

Abstract
Aortic arch malformations are common congenital disorders that are frequently of unknown etiology. To gain insight into the factors that guide branchial aortic arch development, we examined the process by which these vessels assemble in wild type zebrafish embryos and in kurzschlusstr12 (kus tr12) mutants. In wild type embryos, each branchial aortic arch first appears as an island of angioblasts in the lateral pharyngeal mesoderm, then elaborates by angiogenesis to connect to the lateral dorsal aorta and ventral aorta. In kustr12 mutants, angioblast formation and initial sprouting are normal, but aortic arches 5 and 6 fail to form a lumenized connection to the lateral dorsal aorta. Blood enters these blind-ending vessels from the ventral aorta, distending the arteries and precipitating fusion with an adjacent vein. This arteriovenous malformation (AVM), which shunts nearly all blood directly back to the heart, is not genetically programmed, as its formation correlates with blood flow and aortic arch enlargement. By positional cloning, we have identified a nonsense mutation in unc45a in kustr12 mutants. Our results are the first to ascribe a role for Unc45a, a putative myosin chaperone, in vertebrate development, and identify a novel mechanism by which an AVM can form.
Keywords: aortic arches, pharyngeal arches, unc45a, zebrafish, arteriovenous malformation, kurzschluss
INTRODUCTION
The aortic arch system in human embryos consists of paired, bilateral arteries that carry blood from the aortic sac to the dorsal aortae. These arteries, each of which is embedded in its respective pharyngeal arch, form in a cranial-to-caudal sequence but regress or remodel extensively into the definitive vascular pattern. Specifically, aortic arches 1 and 5 regress, whereas 2, 3, 4, and 6 remodel extensively and contribute to the hyoid artery; the common carotid artery and the base of the internal carotid artery; the arch of the aorta and the right subclavian artery; and the ductus arteriosus and pulmonary artery, respectively. While the anatomical transition from embryonic aortic arches to mature vessels is well-documented (Congdon, 1922), the individual steps and molecular cues that orchestrate initial aortic arch formation are poorly understood. While some authors have suggested an angiogenic origin for these vessels (Noden, 1989), others have suggested a vasculogenic origin (DeRuiter et al., 1993; Hiruma and Hirakow, 1995; Waldo et al., 1996).
Several human congenital abnormalities are associated with the aortic arch system, including persistence of a patent ductus arteriosus, aberrant right subclavian artery, right aortic arch, aortic arch interruption, and persistence of normally transient aortic arches. In most cases, a genetic basis for these abnormalities has not been identified. Genes that have been shown to be required for proper aortic arch patterning in humans and/or mice include Tbx1; HoxA3, Pax3, Endothelin-1/Endothelin A receptor, and retinoic acid receptors (Chisaka and Capecchi, 1991; Epstein et al., 1991; Lindsay et al., 2001; Mendelsohn et al., 1994; Yanagisawa et al., 1998). Interestingly, mutations in each of these genes disrupt overall pharyngeal arch patterning but not global blood vessel development, suggesting that effects on aortic arches might be secondary to pharyngeal patterning defects.
The aortic arch system in zebrafish is very similar to the embryonic aortic arch system in birds and mammals (Isogai et al., 2001). Six pairs of vessels, connecting the ventral aorta to the lateral dorsal aortae, emerge in a cranial-to-caudal sequence. Aortic arch 1 comes on-line as circulation begins, about 24 hours post-fertilization (hpf); it is a critical component of the initial circulation loop. In contrast, aortic arch 2 never fully forms and does not contribute to the final vasculature. Aortic arches 3 through 6 arise in pharyngeal arches 3 through 6, respectively, and are collectively known as the branchial aortic arches, as they eventually split into interconnected afferent and efferent vessels and supply blood to the gills (Isogai et al., 2001). This initial pattern of the aortic arches remains intact in zebrafish. The first, third, and fourth aortic arches send blood primarily to the cranial vasculature, whereas the fifth and sixth become the main source of blood for the trunk and tail. Thus, the initial formation and patterning of the aortic arches is very similar across vertebrate classes, although the complicated remodeling of the aortic arches that occurs in air-breathing vertebrates does not occur in zebrafish. Despite this difference, the genetic and embryonic accessibility of the zebrafish make it a superb model with which to identify genetic factors that help to set up and stabilize the initial aortic arch pattern.
Using zebrafish forward genetics, we have uncovered a new player in aortic arch development: Unc45a. unc45a is one of two vertebrate paralogs of C. elegans unc-45, mutations in which lead to a paralyzed or partially paralyzed (“uncoordinated”) phenotype (Epstein and Thomson, 1974; Venolia and Waterston, 1990). UNC-45 protein colocalizes with type II muscle myosin heavy chain B (Ao and Pilgrim, 2000) and, in concert with HSP90, chaperones the motor domains of type II and type V myosins via its C-terminal UCS (UNC-45, Cro1, She4p) domain (Barral et al., 2002; Kachur et al., 2004). Maternal unc-45 is required for myosin-mediated cytokinesis, whereas zygotic unc-45 is critical for incorporation of myosins into thick filaments and thus sarcomere formation (Barral et al., 1998; Kachur et al., 2004; Venolia and Waterston, 1990).
In vertebrates, there are two unc-45 paralogs, Unc45a and Unc45b. Unc45b is expressed only in cardiac muscle in the mouse embryo, but in both skeletal and cardiac muscle in adult mice (Price et al., 2002). Zebrafish unc45b is expressed embryonically in most striated muscles and in the myocardium, and mutation or knockdown results in disrupted sarcomere formation, leading to paralysis and defects in ventricular contraction (Etard et al., 2007; Etheridge et al., 2002; Wohlgemuth et al., 2007). In contrast, Unc45a is expressed ubiquitously in both embryonic and adult mice, with noticeably strong expression in the pharyngeal arches at embryonic day 9.75 (Price et al., 2002). In cultured C2C12 myoblasts and ovarian cancer cells, Unc45a promotes proliferation and/or motility (Bazzaro et al., 2007; Price et al., 2002). Taken together, these results suggest that Unc45b may play a role in thick filament assembly by chaperoning skeletal and cardiac muscle myosin II, similar to C. elegans zygotic UNC-45, whereas Unc45a may function in cytokinesis and motility by chaperoning nonmuscle myosin II, similarly to C. elegans maternal UNC-45. However, no functional studies of vertebrate Unc45a have been performed in vivo. Furthermore, human UNC45a has also been shown to cooperate with HSP90 in chaperoning the progesterone receptor, suggesting that its role in the cell may not be limited to myosin chaperone activity (Chadli et al., 2006).
In this work, we describe for the first time the temporal sequence of events that leads to formation of the branchial aortic arches, and identify a critical role for Unc45a in zebrafish pharyngeal arch and aortic arch development. A complete understanding of the Unc45a-mediated event required for proper pharyngeal arch development may ultimately lead to an appreciation of some of the underlying causes of human congenital aortic arch anomalies and arteriovenous malformations (AVMs).
MATERIALS AND METHODS
Zebrafish lines and maintenance
Adult zebrafish (Danio rerio) were maintained as described (Westerfield, 1995). kustr12 was isolated from an ethylnitrosourea mutagenesis screen on a Tübingen AB background (Chen et al., 1996; Haffter et al., 1996). Mapping lines were generated by outcrossing kustr12 heterozygotes to Hong Kong and WIK strains. Embryos were raised and staged as described (Kimmel et al., 1995; Westerfield, 1995). When appropriate, embryo medium was supplemented with 0.003% phenylthiourea (Sigma) at 24 hpf to prevent melanin formation (Westerfield, 1995).
