

Abstract
Retinal ganglion cells (RGCs) are the first cell type to differentiate during retinal histogenesis. It has been postulated that specified RGCs subsequently influence the number and fate of the remaining progenitors to produce the rest of the retinal cell types. However, several genetic knockout models have argued against this developmental role for RGCs. Although it is known that RGCs secrete cellular factors implicated in cell proliferation, survival, and differentiation, until now, limited publications have shown that reductions in the RGC number cause significant changes in these processes. In this study, we observed that Math5 and Brn3b double null mice exhibited over a 99% reduction in the number of RGCs during development. This severe reduction of RGCs is accompanied by a drastic loss in the number of all other retinal cell types that was never seen before. Unlike Brn3b null or Math5 null animals, mice null for both alleles lack an optic nerve and have severe retinal dysfunction. Results of this study support the hypothesis that RGCs play a pivotal role in the late phase of mammalian retina development.
Keywords: math5, ath5, atoh7, brn3b, POU4f2, RGC, progenitor, retina, development, mouse
INTRODUCTION
The retina serves as an ideal model for studying central nervous system (CNS) development because of its accessibility, well-defined structure, distinct cell types, and the extensive literature relating to its physiology and function. The vertebrate retina contains six major neuronal and one glial cell types that are derived from a common progenitor pool (Turner and Cepko, 1987; Holt et al., 1988; Wetts and Fraser, 1988). In mammals, specification of retinal cell types follows a well-defined temporal sequence. The first few cells to be specified are the retinal ganglion cells (RGCs). Subsequent emerging RGCs are accompanied by the specification of amacrine cells, horizontal cells, and cone photoreceptors. Collectively, the specification of these cells constitutes the early wave of neurogenesis; they are often referred as embryonic or “early” cell types. Rod photoreceptors, bipolar cells, and Müller glia are specified later and constitute the latter wave of cell type specification; they are often referred as postnatal or “late” cell types (Kuwabara & Weidman, 1974; Carter-Dawson & LaVail, 1979; Young, 1985; Cepko, 1996).
In the mouse retina, the first sign of neurogenesis is marked by expression of math5 (also known as atoh7 or ath5), which leads to RGC specification (Brown et al., 1998; Yang et al., 2003). Since RGCs are the first neuronal cell type to be specified, it was postulated that once RGCs are produced, they influence the remaining retinal progenitor cells (RPCs) to produce other retinal cell types (Cepko, 1996; Waid & Mcloon, 1995; 1998; Gonzalez-Hoyuela et al., 2001). For example, RGCs release GDF11 to prevent further RGC production from the progenitor pool (Kim et al., 2005). New born RGCs also release Sonic Hedgehog to stimulate progenitor proliferation (Wang et al., 2002; Moshiri & Reh 2004; Mu et al., 2004; Wang et al., 2005; Agathocleous et al., 2007). Following this line of thoughts, it was predicted that RGC ablation prior to formation of other cell types would result in alterations in retinal differentiation and severe tissue loss because continuing retinal histogenesis and cell differentiation would rely largely on existing RGCs.
However, RGCs’ requirement in the developing mammalian retina became uncertain when results from studies on aquatic vertebrates were interpolated. No RGCs were found in the zebrafish lakritz mutant; yet, the remaining retina appears to be intact. There was no cell loss other than RGCs. Instead, cell number in the inner nuclear layer appeared to be increased (Kay et al., 2001).
The developmental role of RGCs in the mammalian retina was finally reinstated in a recent elegant study. Based on the paradigm that diphtheria toxin A (dta) kills only dta-expressing cells, Klein and colleagues ablated the majority of newborn RGCs by expressing dta at the brn3b locus (Mu et al., 2005). They showed that production of other retinal cell types are affected and concluded that RGCs are required for normal progenitor proliferation but not retinal patterning.
Interestingly, if RGCs were indeed essential for furthering a normal retinal development, their required number for supporting subsequent retinogenesis in the mammalian retina could likely be very small. Earlier attempts in genetically ablating new born RGCs in the mouse retina failed to demonstrate their essential developmental role because there were substantial amount of remaining RGCs. Disruption of the POU domain transcription factor, Brn3b, reduced the RGC number by ~80% without affecting the production of other retinal cell types (Erkman et al., 1996; Gan et al., 1996; Gan et al., 1999; Wang et al., 2001; Lin et al., 2004). The role of RGCs in retinogenesis was not observed in a study that further reduced RGC number by creating a Brn3b/Brn3c double mutant (Wang et al., 2002). A 95% reduction of RGC number as found in Math5-deficient mouse showed merely minor deficiencies of other retinal cell types (Brown et al., 2001; Wang et al., 2001; Lin et al., 2004; Brzezinski et al., 2005; Le et al., 2006). A significant retinal tissue reduction was finally seen when the RGC number was reduced up to 98% (Mu et al., 2005).
Cumulative reports suggest a non-linear relationship between the numbers of ablated RGCs and the numbers of remaining retinal cells. In this hypothetical non-linear curve, up to 80% RGC reduction has little or no effect on the number of other retinal cell types. The curve starts to drop afterwards and shows a detectable total cell number reduction in a 95% RGC depleted retina. The curve drops sharply after that. Therefore, a more severe total cell number reduction can be seen in a 98% RGC ablated retina. If such a non-linear relationship indeed exists, we predict a slightly more than 98% RGC reduction will elicit a much more drastic cell loss in the entire retina.
In this study, we further reduced RGC number to greater than 99%. We circumvented the necessity of using toxins by creating double knockout of genes that are required for RGC formation. We found that mice null for both Math5 and Brn3b have only a handful RGCs. RGC loss in these animals occurs during early retinogenesis. As predicted, the near-complete loss of RGCs leads to a drastic reduction in all other retinal cell types that is much more severe than a 98% RGC ablated retina. In addition, not seen by previous studies, we observed planar patterning defects. Our results also suggest a temporal difference in the response of progenitor cells to RGC shortage: early progenitors increase proliferation when there is a lack of RGCs, whereas late progenitors decrease proliferation.
MATERIALS AND METHODS
Animals and genotyping
Animal use in these experiments was in accordance with the guidelines established by the National Institutes of Health and the local animal welfare committees at the University of Texas-Houston Health Science Center or at the University of Houston. Single knockouts of brn3b and math5 were targeted using embryonic stem cells (AB-1) derived from 129/J mouse strain (Gan et al., 1996; Gan et al., 1999; Wang et al., 2001). Created knockout mice were crossed with the C57BL/6 for more than 11 times. Genotyping was performed using the Southern blotting method described in previous studies (Gan et al., 1999; Wang et al., 2001). Double knockout mice were generated by subsequent crossings of math5−/− (math5lacZ/lacZ) and brn3b−/− (brn3bAP/AP; AP: alkaline phosphatase) lines. Wildtype and math5−/− mice used in the study were derived from the crossings of math5+/−/brn3b+/− x math5+/−/brn3b+/− compound heterozygous mice to ensure similar genome-wide genetic backgrounds. Comparisons within the littermates were not strictly enforced in this study because we were unable to obtain enough embryos or pups with desired genotypes by math5+/−/brn3b+/− x math5+/−/brn3b+/− crossings. Embryos and newborn pups were timed by virginal plugs as well as overall morphology or birth date. Virginal plugs were checked daily in the morning. Positive plugs were designated as E0.5. Occasionally, dated embryos exhibited delayed overall development. These embryos were not included in the study.
Immunohistochemistry
Cryosections were collected at 40 μm thickness on Superfrost slides and incubated with primary antibody in a solution of 0.1% Triton X-100 and 2% bovine serum albumin in PBS for about 2 hours at room temperature or 15 hours at 4°C, followed by incubation with appropriate fluorescent secondary antibody. Primary antibodies used in this study included mouse anti-neurofilament-light chain (NF-L; Zymed 13-0400; 1:500), rabbit anti-phospho-histone 3 (PH3; Upstate 06-570; 1:300), rabbit anti-Pax6 (Chemicon AB5409; 1:300), goat anti-choline-acytel-transferase (ChAT; Chemicon AB144P, 1:100), mouse anti-Calbindin (AbCam ab9481; 1:400), rabbit anti-recoverin (a gift from Alexander Dizhoor; 1:400), rabbit anti-PKCα (Sigma P4334; 1:500), mouse anti-glutamine synthetase (GS; Chemicon MAB302; 1:500), rabbit anti-Sox9 (Chemicon AB5535; 1:200), and mouse anti-Brn3a (SCB SC-8429; 1:200).
Terminal dUTP Nick-End Labeling Assay
TUNEL assay was performed using an APO-BrdU TUNEL Assay Kit (Invitrogen A23210). Cryosections were washed in 1x PBS-T (0.1% Triton X-100 in 1x PBS) three times and stored in 70% ethanol at −20°C overnight. Ethanol-treated sections were washed in 1x PBS-T 3 times and incubated with terminal deoxynucleotidyl transferase (TdT) buffer containing 10 nmol/ml 5-Bromo-2′-deoxyuridine-5′-triphosphate (BrdU) and 600 U/ml TdT at 37°C for 2 hr. The reaction was stopped with 3 PBS washings. Alexa Fluor 488 conjugated anti-BrdU was used to label the nick-end labeled BrdU. Propidium iodide was used to label nuclei and DNA fragments. Tissue sections were mounted with fluoromount-G (EMS 17984-25) and coverslipped for confocal microscopy. Images were collected using a Zeiss 510 Meta laser scanning confocal microscope. Unless otherwise stated, images were projected from 6 optical sections with 1 μm intervals. Color intensity of images was adjusted using Adobe Photoshop. No other image manipulations were used.
