

Abstract
In this study, we present evidence that the Dorsal activator interacts with limiting amounts of the TFIID complex in the Drosophila embryo. In vitro transcription reactions and protein binding assays implicate the TAFII110 and TAFII60 subunits of the TFIID complex in contributing to Dorsal-mediated activation. Mutations in TAFII110 and TAFII60 result in altered patterns of snail and twist transcription in embryos derived from dl/+ females. These results suggest that TAFIIs contribute to the activation of transcription in vivo and support the hypothesis that subunits of TFIID may serve as targets of enhancer binding proteins.
Transcriptional activation via the interplay of enhancer binding proteins with the core machinery has been studied most extensively by in vitro biochemical strategies. A large body of evidence suggests that different activators can contact distinct components of the core machinery, including different subunits of the TFIID complex (1, 2). It has been found that certain classes of Drosophila and human activators require the presence of the TAFII subunits of TFIID to mediate activation in vitro. Moreover, various classes of activation domains have been shown to bind directly to specific TAFII subunits, and these interactions are important for mediating transcriptional activation in vitro (3–5). However, the in vivo role of TAFIIs in metazoan transcription has not been established as firmly.
Early studies of a temperature-sensitive mutant in TAFII250 provided evidence for the function of TAFIIs in the transcription of cell cycle-regulated genes in mammalian tissue culture cells (6). Previously, an attempt was made to determine the effects of TAFII mutations on the expression of hunchback directed by the activator Bicoid in Drosophila embryos (7). However, the embryo staining results of this study recently were found to be incorrect (8) so that the in vivo role of TAFIIs in metazoan development remains obscure.
Here, we have examined the maternal Dorsal gradient to assess the role of TAFIIs in the Drosophila embryo. A nuclear gradient of Dorsal initiates the differentiation of the embryonic mesoderm, neurogenic ectoderm, and dorsal ectoderm (9–11). Dorsal is a member of the Rel-family of transcriptional enhancer factors and is regulated by a highly conserved signaling pathway that includes the Toll receptor and cactus inhibitor (12–14). Dorsal establishes distinct thresholds of gene expression and tissue differentiation through the regulation of different target genes in a concentration-dependent fashion (15). For example, high levels of Dorsal activate two regulatory genes, twist and snail, which initiate the differentiation of the mesoderm in ventral regions of precellular embryos (16–22). Low levels of the Dorsal gradient are insufficient to activate twist or snail but are able to trigger the expression of rhomboid and short gastrulation, which define the limits of the presumptive neurogenic ectoderm in lateral regions (23–25). These different thresholds of gene activity depend on the binding affinities of Dorsal operator sites within the target promoters and synergistic interactions between Dorsal and other adjacently bound activators (26). Dorsal not only functions as a sequence-specific transcriptional activator but also mediates repression by recruiting corepressor proteins to closely linked sites within the zen and dpp promoter regions (27, 28). There are several favorable considerations regarding the analysis of Dorsal–TFIID interactions. For example, dorsal gene activity is limiting in the early embryo, and the expression patterns of Dorsal target genes are relatively stable during cellularization and gastrulation. We have, therefore, chosen the Dorsal-snail system to reevaluate the potential role of TAFIIs in mediating transcriptional activation in vivo.
METHODS
Plasmids.
Expression plasmid encoding FLAG-tagged Dorsal protein was generated by using a NdeI/SpeI (blunt) fragment derived from pKS-Dl678 (10) inserted into pVL1392-FLAG (29) (PharMingen). The reporter plasmid was derived from E1BCAT by insertion of an oligo containing three copies of Dl binding sites of the rho NEE (−1,753 to −1,780) (24).
In Vitro Transcription.
In vitro transcription and primer extension analysis were performed as described (30) except that 100 ng of template DNA was used in 25 μl total volume. All transcription reactions contained 50 ng of dTFIIA, 25 ng of dTFIIB, 3 ng of hTFIIE56, 15 ng of hTFIIE34, and 1 μl of PolII/IIF/IIH-containing S300 fraction supplemented with 1 ng of TBP or 20 ng of peptide-eluted TFIID immunopurified from the Heparin 0.4 M fraction of Drosophila nuclear extracts. Reactions were preincubated for 10 min at 20°C, and transcription was initiated by adding rNTPs to 0.6 mM. In vitro transcription was allowed to proceed for an additional 30 min at 20°C. The mutant TAFs were preincubated with activator and template for 5 min before the addition of the other components of the transcription reaction.
Protein Binding Assays.
Protein binding assays were performed by incubating purified FLAG-Dorsal protein (1 μg) immobilized on FLAG antibody resin (Eastman Kodak) in 0.1 M NaCl HEMG-NCDMP [25 mM Hepes, pH 7.6/12.5 mM MgCl2/1 mM EDTA/10% glycerol/0.1% NP40/0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate/1 mM DTT/1 mM sodium metabisulfite/0.2 mM 4-(2-aminoethyl)-benzene sulfonyl fluoride] with [35S]methionine-labeled wild-type or mutant TAFs produced with the TNT-coupled reticulocyte lysate in vitro transcription/translation system (Promega) for 2 hours at 4°C. The resin then was washed extensively with 0.4 M NaCl HEMG-NCDMP resuspended in SDS loading buffer and was analyzed by SDS/PAGE.
Protein Expression and Purification.