Microangiography and confocal imaging
Microangiography was performed as described (Weinstein et al., 1995). For confocal microscopy, embryos were anesthetized using 0.016% Tricaine (Sigma), mounted on depression slides using 5% methylcellulose (Sigma), and Z-series of frame-averaged optical sections were generated using a FluoView 500 (Olympus) or Radiance 2000 (Bio-Rad) laser scanning confocal microscope. Two-dimensional and three-dimensional projections were generated using FluoView or MetaMorph software (Universal Imaging). Time lapse confocal microscopy was performed as described (Kamei and Weinstein, 2005), and movies generated using Metamorph.
2,3-butanedione monoxime treatment
Embryos were treated with 40 mM 2,3-butanedione monoxime (BDM; Sigma) in 30% Danieu’s [17.4 mM NaCl, 0.2 mM KCl, 0.12 mM MgSO4, 0.18 mM Ca(NO3)2, 1.5 mM HEPES, pH 7.5] for approximately five minutes to quickly stop the heartbeat, then transferred to 20 mM BDM and incubated at 28.5°C (Serluca et al., 2002). After treatment, embryos where washed 3 × 5 minutes with 30% Danieu’s.
Cartilage immunohistochemistry and measurements
To visualize cartilages, embryos were fixed overnight at 4°C with Carnoy’s fixative (ethanol:chloroform:acetic acid, 6:3:1), dehydrated with methanol, and stored at −20°C. Following rehydration, embryos were permeabilized with proteinase-K (10 µg/ml) for 30 min, blocked in 2% BSA/5% normal goat serum/PBS-0.1% Tween (PBT) for 1 hr, then incubated with anti-collagen type-II (II-116B3; DSHB; 1:100 in block) overnight at 4°C. Primary antibody was detected using Alexa Fluor 568-conjugated goat anti-mouse IgG (Invitrogen, 1:200 in block). For double staining, anti-GFP (Invitrogen; 1:200 in block) was added simultaneously with the collagen antibody, and Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen; 1:200 in block) was added simultaneously with the antimouse IgG. Embryos were cleared with and mounted in 50% glycerol, and images acquired using an MVX10 MacroView and DP71 camera (Olympus). Relative measurements were made using the measure tool in Photoshop.
Meiotic and physical mapping
Bulked segregant analysis using SSLP markers was performed as previously described (Knapik et al., 1998; Roman et al., 2002). To construct a physical contig, BAC (Incyte Genomics) and PAC clones (C. Amemiya, Virginia Mason Medical Center) were identified by PCR or filter hybridization using SSLP primers or primers specific to sequenced BAC or PAC ends. Clones were purified using Nucleobond columns (BD-Clontech). BAC/PAC assembly and polymorphism identification were performed as previously described (Roman et al., 2002). For fine meiotic mapping, single nucleotide polymorphisms were assayed by restriction fragment length polymorphism (RFLP) analysis; derived cleaved amplified polymorphic sequence (dCAPS) analysis (Neff et al., 1998); or sequencing of PCR-amplified genomic DNA. To identify genes within the meiotic critical interval, BAC95k20 was shotgunned (TOPO Shotgun Subcloning Kit, Invitrogen), clones were end sequenced, and contigs were generated using SeqMan (Lasergene DNAstar). Candidate genes within the critical interval were PCR amplified from pooled wild type and pooled mutant cDNA using Amplitaq gold (Applied Biosystems), cloned into pCRII-TOPO (Invitrogen), and four to six clones representing each phenotype were sequenced. The T-to-A transversion found in unc45a in kustr12 mutants was assayed by dCAPS analysis (Neff et al., 1998) using the following primers: 5’-CTTTTTTCCTCCTCTTCACAGGGTGACTT-3’ and 5’-TCAGATTTGAGTTTGAGCAGTTGATC-3’. The forward primer contains a single mismatch (underlined) creating a DdeI site in the kustr12 but not the wild type allele.
Expression constructs and morpholinos
Full length unc45a was amplified using platinum Pfx DNA polymerase (Invitrogen) from 24 hpf cDNA using the following primers: 5’-TTATCTAAACAGCGAATACAACG-3’ (−35 to −13) and 5’-ATTGCGGCCTCCATCAAACACTGC-3’ (5 to 28 in 3’UTR). The 3056 nucleotide product was cloned into pCR-BluntII-TOPO (Invitrogen) and sequenced bidirectionally. To generate a native expression construct, the unc45a-TOPO insert was excised with EcoRI and ligated into EcoRI-digested pCS2+ (Rupp et al., 1994). To generate a C-terminally myc-tagged expression construct, the unc45a-TOPO clone was used as template in a PCR reaction using PfuUltra (Stratagene) with the following primers: 5’-TATAGGATCCTTATCTAAACAGCGAATACAACG-3’ (BamHI site underlined; −35 to −13) and 5’-TATAATCGATTAATGGAATTACCATTAACTCCGC-3’ (ClaI site underlined; 2783 to 2805). The 2986 bp PCR product was cloned into pCR-BluntII-TOPO, excised with BamHI/ClaI and cloned into BamHI/ClaI-digested pCS2+myc (Rupp et al., 1994). Fidelity of the PCR reaction and confirmation of in-frame tagging was verified by bidirectional sequencing. A myc-tagged unc45akus construct was generated using the forward primer above and the ClaI-containing reverse primer, 5’-TATAATCGATTAATCACCCTGGCGATGCACTCTC-3’. This primer is complementary to coding nucleotides 1940 to 1962 and generates a product that encodes a 654 amino acid, C-terminally truncated protein corresponding to the kustr12 allele of unc45a. Capped mRNA was synthesized from NotI-digested constructs using mMessage mMachine with SP6 RNA polymerase (Ambion). Morpholino-modified antisense oligonucleotides (GeneTools) used were as follows: cardiac troponin T2 (tnnt2), CATGTTTCGTCTGATCTGACACGCA, targeting −22 to +3 (Sehnert et al., 2002); unc45a ATG, ACATCTTTCTCTGTAGCGTTGTATT, targeting −21 to +4; and unc45a splice, agctttatagaCCTGGCGATGCACT, targeting the exon 13 (capitals)/intron 13 (lower case) boundary. Capped mRNAs and morpholinos were injected into 1 to 4-cell embryos as described previously (Westerfield, 1995).
In situ hybridization
Whole mount in situ hybridization using digoxigenin-labeled riboprobes and alkaline phosphatase-conjugated anti-digoxigenin (Roche) was performed as described (Hauptmann and Gerster, 1994). For unc45a detection, a riboprobe was transcribed from the full length clone (unc45a-TOPO, described above). Embryos were examined as whole mounts or embedded in JB4 and sectioned at 5 µm.
