 minutes at 30°C followed by ± formaldehyde to 3.7% for 15 minutes at RT. In experiments using NAO, the dye was incubated with mitochondria for 5 minutes before synuclein addition. Mitochondria were pelleted at 14,000 × g for 10 minutes. The supernatant was recovered and Tris base (or NH4OH) was added to neutralize the formaldehyde, if necessary. The pellet was rinsed with 0.1 M pH buffer, resuspended in an equal volume of PBS and briefly sonicated. SDS-PAGE buffer was added and solutions were incubated at 30°C for 15 minutes before gel electrophoresis and immunoblotting. Sodium carbonate/proteinase K treated mitochondria were prepared by resuspending 1 mg mitochondria in 1 ml cold 0.1 M Na2CO3, pH 11.5. 20 mg (20 U/ml) washed Proteinase K-Agarose beads (Sigma) were added and rocked for 30 minutes at 4°C. Beads were pelleted at 3,000 × g for 2 minutes. PMSF (2 mM final) was added to the membrane supernatant, incubated on ice for 30 minutes, and membranes pelleted at 100,000 × g for 15 minutes. Pellets were resuspended in 0.1 ml MB buffer plus 2 mM PMSF and sonicated for 5 sec before use. The same total amount of input mitochondria was used in control and carbonate/Proteinase K-treated binding reactions.
Lipid extraction of mitochondria was performed using 3:2 (v/v) hexane:isopropanol, as described [28, 29]. Phosphatidylcholine (POPC; Cat # 850457), phosphatidic acid (POPA; Cat # 840857), heart cardiolipin (CL; Cat # 840012) (all in CHCl3 from Avanti Polar Lipids) and mitochondrially-derived liposomes were prepared by sonication of N2-dried lipid films in 5 mM Tris-Cl, pH 7.4, 100 mM NaCl. PC/PA and PC/CL were prepared with 50% of each lipid by weight. Although the total negative charge in PC/PA and PC/CL-containing liposomes is likely similar, under normal physiological conditions, cardiolipin may carry only one negative charge [30]. Binding reactions contained 2 µg synuclein Syn1-102 and ~500 µM total phopspholipid in liposomes (~1:250 molar ratio; ~1:187 ratio for PC/CL), and were in 0.1 ml 50 mM NaCl, 0.5% BSA, 20 mM Tris-Cl, pH 7.4 (20 mM MES pH 6.0 or MOPS pH 7.2, when performing pH binding experiments) for 30 minutes at room temperature. The molar ratio of synuclein to phospholipid in both types of binding assays (intact mitochondria and liposomes) is approximately equal. Assuming complete lipid recovery, saturation of surface exposed cardiolipin in mitochondrial liposomes should occur at ~50 µM NAO. For sucrose fractionation, 0.11 ml 85% sucrose (w/v) was added to 0.05 ml of the binding reaction, mixed well, then overlaid with 0.1 ml 45% sucrose and 0.05 ml 50 mM NaCl, 20 mM buffer, at the indicated pH. Tubes were spun for 2 h at 47k in an SW55 rotor (Beckman). 0.08 ml from the top and bottom of the tubes was collected and diluted to 0.45 ml with H2O. Assuming complete recovery in the upper fraction and 50% in the lower fraction due to sampling dilution, a maximum of 25 ng Syn1-102 was loaded from each fraction onto SDS-PAGE from immunoblotting.
Proton release with formaldehyde
It is known, but generally unappreciated that fixation with aldehydes causes the release of protons, primarily derived from the reaction with primary amines [31, 32]. The released protons will be partially neutralized by the inherent buffering capacity of cytoplasmic components, although this capacity is relatively low [31]. No effect of formaldehyde on pH was observed during the in vitro mitochondrial binding reactions. However, such a fixation-induced drop in pHi was responsible for synuclein translocation to mitochondria in cells preincubated with high concentrations of primary amines (e.g. 25–50 mM NH4Cl, methylamine, etc) and fixed at neutral pH (unpublished data).
Mitochondria lipid measurements
Mitochondria were suspended in pH 6.0 buffer at a phospholipid concentration of 0.1 mM. 1,6-diphenyl-1,3,5-hexatriene (DPH; Molecular Probes), was dissolved in THF and added at a phospholipid to DPH ratio of 300 to 1. Fluorescence lifetime and differential polarization measurements were performed at 30°C with a K2 multifrequency cross-correlation phase fluorometer (ISS, Urbana, IL) as described [33]. Intensity decay and differential polarization measurements were repeated with each sample a minimum of three times. Total fluorescence intensity decays were analyzed with the sum-of-three exponentials, and the intensity-weighted average lifetime, <τ>, is reported. Fluorescence anisotropy decays were analyzed using sum-of–three exponentials, and the weighted average correlation time, <φ>, and order parameter, S, are reported, where S = (r∞/ro)1/2
Results
Synuclein translocates to mitochondria in response to cellular stress
α-Synuclein is a presynaptic neuronal protein that, when expressed in most non-neuronal cells, is diffusely distributed throughout the cytosol with little accumulation at specific intracellular sites [24, 34, 35]. A number of reports have demonstrated an effect of α-synuclein expression on responses to various stress conditions. Alterations in the subcellular distribution of α-synuclein under these conditions, however, have not been carefully addressed. We therefore examined α-synuclein localization under conditions of oxidative and/or metabolic stress. SK-N-SH cells stably expressing wild-type α-synuclein were treated with various concentrations of hydrogen peroxide (H2O2) and processed for immunofluorescence microscopy. In untreated cells, α-synuclein was diffusely distributed throughout the cytoplasm, as expected (Fig. 1A). However, in cells treated for 2 hours with ≥100 µM H2O2, synuclein relocalized from the cytosol to mitochondria, as indicated by colocalization with mitochondria-specific dyes (data not shown) and mitochondria marker proteins (Fig. 1A). The structure of mitochondria was affected by H2O2, and appeared less reticular and often swollen and collapsed near the center of the cells. In addition, synuclein was clustered into variable numbers of structures that did not appear to associate with any intracellular organelle. A similar phenotype was observed in cells treated with metabolic poisons that reduce ATP levels, including 2-deoxyglucose/sodium azide (DOG/Az), and the mitochondrial uncoupler/protonophore CCCP (Fig. 1A). The effects of these reagents could be observed in as little as 30 minutes. Sodium dithionite, a potent reducing agent and oxygen scavenger, was tested to examine the effects of anoxia on α-synuclein localization in an incubator purged with 95% N2/5% CO2 [36]. Unexpectedly, dithionite alone in an air environment induced synuclein translocation to mitochondria (Fig. 1A); this was presumably due to reactive oxygen species formed in the continued presence of oxygen [37]. Hypoxic conditions alone induced a partial relocation of synuclein to mitochondria (unpublished data). Thus, agents previously shown to cause oxidative or metabolic stress induce the redistribution of α-synuclein from the cytosol to mitochondria.
Figure 1. Oxidative and metabolic stresses induce synuclein translocation to mitochondria.