FLAG-Dorsal, FLAG-TAFII110ΔC, and FLAG-TAFII60YY (7) were expressed in Sf9 cells. Cell extracts were prepared 48 hours after infection by sonicating in 0.8 M NaCl HEG-NCDMP. After a high-speed spin, the extracts were diluted to 0.3 M NaCl and were respun before they were incubated with FLAG-M2 antibody resin (Eastman Kodak) for 2 hours at 4°C. The resin was washed extensively with 1.0 M HEG NCDMP (1% NP40, 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate), and then the proteins either were used directly for protein binding assays or were eluted with 2 mg/ml FLAG peptide in 0.4 M HEG NCDMP. Dorsal was purified further by DNA affinity chromatography. The recombinant and partially purified basal factors were prepared as described (31).
Drosophila Strains.
yw67 flies were used in this study as “wild-type” flies. A null allele of Dorsal, dlI5 (11, 32) in the form of dlI5b/CyO, was used. Two mutant alleles for TAFII110, S-466 and XS-793 (encoding TAFII110ΔC and TAFII110ΔB, respectively), and one mutant allele for TAFII60, XS-922 (7), were maintained over synthetic balancer chromosome T2B (abbreviation for TM3, Sb e ry P[ry sev-RaslV12], (33). Deficiencies for TAFII110 {Df(3L)st-f13,Ki[1]roe[1]P[P]/[TM6B], 2993}, TAFII60 {Df(3L)KT02/TM6B], 3617}, and TBP {DF[2R]PU-D17,NW[D]PIN[YT]/SM1], 2605} were obtained from the Bloomington Stock Center (Bloomington, IN). M(2)53C/SM5 flies were from the Rubin laboratory (Univ. of California, Berkeley). Transgenic strain carrying the 1.6 sna lacZ transgene was as described (19).
Drosophila Crosses.
Virgin females doubly heterozygous for dl and each of the TAFII mutants or deficiencies were obtained by crossing dlI5b/CyO males to virgin TAF/T2B or TAF deficiencies/TM6B females. dl, TAF double heterozygous females were crossed to either wild-type males to assess maternal effects of TAFs or TAF/P[Kr-lacZ] males to measure maternal and zygotic effects. TAF/P[Kr-lacZ] flies were generated from crossing TAF/T2B females to males homozygous for P[Kr-lacZ] on the third chromosome (from Gary Struhl, Columbia Univ., New York). dl, Df(2)TBP trans-heterozygous females were generated by crossing dl/CyO males to Df(2)TBP/SM1 females. In control experiments, dl heterozygous females are generated from mating wild-type males with dlI5b/CyO females.
In Situ Hybridization of Drosophila Embryos.
Embryos were collected and aged at 22–24°C. Embryos 2–4 hours old were hybridized with digoxigenin-UTP-labeled antisense RNA probes as described (16, 34).
RESULTS
Strong gene dosage interactions were observed between dl and genes that encode bHLH activators (21, 35). For example, embryos derived from dl/+ females that were also heterozygous for mutations in twist, daughterless, or scute exhibited severe patterning defects, including a reduction in the mesoderm (35). The sensitization achieved by removing one maternal dose of dl offered the opportunity to investigate gene dosage interactions between Dorsal and core components of the transcription complex. The initial experiments involved the use of chromosomal deletions that remove different basal factors, including TBP, TAFII110, and TAFII60. The expression patterns of the Dorsal target genes snail and twist were analyzed in embryos obtained from trans-heterozygous or double heterozygous females that contained one dose of dl+ gene activity and a deficiency in a given basal factor. Embryos were collected and aged at room temperature (22–24°C), were hybridized with digoxigenin-labeled antisense RNA probes, and subsequently were stained with antidigoxigenin antibodies.
The Early Embryo Contains Limiting Amounts of the TFIID Initiator Complex.
Embryos derived from dl/+ heterozygotes exhibited an essentially normal snail expression pattern, which included a band ≈20 cells in width in the ventral and ventrolateral regions that encompass the presumptive mesoderm (Fig. 1A). Embryos derived from dl/+; Df(3)TAFII110/+ double heterozygotes exhibited a subtle but reproducible change in the snail expression pattern (Fig. 1B). There was a slight narrowing in the pattern particularly in the vicinity of the cephalic furrow located near the head/trunk junction (Fig. 1B, arrow). Embryos derived from dl/+; Df(3)TAFII60/+ double heterozygotes exhibited a similar narrowing of the snail expression pattern (Fig. 1C), although a smaller proportion of the embryos exhibit this defect as compared with the dl/+; Df(3)TAFII110/+ double heterozygotes (see Table 1). The most severe defects were observed in embryos derived from dl/DF(2)TBP trans-heterozygotes (Fig. 1D). There was a dramatic narrowing in the snail expression pattern, and gaps appeared near the cephalic furrow and posterior transverse furrow (Fig. 1D, arrows).
Figure 1.