RESULTS
Branchial aortic arches originate by vasculogenesis and elaborate by angiogenesis
tie1 in situ hybridization (Fig. 1A–D) and time lapse confocal microscopy using TG(flk1:EGFP)la116;TG(gata1:dsRed) embryos (Fig. 1E–L) show that the branchial aortic arches (aortic arches 3–6) form initially by vasculogenesis, the de novo formation of vessels from free angioblast precursors. As early as 28 hpf, angioblasts arise bilaterally in the lateral pharyngeal mesoderm in regions corresponding to the prospective inflection point of aortic arch 3. This angioblast island subsequently sprouts bidirectionally, reaching dorsally and ventromedially. This process is repeated for aortic arches 4–6, with angioblast differentiation and sprouting occurring in a cranial-to-caudal sequence (Fig. 1A–L and Movie 1). Ventrally, these sprouts seem to coalesce to form, de novo, the bilateral ventral aortae, which connect to the outflow tract of the heart (Fig. 1M). By 72 hpf, aortic arches 3 through 6 are rooted in a single midline ventral aorta, with the sixth aortic arches emanating from its most distal end (Fig. 1N). These data suggest that the early bilateral vessels migrate toward the midline and fuse. Dorsally, the aortic arches turn medially and connect to the existing lateral dorsal aortae (LDA): aortic arches 3 and 4 connect independently to the LDA, whereas aortic arch 6 merges with 5 prior to reaching the LDA (Fig. 1O). Specifically, the aortic arch 6 dorsal sprout pauses at the level of the primary head sinus, sending out numerous filopodia before making contact with aortic arch 5 around 49 hpf (Movie 2). Interestingly, aortic arch 5 lumenizes prior to reaching the radix of the LDA: in some embryos, blood can be seen pulsating in aortic arch 5, suggesting that blood can get into this vessel via the ventral aorta, but cannot exit to the LDA (data not shown). Pulsation is transient (and thus not observed in all embryos) and is alleviated concomitant with the connection of aortic arch 5 to the LDA. [It should be noted that the connection between aortic arch 5 and the LDA, which we are calling the aortic arch 5 extension (AA5X), clearly emanates from aortic arch 5 and is not a branch of the lateral dorsal aorta, as previously described (Isogai et al., 2001).] On average, all aortic arches are lumenized and carrying blood flow by 50 hpf.
Fig. 1. Branchial aortic arch and ventral aorta development.

Aortic arches 3–6 first appear in an anterior-to-posterior sequence as angioblast islands (indicated as numbers 3 through 6) in the lateral pharyngeal mesoderm; sprouts emerge from these islands and travel ventromedially and dorsally (A–L; see also Movie 1). By 48 hpf (M), ventral sprouts merge to form bilateral ventral aortae (VA), which fuse into a single midline vessel by 72 hpf (N). Dorsally (O; 72 hpf), branchial aortic arches turn medially beneath the primary head sinus (PHS). Aortic arches 3 and 4 connect independently to the lateral dorsal aortae, whereas aortic arches 5 and 6 merge and extend to the lateral dorsal aortae as a single vessel, the aortic arch 5 extension (AA5X). A–D: tie1 in situ hybridization. E–H and N: 2D confocal projections of TG(flk1:GFPla116;gata1:dsRed) embryos, labeling endothelial and blood cells green and red, respectively. E–H are representative time points taken from Movie 1. M,O: 2D confocal projections of TG(flk1:GFPla116) embryos. I–L: wiring diagrams of images in E–H; vessels were traced using Photoshop. A–L: lateral views, anterior to the left. M,N: ventral views, anterior to the left. O: dorsal view, anterior to the left.
kustr12 mutants exhibit an AVM involving aortic arches 5 and 6 and the primary head sinus
In kustr12 mutants, vessel patterning and circulation are normal before 50 hpf; all aortic arches arise in a spatially and temporally correct manner and extend toward the lateral dorsal aorta and ventral aorta (Movie 3 and data not shown). However, lumenization of aortic arches 5 and 6 is delayed in kustr12 mutants by approximately 2 hours (Fig. 2A–D, Movie 3). Lumenization is followed by accumulation of a bolus of pulsating blood cells in the enlarged distal tip of aortic arch 5, just beneath the primary head sinus (Fig. 2D, Movie 3). The appearance of aortic arch 6 at 52 hpf is variable, with some mutants exhibiting thin vessels containing little to no blood, and some exhibiting enlarged distal tips containing blood received from the distal portion of aortic arch 5 and thus pulsating in a retrograde fashion in aortic arch 6 (Movie 4). Pulsation in aortic arch 5 in kustr12 is considerably different from the pulsation observed in wild type embryos. First, in kustr12, blood cells form a large aggregate at the tip of aortic arch 5, whereas they line up single-file in wild type embryos. Second, pulsation lasts, on average, for 4 hours in kustr12 , and is fleeting in wild type embryos. Confocal microangiograms of kustr12;TG(flk1:EGFP)la116 embryos show that this extended pulsation is the result of failure of AA5X to form a lumenized connection to the radix of the LDA (Fig. 2E,F). Between 60 and 72 hpf, the enlarged, blind-ended aortic arch 5/6 junction fuses with the adjacent primary head sinus, the major cranial drainage vein (Fig. 3A–C, F–H; Movie 5,Movie 6). The resulting AVM shunts the majority of blood directly back to the common cardinal vein, from which it immediately re-enters the heart; frequently, hemorrhage is apparent adjacent to the AVM (Fig. 3D,I; Movie 7). The penetrance of this aortic arch defect is 100%. Laterality is random, with AVMs occurring with nearly equal frequency on the left or right side, or less frequently, on both sides (L: 38%; R: 36%; both: 26%; n = 42).
Fig. 2. Aortic arches 5 and 6 exhibit delayed lumenization and fail to properly connect to the lateral dorsal aortae in kustr12 mutants.

In wild type embryos, aortic arches 5 and 6 are lumenized by 50 hpf (A) and exhibit strong blood flow by 52 hpf (C). In kustr12 (B,D), lumenization is delayed by 2–3 hours and blood cells accumulate in distal portions of aortic arch 5 (arrow). This accumulation of blood cells is accompanied by failure of the AA5X to make a patent connection to the LDA [compare E (WT) to F (kustr12)]. A–F, 2D confocal projections of TG(flk1:GFPla116; gata1:dsRed) embryos at 50 hpf (A,B) or 52 hpf (C–F), labeling endothelial and blood cells green and red, respectively. A–D, lateral views, anterior to the left. E–F, dorsal views, anterior to the left.
Fig. 3. Arteriovenous malformation and ventral aorta defect in kustr12 mutants.
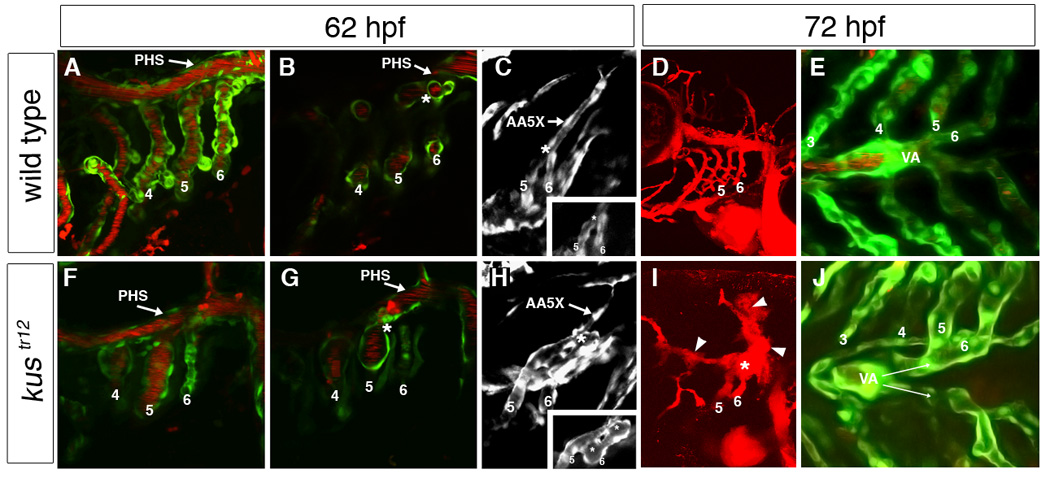
In wild type embryos, aortic arches 5 and 6 fuse just behind the primary head sinus (PHS) and connect to the lateral dorsal aorta via an extension of aortic arch 5 (AA5X); clear demarcation can be seen between the aortic arch 5/6 connection (asterisk) and the PHS (A–C). In contrast, in kustr12 mutants, AA5X is poorly formed, and the enlarged aortic arch 5/6 connection fuses to the adjacent PHS (asterisk), creating an AVM (arrowhead) that shunts blood directly back to the heart (F–H). In addition, the AA5X is poorly formed (H). This AVM (asterisk) carries the majority of blood flow and is typically associated with localized hemorrhage [arrowheads; compare D (WT) to I (kustr12)]. In wild type embryos, the paired ventral aortae (VA) fuse by 72 hpf (E). In contrast, in kustr12, the ventral aortae fail to fuse properly and remain paired at 72 hpf, similar to wild types at 48 hpf (J; compare also to Fig. 1M). 2D confocal projections of TG(flk1:GFPla116;gata1:dsRed) embryos (A,E,F,J); TG(flk1:GFPla116) embryos (C,H); or microangiograms (D,I). B, G and insets in C,H are single planes from confocal Z-stacks represented by A,F,C, and H, respectively. A–C; F–H: 62 hpf. D,E,I,J: 72 hpf. A, B, D, F, G, I: lateral views, anterior to the left. C,H: dorsolateral views, anterior to the left. E,J: ventral views, anterior to the left.