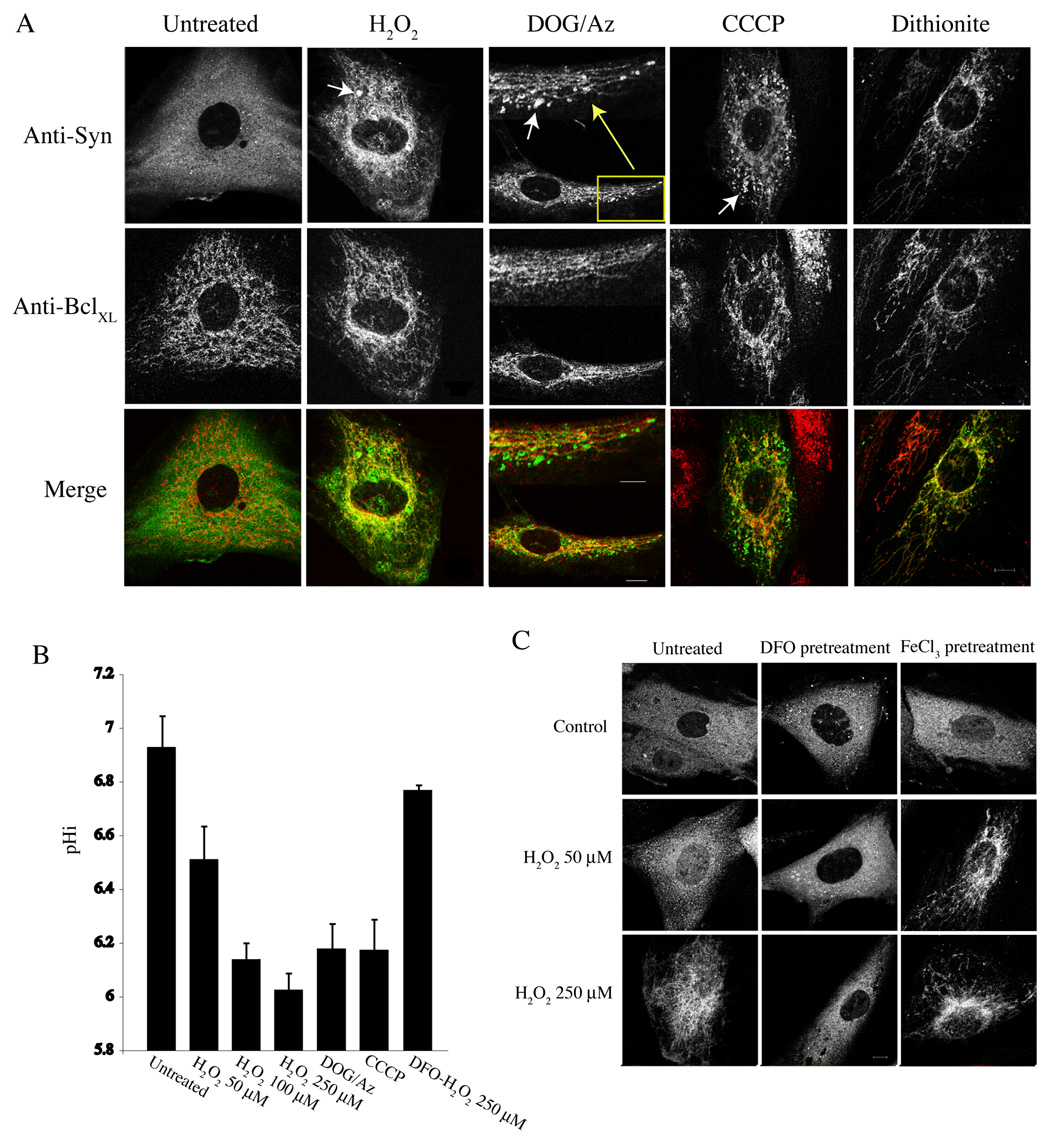
(A) Representitive immunofluorescence images from SK-N-SH cells stably expressing α-synuclein. Cells were untreated or treated for 2 h with 250 µM H2O2, 30 mM 2-deoxyglucose and 0.05% sodium azide (DOG/Az), 1 µM CCCP, or 2 mM sodium dithionite, as indicated, and fixed with 3.7% formaldehyde in 0.1 M PO4 buffer, pH 7.2. Synuclein is labeled with the anti-synuclein antibody 202 and Alexa 488 donkey anti-mouse secondary antibodies. Mitochondria are labeled with antibodies to the outer mitochondrial protein BclXL and Alexa 594 donkey anti-rabbit secondary antibodies. Merged images are shown at the bottom. Arrows indicate synuclein clusters apparently not associated with mitochondria. Bar: 10 µm; inset bar for DOG/Az: 5 µm. (B) pHi measurements determined from treatments in (A) using SNARF 4F (See Materials and Methods). (C) Representitive images showing the effect of DFO and FeCl3 on α-synuclein localization in the presence of H2O2. Note that in cells pretreated with DFO (1 mM for 4 h), synuclein remains diffusely cytosolic at 250 µM H2O2, whereas FeCl3 pretreatment (30 µM for 4 h) sensitizes synuclein translocation to mitochondria at 50 µM H2O2. Not shown are merged images showing colocalization (or lack thereof) of synuclein with mitochondrial markers under these conditions. Bar: 10 µm.
Similar to wild-type synuclein, PD mutant A53T synuclein, β-synuclein, and a Cterminally truncated α-synuclein (Syn1-102) all translocated to mitochondria in the presence of H2O2; however, the PD mutant A30P and γ-synuclein showed dramatically reduced binding (Fig. 2). These results correlate with the ability of these various synucleins to bind intracellular lipid droplets, and further demonstrate that A30P and γ-synuclein are likely membrane-binding deficient [24]. The various synucleins behaved similarly in cells treated with DOG/Az (Supplemental Fig. 1A). Results obtained with HEK293 cell lines stably expressing wild-type and A53T synuclein, but not A30P synuclein, were similar. Low levels of cytoskeletal staining of all of the synucleins were frequently observed under stress conditions, although this phenomenon was not investigated further. Translocation of α-synuclein to the nucleus in cells treated with H2O2 was not observed, as has been reported in MES23.5 cells [38].
Figure 2. Effects of oxidative stress on translocation of different synucleins to mitochondria.
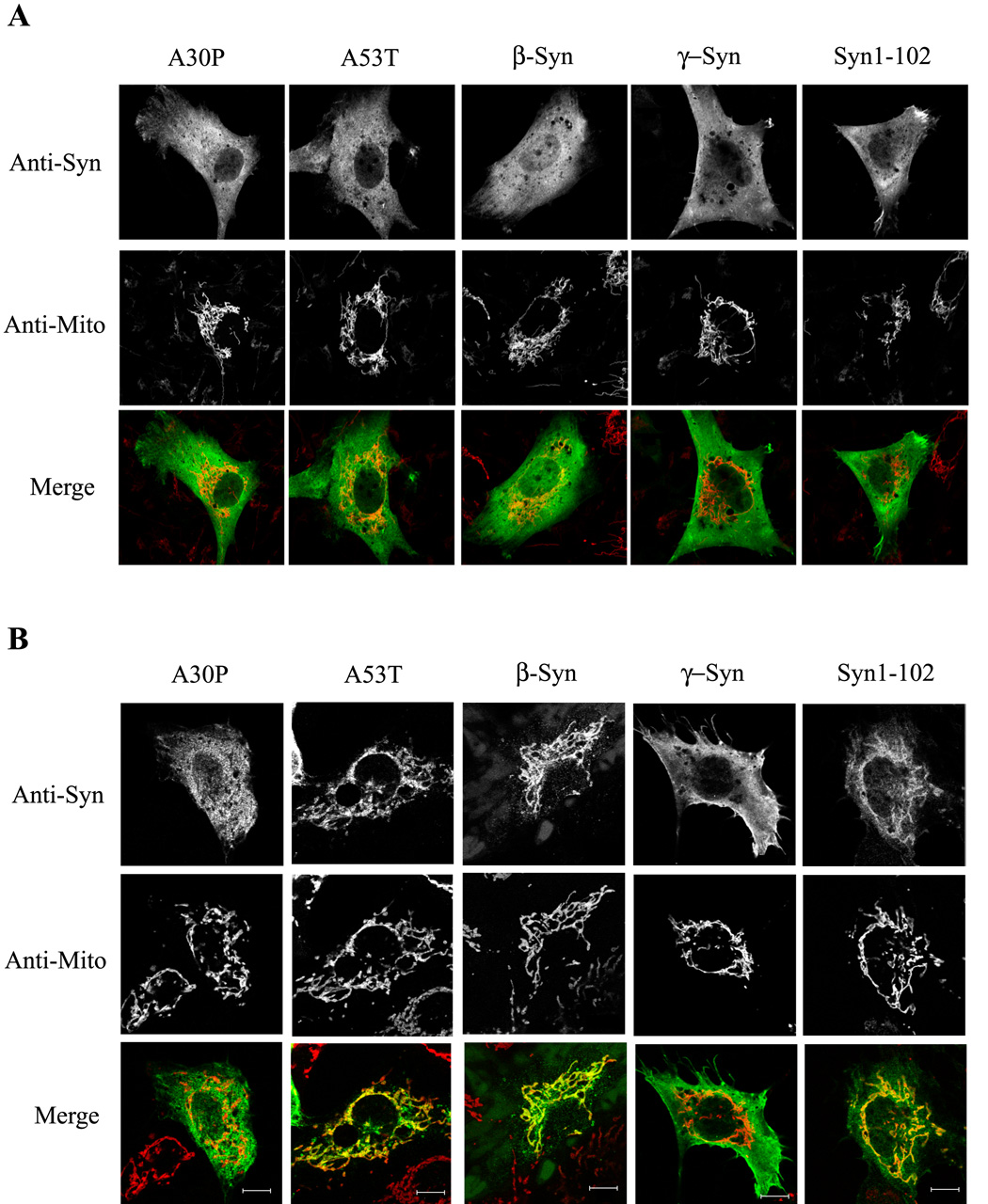
(A) Representative immunofluorescence images of SK-N-SH cells transiently expressing various synucleins. Cells were untreated (A) or treated with 250 µM H2O2 for 2 h (B), before being fixed as in Fig. 1. Note that A53T, β-synuclein, and Syn1-102 translocate to mitochondria with H2O2, whereas translocation was minimal with A30P and γ-synuclein. Mitochondria were labeled either with antibodies to the outer mitochondrial membrane protein Tom20 or to the intermembrane space protein cytochrome c, depending on the species of primary synuclein antibody used. Cells expressing low levels of various synucleins are depicted. Bar: 10 µm.