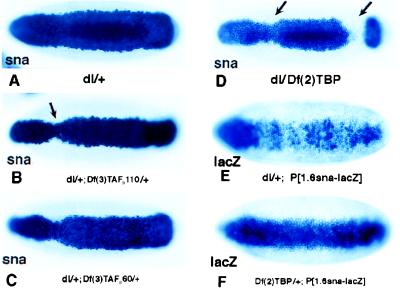
Drosophila embryos deficient in TAFIIs or TBP are impaired for dl-dependent transcription of sna. Whole mount in situ hybridization of stage 5 Drosophila embryos with digoxigenin-UTP-labeled anti-sense RNA probe to sna (A–D) or lacZ gene (E and F) are shown. All embryos are oriented with anterior to the left and ventral surface facing the reader. Female flies with different genotypes were crossed to wild-type males (A–D) or wild-type males carrying a 1.6-kilobase sna-lacZ transgene (E and F) (19). (A) sna expression pattern in embryos from dl/+ mothers. (B) sna expression in dl/+; Df(3)TAF110/+ maternal background. There is a slight narrowing in the pattern, particularly in the cephalic furrow region. (C) sna expression in dl/+; Df(3)TAFII60/+ maternal background. A similar but milder effect is seen. (D) In dl/Df(2)TBP maternal background, the majority of the embryos are severely affected. (E and F) The 1.6-kilobase snail-lacZ transgene directs an abnormal expression pattern in embryos derived from dl/+ females (E); however, it directs a normal expression in embryos Df(2)TBP/+ from females (F).
Table 1.
Phenotypes of sna expression in different genetic background
| Maternal genotypes | Normal, % | Type I, % | Type II, % | n |
|---|---|---|---|---|
| dl/+ | 92.4 | 7.6 | 0.0 | 471 |
| dl/+; Df(3)TAFII110/+ | 42.4 | 43.5 | 14.1 | 177 |
| dl/+; Df(3)TAFII60/+ | 59.0 | 37.8 | 3.2 | 188 |
| dl/Df(2)TBP | 18.6 | 29.4 | 52.0 | 19 |
| dl/M(2)53C | 83.2 | 16.8 | 0.0 | 125 |
“Normal” is defined as sna expression patterns indistinguishable from those of embryos derived from wild-type females. “Type I” embryos displayed a variable narrowing, especially near the cephalic furrow region. “Type II” embryos showed a more severe disruption of the snail staining pattern. snail expression domains were usually 10–15 cells wide around 50% egg length and 5 cells or less around the cephalic region. Embryos from crosses indicated in Fig. 1 (A–D) abd embryos derived from the mating of male M(2)53C/+ X female M(2)53C/dl1 were scored.
The preceding results suggest that components of the TFIID complex, TBP, TAFII110, and TAFII60, are present at limiting concentrations in the early embryo. However, it is conceivable that the altered snail staining patterns result from a reduction in the maternal transcription of dl during oogenesis. That is, the combined reduction in dorsal and TBP gene dose might lead to a severe reduction in the Dorsal nuclear gradient in early embryos. To test this possibility, we analyzed the expression of a sensitized snail-lacZ transgene in embryos derived from Df(2)TBP/+ females. The transgene contains the first 1.6 kilobases of 5′ flanking sequence from the snail gene and normally is expressed in the ventral-most 14–16 cells in response to high levels of the Dorsal gradient. As shown (19), this transgene directs an erratic and abnormal staining pattern in embryos derived from dl/+ heterozygotes (Fig. 1E). These results demonstrate that the snail-lacZ transgene is sensitive to just a 50% reduction in the levels of Dorsal. Nonetheless, a normal staining pattern is observed in embryos derived from Df(2)TBP/+ heterozygotes (Fig. 1F). This result indicates that a reduction in maternal TBP gene dose does not significantly alter the transcription of the dorsal gene during oogenesis. Instead, it would appear that the altered patterns of snail expression seen in the preceding gene dosage assays can be attributed to interactions between Dorsal and the TFIID complex on the snail target promoter (and/or the twist promoter; see below). However, a major limitation of these studies is that relatively large chromosomal deletions were used, so it is uncertain whether the altered snail expression patterns result from reduced levels of TBP, TAFII110, or TAFII60 as opposed to other genes, which are uncovered by the deletions. Both in vitro experiments and gene dosage assays with point mutations were used to investigate this issue.
In Vitro Transcription Activation by Dorsal Requires TAFIIs.
The observed alterations in the pattern of snail expression in embryos with deficiencies in TBP, TAFII110, or TAFII60 suggested that activation by Dorsal may depend on multiple subunits of the TFIID complex. To test directly the TAFII requirements for activation by Dorsal, we have reconstituted in vitro transcription reactions programmed with a DNA template bearing multiple Dorsal binding sites fused to the E1B core promoter. In the presence of purified Drosophila RNA polymerase II and all of the basal factors except for TFIID, no transcription was detectable (Fig. 2A). When purified recombinant Drosophila TBP was added to these reactions, accurate initiation of basal transcription was observed, but no Dorsal-dependent activation of transcription was seen. By contrast, in reactions reconstituted with affinity-purified TFIID complex, a robust Dorsal-dependent enhancement of transcription was obtained. Thus, in vitro activation of transcription by Dorsal appears to be TAFII dependent.
Figure 2.