Ventral aorta development is disrupted in kustr12 mutants
Because the ventral aorta feeds and is contributed to by aortic arches 5 and 6, we hypothesized that ventral aorta defects might accompany aortic arch defects in kustr12 mutants. Both wild type and kustr12 mutants exhibit bilateral ventral aortae at 48 hpf [Figs. 1M (wild type) and data not shown (kustr12)]. At 72 hpf, wild type ventral aortae had merged along nearly their entire lengths into a single midline vessel, with fusion ending just at or slightly anterior to the level of aortic arch 5 (Fig. 3E). In contrast, fusion ended between aortic arches 3 and 4 in all mutants (Fig. 3J). As this vessel configuration looks similar to the wild type 48 hpf embryo (Fig. 1M), these data strongly suggest that development of the ventral aorta is impaired or arrested in kustr12 mutants.
Posterior pharyngeal arch cartilages are mispatterned in kustr12 mutants
The ventral patterns of branchial aortic arches and branchial cartilages bear striking resemblance to one another, as the two tissues interdigitate during development. The ventral aorta lies just ventral to the midline basibranchial cartilages, and each branchial aortic arch (aortic arches 3–6) runs alongside its corresponding ceratobranchial cartilage (pharyngeal cartilages 3–6; Fig. 4A,D). Thus, we hypothesized that defects in pharyngeal cartilage patterning might accompany aortic arch defects in kustr12 mutants. In kustr12 mutants at 3–5 days post-fertilization (dpf), the basibranchial cartilages are compressed along the anterior-posterior axis (Table 1), and the paired ceratobranchial cartilages do not form the characteristic chevron shape (Fig. 4B,E). A similar defect is seen in the more anterior ceratohyal (pharyngeal arch 2) cartilages, and the mandibular palatoquadrate (pharyngeal arch 1) cartilages are significantly shortened (Fig. 4B,E). In addition, the lateral distance between paired ceratobranchial cartilages in pharyngeal arches 5 and 6 is increased in kustr12 mutants (Fig. 4B,E, Table 1). Because kustr12 mutants develop pericardial edema around 3 dpf, we reasoned that the cartilage defects might be secondary to fluid accumulation in this region. Indeed, other embryos that develop edema, including violet beauregarde (acvrl1) mutants (Roman et al., 2002) and tnnt2 morphants (Sehnert et al., 2002), also exhibit compression of the basibranchial cartilages; improper angling of the ceratobranchial and ceratohyal cartilages; and shortening of the palatoquadrates (Fig. 4C,F; Table 1). However, neither of these mutants exhibits increased distance between paired ceratobranchial cartilages in pharyngeal arches 5 and 6. These data suggest that the lateral displacement of these basibranchial cartilages, which occurs with 100% penetrance, is a defect specific to the kustr12 mutation and not secondary to edema. It should be noted that we did not observe any overt patterning defects in 3 dpf kustr12 mutants in pharyngeal endodermal pouches (nkx2.3; bmp2b; Zn8 antibody); pharyngeal muscle (ventricular myosin heavy chain); or pharyngeal neural crest (dlx2; pax3; hand2) (data not shown).
Fig. 4. Increased distance between left and right posterior ceratobranchial cartilages is a specific kustr12 phenotype.

Aortic arches (bright green) develop in close association with pharyngeal cartilages (red) in WT (A) and kustr12 (D). Note the malformed ventral aorta (VA) in kustr12. (The darker green staining is background and represents pharyngeal muscle.) Defects in jaw and pharyngeal cartilages in kustr12 include shortening of the palatoquadrates (PQ); compression of the basibranchial cartilages (horizontal white line); improper angling of the ceratobranchial (asterisks) and ceratohyal (arrowheads) cartilages; and decreased lateral distance (vertical white lines) between left and right ceratobranchial cartilages in pharyngeal arches 5 and 6 [compare B (WT) to E (kustr12)]. Like kustr12, vbgy6 mutants (C) and tnnt2 morphants (F) develop edema and exhibit all described cartilage defects except increased lateral distance between posterior ceratobranchials. Thus, only the latter defect is likely a direct effect of the kustr12 mutation. A,D: 2D confocal projections of TG(flk1:GFPla116; gata1:dsRed) embryos stained with anti-collagen II (red) and anti-GFP (green). B,C,E,F: Macro images of embryos stained with anti-collagen II. All embryos are 4 dpf; ventral views, anterior to the left.
Table 1.
Pharyngeal Cartilage Measurementsa, 4 days post-fertilization
| Ceratobranchial Gap | Basibranchial Length | ||
|---|---|---|---|
| Arch 5 | Arch 6 | ||
| Wild Type n=7 |
62.7 ± 2.8 | 61.6 ± 2.4 | 272.6 ± 5.2 |
|
kustr12 n=7 |
70.0 ± 2.6* | 85.8 ± 5.7 ** | 217.6 ± 6.9 ** |
|
vbgy6 n=10 |
60.3 ± 1.2 | 59.5 ± 1.6 | 240.3 ± 4.9 ** |
|
tnnt2 MO n=10 |
60.7 ± 1.1 | 60.4 ± 2.3 | 218.2 ± 5.0 ** |
Images were captured at 126x using an Olympus MVX10 MacroView; all measurements were made using the “measure” tool in Photoshop and are expressed as arbitrary units ± SE. Data were analyzed using two-tailed Student’s t test.
significantly different from wild type, p < 0.05.
significantly different from wild type, p < 0.005.