Oxidative and metabolic stresses induce cytosolic acidification
One common cellular response to a number of oxidative and metabolic challenges is a reduction of intracellular pH (pHi) [36, 39, 40]; this is likely an early and essential event during apoptosis and/or necrosis [41]. Mechanisms proposed for cytosolic acidification include: dysregulation of ion transport [42]; proton leakage from acidic organelles [43]; and hydrolysis of high energy nucleotides [44]. Notably, the PD toxin 1-methyl-4-phenylpyridinium (MPP+) impairs cellular energy metabolism and leads to a decrease in pHi [45].
To investigate possible alterations in pHi during the above stress conditions, the ratiometric pH indicator SNARF 4F/AM (pKa ~6.4) was used. SK-N-SH cells stably expressing α-synuclein were incubated with SNARF 4F/AM for 1 hour, H2O2 was added at various concentrations, and samples were incubated for 2 hours at 37°C. The pHi was then determined (see Materials and Methods). The pHi in untreated cells incubated in PSS buffer at 37°C was 6.9 ± 0.09 (mean ± s.e.m.) (Fig. 1B). This was slightly lower than parental SK-N-SH cells incubated in the same buffer (7.1 ± 0.08), and may be due to slower growth and less active proton exchange activity [46]. No effect on pHi was observed between α-synuclein and vector-only expressing cells, demonstrating that α-synuclein itself has no effect on pHi (data not shown). Treatment with 50–250 µM H2O2 resulted in a progressively reduced pHi that reached 6.03 ± .06 at 250 µM (Fig. 1B). ATP depletion with DOG/Az reduced the pHi to 6.2 ± 0.09. Cells responded similarly to CCCP (6.2 ± 0.12). The pHi decreased rapidly with DOG/Az and CCCP, with the final pHi reached within 20–30 minutes. These results correlate mitochondrial translocation of α-synuclein with decreased pHi. Interestingly, both synuclein translocation and pHi reduction induced by H2O2 were ameliorated by prior treatment of cells with the intra-lysosomal iron chelator desferroxamine (DFO) (Fig. 1B, C); conversely, ferric chloride (FeCl3) pretreatment sensitized α-synuclein translocation to H2O2 (Fig. 1C). These results are consistent with those of Brunk and colleagues, who proposed that oxidative stress stimulates apoptosis via destabilization of lysosomes through the Fenton reaction [47], and results in cytosolic acidification via proton release [43]. Metabolic inhibition of the vacuolar ATPase or protonophore activity would also be expected to dissipate cellular proton gradients, accelerate the leakage of H+ from acidic organelles, and result in net cytosolic acidification. DFO had no effect on synuclein translocation/pHi reduction with metabolic inhibition (DOG/Az or CCCP) (data not shown), demonstrating that iron-catalyzed oxidative damage to lysosomes was not the cause of intracellular acidification under these conditions, as expected [47].
α-Synuclein translocates to mitochondria directly upon intracellular acidification
The above results correlate stress-induced mitochondrial translocation α-synuclein with decreased pHi. To determine if cytosolic acidification alone is sufficient for this effect, SK-N-SH cells stably expressing α-synuclein were clamped to various pHi for 15 minutes using high K+ buffers and the K+/H+ ionophore nigericin, which equilibrates intracellular with extracellular pH [48]. Cells were then fixed at neutral pH (7.2). As shown in Figure 3, α-synuclein appeared completely cytosolic when the pHi was clamped to pH 7.2, but became increasingly associated with mitochondria at pH 6.0 and below. The translocation of synuclein to mitochondria was complete when the pHi was reduced to 5.0. Thus, direct cytosolic acidification is sufficient for translocation of synuclein to mitochondria. In addition, a number of synuclein clusters were generated at low pHi, similar to those formed under stress conditions (see Fig. 1A), demonstrating that cluster formation can be induced simply by lowering intracellular pH.
Surprisingly, synuclein translocation to mitochondria could be mimicked when cells were fixed directly at low pH. SK-N-SH cells transiently expressing α-synuclein were fixed without prior manipulations at pH 7.2 or at pH 5.0, which resulted in the complete redistribution of synuclein to mitochondria (Fig. 4A). Results obtained with SH-SY5Y, HeLa, CHO, primary human fibroblasts, HEK293, and rho-minus HEK293 cells were similar, irrespective of the mode (transient or stable) or levels of α-synuclein expression (data not shown). Interestingly, prior binding to intracellular lipid droplets did not prevent synuclein from translocating to mitochondria at low pH (see Supplemental Figure 2). These results demonstrate α-synuclein’s selectivity for mitochondria at low pH, and presumably reflect synuclein’s ability to sample various membrane environments under different conditions.
Figure 4. Synuclein translocates to mitochondria during acidic fixation.
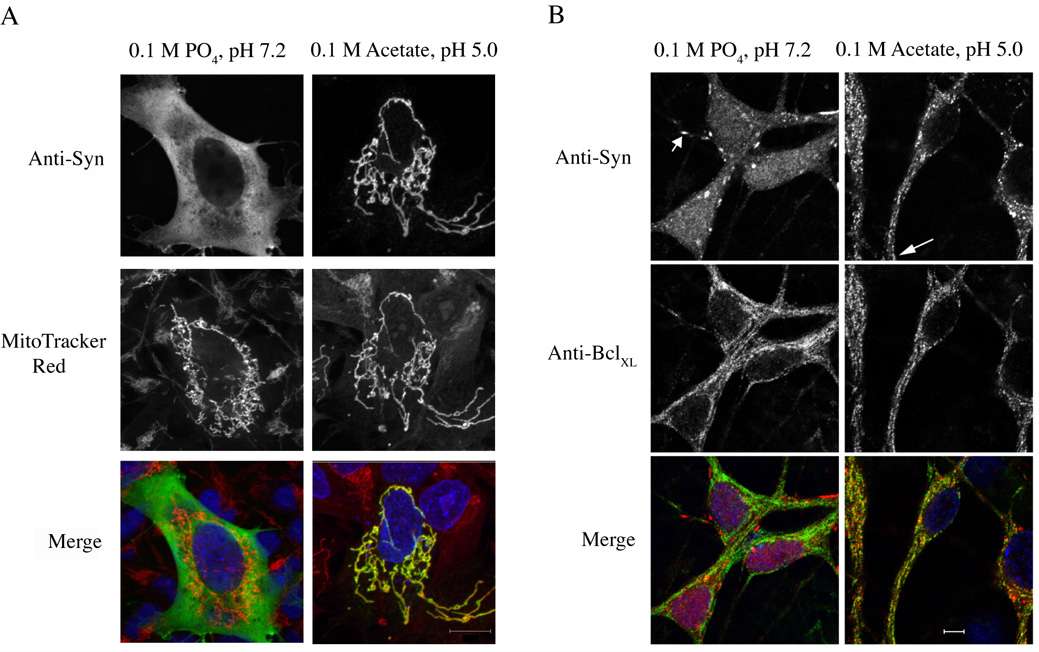
(A) Representative fluorescence micrographs of SK-N-SH cells transiently transfected with α-synuclein. Cells were fixed at pH 7.2 (left panels) or 5.0 (right panels). Synuclein is labeled with the antibody 202 and Alexa 488 donkey anti-mouse secondary antibodies. Mitochondria are labeled with MitoTracker Red. Merged images are shown at the bottom. Nuclei are stained with DAPI (blue). (B) Rat hippocampal neurons (4 div) were fixed at pH 7.2 (left panels) or pH 5.0 (right panels) and stained with antibodies to synuclein or to the mitochondrial protein BclXL. Short arrow indicates a synuclein-positive synaptic bouton; long arrow shows colocalization of synuclein and mitochondria in distal processes. Scale bar: (A) 10 µm; (B) 5 µm.