Cell counts and statistical analyses
Cell counting for mitotic and apoptotic cells was performed on an Olympus IX71 epifluorescent microscope on slide samples that were masked from labeled genotypes. Counting data from two independent individuals were averaged. Data was discarded if the difference between the two individuals was greater than 10%. Four to five different animals of each genotype were used for retina collection. Three 30 μm cryosections from the center part of each retina were collected on one microscopy slide. The center of the retina in the wildtype and Math5-deficient animals was judged by the appearance of the optic disc and the optic nerve. In the Math5/Brn3b-deficient animals, the center of the retina is judge by the largest retinal circumference. TUNEL labeling often revealed scattered small bright spots representing small degraded DNA residues left from dead cells. These small spots were not counted. Only a BrdU positive nucleus or positive aggregates constituting the size of a nucleus were counted as one.
Different retinal cell types from the adult retinas were counted in confocal images of single optical sections containing an area of 150 μm × 150 μm. The right eyes of three different mice of each genotype were collected for fixation and further processes for cryosection. For the wildtype and Math5-deficient retinas, three cryosections that cross the optic disc and optic nerve were collected onto each slide. For the Math5/Brn3b-deficient retinas, three sections with the largest retinal circumference were used because a optic disc could not be found. Six counting results of each genotype were randomly selected for statistical analysis. Data were compared among different genotypes using one-way ANOVA. If a significant difference (P<0.05) in mean was detected, Duncan’s multiple range test was used to further investigate the differences among the groups.
Electroretinogram (ERG) Recording and Analysis
Preparation and recording methods were described previously (Saszik et al. 2002; Robson et al., 2003), and will be described only briefly here. Recordings were made from brn3−/− (n=4), math5−/− (n=3), math5−/−/brn3−/− mice (n=7), and wildtype (n=7) from various litters. Mice ranged in age from 10 to 15 weeks, with most being 12–13 weeks. Mice were initially anesthetized with an intraperitoneal injection of ketamine (60 mg/kg) and xylazine (6 mg/kg), and anesthesia was maintained with ketamine (56 mg/kg) and xylazine (5.6 mg/kg) given every 45–60 minutes via a subcutaneous needle fixed in the flank. The nose was clamped and the two front teeth held in a metal bite bar that also served as the electrical ground. Pupils were fully dilated to 3 mm in diameter with topical atropine (0.5%) and phenylephrine (2.5%). Rectal temperature was maintained at 37–38°C. Recording sessions lasted about 3 hrs for dark-adapted ERGs and 1.5 hours for light-adapted ERGs. The mice were allowed to recover from anesthesia after the session.
ERGs were recorded differentially between silver-impregnated fiber electrodes moistened with methylcellulose sodium (Celluvisc, Allergan Inc, USA), and placed across the center of the cornea of each eye. The cornea of the tested eye was covered with a clear contact lens, the nontested eye with a black contact lens and a black aluminum foil cap that covered both the eye and the skull. For dark-adapted ERGs, animals were dark-adapted overnight and prepared for recording under red illumination (LED, λ >640 nm). For light-adapted ERGs, animals adapted to the rod-suppressing background for at least 15 min before data were collected.
The Ganzfeld stimulus consisted of brief (0.8 μs–4.1 ms) full-field flashes (LEDs, λ max = 462 nm for dark-adapted ERGs from stimuli too weak to produce measurable responses [−6.1 log scotopic Troland seconds (sc Td s)] to strong stimuli (1.5 log sc Td s) that elicited a-waves. The interval between flashes was adjusted so that the ERG had returned to baseline before another stimulus was presented. The Ganzfeld stimulus was produced by rear illumination of a concave white diffuser (35 mm diameter) positioned close to the eye. Time zero was taken as half-way through the flash.
For light-adapted ERGs, the stimulus was a green Ganzfeld flash (λ max = 513 nm) of short duration (<4 ms) on a rod-suppressing background of 2.5 log sc Td also provided by green LEDs (Colorburst mini-Ganzfeld, Espion system, Diagnosys LLC, USA). The luminance (sc cd m−2) and luminous energy (sc cd s m−2) were measured with a scotopically-corrected photometer (International Light model IL1700). Conversions from Troland values to photoisomerizations per rod (R*/rod) assume that 1 sc Td s gives 122 R*/rod (Saszik et al., 2002). Responses were averaged over many trials when stimuli were weak and responses were small, and over fewer trials when responses were larger. Signals were amplified from 0 to 300 Hz, digitized with a resolution of 1 μV at a rate of 1 kHz and digitally filtered offline to remove 60 Hz.
ERG a-wave and b-wave amplitudes were measured at fixed times after the brief stimulus flash, 7 ms for a-waves, and 110 ms for b-waves under dark-adapted conditions, and 65 ms for b-waves under light-adapted conditions; light-adapted a-waves were not measured. Repeated ANOVAs were used to compare transgenic and wildtype results over a range of stimulus energies from 0.3 to 1.8 log sc td for dark-adapted a-waves, from −2.61 to 1.44 log sc td s for dark-adapted b-waves, and from 2.2 to 3.5 log sc td s for light-adapted b-waves.
RESULTS
Math5/Brn3b deficiency has drastically reduced the number of RGCs in the mature retina
To maximally reduce the number of RGCs, Math5 (Atoh7, Ath5) null mice were bred onto a Brn3b (POU4f2) null background to create a Math5/Brn3b double null line. Brn3b null mice have an ~80% RGC reduction (Gan et al., 1996; Erkman et al., 1996; Lin et al., 2004), whereas Math5 null mice have an ~95% RGC reduction (Brown et al., 2001; Wang et al., 2001; Lin et al., 2004). Brn3b is essential for normal RGC axon formation (Erkman et al., 2000; Wang et al., 2000). The 5% remaining RGCs in the Math5-deficient retina require Brn3b to initiate their normal axonal program. Therefore, we predicted a loss of Brn3b would reduce the RGCs in the Math5-deficient retina by an additional 80%. This would leave only 1% of RGCs remaining.
The Math5/Brn3b double null mice are healthy and fertile, but their sexual maturation may be delayed for several weeks; they do not mate before 10 weeks of age. No other physical or behavioral abnormalities were noticed. Their eyes appeared to be normal exteriorly. However, no RGCs were identified on radial sections of the adult (6 to 10 weeks of age) retinas when Brn3a antibody was used. We suspected that radial sections may have missed sparsely distributed RGCs. In addition, even if a section did contain a RGC, it might not be picked up by anti-Brn3a antibody because Brn3a is found only in a subset of mature RGCs (Xiang et al., 1995). We, therefore, used an antibody to Neurofilament-light chain (NF-L), for its large coverage of RGC subtypes (e.g., Wang et al., 2002), to label RGC axons on the flatmount retinas.
Flatmount retinas with NF-L labeling did reveal, by their axons, some RGCs that were not detected in the radial sections (Fig. 1). However, as predicted, the Math5/Brn3b-deficient retinas had much fewer NF-L-positive axons when compared with the Math5-deficient retinas. Only a handful (roughly 50 to 100) of them were detected. These single non-fasciculated axons exhibited much brighter NF-L signals than those of wildtype and each single knockout. They did not converge in a location that we could definitively identify as an optic disc. Often, many axons ran multiple times across the entire retinal surface while making turns at the periphery. We were unable to obtain a precise RGC number in the double knockout retina. The best method would have been a retrograde dye labeling. However, an optic nerve was not identified in all animals examined (n=20) to perform such a task. The best available method left for us to assess RGC numbers in the Math5/Brn3b-deficient retinas was by comparing the numbers of NF-L-positive axons to that of Math5-deficient retinas. The remaining RGC number in the Math5-deficient retina was carefully documented to be approximately 5% of the normal amount (Lin et al., 2004). The NF-L-positive axon number in the double knockout retina was consistently less than one tenth of those found in the Math5-deficient retina. Therefore, it is safe to conclude that the Math5/Brn3b-deficient retina has less than 1% of the normal RGC number (Comparing fig. 1C and 1D).
Fig. 1.
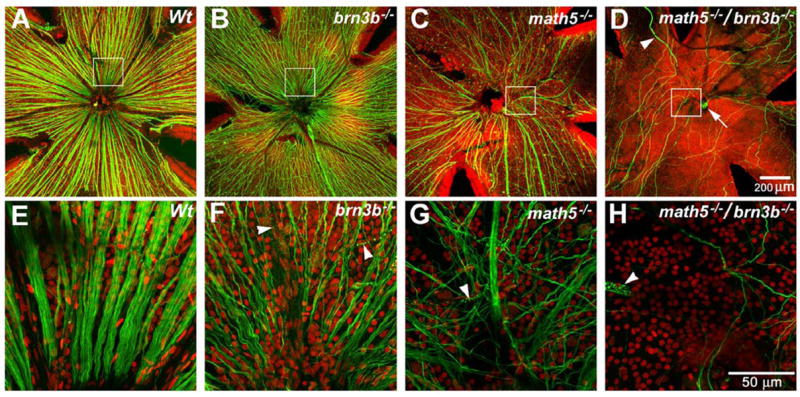
Projections of optical sections of flat-mounted adult retinas. Green: Neurofilament-light chain (NF-L). Red: Propidium Iodide (PI). Panels E, F, G, and H are enlarged from the boxed area of panels A, B, C, and D and rotated so that the optic discs or putative optic discs are at the bottom. A and E, Wildtype NF-L positive axons are abundant and well bundled. B and F, a Brn3b-deficient retina has significant RGC loss, as indicated by the NF-L positive axons. C and G, a Math5-deficient retina has further RGC loss. Many of the RGC axons appear to be disoriented. D and H, only a few NF-L positive axons can be detected. These axons appear to be disoriented and do not enter the optic disc. Many of them traverse the retinal area more than once by turning at the retinal periphery. The putative optic disc, indicated by an arrow in D, contains only capillaries. Other capillaries are indicated by arrowheads.