The AVM and ventral aorta defects in kustr12 mutants are influenced by blood flow
Based on our observation that blood entering blind-ending aortic arches was followed by dilation of these vessels and eventual fusion with an adjacent vein, we reasoned that blood flow might play a major role in both AVM formation as well as ventral aorta disruption in kustr12 mutants. To probe the role of blood flow in development of the kustr12 vascular phenotypes, we exposed kustr12 mutants to 2,3-butanedione monoxime (BDM), a reversible myosin ATPase inhibitor that arrests heartbeat (Serluca et al., 2002). Treatment of wild type and kustr12 embryos for 6 hrs completely stopped heart beat, which recovered fully by 1 hr post-treatment. Treatment between 26 and 32 hpf, or between 54 and 60 hpf, had no effect on any of the kustr12 phenotypes (0/8 and 0/23 rescued, respectively), and wild type embryos developed normally. However, treatment between 47 and 53 hpf, the time during which aortic arches 5 and 6 lumenize, rescued the AVM in 66% of genotypic kustr12 mutants (19/29 rescued; Fig. 5A,B). AVM rescue was accompanied by rescue of the ventral aorta defect, with fusion occurring at or just anterior to aortic arch 5 (Fig. 5 C,D), but distance between posterior ceratobranchial cartilages remained increased in kustr12 mutants (Fig. 5E,F). These data demonstrate that the vascular defects in kustr12 mutants are strongly influenced by blood flow and not solely genetically determined, whereas the posterior cartilage defect is not influenced by vascular defects and/or blood flow.
Fig. 5. Lack of blood flow can rescue AVM formation in kustr12 mutants.

Ninety-eight percent (n = 117) of wild type or heterozygous embryos treated with 2,3-butanedione monoxime (BDM; stops heart beat) between 47 and 53 hpf exhibited normal aortic arches (A), ventral aorta (C), and jaw and pharyngeal cartilages (E). This treatment rescued aortic arch (B) and ventral aorta (D) phenotypes in 67% of kustr12 mutants (n = 29), but had no effect on the basibranchial cartilage phenotype (vertical lines in F). A–D: 2D confocal projections of 74 hpf TG(flk1:GFPla116;gata1:dsRed) embryos, labeling endothelial and blood cells green and red, respectively. G–H: Macro images of 4 dpf embryos stained with anti-collagen II. A,B: lateral views, anterior the left. C,D,E,F: ventral views, anterior to the left. PHS, primary head sinus. VA, ventral aorta.
A nonsense mutation in unc45a causes the kustr12 phenotype
To determine the molecular basis for the vascular defects observed in kustr12 mutants, we performed bulk segregant analysis on a kustr12/HK outcross using simple sequence length polymorphisms (SSLPs) spread throughout all 25 chromosomes. This crude mapping placed kustr12 on chromosome 25, flanked by Z23933 (0.13 cM) and Z15480 (5.1 cM; Fig. 6A). Fine mapping identified a 0.09 cM interval containing two genes, mannosidase2a2 and unc45a. We sequenced the entire man2a2 (XM_684446.1) coding region from wild type and kustr12 cDNA but found no mutations that could account for the kustr12 phenotype (data not shown). Thus, we focused our efforts on unc45a. We identified 5’ and 3’ ends of unc45a by RACE, and verified ends by direct amplification of full length unc45a from 72 hpf cDNA (GenBank accession number DQ003719). The open reading frame predicts a 935-amino acid protein (Fig. 6B). N-terminally, three tetratricopeptide repeat (TPR) motifs [amino acids 5–109, as defined by the Conserved Domain Database, CDD (Marchler-Bauer et al., 2005)] are apparent; these 34-amino acid degenerate motifs are known to mediate protein-protein interactions (Blatch and Lassle, 1999). C-terminally, an UNC-45-Cro1-She4p (UCS) domain (Hutagalung et al., 2002) is present (amino acids 534–935). This domain has been shown to be critical for chaperoning myosin motor domains (Barral et al., 2002). Additionally, four armadillo repeats are found within the UCS domain (amino acids 666–829, as defined by CDD); these 40-amino acid degenerate motifs also mediate protein-protein interactions. It should be noted that while all vertebrate Unc45 paralogs have putative armadillo repeats, C. elegans UNC-45 does not. Genomic structure of zebrafish unc45a reveals a 20-exon gene; the start codon lies within exon 2, and the stop codon within exon 19 (Fig. 6B).
Fig. 6. unc45a is mutated in kustr12 mutants.

(A) Fine mapping placed kustr12 in a 0.09 cM interval on chromosome 25 (blue bar), which contains unc45a (gray shading). (B) Unc45a contains an N-terminal TPR domain, a central domain of unknown function, a C-terminal UCS domain, and armadillo (ARM) repeats. The protein is encoded by 20 exons. (C) Phylogenetic analysis clearly indicates that this gene is homologous to mammalian unc45a, and not the closely related gene, unc45b, which is muscle-specific. (D) Sequencing of unc45a cDNAs in kustr12 mutants and wild type siblings revealed a T-to-A transversion in unc45a, converting a leucine to a stop codon. This mutation truncates the protein within exon 14 (B), deleting the armadillo repeats and a large portion of the UCS domain.
Comparison of the amino acid sequence of zebrafish Unc45a to other Unc45 proteins using ClustalW reveals 31% identity to C. elegans UNC-45; 65% identity to human UNC45a and mouse Unc45a; and 54% identity to human, mouse, and zebrafish Unc45b (Fig. 6C). When compared to human UNC45a, identity is slightly higher in the UCS domain (69%) than in the central region (63%) or the TPR domain (58%).
Comparison of the unc45a coding region between kustr12 mutants and their wild type siblings revealed a nonsense mutation, T1964A, which creates a premature stop codon within the UCS domain at amino acid 655 (Fig. 6B,D). This mutation lies 5’ to the exonic SNP used for mapping (BAC114c19unc45a) and is thus within the critical interval. To provide further evidence that this nonsense mutation in unc45a causes the kustr12 phenotype, we attempted to rescue kustr12 mutants by injecting 100–150 pg C-terminally myc-tagged or GFP-tagged unc45a mRNA into embryos at the one to four-cell stage. unc45a mRNA had no effect on the development of wild type embryos but fully rescued the aberrant circulation and AVM phenotypes in 67% (60/90) of kustr12 embryos, and elicited partial rescue in an additional 17% (15/90) of kustr12 embryos (Fig. 7A,B). Partial rescue was defined as lack of the characteristic AVM, but presence of hemorrhage in cranial vessels near the aortic arches. Importantly, injection of 100 pg mRNA encoding the truncated unc45a allele found in kustr12 mutants (unc45akus) was unable to rescue the AVM phenotype (0/13; Fig. 7C). Failure to rescue was not due to mRNA or protein instability, as the myc tag could be detected by immunohistochemistry as late as 72 hpf (data not shown).
Fig. 7. Rescue and phenocopy experiments confirm the role of unc45a in the kustr12 vascular phenotype.

Injection of full length (B; n = 90) but not C-terminally truncated (C; n = 13) unc45a mRNA (unc45akus, corresponding to the kustr12 allele) restored a wild type vascular phenotype to 67% of kustr12 mutants (compare to uninjected kustr12, A). Conversely, injection of am unc45a translation blocking morpholino (trans MO, E; n = 144) or exon 13/14 splice donor site blocking morpholino (splice MO, F; n = 60) produced a kustr12-like AVM phenotype in 20% and 23%, respectively, of wild type embryos (compare to uninjected wild type, D). A–C, F: 2D confocal projections of 74 hpf TG(flk1:GFPla116;gata1:dsRed) embryos, labeling endothelial cells green and blood cells red. D–E: 2D confocal projections of angiograms of 74 hpf embryos. Lateral views, anterior to the left. PHS, primary head sinus.