Importantly, low pH-induced mitochondrial binding was observed in primary neuronal cells expressing endogenous levels of α-synuclein. Rat hippocampal neurons were grown for 4 days in vitro (div); longer incubations led to a decrease of synuclein within cell bodies and increased amounts in presynaptic terminals. In cells fixed in at pH 7.2, synuclein was distributed diffusely throughout the cell bodies, with a few boutons showing presynaptic staining (Fig. 4B). However, fixation at pH 5.0 induced synuclein translocation to mitochondria, not only in cell bodies, but extending to distal processes (Fig. 4B). These results demonstrate that α-synuclein is highly mobile, even during the initial stages of fixation, with a high selectivity for mitochondria under low pH conditions. Due to diffraction limits and the inability to distinguish synaptic from non-synaptic mitochondria by light microscopy, we could not determine if presynaptic α-synuclein translocated onto presynaptic mitochondria (see below).
Synuclein translocates to the outer mitochondrial membrane at low pH
There is limited literature on an association of α-synuclein with mitochondria, although it has been reported that synuclein may be found within degenerating mitochondria [49]. Since low pH fixation appears to mimic synuclein translocation to mitochondria under stress conditions, we used immunoelectron microscopy to examine the subcellular distribution of synuclein in cells stably expressing wild-type α-synuclein fixed under neutral (pH 7.4) or acidic (pH 5.0) conditions. At neutral pH, synuclein was distributed throughout the cytosol; however, low levels were found on the surface of mitochondria (Fig. 5A, left panel). This was not generally appreciated via immunofluorescence microscopy, most likely due to a higher abundance of cytosolic synuclein. However, under acidic conditions, the proportion of synuclein localized to the outer surface of mitochondria was increased (Fig. 5A, right panel). No synuclein was seen within the mitochondrial matrix or intermembrane space. Quantitation of electron micrograph images indicated that low pH induced a ~3-fold increase in the proportion of synuclein on mitochondria versus the cytosol (Fig. 5B, left graph). The fine structure of the cytoplasm appeared disorganized at low pH. Western blotting demonstrated that a proportion of synuclein was extracted into the fixative in a pH dependent manner (Fig. 5C). Based on total gold particle counts, ~14% of the α-synuclein was extracted from the cytosol at pH 5.0. To compensate for this extraction, we measured the density of gold particles per length of mitochondrial membrane, and found a similar ~3 fold increase of mitochondria-bound synuclein under acidic conditions (Fig. 5B, right graph). Thus, synuclein physically translocates from the cytosol to mitochondrial membranes at low pH. In mature mouse hippocampal neurons (10–21 div), we were unable to determine by immunoelectron microscopy whether synuclein in presynaptic terminals translocated to the surface of presynaptic mitochondria, since synapses containing significant levels of synuclein contained very few mitochondria and, conversely, synapses with mitochondria expressed little or no observable synuclein (unpublished observations). The functional significance of this is unclear.
Figure 5. Synuclein redistributes to the outer membrane of mitochondria.
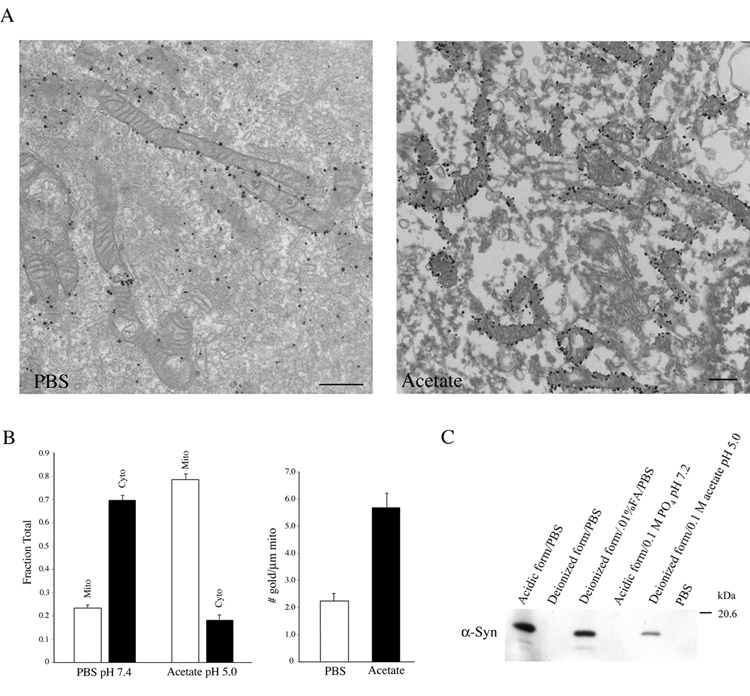
(A) Immunoelectron micrographs of HEK 293 cells stably expressing α-synuclein. Cells were fixed in deionized formaldehyde in PBS (left panel) or 0.1 M acetate buffer, pH 5.0 (right panel) and labeled with the anti-synuclein antibody 202. Scale bar: 500 nm. (B) Quantitative analysis from immunoelectron microscopy in (A). (Left graph) The fraction of total gold particles on mitochondria (mito) versus the cytosol (cyto) is indicated for different fixation conditions. The sum is <100% due to the presence of low levels of gold particles on unidentified organelles. (Right graph) The numbers of gold particles per µm length of individual mitochondria under the indicated fixation conditions. Error bars represent the mean ± s.e.m. (n = ~20 images from 2 independent experiments). (C) Immunoblot of α-synuclein extracted from HEK 293 cells during various fixation conditions. Note that increasing amounts of synuclein are extracted as the pH of the fixative is reduced. Adding 0.01% formic acid to formaldehyde buffered in PBS reduced the pH to 4.5. Increasing the buffering capacity neutralized this effect. Form: formaldehyde. FA: formic acid.
Synuclein binds to purified mitochondria at low pH
We next tested whether purified synuclein could bind to mitochondria in vitro. Recombinant full-length α-synuclein, A30P synuclein, and Syn1-102 were tested for binding to purified mouse brain mitochondria at pH 6.0 and 7.2. There was minimal binding at either pH (Fig. 6A, top). However, in the presence of deionized formaldehyde, similar to conditions used for immunofluorescence, binding of wild-type and Syn1-102, but not A30P was enhanced in a pH-dependent manner (Fig. 6A, bottom). This is consistent with ability of wild-type and Syn1-102, but not A30P, to bind mitochondria in intact cells under low-pHi inducing stress conditions (see Fig. 1 and Suppl. Fig. 1). Prior treatment of mitochondria with formaldehyde did not enhance synuclein binding, and no effect of formaldehyde on the physical properties of mitochondrial membranes themselves could be detected (Table 1). The simplest explanation is that synuclein binding to native mitochondrial membranes is highly dynamic and that formaldehyde stabilizes binding by cross-linking. This is consistent with data suggesting that synuclein binding to native membranes is rapidly reversible [24, 50]. Formaldehyde had no effect on the pH of the binding reaction (see Materials and Methods), demonstrating that reduction to pH 6.0 is sufficient for synuclein binding in vitro. In addition, the proportion of synuclein bound to membranes from a high-speed post mitochondrial pellet (containing primarily endoplasmic reticulum) was significantly reduced when compared with mitochondria (data not shown), thus demonstrating organelle specificity. It is not clear whether the transition from pH 7.2 to 6.0 alters binding via changes in the properties of mitochondria, α-synuclein, or both. Notably, α-synuclein contains a single histidine residue (pKa ~6) at amino acid 50, protonation of which may play a role in synuclein binding at low pH. Given the ability of synuclein to bind acidic phospholipids, the ionic status of the carboxyl group of phosphatidylserine (pKa ~5.5) may also play a role.
Figure 6. Synuclein binds to purified mitochondria at acidic pH.