RGC reduction occurred early during retina development
We wanted to know whether RGC loss in the Math5/Brn3b-deficient retina occurred during early retinogenesis. We examined the retinal properties at P0, a time point when birth of early cell types is declining and birth of late cell types is reaching a peak. Flat-mounted retinas were used to examine the ganglion cell layer and estimate the quantity of NF-L-positive RGC axons (Fig. 2A–F). In the Math5-deficient retinas, despite a dramatic reduction in the number of axons and substantial disorganization of their projections, a significant number of long axons were readily observable. Some axons wandered across the retina, while a few entered the optic disc, thus explaining the thin optic nerve observed in the previous study (Wang et al., 2001). In contrast, virtually no extended NF-L-positive axons were observed in any of the Math5/Brn3b-deficient retinas examined (n=6) at this stage. Scattered NF-L positive cells were observed. These cells had extremely bright NF-L labeling in the soma, but their axons appeared to be underdeveloped and misoriented (Fig. 2F). To ascertain our result, anti-Brn3a antibody was used to label the P0 flatmount retinas. The result confirmed that further RGC loss in the Math5/Brn3b-deficient retina occurred before P0 (Suppl. Fig. 7 & Suppl. Fig. 8).
Fig. 2.
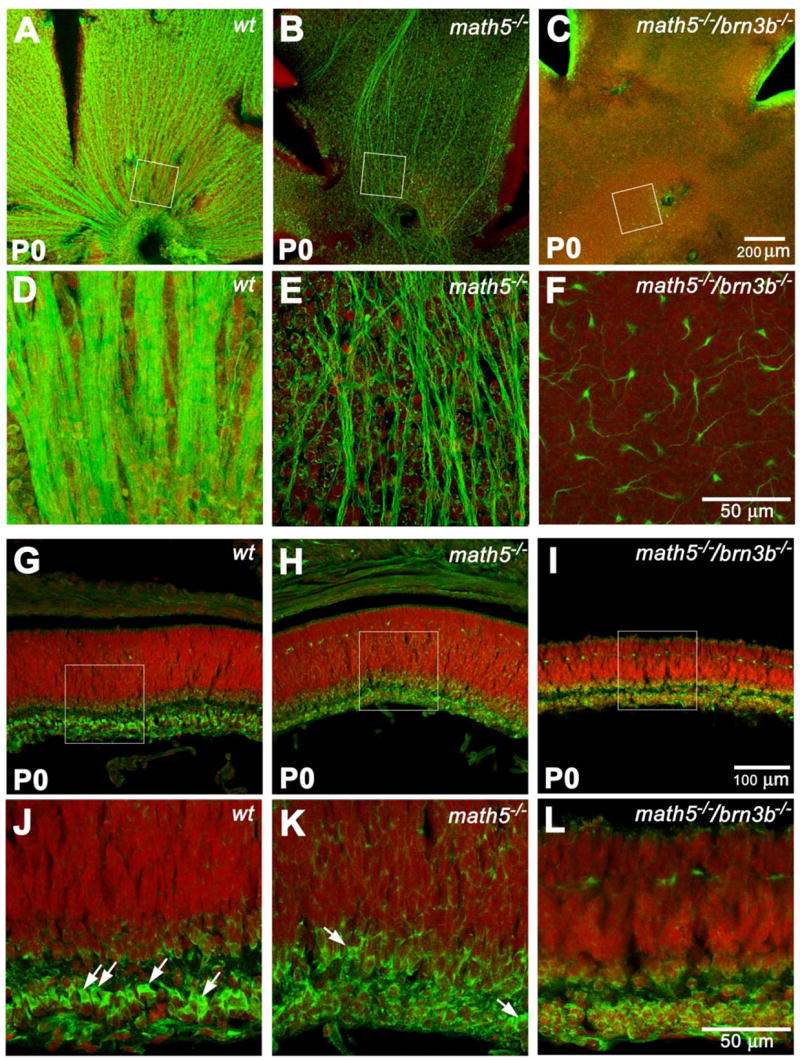
Projections of optical sections of flat-mounted and cross sectioned retinas at P0. Green: NF-L. Red: PI. Panels D, E, and F are enlarged from the boxed area of panels A, B, and C and rotated so that the optic discs or putative optics disc are at the bottom. Panels J, K, and L are enlarged from the boxed area of panels G, H, and I. A and D, well organized wildtype RGC axons can be seen at this stage. B and E, the number of NF-L-positive axons is drastically reduced. However, a significant amount of unorganized ones can be readily detected. C and F, scattered NF-L-positive cell bodies with short neurites can only be detected with high magnification. G and J, a cross sectioned wildtype retina showing newly-separated RGC layer. RGCs have distinct polygonal shapes of NF-L labeling, as indicated by the arrows. Amacrine cells above the emerging inner plexiform layer are also NF-L positive at this stage. The NF-L signal forms a thin circle, suggesting a unique cytoplasmic shape of amacrine cells. H and K, a Math5-deficient retina does not appear to be thinner than a wildtype control at this stage. However, besides a few possible RGCs (arrows), most of the cells in the INL have the shape of amacrine cells. I and L, the Math5/Brn3b-deficient retina is obviously thinner than the wildtype control. Cells exhibiting RGC shape cannot be found. Cell number in the RGC layer and NF-L positive cell number above the inner plexiform layer are severely reduced.
Distributions of RGCs were further examined on radial sections through the retinas immunostained with the anti-NF-L antibody (Fig. 2G–L). In the wildtype retina, NF-L distinctively labeled RGCs, amacrine cells, and horizontal cells (Fig. 2G and J). At this stage, cells in the newly-laminated ganglion cell layer (GCL) were separated from the remainder of the retina by the nascent inner plexiform layer (IPL). The majority of cells in the GCL exhibited a unique morphology that can be easily distinguished from others. They had a prominent polygonal NF-L-positive cytoplasm that was polarized to the apical side of the nucleus. We wanted to know whether such a distinct morphology is characteristic to RGCs at P0. Next, we co-labeled the sections with an antibody to Brn3a, a member of Brn3 (POU4) family that is found exclusively in the RGCs in the retina. With rare exceptions (1 in every 150 μm of retinal length), virtually all these cells are positive of Brn3a (Suppl. Fig. 6). At this stage, most of NF-L positive amacrine cells were separated from the GCL by the IPL and were identifiable by their location. Unlike the polygonal shape seen in RGCs, the amacrine cells had a thin, round, NF-L-positive signal surrounding their nuclei. Therefore, RGCs and amacrine cells could be easily distinguished at P0 by their NF-L staining pattern. Horizontal cells were detected closer to the ventricular margin, presaging the position of the future outer plexiform layer (OPL).
At P0, the Math5/Brn3b null retina exhibited a drastic reduction in cell number, resulting in an ~50% decrease in thickness compared to wildtype, while Math5 null alone did not show any retinal thickness reduction. Also, the number of NF-L-positive cells in the GCL of the Math5/Brn3b-deficient retina was significantly less (~50% reduction) than in Math5-deficient retina despite a similar distribution pattern (compare Fig. 2L to 2K and suppl. Fig. 1). Based on the pattern of NF-L, these cells had the morphology of amacrine cells. The polygonal shape and polarized immunoreactivity characteristic of the RGC cytoplasm were never observed nor were Brn3a positive nuclei detected on radial sections, suggesting an absence of RGCs. Horizontal cell numbers in the double-mutants were comparable to the Math5-deficient retina at this stage. The primary difference between the math5−/− retina and the math5−/−/brn3b−/− retina was that the latter had fewer RGCs. Our results showed that RGC reduction from 95% (math5−/−) to >99% (math5−/−/brn3b−/−) occurred prior to P0 (Suppl. Fig. 7) and resulted in a dramatic decrease in overall cell number during retinogenesis.
Math5/Brn3b deficiency resulted in drastic reduction of all retinal cell types
We were convinced that Math5/Brn3b deficiency resulted in a severe RGC reduction during retinal development that was never seen before. Next, we wanted to know the effect of this much RGC reduction on the quantity and distribution of other retinal cell types. We compared cryosections from adult math5−/−/brn3b−/− retinas to that of math5−/− and wildtype (6 to 10 weeks of age). Labeling with cell type-specific markers revealed that Math5/Brn3b deficiency disproportionately impacted certain cell types over others (Suppl. Fig. 2, 3, & 4), a result distinct from that of Mu et al. (2005). The Math5/Brn3b-deficient retinas exhibited a striking loss of cells in both the ONL (~50%) and INL (~65%) when compared to the wildtype. Horizontal cell number did not show significant change, whereas Pax6-positive amacrine cells in the INL showed an approximate 70% reduction. Rod bipolar cells were reduced by 45%. They appeared to be evenly distributed in the wildtype and math5−/−. However, they form patches along the retinal plane in the math5−/−/brn3b−/−. Recoverin-positive cone bipolar cells were only seen occasionally in the central retina while small aggregates were detected at the retinal periphery. Conservatively, we estimated that cone-bipolar cells were reduced by approximately 85% (Fig. 3 and Suppl. Table 2). The result indicated that the planar distributions of both rod and cone bipolar cells were severely altered.