Effect of TAFIIs on transcriptional activation by Dorsal in vitro. (A) Ability of TBP versus TFIID to mediate activation by Dorsal in a purified in vitro transcription system. (Left) In vitro transcription reactions containing the basal factors and RNA polymerase II but lacking TBP or IID (lanes 1–3) were programmed with 3 × DlTwiE1BCAT as template in the absence (lanes 1, 4, and 7) or presence of Dorsal (10 ng, lanes 2, 5, and 8 or 40 ng, lanes 3, 6, and 9) supplemented with TBP (lanes 4–6) or TFIID (lanes 7–9) and were assayed by primer extension. (Center) Coomassie blue-stained SDS polyacrylamide gel of purified Dorsal (lane 11). (Right) Silver-stained SDS polyacrylamide gel of purified TFIID (lane 12). The molecular sizes of protein standards (in kilodaltons) are indicated on the left. The identity of the TAF subunits is indicated on the right of lane 12. (B) Dorsal interacts with dTAFII110 and dTAFII60. Anti-FLAG antibody resin (lanes 2, 5, 8, 11, 14, and 17) or beads loaded with Dorsal (lanes 3, 6, 9, 12, 15, and 18) were incubated with the [35S]methionine-labeled, in vitro-translated TAFs indicated at the bottom of each panel. Protein complexes were analyzed by SDS/PAGE. Bound TAFs were identified by autoradiography. Lanes 1, 4, 7, 10, 13, and 16 represent 10% of the input material for each binding reaction.
To determine which of the TAFII subunits of TFIID might serve as targets for interaction with Dorsal, we have carried out a series of activator:TAFII binding assays. Affinity resin containing either control beads or a FLAG–Dorsal fusion protein was used to bind individual in vitro-labeled TAFII subunits. Dorsal bound most efficiently to TAFII110 and moderately well to TAFII60 (Fig. 2B). We also observed weak binding of TAFII40 to FLAG-Dorsal whereas no significant interaction was detected for TAFIIs 80, 30α, or 30β (Fig. 2B). These results suggest that at least TAFII110 and possibly TAFII60 and TAFII40 can provide specific interfaces for interaction between Dorsal and subunits of TFIID.
Mutant TAFIIs Squelch Dorsal-Dependent Activation in Vitro.
Mutant alleles for TAFII110 and TAFII60 (TAFII110ΔC, TAFII110ΔB, and TAFII60YY) were isolated in a dominant modifier screen sensitized to transcription levels (7, 33). The two point mutations in TAFII110 give rise to C-terminally truncated products whereas the TAFII60 mutant bears an insertion of two tyrosine residues (Fig. 3A). Biochemical analysis of these mutant TAFII proteins revealed that they fail to bind the core subunit (TAFII250) of TFIID and most likely do not incorporate into stable TFIID complexes (7). Consequently, these mutant proteins are expected to behave as transdominant negative inhibitors that can squelch activation. Because TAFII110 and TAFII60 interact with Dorsal, it should be possible to test, both in vitro and in vivo, whether these mutant TAFIIs play a role in mediating activation by Dorsal. First, we determined whether the mutant TAFII proteins retained the ability to bind the activator. Direct protein binding experiments revealed that FLAG–Dorsal interacts efficiently with all three of these mutant TAFII proteins (Fig. 3B). Next, we tested the ability of TAFII110ΔC and TAFII60YY to inhibit activation of transcription by Dorsal. Fully reconstituted transcription reactions supplemented with purified Dorsal protein and affinity-purified TFIID can be significantly inhibited by the addition of purified TAFII110ΔC or TAFII60YY protein (Fig. 3C). As expected, addition of increasing amounts of mutant TAFII protein reproducibly showed no effect on basal transcription but inhibited Dorsal activation (Fig. 3C, compare lanes 2, 4, and 6 to 7, 9, and 11). These results are consistent with the notion that the mutant TAFIIs are unable to incorporate stably into the TFIID complex but retain one or more coactivator domains that can squelch activation but do not inhibit basal transcription. These data suggest that the in vitro activation of transcription by Dorsal depends on TAFII110 and TAFII60.
Figure 3.

(A) Schematic diagram of the domain structure of wild-type and mutant dTAFII110 and dTAFII60. The black boxed area represents the dTAFII250 interaction domain of each TAF. The dark gray area (marked coactivator) delineates the domains that are thought to interact with activator proteins. (B) TAFII110ΔC, TAFII110ΔB, and TAFII60YY retain the ability to bind selectively to Dorsal. Anti-FLAG antibody resin (lanes 2, 5, and 8) or beads loaded with Dorsal (lanes 3, 6, and 9) were incubated with [35S]methionine-labeled, in vitro-translated mutant TAFs indicated at the bottom of each panel. Protein complexes were separated by SDS/PAGE. Bound mutant TAFs were identified by autoradiography. Lanes 1, 4, and 7 represent 10% of the input material for each binding reaction. (C) Mutant dTAFII110 and dTAFII60 squelch activation by Dorsal but not basal transcription. Reactions containing the basal factors, RNA polII, 3 × DlTwiE1BCAT DNA template, and 0 ng of Dorsal (lane 1 and lanes 7–11) or 40 ng of Dorsal (lanes 2–6) were assayed in the presence of increasing amounts (30 ng or 100 ng) of either TAFII110ΔC (lanes 3 and 8 and lanes 4 and 9, respectively) or TAFII60YY (lanes 5 and 10 and lanes 6 and 11, respectively). Coomassie blue-stained SDS/PAGE gels of mutant TAFII110ΔC (lane 13) and TAFII60YY (lane 15) as well as molecular weight markers (lanes 12 and 14) are shown.
TAFII110 and TAFII60 Are Required for snail and twist Transcription in Vivo.