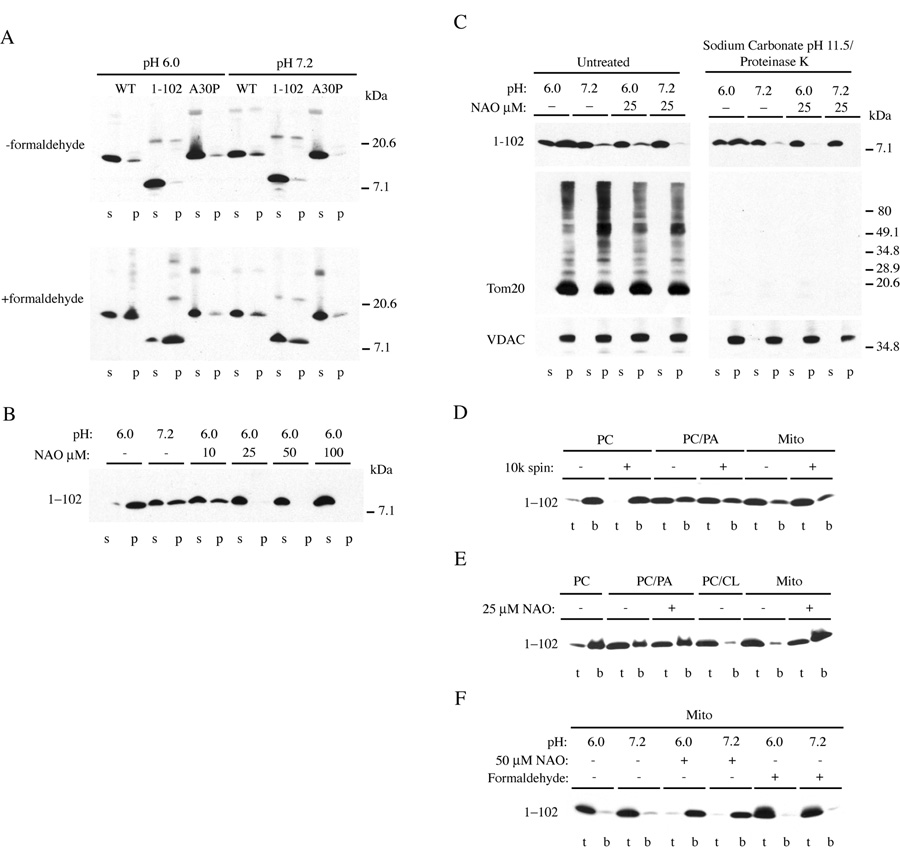
Synuclein binding assays from native mitochondrial membranes (A–C) and liposomes (D–F). (A) Recombinant synuclein proteins were incubated with intact mitochondria at the indicated pH in the absence (top panel) or presence (bottom panel) of 3.7% deionized formaldehyde before pelleting. S, supernatant; p, pellet. Note that formaldehyde treatment enhances pH-dependent binding of wild-type and Syn1-102, but not A30P synucleins. (B) Effect of nonyl acridine orange (NAO) on Syn1-102 binding in the presence of formaldehyde. (C) Effect of alkaline carbonate/proteinase K treatment on Syn1-102 binding (right panels). Immunoblots show the presence or absence of the outer mitochondrial membrane protein Tom20 and membrane embedded VDAC. Note the presence of higher molecular mass forms of Tom20 cross-linked with formaldehyde. No effect of formaldehyde on the mobility of synuclein was observed. All cytochrome c was released from mitochondria during carbonate treatment (not shown). (D–F) Flotation gradients of Syn1-102 binding to liposomes composed of PC, PC/PA, PC/CL, or from purified mitochondria (Mito). T, top of gradient; b, bottom of gradient. (D) Binding was without or with a prior 10,000 × g spin to pellet large lipid aggregates. (E) Specificity of NAO with PC/PA versus mitochondrial liposomes. (F) Formaldehyde- and pH-independent binding to mitochondrial liposomes. NAO inhibits binding.
Table 1.
Summary of DPH fluorescence dynamics in mitochondrial membranes before and after treatment with formaldehyde.
| Florescence Lifetime, <τ> | Average Rotational correlation time, <φ> | Order parameter, S | |
|---|---|---|---|
| Control, 30°C, pH 6.0 | 11.2 ± 0.4 ns | 2.67 ± 0.2 ns | 0.623 ± 0.005 |
| + 3.7 % Formaldehyde | 11.1 ± 0.4 | 2.92 ± 0.23 | 0.626 ± 0.01 |
Parameters obtained from time-resolved fluorescence measurements of DPH in mitochondrial membranes demonstrate that both acyl chain and head group packing are highly ordered in these membranes. The long fluorescence lifetime shows that these membranes are relatively well sealed with respect to water penetration. The high value of the order parameter, S, and long rotational correlation time show that DPH is highly ordered and rotates relatively slowly in the hydrophobic core of the membrane. The values of all 3 parameters are consistent with a membrane with high protein content. Addition of 3.7% formaldehyde at pH 6.0, 30°C, caused no significant changes in the measured properties of the DPH fluorescence dynamics, suggesting that this treatment has no effect on acyl chain or phospholipid headgroup packing. Values are mean ± SD.
The well-known lipid binding properties of α-synuclein [51], led us to investigate whether binding at acidic pH is mediated by mitochondria-specific lipids. Mitochondrial membranes differ from other cellular membranes in that they contain a high proportion of cardiolipin (CL), about 10–20% [52]. Cardiolipin provides essential structural and functional support to a number of proteins involved in mitochondrial bioenergetics, and confers fluidity and stability to the mitochondrial membrane [53, 54]. Cardiolipin levels are highest in the inner mitochondrial membrane, but significant amounts have been reported in the outer membrane or associated with inner- and outer-membrane connections [55]. In addition, translocation of cardiolipin from the inner to outer membranes may occur as an early event in apoptosis [55, 56]. Interestingly, brain cardiolipin levels are decreased by 22% in synuclein knockout mice [28]. We therefore tested the effects of the cardiolipin-binding dye nonyl acridine orange (NAO) on synuclein binding. NAO has a high affinity for cardiolipin, and binds via both electrostatic and hydrophobic interactions with a stoichiometry of 2 mol NAO/mol cardiolipin [57]. Based on phosphorous analysis, 1 mg/ml of total mitochondrial protein contained ~500 µM total phospholipid. Using a spectrophotometric assay that measures binding of NAO to mitochondrial membranes [57], ~100 nmol NAO bound per mg total protein, which corresponds to ~50 nmol cardiolipin/mg protein, or ~10% cardiolipin/mitochondrial phospholipid. Using Syn1-102 in the mitochondrial binding reaction, NAO reduced synuclein binding to mitochondria at pH 6.0 in a concentration-dependent manner (Fig. 6B); full-length α-synuclein gave similar results (unpublished data). Since it is unclear what proportion of cardiolipin is exposed on the outer membrane, the strong inhibitory effect of NAO (10 µM NAO is predicted to bind to ~10% total cardiolipin) suggests that saturation of surface-exposed cardiolipin may be sufficient to prevent synuclein binding at low pH.
To determine if mitochondrial proteins facilitate synuclein binding, intact mitochondria were treated with both sodium carbonate (pH 11.5) [58] and proteinase K. When synuclein was added to treated, re-purified membranes, no difference in pH or formaldehyde-dependent binding was observed compared with untreated mitochondria, nor in the binding sensitivity to NAO (Fig. 6C). The mitochondrial outer membrane protein Tom20 was efficiently degraded by proteinase K under these conditions, whereas the primarily membrane-embedded voltage dependent anion channel (VDAC) remained undigested. Although undigested embedded proteins or protein fragments may remain to facilitate synuclein binding, these results indicate that the low pH-stimulated association of synuclein with mitochondria is likely mediated by direct lipid binding. This is consistent with the general lack of higher molecular mass synuclein cross-links formed with mitochondrial proteins upon formaldehyde treatment (see Fig. 6A, bottom).