Fig. 3.
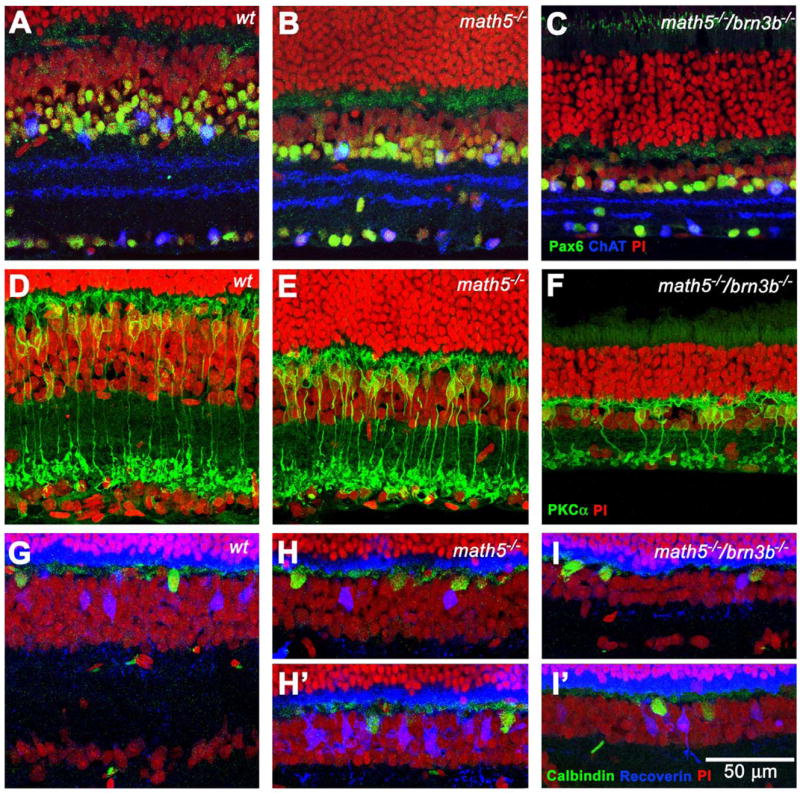
Cross sections showing different cell populations in the inner nuclear layer (INL) in the adult retinas. A, B, and C, Pax6 and ChAT labeling reveals an amacrine cell population in the INL of three different genotypes. There is a significant reduction of Pax6-positive cells in the Math5-deficient retina. These cells are further reduced in number in the Math5/Brn3b-deficient retina. However, the number of ChAT-positive cells is slightly increased. These cells are unevenly distributed in the Math5/Brn3b-deficient retina. D, E, and F, PKCα labeling reveals a rod bipolar cell population. There is no significant difference of rod bipolar cells between Math5-deficient and wildtype retinas. In the double null retina, not only is the rod bipolar cell number significantly reduced, they also form small aggregates. G, H, and I, Calbindin and recoverin labeling show horizontal cells and cone bipolar cells, respectively. H & I were collected from the central retina. H′ & I′ were collected from the peripheral retina. Uneven planar distribution of both cell types can be seen in both Math5- and Math5/Brn3b-deficient retinas.
Significant cell loss was also apparent when the math5−/−/brn3b−/− retinas were compared to the math5−/− retinas. Overall, the INL had 40% less and the ONL had almost 50% less cells (Fig. 3 and suppl. Tab. 2).
Stimulated by a recent report that precocious Müller glia can form in a math5−/− retina (Le et al., 2006), we paid special attention to the distribution of Müller cells in the Math5/Brn3b- and Math5-deficient retinas. Glutamine Synthetase (GS) or vimentin are commonly used to label Müller cells, but they cannot unambiguously locate the cell body. A nuclear localized maker would have to be used to locate the Müller cell body. Among different markers we tested, we found that the Sox9 antibody (a gift from Richard Behringer) matched the best with GS signals and clearly marked the nuclei of GS positive cells of all wildtype retinas we have tested (Fig. 4D). Using Sox9 as a marker, we observed a slight, but not statistically significant, overproduction of Müller cells in the Math5-deficient retina. Surprisingly, many of the Müller cells were mislocalized to the GCL and ONL - a phenomenon not detected in earlier studies (Fig. 4; an extreme case is given in Suppl. Fig. 2). These mislocalized Sox9 positive cells are included in our Müller cell counting, which may explain why the result is slightly different from an earlier report (Brown et al., 2001). However, we were unable to determine whether these mislocalized Müller cells were a result of RGC loss or Math5 deficiency. In the Math5/Brn3b-deficient retina, the number of Müller cells was significantly decreased when compared to either wildtype or math5−/− retinas (~30%), suggesting insufficient RGC number can also lead to reduction of the final number of Müller cells.
Fig. 4.
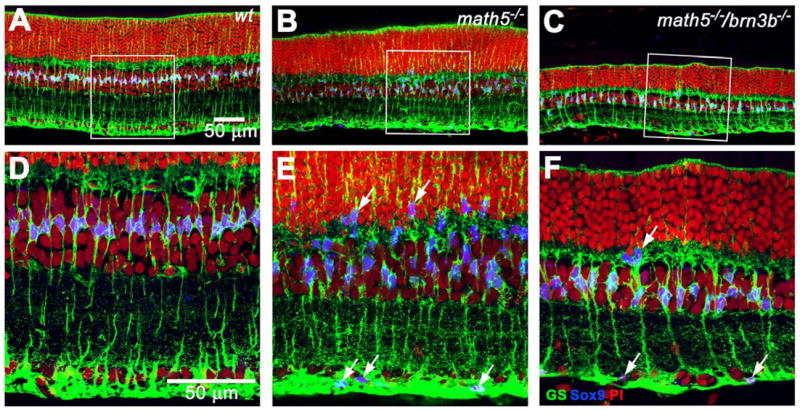
Projections of optical sections labeled with glutamine synthetase (GS) and Sox9 revealing distribution of Müller cells. Panels D, E, and F are enlarged from the boxed area of panels A, B, and C. A and D, Sox9 labels the nuclei of Müller cells, as indicated by positive GS signals. Sox9-positive nuclei are nicely aligned in the middle of the INL in the wildtype retina. Sox9-positive cells are not found in other locations. B and E, the distribution of Müller cells is disturbed. Sox9-positive cells are scattered throughout the outer nuclear layer and ganglion cell layer (arrows). C and F, the number of Müller cells is reduced in the Math5/Brn3b-deficient retina. Müller cells can still be found in the ONL and GCL (arrows).
Total retinal cell loss in the Math5/Brn3b-deficient retina was a result of altered progenitor cell proliferation and increased apoptosis
The thinner retina that we observed in the Math5/Brn3b-deficient mice could be explained by decreased progenitor cell proliferation or by increased apoptosis. To distinguish the contributions of these two processes, we compared the number of dividing cells and the number of dying cells at embryonic day 16 (E16) and postnatal day 0 (P0).
Phosphohistone-3 (PH-3) was chosen as a cell proliferation marker because its narrow expression window at metaphase allows a more accurate assessment of cell numbers. Our results showed that at E16.5, there were more PH-3 positive cells in both Math5/Brn3b double null and Math5 null retinas as compared to their wildtype counterparts (Fig. 5 & Suppl. Fig. 5). This is not surprising because it has been reported that math5−/−-expressing cells may reenter their cell cycle and be accounted for the increased PH-3 positive cells (Kay et al., 2001; 2005). However, for reasons given later in the DISCUSSION section, we believe increased cell proliferation observed in the math5−/−/brn3b−/− and math5−/− retinas was a response of early progenitors to RGC shortage.
Fig. 5.
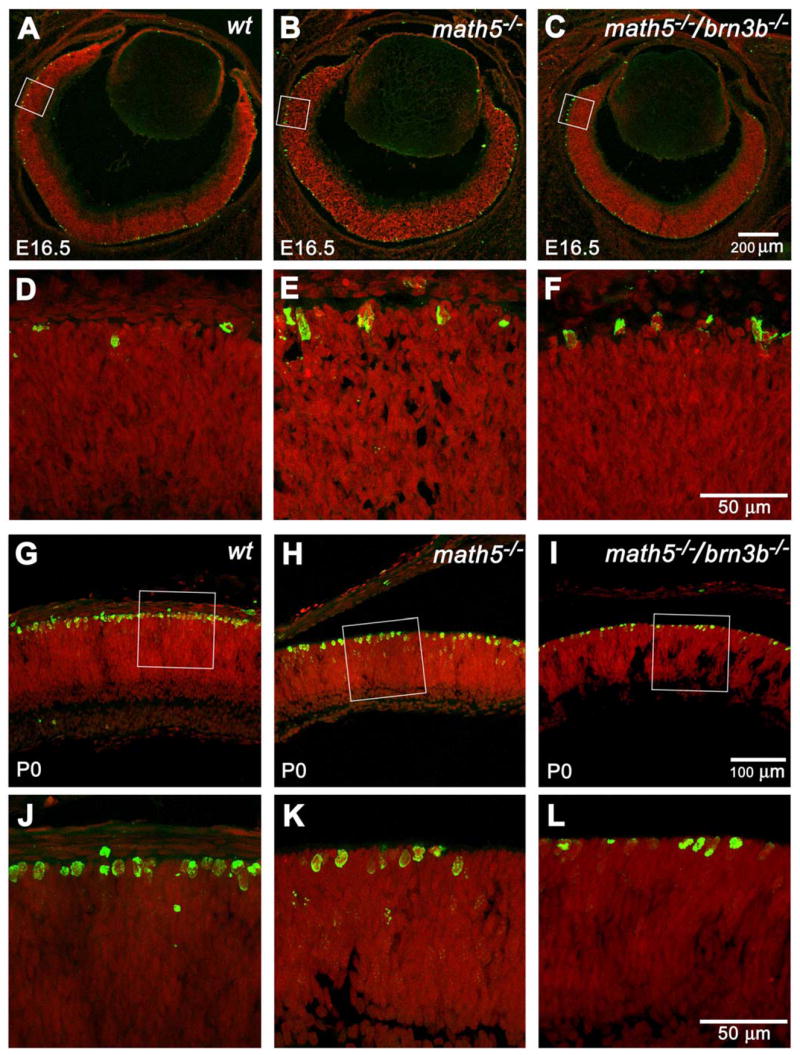
Representative optical sections of phosphohistone-3 (PH3) labeled retinas at E16.5 and P0. Green: PH3. Red: PI. Panels D, E, and F are enlarged from the boxed area of panels A, B, and C and rotated so that the ganglion cell layers are at the bottom. All PH3-positive cells are detected at the ventricular margin at the mitotic zone. At E16.5, both Math5- and Math5/Brn3b-deficient mice have higher numbers of PH3 positive cells than that of a wildtype control. Panels J, K, and L are enlarged from the boxed area of panels G, H, and I. At P0, a Math5-deficient retina has fewer PH3-positive cells than a wildtype control. A Math5/Brn3b-deficient retina has a further reduction in PH3-positive cells.