In a separate unc45a mRNA rescue experiment, we assessed the relationship between AVM, ventral aorta, and cartilage rescue (supplementary Fig. 1). Interestingly, we found that ventral aorta rescue was necessary but not sufficient for AVM rescue. Furthermore, the ventral aorta could be rescued in the presence of cartilage defects, and cartilage defects could be rescued in the presence ventral aorta defects, though most frequently, ventral aorta and cartilage rescue went hand in hand (11/17 rescued mutants). Taken together, these data suggest that there may be both ventral and dorsal requirements for unc45a in the pharyngeal arches, and that cartilage and vessel defects can be uncoupled.
In the converse experiment, we phenocopied kustr12 by injecting either translation-blocking or splice-blocking unc45a morpholinos. Fifteen ng translation-blocking unc45a morpholino (ATG MO) phenocopied the kustr12 vascular defects in 20% (29/144) of injected wild type embryos (Fig. 7D,E) but had no effect on early cleavage, which was somewhat surprising given the importance of maternal unc-45 in cytokinesis in C. elegans (Kachur et al., 2004). The splice-blocking morpholino (splice MO) resulted in insertion of intron 13 into the unc45a transcript, appending 43 amino acids encoded by intron 13 sequence before ending in a stop codon (data not shown). This truncates Unc45a within the UCS domain, in a similar position to the kustr12 mutation. Injection of 20 ng splice blocking morpholino phenocopied the kustr12 vascular defects in 23% (14/60) of injected embryos (Fig. 7F). Based on these results, we conclude that the gene mutated in kustr12 is undoubtedly unc45a.
unc45a is expressed in the pharyngeal arches from the time of onset of kustr12 vascular defects
To gain insight into the cell-autonomous functions of unc45a during vertebrate development, we examined mRNA expression pattern at various time points by whole mount in situ hybridization (Fig. 8). unc45a is expressed globally at the 64-cell stage, prior to the mid-blastula transition, thus representing maternal transcript (Fig. 8A). At shield stage (6 hpf), weak global expression is observed (Fig. 8B), whereas at the 3-somite stage (11 hpf), expression is strongest in the polster, an anterior mesendodermal structure that gives rise to the hatching gland (Fig. 8C). By 24 hpf, expression is concentrated in the midbrain, hindbrain, and retina (Fig. 8D), whereas by 46 hpf, additional expression domains arise in the pharyngeal arches, pharynx, liver, gut, and otic vesicle (Fig. 8E). Expression in the pharynx is quite strong at this time (Fig. 8F), whereas expression in the pharyngeal arches predominates laterally and is highly mosaic, suggesting expression in only a few cells potentially representing multiple cell types (Fig. 8G). unc45a is not obviously expressed in the aortic arch endothelium, but weak mosaic expression cannot be ruled out. By 5 dpf, the pharyngeal arch expression pattern becomes laterally restricted and somewhat segmental (Fig. 8H). It should be noted that unc45a expression is significantly downregulated in kustr12 mutants, suggesting that the premature termination codon might cause nonsense-mediated mRNA decay (data not shown). Taken together, these results demonstrate that unc45a is expressed in the pharyngeal arches at the time that the kustr12 phenotypes develop.
Fig. 8. Developmental expression patterns of unc45a mRNA.

In situ hybridization reveals diffuse, ubiquitous unc45a expression at 64-cell (A) and shield stage (B). By the three somite stage (C), there is specific expression in the polster (p). At 24 hpf (D), additional expression domains arise, including retina (r), midbrain (m), and hindbrain (h). By 46 hpf (E–G), expression continues in retina and brain, and is also strong in the liver (l, and arrow in E inset), pharynx (ph), gut (not shown), and pharyngeal arches (pa). The mosaic expression pattern in the pharyngeal arches is illustrated in G. This general pattern continues up to 5 dpf (H), with expression becoming more laterally restricted and segmented in the pharyngeal arches. A–B: animal pole at top. C–E, H: lateral views, anterior to the left. Inset in E: dorsal view, anterior to the left. F: transverse section. G: coronal section.
DISCUSSION
The aortic arch primordia arise by vasculogenesis and extend via angiogenesis
In most vertebrates, it is difficult if not impossible to make real-time observations of embryonic vessel development in the live animal. Thus, inferences are typically made from a series of static observations of independent embryos. With respect to the posterior aortic arches, immunohistochemical studies of staged quail embryos revealed the presence of endothelial cords situated between the aortic sac and the dorsal aorta prior to lumenization of these vessels, suggesting a vasculogenic origin (DeRuiter et al., 1993; Waldo et al., 1996). However, the precise origin of these endothelial cells could not be determined from these studies. In contrast, corrosion casts of staged chick and mouse embryos suggested that the posterior aortic arches might be angiogenic vessels. Using this technique, which allows visualization of only patent vessels, the posterior aortic arches are first evident as short ventral and dorsal segments extending from the aortic sac and dorsal aorta, respectively (Hiruma and Hirakow, 1995; Hiruma et al., 2002). None of these observations contrasts with our data. Using the externally fertilized, optically transparent zebrafish embryo, we have succeeded in dynamic tracking of branchial aortic arch development in live embryos, and describe for the first time the vasculogenic origin and subsequent angiogenic elaboration of these vessels. These eight vessels first appear as four pairs of bilateral, isolated islands of angioblasts in the lateral pharyngeal mesoderm, between the preformed lateral dorsal aorta and future site of the ventral aorta. Endothelial cells from these primodia migrate both dorsally and ventromedially as what we believe to be true angiogenic sprouts. Time lapse confocal microscopy studies show filopodial activity at the leading edge of the sprout; however, we have not formally proven that a dynamic non-dividing tip cell leads trailing, dividing stalk cells (Gerhardt et al., 2003). Dorsally, aortic arches 3 and 4 connect independently to the LDA, whereas aortic arch 6 fuses with aortic arch 5 lateral to the LDA, and the two vessels connect to the LDA via a single conduit. A similar configuration has been reported in the chick embryo (Hiruma and Hirakow, 1995). Ventrally, the ventral-most portions of the branchial aortic arches seem to coalesce bilaterally to form primitive left and right ventral aortae that eventually fuse at the midline into a single vessel. While these vessels form via bidirectional sprouting from a central location, lumenization occurs first at connections with the ventral aorta and dorsal aorta, which explains the results of corrosion casting experiments. This interesting mechanism of blood vessel formation, a combination of vasculogenesis and angiogenesis, has not been previously described.
An AVM forms in kustr12 mutants as a result of artery/vein fusion
AVMs are fairly common human vessel malformations characterized by a direct connection between a high-pressure artery and a vein. In the absence of an intervening capillary network to facilitate gas exchange and to step down the pressure, the surrounding tissue becomes ischemic, and the involved vessels become enlarged, fragile and tortuous. These events can lead to localized or systemic hypoxia, hemorrhage, or stroke, depending on the location and size of the lesion. While it is generally accepted that AVMs form during embryogenesis, the mechanisms by which they form are not understood. There are several reports in the literature demonstrating the importance of particular proteins, such as ACVRL1, Dll4, Endoglin, Notch, and VEGF, in maintaining the separation between arteries and veins (Carlson et al., 2005; Gale et al., 2004; Krebs et al., 2004; Lawson et al., 2001; Lawson et al., 2002; Oh et al., 2000; Sorensen et al., 2003). However, none of these studies describes the cellular mechanism by which AVMs form. The embryonic and genetic accessibility of the zebrafish embryo allow us to literally watch AVMs form in real time, and to elucidate cellular events and environmental forces associated with their formation. We have found that in kustr12 embryos, failure to make a timely dorsal connection between the AA5X and the LDA results in formation of a blind-ended aortic arch that distends with blood received from the ventral aorta. Distension brings this artery into contact with an adjacent vein, the primary head sinus, and these two vessels fuse. In support of this mechanism, we found that in the absence of blood flow, the aortic arches do not dilate and are less likely to fuse with the primary head sinus. Taken together, these findings demonstrate that kustr12 AVM formation is under both genetic and environmental control, and that flow-based enlargement of a blind-ended artery can precipitate fusion with an adjacent vein. It will be interesting to determine whether there are any molecular deficits in artery/vein specification (Lawson and Weinstein, 2002) during the course of AVM formation in kustr12.