Given α-synuclein’s capacity to bind liposomes containing acidic phospholipids [51], the pH and formaldehyde dependence of synuclein binding to synthetic liposomes and to liposomes generated from lipid-extracted mitochondria was examined. As expected, synuclein bound to liposomes containing equal (weight) percentages of phosphatidylcholine (PC) and phosphatidic acid (PA), but not to PC itself (Fig. 6D). A greater proportion of synuclein bound to liposomes made from mitochondrial lipids (Fig. 6D), and liposomes containing PC and cardiolipin (CL) (Fig. 6E), establishing synuclein’s affinity for cardiolipin. The specificity of NAO for cardiolipin versus other anionic phospholipids has been questioned [59]. We found, however, that synuclein binding was less sensitive to the effects of NAO with liposomes composed of PC/PA than those from CL-containing mitochondria (Fig. 6E), indicating a reasonable degree of dye specificity in this assay. NAO also blocked synuclein binding to PC/CL liposomes (not shown), Unlike binding to native mitochondria, binding of synuclein to mitochondrial liposomes was no longer pH- or formaldehyde-dependent, although NAO still inhibited binding (Fig. 6F). It is unclear whether the loss of pH/formaldehyde sensitivity was due to extraction of an important protein component from the membranes, to changes in composition or shape of the exposed liposome surface, or to the dynamic nature of synuclein binding to native versus synthetic membranes [50]. Nevertheless, these results suggest that low pH may increase cardiolipin levels exposed to the outer mitochondrial membrane and facilitate synuclein binding.
Discussion
Oxidative stress and mitochondrial dysfunction are common features in virtually all neurodegenerative diseases, and there is increasing evidence for a causal role of these stresses in disease pathogenesis. In PD, identification of disease-specific genes that influence mitochondrial physiology, either directly or indirectly, have contributed greatly to our understanding of the role of mitochondrial impairment in the etiology of this disease [1, 2]. For example, mitochondrial pathology and oxidative stress are prominent in Drosophila parkin null mutants, and are associated with degeneration of a subset of dopaminergic neurons in the brain [11, 60, 61]. Although nigral degeneration is absent in parkin-deficient mice, they exhibit decreased striatal mitochondrial respiratory capacity and decreased levels of proteins involved in protection from oxidative stress [12]. In Drosophila, deficiency in the mitochondrial kinase PINK1 resembles the pathology associated with mutant parkin, and notably, PINK1 defects can be rescued by wild-type parkin overexpression [62–64]. This implies that these proteins function in a common pathway to maintain mitochondrial integrity and function. Although the precise cellular distribution of the PD protein DJ-1 is unsettled, it has been shown to protect against neuronal apoptosis associated with oxidative stress, possibly by functioning as a redox-sensitive co-transcriptional activator [1, 65]. DJ-1-deficient mice are hypersensitive to MPTP and oxidative stress [10], and inhibition of DJ-1A function in Drosophila by RNA interference (RNAi) results in a number of defects, including hypersensitivity to oxidative stress, degeneration of dopaminergic neurons and impaired phosphatidylinositol 3-kinase (PI3K)/Akt signaling [66]. Interestingly, the same authors also observed impaired PI3K/Akt signaling in parkin RNAi transgenic flies, hinting at a common pathway in the pathogenesis of these PD genes [66].
Similar to other PD genes, α-synuclein has been shown to variably influence cellular responses to a number of oxidative and metabolic stresses [16–19]. The mechanisms responsible for these effects have not been firmly established. Here, we have found that one common feature to a number of cell stresses is the translocation of α-synuclein (wild-type and A53T, but not A30P) from the cytosol onto the mitochondrial surface. In each case, this was induced by a reduction in intracellular pH. Surprisingly, cytosolic acidification alone mimicked these effects, which occurred rapidly (within ~15 minutes) under artificially-induced low pH conditions. The decreases in pHi associated with cellular stresses, however, were generally less than that required for synuclein translocation via direct acidification (by ~1 pH unit; see Fig. 1, Fig. 3). They were, however, similar to the pH decrease required for binding to purified mitochondrial membranes (Fig 6). It is unclear why, in cells, direct acidification required a lower pHi than that generated by oxidative and metabolic stresses. Perhaps, these stresses induce additional cytosolic or mitochondrial signals or biophysical changes in the mitochondrial membrane that enhance synuclein binding. At pHi 5.0, these signals may be artificially bypassed. Nevertheless, reduction of pHi is an essential component of synuclein translocation. Since low levels of synuclein were observed on the mitochondrial surface at neutral pH (Fig. 5 A, left panel), synuclein binding to mitochondria may be a physiological important component of its normal function.
The requirement for formaldehyde (this study) or other cross-linking reagents in stabilizing these interactions underscores the dynamic nature of synuclein binding to native membranes [24, 50], and likely explains our inability to observe significant mitochondrial binding at low pH by subcellular fractionation in the absence of cross-linking (data not shown). It can be questioned whether the observed binding of synuclein to mitochondria is physiologically relevant. Although treatment of cells at low pH (see Fig. 3, Fig. 4) represents a non-physiological manipulation of the cellular environment, we do observe synuclein translocation to the surface of mitochondria in the absence of low-pH fixation under conditions of oxidative stress and metabolic dysfunction (see Fig. 1). In addition, pHi decreases of similar magnitude (0.6 to 1.4 pH units) can occur under pathophysiological conditions, such as during ischemia and hypoxia [67]. Thus, the acute manipulations described here may expose a pathway that occurs minimally under normal conditions but is enhanced during pathological situations.
The consequences of α-synuclein translocation to mitochondria are unclear. Binding under stress conditions may protect mitochondria from subsequent damage (e.g during apoptosis), or alternatively, target defective mitochondria for degradation. Conversely, based on the reduced sensitivity of α-synuclein knockout mice to mitochondrial toxins [20], α-synuclein binding may actually enhance mitochondrial dysfunction and/or result in the accumulation of defective mitochondria. We were unable to detect significant differences in apoptosis (as measured by cytochrome c release from mitochondria or caspase 3 activation) in H2O2-treated HEK293 or HeLa cells expressing the various synucleins (data not shown). Whether use of dopaminergic neuronal cells would reveal a distinction is currently being tested [17]. As α-synuclein is normally degraded, in part, by autophagy [68], defective mitochondrial turnover may also result in the accumulation of mitochondrially-bound synuclein, with subsequent synuclein aggregation/fibrillization leading ultimately to Lewy body formation. Our results suggest that α-synuclein may be linked directly to mitochondrial physiology, and shed light on what may be an underlying signaling network that links cell stress to functional interactions between the various familial PD genes.
Supplementary Material
Supplemental Figure 1. Effect of energy depletion and low pH fixation on synuclein translocation to mitochoodria. Representative immunofluorescence images of SK-N-SH cells transiently expressing various synucleins. (A) Cells were treated with DOG/Az for 2 h then fixed with 3.7% formaldehyde in 0.1 M PO4 at pH 7.2. In (B), cells were directly fixed with 3.7% formaldehyde in 0.1 M acetate at pH 5.0. Note that A53T, β-synuclein, and Syn1-102 translocate to mitochondria in both DOG/Az-treated cells and when cells were directly fixed at low pH; translocation was minimal with A30P and γ-synuclein, although some staining was observed with low pH fixation. Mitochondria were labeled with antibodies to Tom20 or cytochrome c, depending on the primary synuclein antibody. Bar: 10 µm.
Supplemental Figure 2. Effect of low pH fixation on synuclein binding to lipid droplets. SK-N-SH cells transiently expressing wild-type α-synuclein were incubated overnight with 300 µM oleic acid to generate lipid droplets [24]. Cells were incubated for 1 h with MitoTracker Deep Red, and then fixed at neutral (left panel) or acidic (right two panels) pH. Cells were incubated with antibodies to synuclein or the lipid droplet binding protein ADRP, as indicated. Alexa 546 conjugated anti-mouse secondary antibodies were used to stain for synuclein and ADRP, and Bodipy 493/503 was used to label lipid droplets. Mitochondria were imaged at 633 nm (HeNe laser). Note (see arrows) that synuclein surrounds lipid droplets in cells fixed at neutral pH (left panels) but translocates to mitochondria when cells are fixed at pH 5.0 (middle panels). ADRP remains on lipid droplets under these conditions (right panels). Bar:10 µm.
Acknowledgements
We thank the NINDS electron microscopy facility for sample preparation, C. Winters (NINDS) for providing rat primary hippocampal neurons, and C. Ellis (NHGRI) for purified mitochondria. We also thank John Church (Department of Physiology, Univ. of British Columbia) for advice on intracellular pH measurements, and Rod Levine (NHLBI) for many helpful discussions. Part of this work was funded by the NINDS intramural program (to N.B.C.).