In contrast to E16.5, Math5/Brn3b double null retinas (Fig. 5 I, L) had significantly fewer metaphase cells than their wildtype counterparts at P0 (Fig. 5 G, J). Math5 null (Fig. 5 H, K) retinas had an intermediate number of metaphase cells at this stage (n=4). Our result showed two distinct cell proliferation responses to RGC shortage during early and late retinogenesis, but was unable to explain the thinner retina that was observed in the double mutant at P0. We suspected it could be increased cell death.
To ascertain the contribution of cell death to the reduced retinal thickness and the difference in cell number observed in Math5/Brn3b-deficient mice, terminal UTPase nick end labeling (TUNEL) was used to mark apoptotic cells. The same two stages that were analyzed in the cell proliferation assay, E16.5 and P0, were chosen. We found that at E16.5, both Math5/Brn3b double null and Math5 null retinas had an ~65% increase in the number of apoptotic cells when compared to wildtype retinas (Fig. 6A–F & suppl. Fig. 5). At P0, Math5/Brn3b double null mice had 30% more TUNEL-positive nuclear profiles than Math5 null animals, and 500% more than the wildtype mice (Fig. 6 G–L & suppl. Fig. 5).
Fig. 6.
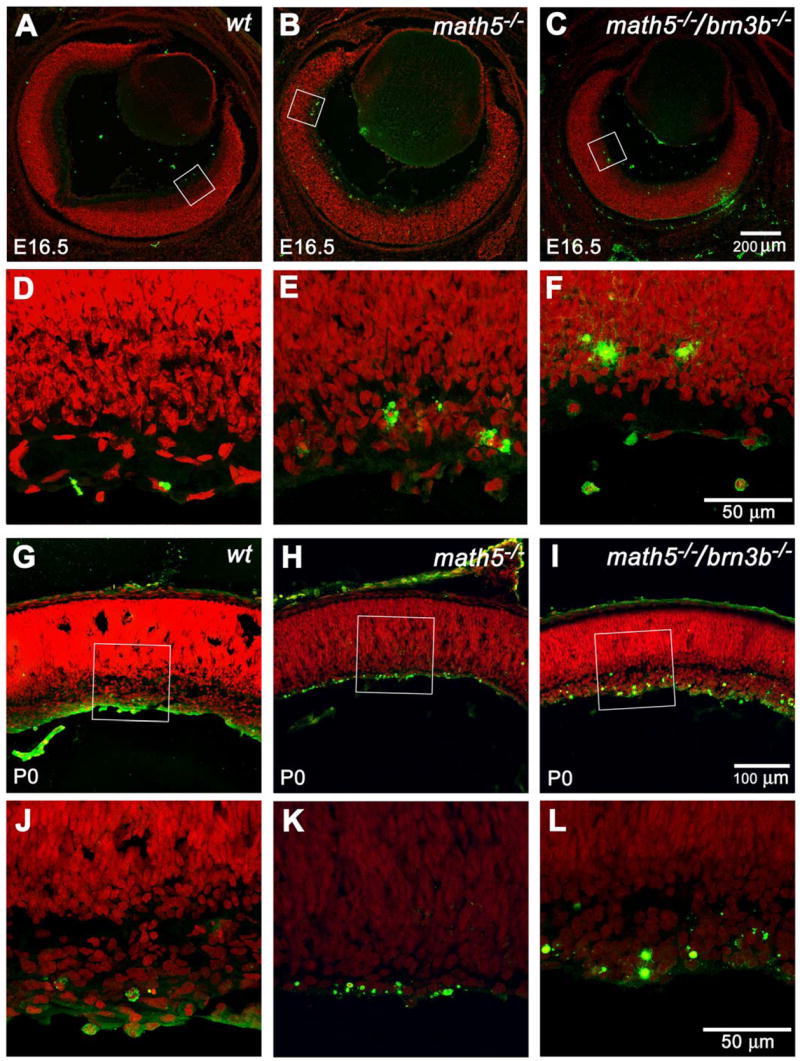
Representative optical sections of TUNEL labeled retinas at E16.5 and P0. Green: TUNEL signal. Red: PI. Panels D, E, and F are enlarged from the boxed area of panels A, B, and C and rotated so that the putative ganglion cell layers are at the bottom. At E16.5, most of the cell death appears in the putative ganglion cell layer. The Math5-deficient retina has more apparent apoptotic cells than the wildtype retina. The Math5/Brn3b-deficient retina has even more apoptotic cells than the Math5-deficient retina. Panels J, K, and L are enlarged from the boxed area of panels G, H, and I. The overall trend of cell death among the three genotypes persisted at P0. In the Math5/Brn3b-deficient retina, cell death in the newly-separated ganglion cell layer appears to be intensified.
Collectively, these results show that, despite a higher cell proliferation rate during early retinogenesis, increased cell apoptosis at all stages combined with reduced cell proliferation during later retinogenesis resulted in the thinner retina observed in the Math5/Brn3b double null mice.
Inner nuclear layer function of the Math5/Brn3b-deficient retina was drastically reduced
To determine the influence of RGC shortage on retinal function, we recorded full field scotopic (dark-adapted) and photopic (light-adapted) electroretinograms (ERGs) for each genotype. Brn3b-deficient mice (n=4) had close to normal scotopic and photopic ERGs (Fig. 7A). No statistically significant differences from the wild type group were found for a-wave or b-wave amplitudes under either adaptation condition (F=8.7, p=0.02, repeated ANOVA; Suppl. Fig. 10), except that the saturated light-adapted b-wave elicited with a xenon flash was lower in the Brn3b-deficient retinas. However, for the low energy stimuli (less than −3.2 log sc td s), the positive STR (pSTR), a small positive potential known to originate from ganglion cells in rodents (Bui and Fortune, 2004), was absent in all Brn3b-deficient mice, consistent with reduced numbers of RGCs in these animals. In these animals and the other mice lacking ganglion cells, a fairly sensitive negative wave remained in the ERG when the pSTR was removed. Presumably this remaining component was akin to the negative STR, likely generated by AII amacrine cells in normal retinas (Saszik et al., 2002), but it was of slightly faster time course than in normal retinas, perhaps due loss of some slow ganglion-cell generated portion of the negative response. Oscillatory potentials (OPs) that arise from inner retina, probably amacrine cells, were slightly low (not significant) in photopic ERGs of Brn3b-deficient mice; scotopic OPs were minimal even in control eyes for the stimuli that we used.
Fig. 7.