We investigated gene dosage interaction between dorsal and the mutant TAFIIs in the early embryo. The initial series of experiments involved the mating of dl/+; TAFII110/+ or dl/+; TAFII60/+ double heterozygous females with wild-type males to assess potential interactions between Dorsal and maternally deposited TAFIIs (Fig. 4 A–D). The mutations in TAFII110 (ΔC and, less frequently, ΔB) exhibited similar genetic interactions with Dorsal (Fig. 4 B and C, respectively, and Table 2). There was a narrowing of the overall snail expression pattern; this narrowing was most severe in the cephalic region (Fig. 4 B and C, arrows; compare with Fig. 4A). A similar, but somewhat less frequent, change in the snail pattern was observed in embryos derived from dl/+; TAFII60YY/+ double heterozygotes (Fig. 4D and Table 2). In all cases, the alterations of snail expression patterns were more severe than those observed with the chromosomal deficiencies (Fig. 1 B and C). This observation is consistent with the finding that TAFII110 and TAFII60 mutants encode dominant negative proteins (see above and Discussion).
Figure 4.
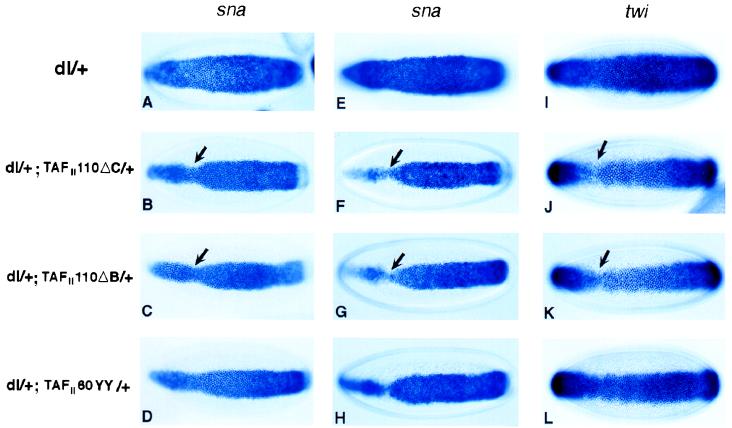
TAFII110 and TAFII60 are required for Dorsal-dependent transcriptional activation of twist and snail in the early embryos. In situ hybridizations of mid-stage five embryos by using snail (A–H) or twist (I–L) antisense RNA probe are shown. Embryos from dl/+ (A, E, and I), dl/+; TAFII110ΔC/+ (B, F, and J), dl/+; TAFII110ΔB/+ (C, G, and K), or dl/+; TAFII60YY/+ (D, H, and L) females that are mated with either wild-type males (A, B, C, D, E, and I), TAFII110ΔC/P[Kr-lacZ] males (F and J), TAFII110ΔB/P[Kr-lacZ] males (G and K), or TAFII60YY/P[Kr-lacZ] males (H and L) are shown.
Table 2.
Phenotypes of sna expression in different genetic background
| Maternal genotypes | Kr-lacZ embryos, +/+ or TAF/+
|
Non-Kr-lacZ embryos, TAF/+ or TAF/TAF
|
||||||
|---|---|---|---|---|---|---|---|---|
| Normal, % | Type I, % | Type II, % | n | Normal, % | Type I, % | Type II, % | n | |
| wild-type | 96.2 | 3.8 | 0.0 | 400 | ||||
| TAFII110ΔC/+ | 94.7 | 5.3 | 0.0 | 337 | ||||
| TAFII110ΔB/+ | 94.4 | 5.6 | 0.0 | 285 | ||||
| TAFII60YY/+ | 96.3 | 3.7 | 0.0 | 351 | ||||
| dl/+; TAFII110ΔC/+ | 23.2 | 57.1 | 19.7 | 452 | 8.1 | 55.1 | 36.8 | 494 |
| dl/+; TAFII110ΔB/+ | 22.4 | 63.0 | 14.6 | 343 | 6.9 | 64.8 | 28.3 | 361 |
| dl/+; TAFII60YY/+ | 21.7 | 65.0 | 13.3 | 309 | 11.8 | 58.1 | 30.1 | 356 |
Normal, Type I, and Type II are as in Table 1. Wild-type females or female flies with designated genotype were mated with wild-type males or male flies with the corresponding TAFIImutations over the Kr-lacZ-marked chromosome. Embryos with Kr-lacZ expression were either wild-type or contained one mutant copy of the indicated TAF gene; embryos without Kr-lacZ expression contained either one copy of mutant TAFs or were homozygous for the TAF mutation.
It is conceivable that the most severely affected embryos are TAFII/+ heterozygotes. In this regard, we note that in situ hybridization assays suggest that the TAFII110 and TAFII60 genes are transcribed quite early, during nuclear cleavage cycle 12/13 (data not shown). To investigate this issue of zygotic interactions, additional experiments were done with embryos derived from the mating of dl/TAFII double heterozygous females and TAFII/P[Kr-lacZ] males. One-fourth of the resulting embryos corresponded to TAFII/TAFII homozygotes. The males used in these matings carried a Kr-lacZ transgene so that one-half of the embryos lacking lacZ staining corresponded to TAFII homozygotes.