While this manuscript was under revision, two papers [69, 70] reported the import of synuclein into mitochondria and its effect on mitochondrial physiology. Under our conditions, however, we are able to detect synuclein only on the outer mitochondrial membrane.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci. 2006;7:207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 2.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 3.Betarbet R, Sherer TB, Di Monte DA, Greenamyre JT. Mechanistic approaches to Parkinson's disease pathogenesis. Brain Pathol. 2002;12:499–510. doi: 10.1111/j.1750-3639.2002.tb00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Przedborski S, Tieu K, Perier C, Vila M. MPTP as a mitochondrial neurotoxic model of Parkinson's disease. J Bioenerg Biomembr. 2004;36:375–379. doi: 10.1023/B:JOBB.0000041771.66775.d5. [DOI] [PubMed] [Google Scholar]
- 5.Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T, Matsuno-Yagi A, Miller GW, Greenamyre JT. Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson's disease. J Neurochem. 2007;100:1469–1479. doi: 10.1111/j.1471-4159.2006.04333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoglinger GU, Carrard G, Michel PP, Medja F, Lombes A, Ruberg M, Friguet B, Hirsch EC. Dysfunction of mitochondrial complex I and the proteasome: interactions between two biochemical deficits in a cellular model of Parkinson's disease. J Neurochem. 2003;86:1297–1307. doi: 10.1046/j.1471-4159.2003.01952.x. [DOI] [PubMed] [Google Scholar]
- 7.Sullivan PG, Dragicevic NB, Deng JH, Bai Y, Dimayuga E, Ding Q, Chen Q, Bruce-Keller AJ, Keller JN. Proteasome inhibition alters neural mitochondrial homeostasis and mitochondria turnover. J Biol Chem. 2004;279:20699–20707. doi: 10.1074/jbc.M313579200. [DOI] [PubMed] [Google Scholar]
- 8.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 9.Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A. 2004;101:9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, Wakeham A, You-Ten AJ, Kalia SK, Horne P, Westaway D, Lozano AM, Anisman H, Park DS, Mak TW. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci U S A. 2005;102:5215–5220. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 13.Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, Gasser T, Wszolek Z, Muller T, Bornemann A, Wolburg H, Downward J, Riess O, Schulz JB, Kruger R. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum Mol Genet. 2005;14:2099–2111. doi: 10.1093/hmg/ddi215. [DOI] [PubMed] [Google Scholar]
- 14.Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem. 1990;54:823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- 15.Mizuno Y, Yoshino H, Ikebe S, Hattori N, Kobayashi T, Shimoda-Matsubayashi S, Matsumine H, Kondo T. Mitochondrial dysfunction in Parkinson's disease. Ann Neurol. 1998;44:S99–S109. doi: 10.1002/ana.410440715. [DOI] [PubMed] [Google Scholar]
- 16.Lee M, Hyun D, Halliwell B, Jenner P. Effect of the overexpression of wild-type or mutant alpha-synuclein on cell susceptibility to insult. J Neurochem. 2001;76:998–1009. doi: 10.1046/j.1471-4159.2001.00149.x. [DOI] [PubMed] [Google Scholar]
- 17.Li W, Lee MK. Antiapoptotic property of human alpha-synuclein in neuronal cell lines is associated with the inhibition of caspase-3 but not caspase-9 activity. J Neurochem. 2005;93:1542–1550. doi: 10.1111/j.1471-4159.2005.03146.x. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa A, V LD, Dawson TM, Ross CA. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001;10:919–926. doi: 10.1093/hmg/10.9.919. [DOI] [PubMed] [Google Scholar]
- 19.Jiang H, Wu YC, Nakamura M, Liang Y, Tanaka Y, Holmes S, Dawson VL, Dawson TM, Ross CA, Smith WW. Parkinson's disease genetic mutations increase cell susceptibility to stress: Mutant alpha-synuclein enhances H(2)O(2)- and Sin-1-induced cell death. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 20.Klivenyi P, Siwek D, Gardian G, Yang L, Starkov A, Cleren C, Ferrante RJ, Kowall NW, Abeliovich A, Beal MF. Mice lacking alpha-synuclein are resistant to mitochondrial toxins. Neurobiol Dis. 2006;21:541–548. doi: 10.1016/j.nbd.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 21.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, Dawson VL, Dawson TM, Ross CA. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet. 2005;14:3801–3811. doi: 10.1093/hmg/ddi396. [DOI] [PubMed] [Google Scholar]
- 23.Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp Neurol. 2004;186:158–172. doi: 10.1016/S0014-4886(03)00342-X. [DOI] [PubMed] [Google Scholar]
- 24.Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL. Lipid droplet binding and oligomerization properties of the Parkinson's disease protein alpha-synuclein. J Biol Chem. 2002;277:6344–6352. doi: 10.1074/jbc.M108414200. [DOI] [PubMed] [Google Scholar]
- 25.Cole NB, Murphy DD, Lebowitz J, Di Noto L, Levine RL, Nussbaum RL. Metal-catalyzed oxidation of alpha-synuclein: helping to define the relationship between oligomers, protofibrils, and filaments. J Biol Chem. 2005;280:9678–9690. doi: 10.1074/jbc.M409946200. [DOI] [PubMed] [Google Scholar]
- 26.Tanner VA, Ploug T, Tao-Cheng JH. Subcellular localization of SV2 and other secretory vesicle components in PC12 cells by an efficient method of preembedding EM immunocytochemistry for cell cultures. J Histochem Cytochem. 1996;44:1481–1488. doi: 10.1177/44.12.8985140. [DOI] [PubMed] [Google Scholar]
- 27.Sheldon C, Cheng YM, Church J. Concurrent measurements of the free cytosolic concentrations of H+ and Na+ ions with fluorescent indicators. Pflugers Arch. 2004;449:307–318. doi: 10.1007/s00424-004-1344-8. [DOI] [PubMed] [Google Scholar]
- 28.Ellis CE, Murphy EJ, Mitchell DC, Golovko MY, Scaglia F, Barcelo-Coblijn GC, Nussbaum RL. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol Cell Biol. 2005;25:10190–10201. doi: 10.1128/MCB.25.22.10190-10201.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hara A, Radin NS. Lipid extraction of tissues with a low-toxicity solvent. Anal Biochem. 1978;90:420–426. doi: 10.1016/0003-2697(78)90046-5. [DOI] [PubMed] [Google Scholar]
- 30.Schlame M, Rua D, Greenberg ML. The biosynthesis and functional role of cardiolipin. Prog Lipid Res. 2000;39:257–288. doi: 10.1016/s0163-7827(00)00005-9. [DOI] [PubMed] [Google Scholar]
- 31.Johnson TJ. Aldehyde fixatives:quantification of acid-producing reactions. Journal of Electron Microscopy Technique. 1985;2:129–138. [Google Scholar]
- 32.Griffiths G. Fine Structure Immunocytochemistry. Heidelberg: Springer-Verlag; 1993. [Google Scholar]
- 33.Mitchell DC, Litman BJ. Molecular order and dynamics in bilayers consisting of highly polyunsaturated phospholipids. Biophys J. 1998;74:879–891. doi: 10.1016/S0006-3495(98)74011-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. Lipid rafts mediate the synaptic localization of alpha-synuclein. J Neurosci. 2004;24:6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mazzulli JR, Mishizen AJ, Giasson BI, Lynch DR, Thomas SA, Nakashima A, Nagatsu T, Ota A, Ischiropoulos H. Cytosolic catechols inhibit alpha-synuclein aggregation and facilitate the formation of intracellular soluble oligomeric intermediates. J Neurosci. 2006;26:10068–10078. doi: 10.1523/JNEUROSCI.0896-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheldon C, Church J. Intracellular pH response to anoxia in acutely dissociated adult rat hippocampal CA1 neurons. J Neurophysiol. 2002;87:2209–2224. doi: 10.1152/jn.2002.87.5.2209. [DOI] [PubMed] [Google Scholar]
- 37.Archer SL, Hampl V, Nelson DP, Sidney E, Peterson DA, Weir EK. Dithionite increases radical formation and decreases vasoconstriction in the lung. Evidence that dithionite does not mimic alveolar hypoxia. Circ Res. 1995;77:174–181. doi: 10.1161/01.res.77.1.174. [DOI] [PubMed] [Google Scholar]
- 38.Xu S, Zhou M, Yu S, Cai Y, Zhang A, Ueda K, Chan P. Oxidative stress induces nuclear translocation of C-terminus of alpha-synuclein in dopaminergic cells. Biochem Biophys Res Commun. 2006;342:330–335. doi: 10.1016/j.bbrc.2006.01.148. [DOI] [PubMed] [Google Scholar]