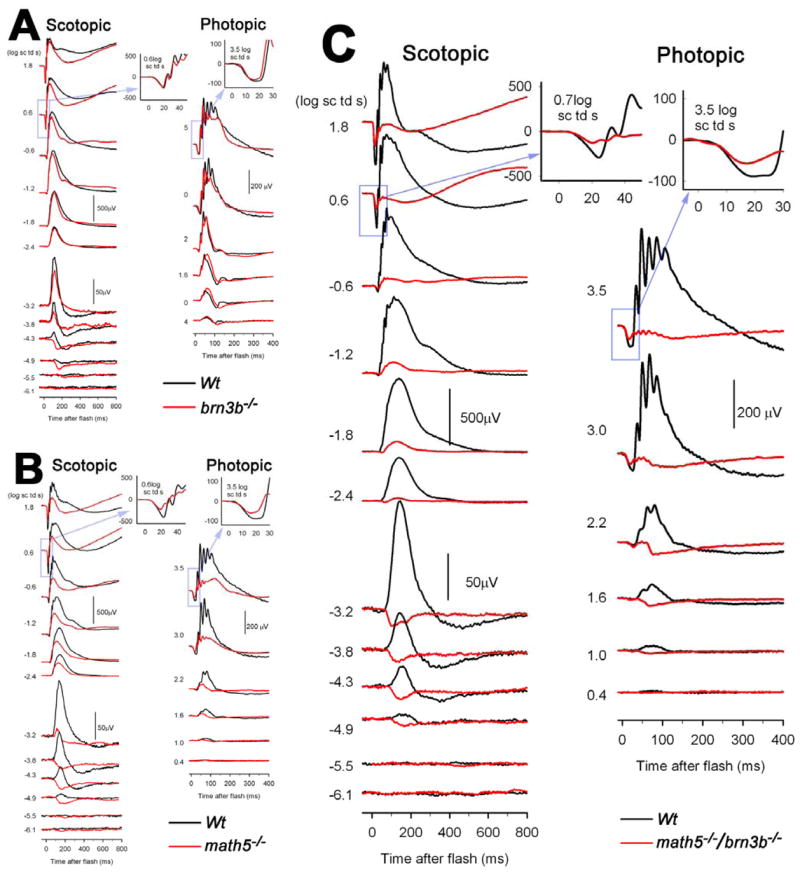
Electroretinograms (ERGs) of brn3b−/−, math5−/−, and brn3b−/−/math5−/− mice, compared to ERGs from age-matched wildtype mice. A) The a-waves and b-waves of scotopic (dark-adapted: left) and photopic (light-adapted: right) full field ERGs of Brn3b-deficient mice were hardly different from those of the wildtype mice, except for very weak stimuli under dark-adapted conditions where the positive scotopic threshold response (pSTR), thought to be generated by retinal ganglion cells, was not detectable in the Brn3b-deficient mice. The insets show a-waves on an expanded time scale in response to a high energy flash. B) Both a-waves and b-waves were reduced in the dark-adapted and light-adapted ERGs of the Math5-deficient mice. The change in dark-adapted a-waves was not significant for the entire group. C) In the Math5/Brn3b-deficient mice, a-waves and b-waves of both light-adapted and dark adapted ERGs were greatly reduced. Under dark-adapted conditions, the a-wave was reduced at times in the leading edge when the response was dominated by rod photoreceptor activity, indicating effects on the rods. The photopic a-wave for both Math5-deficient and Math5/Brn3b-deficient mice was affected at times when Off-pathway neurons were contributing to the response.
In contrast to the Brn3b-deficient mice, and similar to that described previously by Brzezinski et al. (2005), reduction in function was observed in the Math5-deficient mice (n=4). In addition to the loss of the pSTR, the amplitudes of b-waves were significantly reduced in both scotopic (F=28.913, p=0.0007) and photopic (F=18.486, p=0,077, repeated ANOVA) ERGs. An example is given in Fig. 7B, and group data are shown in suppl. Fig. 10. The relatively greater effect on photopic b-waves could reflect a greater loss of cone versus rod bipolar cells.
The Math5/Brn3b double null animals (n=7), had significantly reduced b-waves (F=100.45, p=0.0001, repeated ANOVA) under scotopic conditions (Fig. 7C), and virtually no b-waves under photopic conditions (F=31.412, p=0.0008). The scotopic results were more variable than those under photopic conditions. Two animals had scotopic ERGs with more prominent b-waves than those in Fig. 7C while others had almost no b-wave at all.
These ERG results were consistent with our histological data. They revealed that in animals having only 20% of the normal RGC number (brn3b−/−), electrophysiological function of the retina proximal to ganglion cells was normal. Moreover, approximately 50% of the photopic ERG, and an even higher percent of scotopic ERG reflecting INL function was sustained when merely 5% (math5−/−) of the RGCs were present. However, in the double mutant, when the RGC number dropped to lower than 1%, b-waves became minimal.
DISCUSSION
Based on cumulative publications, we hypothesized that there is a non-linear relationship between depleted newborn RGC numbers and total cell numbers in the remaining mammalian retina. This hypothesis predicted a much greater total retinal cell loss if the RGC number were reduced slightly more than 98%. We showed that the combined deficiency of both Math5 and Brn3b resulted in a >99% RGC loss. RGC reduction at this number led to changes in the retina that were much greater than those previously observed in mice with major ganglion cell loss (Erkman et al., 1996; Gan et al., 1996; Brown et al., 2001; Wang et al. 2001; Wang et al., 2002; Mu et al., 2005). The changes include: 1) severe reduction in the numbers of cone bipolar and amacrine cells, 2) a less severe, but significant reduction in the numbers of rod bipolar cells, photoreceptors, and Müller glia, 3) altered planar distribution of cone and rod bipolar cells, and, 4) greatly reduced b-waves of the scotopic ERGs and nearly abolished b-waves of the photopic ERGs. In this study, we also found that early and late retinal progenitor cells respond differently to RGC shortage. Collectively, we demonstrated that RGCs are not only essential for retinal histogenesis, but also play an essential patterning role that affects the final structure of the mature retina. Our results support our hypothesis that a non-linear relationship exists between depleted newborn RGC numbers and total cell numbers in the mouse retina (Fig. 8).
Fig. 8.
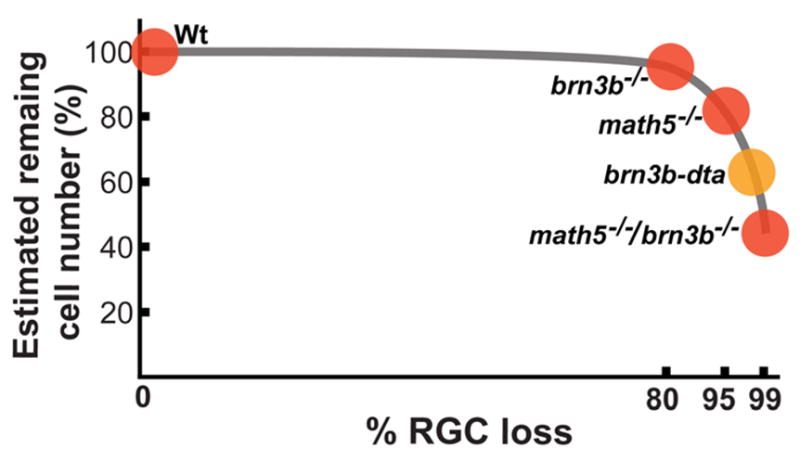
The non-linear relationship between percentage of RGC loss and percentage of remaining retinal cell number. The Brn3b-deficient retina has ~80% RGC loss. Reduction of other retinal cell types may be too trivial to be detected. A minor, but significant, reduction of INL cells can be detected in the Math5-deficient retina that 95% of RGCs being depleted. Total retinal cell loss becomes obvious in the 98% RGC depleted, brn3b-dta, retina. This data point is a rough estimation according to the result of Mu et al., (2005). A slight more RGC depletion (>99%) results in much more dramatic total retinal cell loss (~55%). The highest amount of retinal cell loss observed to date.
Were the observed defects in the Math5/Brn3b-deficient retina a direct result of gene loss or an indirect result of RGC loss?
The study of Klein and colleagues using diphtheria toxin A (dta) to ablate 98% new born RGCs provided first clear in vivo evidence that RGC is required for retinal histogenesis (Mu et al., 2005). This novel approach was highly regarded in the field. However, the methodology was occasionally criticized that leaking dta might have killed neighboring non-RGCs. In addition, Cre activity, that was required for dta activation, might vary among individuals creating further difficulties for data interpretation.
In this study, we bypassed the requirement of Cre and dta by deleting two genes that are essential for RGC formation. Although this approach was able to achieve a higher RGC deletion than the Cre-dta method, it might also invite a different set of criticisms. The major concern is that deleted math5 and/or brn3b genes may be required for cells other than RGCs. In this case, observed phenotype could be a direct impact of gene loss rather than the indirect result of RGC loss. However, after careful analyses on data from previous publications, we believe neither Math5 nor Brn3b is essential for cell formation other than RGCs. Our reasons are listed below.
First, Math5 is not required for the formation of non-RGCs. In fact, Math5 deficiency can actually promote formation of non-RGCs. Gan and colleagues, using a Cre activated reporter system, has clearly demonstrated that math5 expression occurs in the precursors of all “early” cell types but not “late” cell types (Yang et al., 2003). This result may lead us to question whether formation of amacrine cells, horizontal cells, and cone cells require Math5. However, three independent groups have demonstrated that Math5 deficiency actually leads to overproduction of amacrine cells and cone cells at the expense of RGCs (Brown et al., 2001; Wang et al., 2001; Le et al., 2006). In this report, we showed that horizontal cells are also overproduced in the Math5 null retina. Therefore, despite math5 being expressed in all early cell types, Math5 deficiency actually leads to over production of early cell types at RGCs’ expense. Clearly, Math5 is not required for the formation of retinal cells other than RGCs. Kageyama and his group hypothesized a model that competitions among different bHLH factors in combination with different homeodomain factors determine the final retinal cell fate (Hatakeyama et al., 2001; Inoue et al., 2002; Hatakeyama & Kageyama, 2004). This model can reasonably explain why math5 is expressed in all early retinal cell types but is only essential for RGC formation.
Second, Brn3b is not required for the formation of non-RGCs in the retina. Expression of brn3b is well documented to be restricted to RGCs in the retina (Xiang et al., 1993; Xiang et al., 1995; Gan et al., 1999; Erkman et al., 2000). If brn3b was expressed in other retinal cells, results from Mu et al., (2005), which using brn3b locus to express dta toxin, would have to be reevaluated. Deletion of brn3b results in 80% of RGC reduction but has no detectable effects on the remaining cell types indicating Brn3b is not essential for the formation of non-RGC retinal cells.