Although the putative TAFII homozygous embryos from dl+ females displayed normal snail staining, the putative TAFII homozygous embryos exhibited severe changes in the snail expression pattern (Fig. 4 F–H and Table 2). In early embryos (at the midpoint of cellularization), there was a particularly marked narrowing of the snail expression pattern in anterior regions (Fig. 4 F and G, arrows). We also observed a significant number of embryos (4–8%) displaying even more severe phenotypes. For example, 8% of the TAFII110ΔC mutants displayed either almost no snail staining in the anterior third of the embryo at early stage 5, or there was a pronounced gap near the cephalic furrow at mid- or late stage 5 (data not shown). These alterations in the snail pattern were more severe than those observed in embryos derived from double heterozygotes mated with wild-type males (Fig. 4 B and C). As seen for the chromosomal deletions, the TAFII110 mutants, especially TAFII110ΔC, exhibited a somewhat more severe disruption in the snail pattern as compared with the TAFII60 mutant (compare Fig. 4H with Fig. 4 F and G; Tables 1 and 2).
Defects in the snail expression pattern were more pronounced in anterior regions of mutant embryos, particularly in the region spanning the presumptive cephalic furrow. These defects were similar to those observed in embryos containing reduced levels of Dorsal and twist (19, 35). It is conceivable that one or more repressors are required for the establishment of the cephalic furrow, thereby sensitizing the twist and snail expression patterns in this specific region of the embryo. We suggest that the residual twist and snail expression patterns observed in dl;TAFII mutant embryos resulted from the perdurance of normal TFIID initiator complexes present in the germline. It has not been possible to obtain germline clones for TAFII mutants, so that the TAF/+ heterozygous females used in this study always contained a wild-type copy of the gene.
The specificity of dl/TAFII interactions was tested by analyzing a minute mutation, M53(C), which results in a general reduction in the rates of translation and embryogenesis (45). Embryos derived from the mating of M53(C)/dl females and M53(C)/+ males exhibited relatively minor effects on snail expression, as compared with the disruptions observed in embryos derived from dl;TAFII females (see Table 2).
The preceding analysis raises the possibility that Dorsal-TAFII interactions are important for the transcriptional activation of the snail promoter. However, it is also possible that such interactions occur on the twist promoter, which can be thought of as an immediate early target of the Dorsal gradient. Previous studies have shown that the Dorsal gradient activates twist and that the two proteins then work synergistically to activate snail (16–19). Thus, the disruptions we observed in the snail expression pattern could result, indirectly, from reduced levels of Twist. To test this possibility, embryos derived from the mating of dl;TAFII females and TAFII/P[Kr-lacZ] males were hybridized with a twist antisense RNA probe (Fig. 4 I–L).
The twist staining pattern observed in embryos derived from dl/+ heterozygotes was essentially normal and encompassed the ventral-most 20–22 cells (Fig. 4I). The twist and snail patterns were similar, except that snail exhibited sharper borders of expression. Mutant embryos that were presumably homozygous for TAFII110 exhibited a narrowing in the twist pattern, particularly in anterior regions (Fig. 4 J and K, arrows). A similar, but somewhat less severe, alteration also was observed in TAFII60 mutants (Fig. 4H). In general, the changes in the snail patterns were more severe than those observed for twist (e.g., compare Fig. 4F with Fig. 4J). The simplest interpretation of these results is that Dorsal-TAFII interactions are important for the activation of both twist and snail. Reduced levels of Twist, together with a breakdown in Dorsal-TAFII interactions, resulted in a relatively severe disruption in the snail pattern.
DISCUSSION
We have presented evidence that TFIID complexes are limiting in the early embryo and that two specific TAFIIs (TAFII110 and TAFII60) are required for Dorsal-mediated activation. One can imagine several nonexclusive models for Dorsal–TAF interactions in the Drosophila embryo. First, Dorsal may activate transcription through direct protein–protein interactions with TAFII110 and/or TAFII60. Second, mutations in TAFII110 and TAFII60 may alter TFIID structure or function and indirectly disrupt activation of the twist and snail target genes in sensitized dl/+ embryos. Perhaps, mutant TAF proteins impair the recognition of the twist and snail promoters by TFIID; alternatively, the defective TFIID might affect activator interactions with other components of the transcription machinery. Finally, it is conceivable that dl/+ embryos are sensitized to the point that any perturbation in the core transcription complex may disrupt snail and twist expression. At present, it is difficult to discriminate between these potential mechanisms.
Studies of yeast TAFII mutants suggested that disrupting the TFIID complex does not lead to detectable transcriptional defects (36, 37). However, more recent analysis of yeast mutant TAFIIs confirm earlier studies of mammalian TAFIIs (6) that transcription of several genes involved in cell cycle control, in fact, depend on yeast TAFII145 (38, 39). The yeast studies have been interpreted to indicate that TAFII145 plays a role in promoter recognition rather than mediating activation. It was, however, unclear whether mutations in TAFII145, the homolog of Drosophila, and human TAFII250 affected activation, promoter selectivity, or both because neither the identity of the activator nor the location of the critical cis-controlling elements responsive to TAFII145 have been identified. By contrast, studies of ts mutants of human TAFII250 established that the core subunit of TFIID can serve as a target for activation and also can help direct promoter recognition at the cyclin A gene (40).
The promoter selectivity function of TAFII250 is consistent with previous biochemical studies demonstrating the ability of certain TFIID subunits to contact core promoter DNA sequences directly (4, 41–43). In vitro transcription assays also indicated that TAFII250, TAFII150, and TAFII60 may contribute to the binding of TFIID to specific core promoter sequences. However, these DNA binding functions of TAFIIs appear to be independent of activators, and, thus far, only three of nine TAFIIs have been found to bind DNA in a sequence specific manner. At present, there is no evidence to indicate that Drosophila TAFII110 interacts with DNA.