unc45a is necessary for proper vessel and cartilage morphogenesis in the fifth and sixth pharyngeal arches
In kustr12 mutants, a nonsense mutation in unc45a is the underlying cause of the AVM involving aortic arches 5 and 6; the accompanying ventral aorta defect; as well as the subtle defect in ceratobranchial cartilages of pharyngeal arches 5 and 6. The reason for the highly specific spatial location of these lesions is not clear, as unc45a is expressed in a mosaic, unstructured pattern throughout the pharyngeal arches, as well as in other tissues such as brain, eye, liver, and gut that are not overtly affected by loss of function. While the kustr12 phenotype may reflect partial maternal compensation for loss of zygotic unc45a mRNA, we consider this possibility unlikely given that a translation blocking morpholino, which targets both maternal and zygotic mRNA, produces a phenotype identical to that seen in kustr12. Redundancy with unc45b is also unlikely, given that this paralog is expressed exclusively in muscle tissue (Etheridge et al., 2002; Wohlgemuth et al., 2007) and thus could not compensate for loss of unc45a in nonmuscle tissues. Given that Unc45a is likely a myosin co-chaperone protein (Barral et al., 2002), it is possible that a client myosin is expressed specifically within this particular region of the embryo. While skeletal muscle structure and function are unaffected in kustr12, the relevant client might be a cytoskeletal type II myosin or an unconventional myosin (Kachur et al., 2004) important for cytokinesis, cell migration, or vesicle trafficking. Given that the progesterone receptor is also an UNC45a client (Chadli et al., 2006), it is very possible that this protein or another non-myosin protein might be the critical client whose loss of function leads to the kustr12 phenotype. Current studies are aimed at identification of spatially relevant Unc45a clients, as well as determination of cell autonomy of the kustr12 mutation. These findings will greatly aid our understanding of the role of Unc45a in vessel and cartilage development in pharyngeal arches 5 and 6.
Defects in aortic arches and pharyngeal arches can be uncoupled in kustr12
It is not surprising that aortic arch defects are often associated with other defects in pharyngeal arch-derived structures. For example, DiGeorge syndrome involves aortic arch malformations as well as defects in pharyngeal endodermal pouch-derived organs (thymus, parathyroid) and neural crest-derived craniofacial structures (Wurdak et al., 2006). Similarly, mutations in Endothelin-1 result in aortic arch malformations and craniofacial abnormalities (Kurihara et al., 1995; Kurihara et al., 1994). Pharyngeal arch structure is highly complex, with contributions from endoderm, mesoderm, ectoderm, and neural crest. These different lineages depend upon one another for proper tissue organization and function. kustr12 mutants exhibit defects in ceratobranchial cartilage patterning along with defects in the aortic arches only in pharyngeal arches 5 and 6, suggesting that a patterning defect confined to this region may underlie both phenotypes. However, by injecting unc45a mRNA into kustr12 embryos at the 1 to 4-cell stage, we have been able to achieve vascular rescue in the absence of cartilage rescue, and vice versa. This result demonstrates that vessel defects do not cause cartilage defects, and that cartilage defects do not cause vessel defects, suggesting that the primary lesion in kustr12 may reside in a cell type that independently supports or patterns both vessels and cartilage.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank C. Nüsslein-Volhard (Max-Planck-Institute for Developmental Biology, Tübingen, Germany) for kustr12; D. Elson, S.-H. Hsieh, A. Park and A. Potocki for technical assistance; A. Davis and T. Simmons for fish care; J.-N. Chen (University of California-Los Angeles) for the TG(flk1:GFP)la116 line; D. Traver (University of California-San Diego) for the TG(gata1:dsRed) line; M. Tsang (University of Pittsburgh) for bmp2b plasmid; P. Yelick (Tufts University) for nkx2.3 plasmid; and D. Yelon (New York University Medical Center) for vmhc plasmid. This work was supported by AHA SDG 0335202N to BLR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ao W, Pilgrim D. Caenorhabditis elegans UNC-45 is a component of muscle thick filaments and colocalizes with myosin heavy chain B, but not myosin heavy chain A. J Cell Biol. 2000;148:375–384. doi: 10.1083/jcb.148.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral JM, et al. Unc-45 mutations in Caenorhabditis elegans implicate a CRO1/She4p-like domain in myosin assembly. J Cell Biol. 1998;143:1215–1225. doi: 10.1083/jcb.143.5.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral JM, et al. Role of the myosin assembly protein UNC-45 as a molecular chaperone for myosin. Science. 2002;295:669–671. doi: 10.1126/science.1066648. [DOI] [PubMed] [Google Scholar]
- Bazzaro M, et al. Myosin II Co-Chaperone General Cell UNC-45 Overexpression Is Associated with Ovarian Cancer, Rapid Proliferation, and Motility. Am J Pathol. 2007;171:1640–1649. doi: 10.2353/ajpath.2007.070325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatch GL, Lassle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays. 1999;21:932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Carlson TR, et al. Endothelial expression of constitutively active Notch4 elicits reversible arteriovenous malformations in adult mice. Proc Natl Acad Sci U S A. 2005;102:9884–9889. doi: 10.1073/pnas.0504391102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadli A, et al. GCUNC-45 is a novel regulator for the progesterone receptor/hsp90 chaperoning pathway. Mol Cell Biol. 2006;26:1722–1730. doi: 10.1128/MCB.26.5.1722-1730.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JN, et al. Mutations affecting the cardiovascular system and other internal organs in zebrafish. Development. 1996;123:293–302. doi: 10.1242/dev.123.1.293. [DOI] [PubMed] [Google Scholar]
- Chisaka O, Capecchi MR. Regionally restricted developmental defects resulting from targeted disruption of the mouse homeobox gene hox-1.5. Nature. 1991;350:473–479. doi: 10.1038/350473a0. [DOI] [PubMed] [Google Scholar]
- Congdon ED. Transformation of the aortic arch system during the development of the human embryo. Contrib. Embryol. 1922;14:47–110. [Google Scholar]
- DeRuiter MC, et al. Development of the pharyngeal arch system related to the pulmonary and bronchial vessels in the avian embryo. With a concept on systemic-pulmonary collateral artery formation. Circulation. 1993;87:1306–1319. doi: 10.1161/01.cir.87.4.1306. [DOI] [PubMed] [Google Scholar]
- Epstein DJ, et al. Splotch (Sp2H), a mutation affecting development of the mouse neural tube, shows a deletion within the paired homeodomain of Pax-3. Cell. 1991;67:767–774. doi: 10.1016/0092-8674(91)90071-6. [DOI] [PubMed] [Google Scholar]
- Epstein HF, Thomson JN. Temperature-sensitive mutation affecting myofilament assembly in Caenorhabditis elegans. Nature. 1974;250:579–580. doi: 10.1038/250579a0. [DOI] [PubMed] [Google Scholar]