Third, brn3b is neither activated in, nor required for, non-RGCs in the math5−/− retina. Another valid concern is that Brn3b may become important for non-RGC retinal cells in a math5−/− environment. This concern can be discounted by the result that brn3b is only expressed in sparsely distributed RGCs but not in other cell types of the math5−/− retina (Wang et al., 2001). Expression of brn3b is linked to Math5 (Liu et al., 2001; Mu et al., 2005). Activities of math5 can be detected in all early retinal cell types (Yang et al., 2003). In a math5−/− retina the numbers of all non-RGC early cell types have increased. Therefore formation of non-RGCs does not require Brn3b in a math5−/− environment.
In summary, observed phenotype in the Math5/Brn3b-deficient retina is not possible to be a direct impact of gene loss. It is rather resulted from an indirect influence of RGC loss. This result shows that the amount of RGC reduction is related to the amount of retinal tissue loss and supports our hypothesis that a non-linear relationship exists between depleted RGC numbers and remaining retinal cell numbers (Fig. 8). A well documented work from Robert Williams’ group showed that RGC number in wildtype C57BL/6J mice can vary up to almost 30% (Williams et al., 1996). Our hypothesis predicts normal functioning retinas in these wildtype mice despite a large difference in their RGC numbers.
Do Early and late retinal progenitor cells respond differently to RGC shortage?
We observed increased cell proliferation at E16 and decreased cell proliferation at P0 in both Math5/Brn3b double null and Math5 null retinas. It has been reported in fish and mouse that Ath5-deficinet cells can reenter the cell cycle or delay cell differentiation (Kay et al., 2001; Kay et al., 2005; Le et al., 2006). Therefore, ectopic proliferating cells observed in this study could be cells that expressing math−/−, i.e., a direct effect of math5 gene loss. However, the following three observations led us to propose otherwise.
First, math5 expression in most parts of the retina is normally down-regulated at E16 (Brown et al., 1998; Brown et al., 2001; Wang et al., 2001). There are unlikely enough math5−/− cells at this stage to account for all the increased proliferating cells. Second, it has been shown that competitive bHLH factors are usually up regulated when others are inactivated (e.g. Hatakeyama et al., 2001; Inoue et al., 2002). For example, math3 is usually activated in the absence of Math5 (Le et al., 2006). In the place of Math5, these bHLH factors also promote neuronal differentiation and prevent precursor cells from reentering the cell cycle. Therefore, the probability of Math5-deficient cells reentering the cell cycle is further diluted at E16. Third, an observation made by Klein and colleagues has indirectly implied that early progenitor cells accelerate proliferation in response to RGC shortage (Mu et al., 2005). They showed an increase in the number of math5-expressing cells at E14.5 when most of the RGCs were ablated. Since math5 is expressed during or immediately after terminal mitosis (Brown et al., 1998; Brown et al., 2001; Le et al., 2006), proliferation of progenitor cells would have to be increased to produce more math5-expressing cells.
In contrast to E16, the numbers of mitotic cells at P0 were greatly reduced in the Math5/Brn3b- and Math5-deficient retinas. Therefore we propose that early and late retinal progenitors respond differently to RGC shortage.
Is Sonic hedgehog secreted by RGCs the major player in regulating progenitor cell proliferation and retinal patterning?
Results in this study defend RGCs’ general role in retinogenesis including regulating retinal progenitor cell (RPC) proliferation and affecting the planar distribution of other retinal cell types. Our results do not allow us to pinpoint the function of a specific molecule that is secreted by the RGCs. One may exert a reasonable speculation that, out of all possible factors secreted by the RGCs, Sonic hedgehog (Shh) may play the major role in regulating RPC proliferation and patterning the retina. It has been shown that disruption of Shh pathway affects RPC proliferation and retina patterning (Wang et al., 2002; 2005; Moshiri et al., 2004). However, Shh is not solely produced by RGCs. Cepko and colleagues showed that amacrine cells and cone cells in the mouse retina can produce Shh (Jadhav et al., 2006). Production of Shh by non-RGCs was also reported in Zebrafish (Neumann & Nuesslein-Volhard, 2000; Shkumatava et al., 2004). This may explain why disruption of Shh pathway resulted in rosette patterns in the photoreceptor layer (Wang et al., 2002; 2005), but depletion of newborn RGCs in this study resulted only minor planar patterning defects.
Is there a difference between mammals and aquatic vertebrates in their RGC requirement for retinal histogenesis?
Complete depletion of RGCs was reported in the lakritz mutant of Zebrafish (Kay et al., 2001). However, retinal histogenesis did not seem to be affected in the fish. Instead, increase of bipolar cell number was detected. Are RGCs not required for retinal histogenesis in Zebrafish? Alternatively, can new neurons continuously generated from the ciliary margin zone, a property that does not exist in mammals, mask the original phenotype in the lakritz mutant? It was shown that amacrine cells were overproduced from the periphery of goldfish and frog retinas after they were selectively destroyed (Nagishi et al., 1982; Reh & Tully, 1986). Could the increased bipolar cells observed in the lakritz mutant a response to the initially insufficient bipolar cell number due to RGC shortage?
Baier and colleagues showed that lakritz mutant is a null mutation of the zebrafish ath5, an orthologue of the mouse math5 (Kay et al., 2001). Interestingly, there are 5% RGCs remaining in the Math5 null mouse retina but none were detected in the Ath5 null zebrafish retina. Therefore, there may be subtle differences in the requirement of Ath5 on RGC formation between mouse and Zebrafish. Conversely, there is also a possibility that a diminutive number of RGCs in the lakritz mutant was not detected. In fact, in the early phase of our study, we were unable to detect any RGCs on all sections of the math5−/−/brn3b−/− retinas. A small number of RGC axons were detected only when the specific marker, NF-L, was used on the flat-mounted samples. Therefore, whether there is a difference in RGC requirement for retinal histogenesis between Zebrafish and mouse remains to be elucidated.
Supplementary Material
Acknowledgments
We thank Drs. Alice Z. Chuang and Wenyaw Chan for their assistance with the statistical analyses. We thank Dr. Debra Otteson, Dr. Stephen Massey, and Michael Wise for their critical review of the manuscript. Recoverin antibody was kindly provided by Dr. Alexander Dizhoor 5 years ago. This manuscript was supported by Vision to Prevent Blindness, the Herman Eye Fund, NIH grant EY06671 (LJF), and an NEI Vision core grant EY10608.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agathocleous M, Locker M, Harris WA, Perron M. A general role of hedgehog in the regulation of proliferation. Cell Cycle. 2007;6:156–9. doi: 10.4161/cc.6.2.3745. [DOI] [PubMed] [Google Scholar]
- Brown NL, Kanekar S, Vetter ML, Tucker PK, Gemza DL, Glaser T. Math5 encodes a murine basic helix-loop-helix transcription factor expressed during early stages of retinal neurogenesis. Development. 1998;125:4821–33. doi: 10.1242/dev.125.23.4821. [DOI] [PubMed] [Google Scholar]
- Brown NL, Patel S, Brzezinski J, Glaser T. Math5 is required for retinal ganglion cell and optic nerve formation. Development - Supplement. 2001;128:2497–508. doi: 10.1242/dev.128.13.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzezinski JAt, Brown NL, Tanikawa A, Bush RA, Sieving PA, Vitaterna MH, Takahashi JS, Glaser T. Loss of circadian photoentrainment and abnormal retinal electrophysiology in Math5 mutant mice. Investigative Ophthalmology & Visual Science. 2005;46:2540–51. doi: 10.1167/iovs.04-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui BV, Fortune B. Ganglion cell contributions to the rat full-field electroretinogram. Journal of Physiology. 2004;555:153–73. doi: 10.1113/jphysiol.2003.052738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-Dawson LD, LaVail MM. Rods and cones in the mouse retina. II. Autoradiographic analysis of cell generation using tritiated thymidine. Journal of Comparative Neurology. 1979;188:263–72. doi: 10.1002/cne.901880205. [DOI] [PubMed] [Google Scholar]
- Cepko CL, Austin CP, Yang X, Alexiades M, Ezzeddine D. Cell fate determination in the vertebrate retina. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:589–95. doi: 10.1073/pnas.93.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkman L, McEvilly RJ, Luo L, Ryan AK, Hooshmand F, O’Connell SM, Keithley EM, Rapaport DH, Ryan AF, Rosenfeld MG. Role of transcription factors Brn-3.1 and Brn-3.2 in auditory and visual system development. Nature. 1996;381:603–6. doi: 10.1038/381603a0. [DOI] [PubMed] [Google Scholar]