It is possible that dorsal/TAFII dosage-sensitive interactions reflect a failure of TFIID complexes lacking TAFII110 and TAFII60 to perform core promoter recognition at the snail and twist gene sequences. However, Dorsal has been shown to be promiscuous in its ability to activate distinct core promoters, suggesting that promoter selectivity may not be the basis for the observed Dorsal/TAFII interactions (15). Indeed the twist and snail promoters appear to represent two distinct classes of core promoters (17, 19, 44) that are fully responsive to Dorsal, and both are affected by mutations in TAFII110 and TAFII60. Thus, it seems unlikely that the TAFII-mediated activation of snail and twist by Dorsal depends solely on promoter selectivity.
Although the in vivo basis for Dorsal-TAFII interactions is uncertain, there are several arguments for direct protein–protein interactions. First, the Dorsal activator can bind selectively to at least two TAFIIs in vitro, and Dorsal is able to interact directly with purified TFIID in a protein binding assay in the absence of promoter DNA (data not included). Moreover, activation by Dorsal in reconstituted transcription reactions is TAFII-dependent and is squelched by transdominant negative versions of these two TAFIIs. The mutant TAFs retain their ability to bind Dorsal but fail to incorporate into a stable TFIID (Fig. 3B and ref. 7) and therefore interfere with transcription when present in the embryo with wild-type TAFIIs. Consistent with this possibility is the finding that the mutant TAFIIs produce stronger effects than their corresponding deficiencies in gene dosage assays (Tables 1 and 2).
Acknowledgments
We thank G. Struhl for fly stocks and helpful advice, F. Sauer for FLAG-TAFII60YY and FLAG-TAFII110ΔC recombinant baculoviruses, D. Wasserman for stocks of TAFII mutant flies, M. Brodsky for Minute flies, M. Holmes for the generous gift of recombinant dTBP and hTFIIE protein, as well as a successful collaboration to set up a purified Drosophila in vitro transcription system. We also thank Don Rio, Tom Cline, Steve McKnight, and Mattias Mannervik for critical reading of this manuscript, Jennifer Gin for technical support, and Janeen Lim for preparation of this manuscript. This work was funded in part by National Institutes of Health grants for R.T. (Grant CA25417) and M.L. (Grant GM46638). J.Z. is supported by a National Institutes of Health postdoctoral fellowship (GM19329-01), and Jörk Zwicker is supported by a Deutsche Forschungs Gemeinschaft postdoctoral fellowship.
References
- 1.Tjian R, Maniatis T. Cell. 1994;77:5–8. doi: 10.1016/0092-8674(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 2.Roeder R G. Trends Biochem Sci. 1996;21:327–335. [PubMed] [Google Scholar]
- 3.Chen J L, Attardi L D, Verrijzer C P, Yokomori K, Tjian R. Cell. 1994;79:93–105. doi: 10.1016/0092-8674(94)90403-0. [DOI] [PubMed] [Google Scholar]
- 4.Verrijzer C P, Tjian R. Trends Biochem Sci. 1996;21:338–342. [PubMed] [Google Scholar]
- 5.Goodrich J A, Cutler G, Tjian R. Cell. 1996;84:825–830. doi: 10.1016/s0092-8674(00)81061-2. [DOI] [PubMed] [Google Scholar]
- 6.Wang E H, Tjian R. Science. 1994;263:811–814. doi: 10.1126/science.8303298. [DOI] [PubMed] [Google Scholar]
- 7.Sauer F, Wassarman D A, Rubin G M, Tjian R. Cell. 1996;87:1271–1284. doi: 10.1016/s0092-8674(00)81822-x. [DOI] [PubMed] [Google Scholar]
- 8.Sauer, F., Wassarman, D., Rubin G. & Tjian R. (1998) Cell 95, in press. [DOI] [PubMed]
- 9.Roth S, Stein D, Nüsslein-Volhard C. Cell. 1989;59:1189–1202. doi: 10.1016/0092-8674(89)90774-5. [DOI] [PubMed] [Google Scholar]
- 10.Rushlow C A, Han K, Manley J L, Levine M. Cell. 1989;59:1165–1177. doi: 10.1016/0092-8674(89)90772-1. [DOI] [PubMed] [Google Scholar]
- 11.Steward R, Nüsslein-Volhard C. Genetics. 1986;113:665–678. doi: 10.1093/genetics/113.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wasserman S A. Mol Biol Cell. 1993;4:767–771. doi: 10.1091/mbc.4.8.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steward R, Govind S. Curr Opin Genet Dev. 1993;3:556–561. doi: 10.1016/0959-437x(93)90090-c. [DOI] [PubMed] [Google Scholar]