- Etard C, et al. The UCS factor Steif/Unc-45b interacts with the heat shock protein Hsp90a during myofibrillogenesis. Dev Biol. 2007;308:133–143. doi: 10.1016/j.ydbio.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Etheridge L, et al. A zebrafish unc-45-related gene expressed during muscle development. Dev Dyn. 2002;224:457–460. doi: 10.1002/dvdy.10123. [DOI] [PubMed] [Google Scholar]
- Gale NW, et al. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci U S A. 2004;101:15949–15954. doi: 10.1073/pnas.0407290101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt H, et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161:1163–1177. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffter P, et al. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development. 1996;123:1–36. doi: 10.1242/dev.123.1.1. [DOI] [PubMed] [Google Scholar]
- Hauptmann G, Gerster T. Two-color whole-mount in situ hybridization to vertebrate and Drosophila embryos. Trends Genet. 1994;10:266. doi: 10.1016/0168-9525(90)90008-t. [DOI] [PubMed] [Google Scholar]
- Heuser CH. The branchial vessels and their derivatives in the pig. Contrib. Embryol. 1923;15:121–139. [Google Scholar]
- Hiruma T, Hirakow R. Formation of the pharyngeal arch arteries in the chick embryo. Observations of corrosion casts by scanning electron microscopy. Anat Embryol (Berl) 1995;191:415–423. doi: 10.1007/BF00304427. [DOI] [PubMed] [Google Scholar]
- Hiruma T, et al. Development of pharyngeal arch arteries in early mouse embryo. J Anat. 2002;201:15–29. doi: 10.1046/j.1469-7580.2002.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutagalung A, H , et al. The UCS family of myosin chaperones. J Cell Sci. 2002;115:3983–3990. doi: 10.1242/jcs.00107. [DOI] [PubMed] [Google Scholar]
- Isogai S, et al. The vascular anatomy of the developing zebrafish: an atlas of embryonic and early larval development. Dev. Biol. 2001;230:278–301. doi: 10.1006/dbio.2000.9995. [DOI] [PubMed] [Google Scholar]
- Kachur T, et al. Maternal UNC-45 is involved in cytokinesis and colocalizes with non-muscle myosin in the early Caenorhabditis elegans embryo. J Cell Sci. 2004;117:5313–5321. doi: 10.1242/jcs.01389. [DOI] [PubMed] [Google Scholar]
- Kamei M, Weinstein BM. Long-term time-lapse fluorescence imaging of developing zebrafish. Zebrafish. 2005;2:113–123. doi: 10.1089/zeb.2005.2.113. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, et al. Stages of embryonic development in the zebrafish. Dev. Dynam. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Knapik EW, et al. A microsatellite genetic linkage map for zebrafish (Danio rerio) Nature Genet. 1998;18:338–343. doi: 10.1038/ng0498-338. [DOI] [PubMed] [Google Scholar]
- Krebs LT, et al. Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 2004;18:2469–2473. doi: 10.1101/gad.1239204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara Y, et al. Aortic arch malformations and ventricular septal defect in mice deficient in endothelin-1. J Clin Invest. 1995;96:293–300. doi: 10.1172/JCI118033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara Y, et al. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature. 1994;368:703–710. doi: 10.1038/368703a0. [DOI] [PubMed] [Google Scholar]
- Lawson ND, et al. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development. 2001;128:3675–3683. doi: 10.1242/dev.128.19.3675. [DOI] [PubMed] [Google Scholar]
- Lawson ND, et al. sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev Cell. 2002;3:127–136. doi: 10.1016/s1534-5807(02)00198-3. [DOI] [PubMed] [Google Scholar]
- Lawson ND, Weinstein BM. Arteries and veins: making a difference with zebrafish. Nat Rev Genet. 2002;3:674–682. doi: 10.1038/nrg888. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, et al. CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. 2005;33:D192–D196. doi: 10.1093/nar/gki069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn C, et al. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development. 1994;120:2749–2771. doi: 10.1242/dev.120.10.2749. [DOI] [PubMed] [Google Scholar]
- Neff MM, et al. dCAPS, a simple technique for the genetic analysis of single nucleotide polymorphisms: experimental applications in Arabidopsis thaliana genetics. Plant J. 1998;14:387–392. doi: 10.1046/j.1365-313x.1998.00124.x. [DOI] [PubMed] [Google Scholar]
- Noden DM. Origins and assembly of avian embryonic blood vessels. Ann NY Acad Sci. 1990;588:236–249. doi: 10.1111/j.1749-6632.1990.tb13214.x. [DOI] [PubMed] [Google Scholar]
- Oh SP, et al. Activin receptor-like kinase 1 modulates transforming growth factor-b1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. USA. 2000;97:2626–2631. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MG, et al. Two mammalian UNC-45 isoforms are related to distinct cytoskeletal and muscle-specific functions. J Cell Sci. 2002;115:4013–4023. doi: 10.1242/jcs.00108. [DOI] [PubMed] [Google Scholar]
- Roman BL, et al. Disruption of acvrl1 increases endothelial cell number in zebrafish cranial vessels. Development. 2002;129:3009–3019. doi: 10.1242/dev.129.12.3009. [DOI] [PubMed] [Google Scholar]
- Rupp RA, et al. Xenopus embryos regulate the nuclear localization of XMyoD. Genes Dev. 1994;8:1311–1323. doi: 10.1101/gad.8.11.1311. [DOI] [PubMed] [Google Scholar]
- Sehnert AJ, et al. Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat Genet. 2002;31:106–110. doi: 10.1038/ng875. [DOI] [PubMed] [Google Scholar]
- Serluca FC, et al. Endothelial signaling in kidney morphogenesis: a role for hemodynamic forces. Curr Biol. 2002;12:492–497. doi: 10.1016/s0960-9822(02)00694-2. [DOI] [PubMed] [Google Scholar]
- Sorensen LK, et al. Loss of distinct arterial and venous boundaries in mice lacking endoglin, a vascular-specific TGFbeta coreceptor. Dev Biol. 2003;261:235–250. doi: 10.1016/s0012-1606(03)00158-1. [DOI] [PubMed] [Google Scholar]
- Venolia L, Waterston RH. The unc-45 gene of Caenorhabditis elegans is an essential muscle-affecting gene with maternal expression. Genetics. 1990;126:345–353. doi: 10.1093/genetics/126.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldo KL, Kumiski D, Kirby ML. Cardiac neural crest is essential for the persistence rather than the formation of an arch artery. Devel Dyn. 1996;205:281–292. doi: 10.1002/(SICI)1097-0177(199603)205:3<281::AID-AJA8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Weinstein BM, et al. gridlock, a localized heritable vascular patterning defect in the zebrafish. Nature Med. 1995;11:1143–1147. doi: 10.1038/nm1195-1143. [DOI] [PubMed] [Google Scholar]
- Westerfield M. The zebrafish book. Eugene: University of Oregon Press; 1995. [Google Scholar]
- Wohlgemuth SL, et al. The myosin co-chaperone UNC-45 is required for skeletal and cardiac muscle function in zebrafish. Dev Biol. 2007;303:483–492. doi: 10.1016/j.ydbio.2006.11.027. [DOI] [PubMed] [Google Scholar]
- Wurdak H, et al. DiGeorge syndrome and pharyngeal apparatus development. Bioessays. 2006;28:1078–1086. doi: 10.1002/bies.20484. [DOI] [PubMed] [Google Scholar]
- Yanagisawa H, et al. Role of Endothelin-1/Endothelin-A receptor-mediated signaling pathway in the aortic arch patterning in mice. J Clin Invest. 1998;102:22–33. doi: 10.1172/JCI2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.