- Erkman L, Yates PA, McLaughlin T, McEvilly RJ, Whisenhunt T, O’Connell SM, Krones AI, Kirby MA, Rapaport DH, Bermingham JR, O’Leary DD, Rosenfeld MG. A POU domain transcription factor-dependent program regulates axon pathfinding in the vertebrate visual system. Neuron. 2000;28:779–92. doi: 10.1016/s0896-6273(00)00153-7. [DOI] [PubMed] [Google Scholar]
- Gan L, Wang SW, Huang Z, Klein WH. POU domain factor Brn-3b is essential for retinal ganglion cell differentiation and survival but not for initial cell fate specification. Developmental Biology. 1999;210:469–80. doi: 10.1006/dbio.1999.9280. [DOI] [PubMed] [Google Scholar]
- Gan L, Xiang M, Zhou L, Wagner DS, Klein WH, Nathans J. POU domain factor Brn-3b is required for the development of a large set of retinal ganglion cells. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:3920–5. doi: 10.1073/pnas.93.9.3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Hoyuela M, Barbas JA, Rodriguez-Tebar A. The autoregulation of retinal ganglion cell number. Development. 2001;128:117–24. doi: 10.1242/dev.128.1.117. [DOI] [PubMed] [Google Scholar]
- Hatakeyama J, Kageyama R. Retinal cell fate determination and bHLH factors. Seminars in Cell & Developmental Biology. 2004;15:83–9. doi: 10.1016/j.semcdb.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Hatakeyama J, Tomita K, Inoue T, Kageyama R. Roles of homeobox and bHLH genes in specification of a retinal cell type. Development - Supplement. 2001;128:1313–22. doi: 10.1242/dev.128.8.1313. [DOI] [PubMed] [Google Scholar]
- Hinds JW, Hinds PL. Differentiation of photoreceptors and horizontal cells in the embryonic mouse retina: an electron microscopic, serial section analysis. Journal of Comparative Neurology. 1979;187:495–511. doi: 10.1002/cne.901870303. [DOI] [PubMed] [Google Scholar]
- Holt CE, Bertsch TW, Ellis HM, Harris WA. Cellular determination in the Xenopus retina is independent of lineage and birth date. Neuron. 1988;1:15–26. doi: 10.1016/0896-6273(88)90205-x. [DOI] [PubMed] [Google Scholar]
- Inoue T, Hojo M, Bessho Y, Tano Y, Lee JE, Kageyama R. Math3 and NeuroD regulate amacrine cell fate specification in the retina. Development - Supplement. 2002;129:831–42. doi: 10.1242/dev.129.4.831. [DOI] [PubMed] [Google Scholar]
- Jadhav AP, Cho SH, Cepko CL. Notch activity permits retinal cells to progress through multiple progenitor states and acquire a stem cell property. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:18998–9003. doi: 10.1073/pnas.0608155103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay JN, Finger-Baier KC, Roeser T, Staub W, Baier H. Retinal ganglion cell genesis requires lakritz, a Zebrafish atonal Homolog. Neuron. 2001;30:725–36. doi: 10.1016/s0896-6273(01)00312-9. [DOI] [PubMed] [Google Scholar]
- Kay JN, Link BA, Baier H. Staggered cell-intrinsic timing of ath5 expression underlies the wave of ganglion cell neurogenesis in the zebrafish retina. Development. 2005;132:2573–85. doi: 10.1242/dev.01831. [DOI] [PubMed] [Google Scholar]
- Kim J, Wu HH, Lander AD, Lyons KM, Matzuk MM, Calof AL. GDF11 controls the timing of progenitor cell competence in developing retina. Science. 2005;308:1927–30. doi: 10.1126/science.1110175. [DOI] [PubMed] [Google Scholar]
- Kuwabara T, Weidman TA. Development of the prenatal rat retina. Investigative Ophthalmology. 1974;13:725–39. [PubMed] [Google Scholar]
- Le TT, Wroblewski E, Patel S, Riesenberg AN, Brown NL. Math5 is required for both early retinal neuron differentiation and cell cycle progression. Developmental Biology. 2006;295:764–78. doi: 10.1016/j.ydbio.2006.03.055. [DOI] [PubMed] [Google Scholar]
- Lin B, Wang SW, Masland RH. Retinal ganglion cell type, size, and spacing can be specified independent of homotypic dendritic contacts.[see comment] Neuron. 2004;43:475–85. doi: 10.1016/j.neuron.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Liu W, Mo Z, Xiang M. The Ath5 proneural genes function upstream of Brn3 POU domain transcription factor genes to promote retinal ganglion cell development. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:1649–54. doi: 10.1073/pnas.98.4.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moshiri A, Reh TA. Persistent progenitors at the retinal margin of ptc+/− mice. Journal of Neuroscience. 2004;24:229–37. doi: 10.1523/JNEUROSCI.2980-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X, Beremand PD, Zhao S, Pershad R, Sun H, Scarpa A, Liang S, Thomas TL, Klein WH. Discrete gene sets depend on POU domain transcription factor Brn3b/Brn-3.2/POU4f2 for their expression in the mouse embryonic retina. Development. 2004;131:1197–210. doi: 10.1242/dev.01010. [DOI] [PubMed] [Google Scholar]
- Mu X, Fu X, Sun H, Beremand PD, Thomas TL, Klein WH. A gene network downstream of transcription factor Math5 regulates retinal progenitor cell competence and ganglion cell fate. Developmental Biology. 2005a;280:467–81. doi: 10.1016/j.ydbio.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Mu X, Fu X, Sun H, Liang S, Maeda H, Frishman LJ, Klein WH. Ganglion cells are required for normal progenitor- cell proliferation but not cell-fate determination or patterning in the developing mouse retina. Current Biology. 2005b;15:525–30. doi: 10.1016/j.cub.2005.01.043. [DOI] [PubMed] [Google Scholar]
- Negishi K, Teranishi T, Kato S. New dopaminergic and indoleamine-accumulating cells in the growth zone of goldfish retinas after neurotoxic destruction. Science. 1982;216:747–9. doi: 10.1126/science.7079736. [DOI] [PubMed] [Google Scholar]
- Reh TA, Tully T. Regulation of tyrosine hydroxylase-containing amacrine cell number in larval frog retina. Developmental Biology. 1986;114:463–9. doi: 10.1016/0012-1606(86)90210-1. [DOI] [PubMed] [Google Scholar]
- Shkumatava A, Fischer S, Muller F, Strahle U, Neumann CJ. Sonic hedgehog, secreted by amacrine cells, acts as a short-range signal to direct differentiation and lamination in the zebrafish retina. Development. 2004;131:3849–58. doi: 10.1242/dev.01247. [DOI] [PubMed] [Google Scholar]
- Turner DL, Cepko CL. A common progenitor for neurons and glia persists in rat retina late in development. Nature. 1987;328:131–6. doi: 10.1038/328131a0. [DOI] [PubMed] [Google Scholar]
- Waid DK, McLoon SC. Immediate differentiation of ganglion cells following mitosis in the developing retina. Neuron. 1995;14:117–24. doi: 10.1016/0896-6273(95)90245-7. [DOI] [PubMed] [Google Scholar]
- Waid DK, McLoon SC. Ganglion cells influence the fate of dividing retinal cells in culture. Development. 1998;125:1059–66. doi: 10.1242/dev.125.6.1059. [DOI] [PubMed] [Google Scholar]
- Wang SW, Gan L, Martin SE, Klein WH. Abnormal polarization and axon outgrowth in retinal ganglion cells lacking the POU-domain transcription factor Brn-3b. Molecular & Cellular Neurosciences. 2000;16:141–56. doi: 10.1006/mcne.2000.0860. [DOI] [PubMed] [Google Scholar]
- Wang SW, Kim BS, Ding K, Wang H, Sun D, Johnson RL, Klein WH, Gan L. Requirement for math5 in the development of retinal ganglion cells. Genes & Development. 2001;15:24–9. doi: 10.1101/gad.855301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SW, Mu X, Bowers WJ, Kim DS, Plas DJ, Crair MC, Federoff HJ, Gan L, Klein WH. Brn3b/Brn3c double knockout mice reveal an unsuspected role for Brn3c in retinal ganglion cell axon outgrowth. Integrated Annual Indexes. 2002;129:467–77. doi: 10.1242/dev.129.2.467. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dakubo GD, Thurig S, Mazerolle CJ, Wallace VA. Retinal ganglion cell-derived sonic hedgehog locally controls proliferation and the timing of RGC development in the embryonic mouse retina. Development. 2005;132:5103–13. doi: 10.1242/dev.02096. [DOI] [PubMed] [Google Scholar]
- Wang YP, Dakubo G, Howley P, Campsall KD, Mazarolle CJ, Shiga SA, Lewis PM, McMahon AP, Wallace VA. Development of normal retinal organization depends on Sonic hedgehog signaling from ganglion cells. Nature Neuroscience. 2002;5:831–2. doi: 10.1038/nn911. [DOI] [PubMed] [Google Scholar]
- Wetts R, Fraser SE. Multipotent precursors can give rise to all major cell types of the frog retina. Science. 1988;239:1142–5. doi: 10.1126/science.2449732. [DOI] [PubMed] [Google Scholar]
- Williams RW, Strom RC, Rice DS, Goldowitz D. Genetic and environmental control of variation in retinal ganglion cell number in mice. Journal of Neuroscience. 1996;16:7193–205. doi: 10.1523/JNEUROSCI.16-22-07193.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M, Zhou L, Macke JP, Yoshioka T, Hendry SH, Eddy RL, Shows TB, Nathans J. The Brn-3 family of POU-domain factors: primary structure, binding specificity, and expression in subsets of retinal ganglion cells and somatosensory neurons. Journal of Neuroscience. 1995;15:4762–85. doi: 10.1523/JNEUROSCI.15-07-04762.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M, Zhou L, Peng YW, Eddy RL, Shows TB, Nathans J. Brn-3b: a POU domain gene expressed in a subset of retinal ganglion cells. Neuron. 1993;11:689–701. doi: 10.1016/0896-6273(93)90079-7. [DOI] [PubMed] [Google Scholar]
- Yang Z, Ding K, Pan L, Deng M, Gan L. Math5 determines the competence state of retinal ganglion cell progenitors. Developmental Biology. 2003;264:240–54. doi: 10.1016/j.ydbio.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Young RW. Cell differentiation in the retina of the mouse. Anatomical Record. 1985;212:199–205. doi: 10.1002/ar.1092120215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.