- 39.Gores GJ, Nieminen AL, Wray BE, Herman B, Lemasters JJ. Intracellular pH during "chemical hypoxia" in cultured rat hepatocytes. Protection by intracellular acidosis against the onset of cell death. J Clin Invest. 1989;83:386–396. doi: 10.1172/JCI113896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahmad KA, Iskandar KB, Hirpara JL, Clement MV, Pervaiz S. Hydrogen peroxide-mediated cytosolic acidification is a signal for mitochondrial translocation of Bax during drug-induced apoptosis of tumor cells. Cancer Res. 2004;64:7867–7878. doi: 10.1158/0008-5472.CAN-04-0648. [DOI] [PubMed] [Google Scholar]
- 41.Harguindey S, Reshkin SJ, Orive G, Arranz JL, Anitua E. Growth and trophic factors, pH and the Na+/H+ exchanger in Alzheimer's disease, other neurodegenerative diseases and cancer: new therapeutic possibilities and potential dangers. Curr Alzheimer Res. 2007;4:53–65. doi: 10.2174/156720507779939841. [DOI] [PubMed] [Google Scholar]
- 42.Lagadic-Gossmann D, Huc L, Lecureur V. Alterations of intracellular pH homeostasis in apoptosis: origins and roles. Cell Death Differ. 2004;11:953–961. doi: 10.1038/sj.cdd.4401466. [DOI] [PubMed] [Google Scholar]
- 43.Nilsson C, Johansson U, Johansson AC, Kagedal K, Ollinger K. Cytosolic acidification and lysosomal alkalinization during TNF-alpha induced apoptosis in U937 cells. Apoptosis. 2006;11:1149–1159. doi: 10.1007/s10495-006-7108-5. [DOI] [PubMed] [Google Scholar]
- 44.Tsai KL, Wang SM, Chen CC, Fong TH, Wu ML. Mechanism of oxidative stress-induced intracellular acidosis in rat cerebellar astrocytes and C6 glioma cells. J Physiol. 1997;502(Pt 1):161–174. doi: 10.1111/j.1469-7793.1997.161bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marini AM, Nowak TS., Jr Metabolic effects of 1-methyl-4-phenylpyridinium (MPP(+)) in primary neuron cultures. J Neurosci Res. 2000;62:814–820. doi: 10.1002/1097-4547(20001215)62:6<814::AID-JNR8>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 46.Hagag N, Lacal JC, Graber M, Aaronson S, Viola MV. Microinjection of ras p21 induces a rapid rise in intracellular pH. Mol Cell Biol. 1987;7:1984–1988. doi: 10.1128/mcb.7.5.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Terman A, Gustafsson B, Brunk UT. The lysosomal-mitochondrial axis theory of postmitotic aging and cell death. Chem Biol Interact. 2006;163:29–37. doi: 10.1016/j.cbi.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 48.Thomas JA, Buchsbaum RN, Zimniak A, Racker E. Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry. 1979;18:2210–2218. doi: 10.1021/bi00578a012. [DOI] [PubMed] [Google Scholar]
- 49.Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson's disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26:41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim YS, Laurine E, Woods W, Lee SJ. A novel mechanism of interaction between alpha-synuclein and biological membranes. J Mol Biol. 2006;360:386–397. doi: 10.1016/j.jmb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 51.Rhoades E, Ramlall TF, Webb WW, Eliezer D. Quantification of alpha-synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys J. 2006;90:4692–4700. doi: 10.1529/biophysj.105.079251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daum G. Lipids of mitochondria. Biochim Biophys Acta. 1985;822:1–42. doi: 10.1016/0304-4157(85)90002-4. [DOI] [PubMed] [Google Scholar]
- 53.Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol. 2007;292:C33–C44. doi: 10.1152/ajpcell.00243.2006. [DOI] [PubMed] [Google Scholar]
- 54.Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007;47:143–183. doi: 10.1146/annurev.pharmtox.47.120505.105122. [DOI] [PubMed] [Google Scholar]
- 55.Kim TH, Zhao Y, Ding WX, Shin JN, He X, Seo YW, Chen J, Rabinowich H, Amoscato AA, Yin XM. Bid-cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome C release. Mol Biol Cell. 2004;15:3061–3072. doi: 10.1091/mbc.E03-12-0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garcia Fernandez M, Troiano L, Moretti L, Nasi M, Pinti M, Salvioli S, Dobrucki J, Cossarizza A. Early changes in intramitochondrial cardiolipin distribution during apoptosis. Cell Growth Differ. 2002;13:449–455. [PubMed] [Google Scholar]
- 57.Petit JM, Huet O, Gallet PF, Maftah A, Ratinaud MH, Julien R. Direct analysis and significance of cardiolipin transverse distribution in mitochondrial inner membranes. Eur J Biochem. 1994;220:871–879. doi: 10.1111/j.1432-1033.1994.tb18690.x. [DOI] [PubMed] [Google Scholar]
- 58.Fujiki Y, Fowler S, Shio H, Hubbard AL, Lazarow PB. Polypeptide and phospholipid composition of the membrane of rat liver peroxisomes: comparison with endoplasmic reticulum and mitochondrial membranes. J Cell Biol. 1982;93:103–110. doi: 10.1083/jcb.93.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gohil VM, Gvozdenovic-Jeremic J, Schlame M, Greenberg ML. Binding of 10-N-nonyl acridine orange to cardiolipin-deficient yeast cells: implications for assay of cardiolipin. Anal Biochem. 2005;343:350–352. doi: 10.1016/j.ab.2005.04.039. [DOI] [PubMed] [Google Scholar]
- 60.Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–2194. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- 61.Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proc Natl Acad Sci U S A. 2005;102:8024–8029. doi: 10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 63.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 64.Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP. DJ-1, a cancer- and Parkinson's disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc Natl Acad Sci U S A. 2006;103:15091–15096. doi: 10.1073/pnas.0607260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang Y, Gehrke S, Haque ME, Imai Y, Kosek J, Yang L, Beal MF, Nishimura I, Wakamatsu K, Ito S, Takahashi R, Lu B. Inactivation of Drosophila DJ-1 leads to impairments of oxidative stress response and phosphatidylinositol 3-kinase/Akt signaling. Proc Natl Acad Sci U S A. 2005;102:13670–13675. doi: 10.1073/pnas.0504610102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Allen DG, Orchard CH. Myocardial contractile function during ischemia and hypoxia. Circ Res. 1987;60:153–168. doi: 10.1161/01.res.60.2.153. [DOI] [PubMed] [Google Scholar]
- 68.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 69.Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha -synuclein impairs complex I in human dopaminergic neuronal cultures and Parkinson's disease brain. J Biol Chem. 2008 doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci. 2008 doi: 10.1007/s00018-008-7589-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Effect of energy depletion and low pH fixation on synuclein translocation to mitochoodria. Representative immunofluorescence images of SK-N-SH cells transiently expressing various synucleins. (A) Cells were treated with DOG/Az for 2 h then fixed with 3.7% formaldehyde in 0.1 M PO4 at pH 7.2. In (B), cells were directly fixed with 3.7% formaldehyde in 0.1 M acetate at pH 5.0. Note that A53T, β-synuclein, and Syn1-102 translocate to mitochondria in both DOG/Az-treated cells and when cells were directly fixed at low pH; translocation was minimal with A30P and γ-synuclein, although some staining was observed with low pH fixation. Mitochondria were labeled with antibodies to Tom20 or cytochrome c, depending on the primary synuclein antibody. Bar: 10 µm.
Supplemental Figure 2. Effect of low pH fixation on synuclein binding to lipid droplets. SK-N-SH cells transiently expressing wild-type α-synuclein were incubated overnight with 300 µM oleic acid to generate lipid droplets [24]. Cells were incubated for 1 h with MitoTracker Deep Red, and then fixed at neutral (left panel) or acidic (right two panels) pH. Cells were incubated with antibodies to synuclein or the lipid droplet binding protein ADRP, as indicated. Alexa 546 conjugated anti-mouse secondary antibodies were used to stain for synuclein and ADRP, and Bodipy 493/503 was used to label lipid droplets. Mitochondria were imaged at 633 nm (HeNe laser). Note (see arrows) that synuclein surrounds lipid droplets in cells fixed at neutral pH (left panels) but translocates to mitochondria when cells are fixed at pH 5.0 (middle panels). ADRP remains on lipid droplets under these conditions (right panels). Bar:10 µm.