- 14.Belvin M P, Jin Y, Anderson K V. Genes Dev. 1995;9:783–793. doi: 10.1101/gad.9.7.783. [DOI] [PubMed] [Google Scholar]
- 15.Huang A M, Rusch J, Levine M. Genes Dev. 1997;11:1963–1973. doi: 10.1101/gad.11.15.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang J, Kosman D, Ip Y T, Levine M. Genes Dev. 1991;5:1881–1891. doi: 10.1101/gad.5.10.1881. [DOI] [PubMed] [Google Scholar]
- 17.Pan D J, Huang J D, Courey A J. Genes Dev. 1991;5:1892–1901. doi: 10.1101/gad.5.10.1892. [DOI] [PubMed] [Google Scholar]
- 18.Thisse C, Perrin-Schmitt F, Stoetzel C, Thisse B. Cell. 1991;65:1191–1201. doi: 10.1016/0092-8674(91)90014-p. [DOI] [PubMed] [Google Scholar]
- 19.Ip Y T, Park R E, Kosman D, Yazdanbakhsh K, Levine M. Genes Dev. 1992;6:1518–1530. doi: 10.1101/gad.6.8.1518. [DOI] [PubMed] [Google Scholar]
- 20.Leptin M. Genes Dev. 1991;5:1568–1576. doi: 10.1101/gad.5.9.1568. [DOI] [PubMed] [Google Scholar]
- 21.Kosman D, Ip Y T, Levine M, Arora K. Science. 1991;254:118–122. doi: 10.1126/science.1925551. [DOI] [PubMed] [Google Scholar]
- 22.Alberga A, Boulay J L, Kempe E, Dennefeld C, Haenlin M. Development (Cambridge, UK) 1991;111:983–992. doi: 10.1242/dev.111.4.983. [DOI] [PubMed] [Google Scholar]
- 23.Bier E, Jan L Y, Jan Y N. Genes Dev. 1990;4:190–203. doi: 10.1101/gad.4.2.190. [DOI] [PubMed] [Google Scholar]
- 24.Ip Y T, Park R E, Kosman D, Bier E, Levine M. Genes Dev. 1992;6:1728–39. doi: 10.1101/gad.6.9.1728. [DOI] [PubMed] [Google Scholar]
- 25.Francois V, Solloway M, O’Neill J W, Emery J, Bier E. Genes Dev. 1994;8:2602–2616. doi: 10.1101/gad.8.21.2602. [DOI] [PubMed] [Google Scholar]
- 26.Jiang J, Levine M. Cell. 1993;72:741–752. doi: 10.1016/0092-8674(93)90402-c. [DOI] [PubMed] [Google Scholar]
- 27.Jiang J, Cai H, Zhou Q, Levine M. EMBO J. 1993;12:3201–3209. doi: 10.1002/j.1460-2075.1993.tb05989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang J D, Schwyter D H, Shirokawa J M, Courey A J. Genes Dev. 1993;7:694–704. doi: 10.1101/gad.7.4.694. [DOI] [PubMed] [Google Scholar]
- 29.Ruppert S, Wang E H, Tjian R. Nature (London) 1993;362:175–179. doi: 10.1038/362175a0. [DOI] [PubMed] [Google Scholar]
- 30.Heberlein U, England B, Tjian R. Cell. 1985;41:965–977. doi: 10.1016/s0092-8674(85)80077-5. [DOI] [PubMed] [Google Scholar]
- 31.Hansen S K, Tjian R. Cell. 1995;82:565–575. doi: 10.1016/0092-8674(95)90029-2. [DOI] [PubMed] [Google Scholar]
- 32.Isoda K, Roth S, Nüsslein-Volhard C. Genes Dev. 1992;6:619–630. doi: 10.1101/gad.6.4.619. [DOI] [PubMed] [Google Scholar]
- 33.Karim F D, Chang H C, Therrien M, Wassarman D A, Laverty T, Rubin G M. Genetics. 1996;143:315–329. doi: 10.1093/genetics/143.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tautz D, Pfeifle C. Chromosoma. 1989;98:81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- 35.Gonzalez-Crespo S, Levine M. Genes Dev. 1993;7:1703–1713. doi: 10.1101/gad.7.9.1703. [DOI] [PubMed] [Google Scholar]
- 36.Moqtaderi Z, Bai Y, Poon D, Weil P A, Struhl K. Nature (London) 1996;383:188–191. doi: 10.1038/383188a0. [DOI] [PubMed] [Google Scholar]
- 37.Walker S S, Reese J C, Apone L M, Green M R. Nature (London) 1996;383:185–188. doi: 10.1038/383185a0. [DOI] [PubMed] [Google Scholar]
- 38.Shen W C, Green M R. Cell. 1997;90:615–624. doi: 10.1016/s0092-8674(00)80523-1. [DOI] [PubMed] [Google Scholar]
- 39.Walker S S, Shen W C, Reese J C, Apone L M, Green M R. Cell. 1997;90:607–614. doi: 10.1016/s0092-8674(00)80522-x. [DOI] [PubMed] [Google Scholar]
- 40.Wang E H, Zou S, Tjian R. Genes Dev. 1997;11:2658–2669. doi: 10.1101/gad.11.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verrijzer C P, Chen J L, Yokomori K, Tjian R. Cell. 1995;81:1115–1125. doi: 10.1016/s0092-8674(05)80016-9. [DOI] [PubMed] [Google Scholar]
- 42.Burke T W, Kadonaga J T. Genes Dev. 1997;11:3020–3031. doi: 10.1101/gad.11.22.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilmour D S, Dietz T J, Elgin S C. Mol Cell Biol. 1990;10:4233–4238. doi: 10.1128/mcb.10.8.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohtsuki S, Levine M, Cai H N. Genes Dev. 1998;12:547–556. doi: 10.1101/gad.12.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lindsley D L, Zimm G G. The Genome of Drosophila melanogaster. New York: Academic; 1992. pp. 433–443. [Google Scholar]